
Thermally induced N–S bond insertion reaction of diazo compounds with N-sulfenylsuccinimides: synthesis of sulfides and mechanism studies†
Xiaolu
Zhang
a,
Yang
Zheng
a,
Lihua
Qiu
*a and
Xinfang
Xu
 *ab
*ab
aKey Laboratory of Organic Synthesis of Jiangsu Province, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou 215123, P. R. China. E-mail: qiulihua@suda.edu.cn; xinfangxu@suda.edu.cn
bState Key Laboratory of Elemento-Organic Chemistry, Nankai University, Tianjin 300071, China
First published on 13th November 2017
Abstract
A metal-free gem-functionalization reaction of diazo compounds with N-sulfenylsuccinimides via a formal N–S bond insertion process has been reported. The transformation features mild reaction conditions, broad substrate scope and functional group tolerance, offering an efficient approach to sulfide synthesis in moderate to high yields. In addition, the mechanism studies indicate the formation of free carbene under thermal conditions followed by ylide generation, then N–S bond cleavage and C–N bond formation occurred in sequence to give the sulfide products.
Introduction
Diazo compounds, which are pervasively used as carbene precursors,1 are versatile and powerful reagents, and have been extensively applied in modern synthetic organic chemistry.2 Among these transformations, the direct introduction of two substituents onto the carbon center of the diazo group, namely gem-difunctionalization, has attracted much attention recently.3 Three major approaches of the dinitrogen extrusion processes are widely acknowledged through different reactive intermediates: (1) metal carbene mediated reactions, which include multi-component reactions (MCRs),4 X–X (X = Si, Sn or C) insertions,5 X–Y insertion reactions (Scheme 1a),6 and others;7 (2) Lewis acid8 or substrate promoted9 transformations via procarbenium ion intermediates (Scheme 1b); and (3) thermolysis or photolysis induced free carbene processes.10–14 However, compared to the other two approaches, selective transformation via a free carbene intermediate is more challenging, as the reactivity and selectivity of free carbenes are more difficult to tune. Recent advances include cross-coupling reactions,10 cyclopropenation and cyclopropanation,11 X–H and C–H insertions,12 S–S σ-bond insertions (Scheme 1c),13 and others.14 Inspired by these works, together with our continuing interest in the reactions of free carbenes derived from diazo coumpounds,15 we envisaged that N-sulfenylsuccinimide, a good sulfur electrophile, could be easily attacked by the free carbene generated in situ from the diazo compounds under thermal conditions.6a,13a Herein, we describe a thermally induced formal S–N bond insertion reaction of free carbene with N-sulfenylsuccinimides through radical intermediates, and provide an efficient approach to sulfide synthesis in moderate to high yields (Scheme 1d).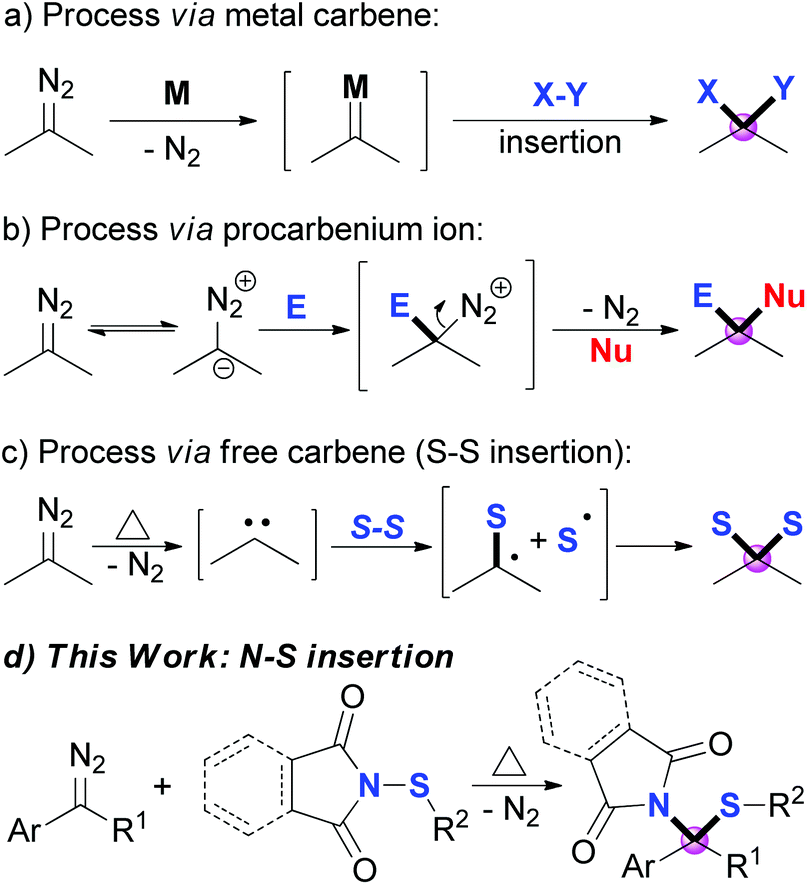 | ||
| Scheme 1 Diazo compound gem-difunctionalization. | ||
Results and discussion
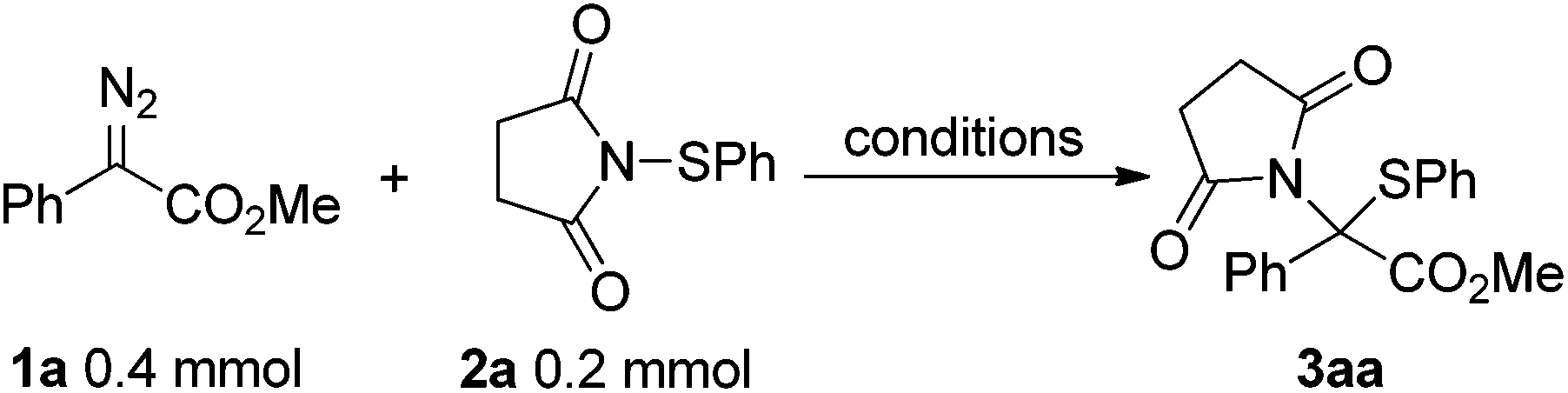
We commenced the investigation by inducing the thermal decomposition of phenyl diazoacetate 1a in the presence of N-phenylsulfenylsuccinimide 2a in DCE (Table 1) at 80 °C for 24 h and the expected product 3aa was afforded in 27% yield (entry 1). Encouraged by these results, other solvents, including benzene, toluene, and trifluoromethylbenzene, were investigated (entries 2–4), and the best results were obtained with trifluoromethylbenzene at 105 °C for 12 h (entry 4, 85% isolated yield). Reducing the reaction temperature to 90 °C slowed down the reactivity drastically (entry 5) and <5% conversion was found at 70 °C or below (entry 6).|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Solvent | Temp. (°C) | Yieldb (%) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a All the reactions were carried out on a 0.2 mmol scale in 2.0 mL of solvent in a sealed tube under an argon atmosphere at the indicated temperature, and the diazo compound was added via a syringe pump over 5 h followed by stirring for an additional 7 h under these conditions. b Isolated yields. c In 24 h, all of the diazo compound 1a was consumed and the rest of 2a was recovered. DCE = 1,2-dichloroethane. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | DCE | 80 | 27c | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | Benzene | 80 | 44c | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | PhCH3 | 110 | 48 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | PhCF 3 | 105 | 85 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | PhCF3 | 90 | 26c | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | PhCF3 | 70 | Trace | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
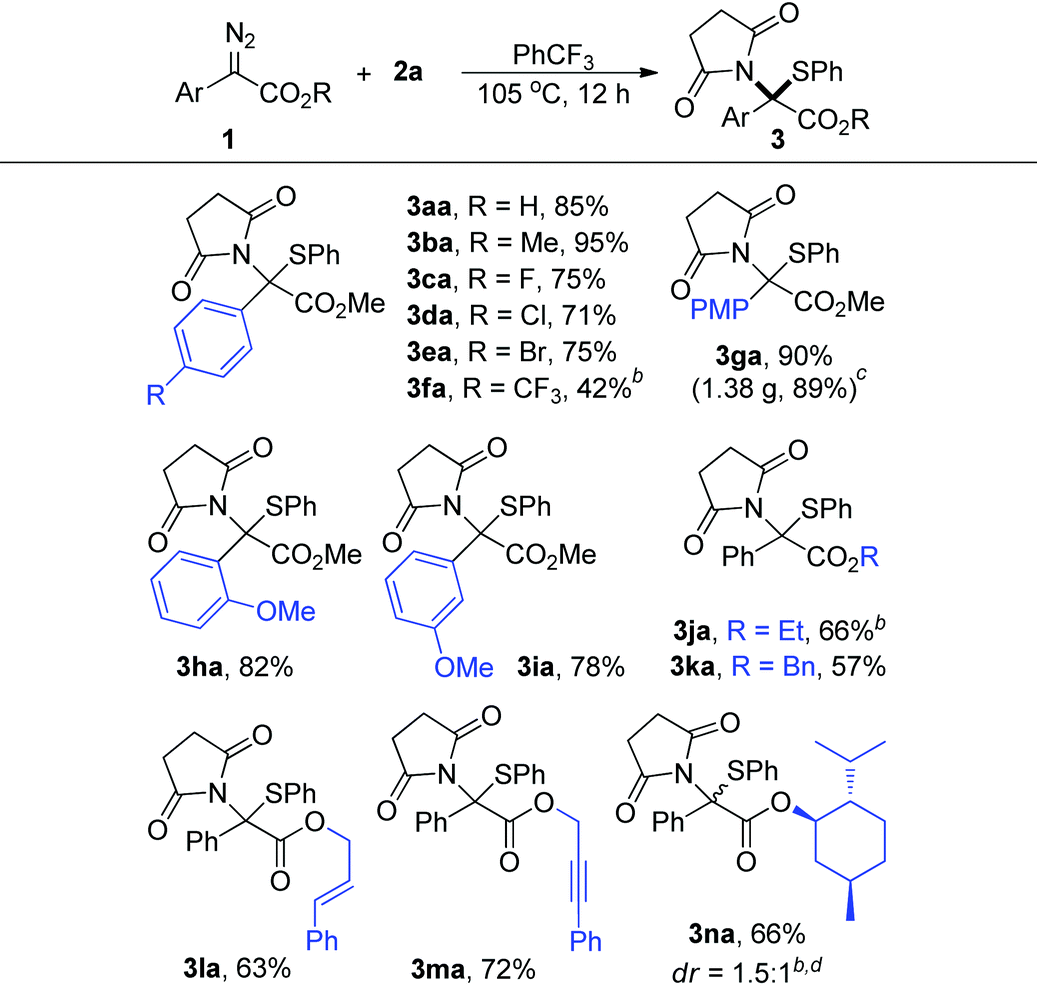
With the optimal reaction conditions in hand, we subsequently investigated the reactions between a series of aryl diazo esters 1 with 2a (Table 2). Most diazo compounds being tested reacted with 2a smoothly to give high yields (>71%, 3aa–3ea and 3ga–3ia). In the case of 1f with a strong EWG such as CF3, the substrate showed lower reactivity under the optimized conditions and 3fa was obtained in 42% yield when the reaction was carried out at 110 °C. The diazoacetates with various ester groups (R) were reliably accommodated, producing the corresponding products in 57–72% yields (3ja–3na). It is worth mentioning that these groups, especially the benzyl (3ka), alkenyl (3la), and alkynyl (3ma) groups which were recognized as reactive functional groups towards metal carbene for C–H insertion, cyclopropanation, and cyclopropenation reactions, respectively, were all well-tolerated in this reaction. However, for the α-styryl16 and α-benzyl17 diazo compounds, due to the inherent instability of these materials under thermal conditions, only the intramolecular cyclization product and the β-H shift product were detected, respectively, under the current conditions.18 Moreover, 1n derived from (−)-menthol reacted well with 2a, producing 3na in 66% yield and 1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr under briefly optimized conditions. In addition, the reaction proceeded smoothly on a gram scale, and 3ga was obtained in 89% yield when the reaction was carried out on a 4.0 mmol scale (note c).
:1 dr under briefly optimized conditions. In addition, the reaction proceeded smoothly on a gram scale, and 3ga was obtained in 89% yield when the reaction was carried out on a 4.0 mmol scale (note c).
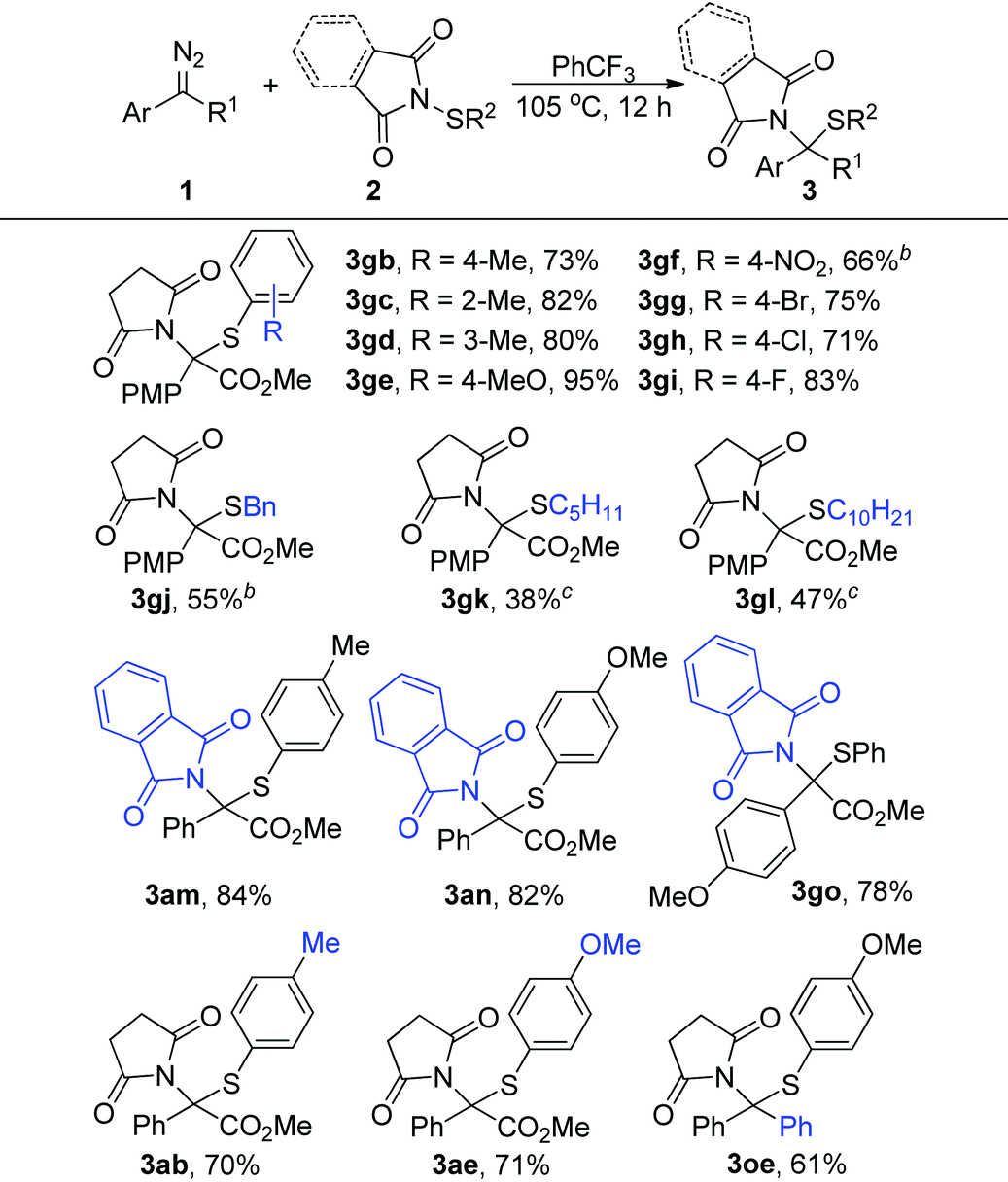
We further evaluated the generality of this reaction regarding N-sulfenylsuccinimides 2. As shown in Table 3, the electric properties and position of the substituent groups on the phenyl group of N-sulfenylsuccinimides 2 had little influence on the reactivity, generating the corresponding products in high to excellent yields (3gb–3gi, 3ab, and 3ae). Moreover, the alkyl substituted substrates 2j–2l were quite compatible in this transformation, which afforded the products 3gj–3gl in synthetically useful yields. Furthermore, this method was also amenable to substrates derived from phthalimides, and the corresponding products were generated in >78% yields (3am, 3an, and 3go). Notably, the diphenyl diazo compound, which is a precursor of donor/donor-free carbene, could also participate in the reactions and afforded the product 3oe in 61% yield.
To shed light on the reaction mechanism, control experiments and a crossover reaction were carried out (Scheme 2). Firstly, a competition experiment was performed: aryl diazo ester 1l and sulfenylsuccinimide 2o were subjected to a reaction in the presence of Rh2(OAc)4,19 and only cyclopropanation product 8 was isolated in >90% yield instead of delivering the corresponding N–S insertion product (Scheme 2a). Moreover, according to the reported dirhodium-catalyzed approach, the N–S insertion product 3qo was isolated only in 44% yield due to the steric effect,6a while this is not a problem at all under current metal-free conditions and the same product was obtained in 80% yield (Scheme 2b). These results clearly illustrated the distinction between the metal carbene reaction and the current method, which has shown a preference to react with the electrophilic sulfur. Then a control experiment in the presence of 5.0 equivalents of TEMPO was carried out under standard conditions, and the reaction was inhibited to give the insertion product only in 30% yield with 4 as the major product via direct oxidation (Scheme 2c). Moreover, the free radical intermediate was intercepted and the corresponding product 5 was detected by LC-MS (Scheme 2d, and see Fig. S5† for details). The crossover reaction was performed by subjecting an equivalent amount of 2b and 2n with 1a under the optimal reaction conditions, and four products 3ab, 3ae, 3am, and 3an were generated in a 26:20:24:30 ratio (Scheme 2e, and see Fig. S6–S10† for details).18 These results suggested that free radical intermediates are involved in this transformation. Based on these and previous works,13 a plausible reaction mechanism was proposed. Initially, the diazoacetate 1 decomposed to generate free carbene under thermal conditions and combined with electrophilic N-sulfenylsuccinimide 2 to form the ylide intermediate. Then N–S bond cleavage and C–N bond formation occurred successively to generate the formal N–S insertion product 3. However, further studies are needed to unambiguously establish the reaction transformation.
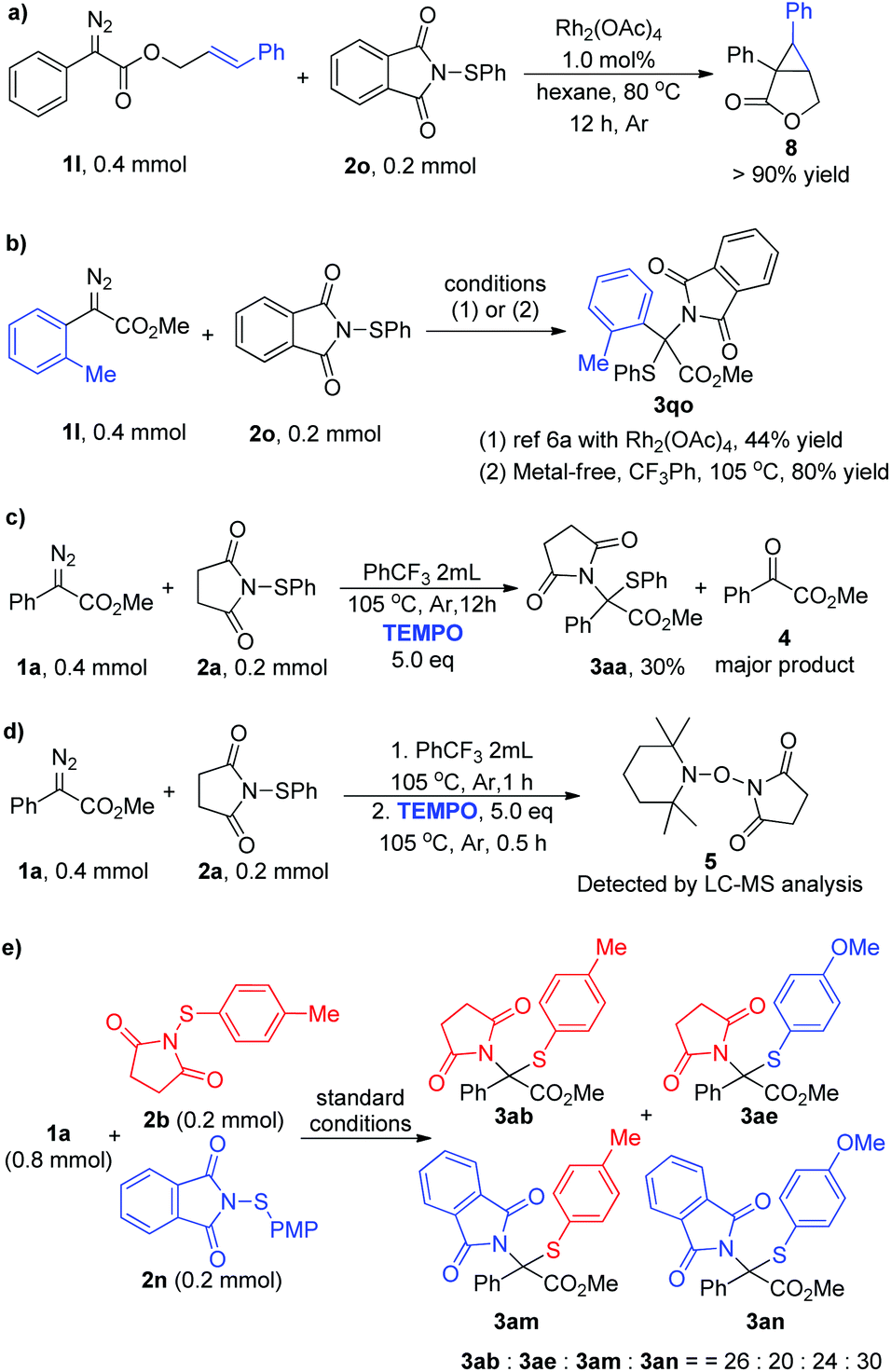 | ||
| Scheme 2 Control reactions and crossover experiment. | ||
As shown in eqn (1), to demonstrate the utility of these sulfide products, the imine derivative 6 could be obtained in 73% yield under slightly modified reported conditions.6a
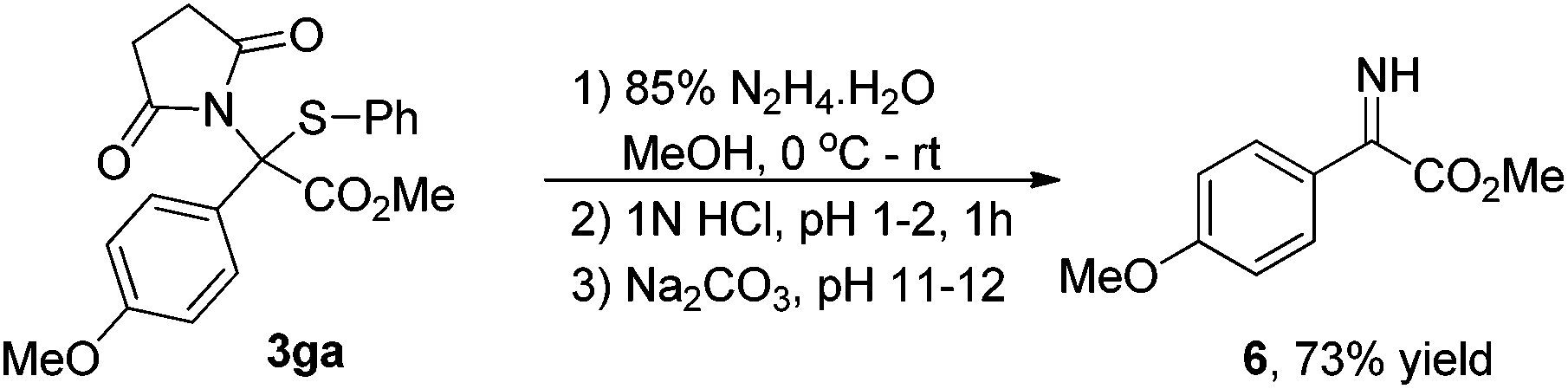 | (1) |
Conclusions
In conclusion, we have developed a thermally induced gem-functionalization reaction of diazo compounds with N-sulfenylimides via a formal N–S bond insertion process, which provided an efficient approach to sulfide synthesis in moderate to high yields under mild reaction conditions with broad substrate scope and functional group tolerance. A reaction pathway involving a sequential N–S bond cleavage and C–N bond formation process was proposed and verified experimentally via crossover reactions. Further exploration of free carbene transformations and mechanism studies are ongoing in our laboratory.Experimental section
General remarks
All reactions are carried out in oven-dried (140 °C) glassware under an atmosphere of argon. Solvents are dried and degassed using standard methods. Flash column chromatography is performed using silica gel (300–400 mesh). Analytical thin-layer chromatography is performed using glass plates pre-coated with 300–400 mesh silica gel impregnated with a fluorescent indicator (254 nm). 1H NMR and 13C NMR spectra are recorded in CDCl3 on a 400 MHz spectrometer; chemical shifts are reported in ppm using the solvent signals as a reference, and coupling constants (J) are given in Hertz. The peak information is described as: br = broad, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, comp = composite. High-resolution mass spectra (HRMS) are recorded on a commercial apparatus (ESI or CI source).General procedure for the N–S bond insertion reaction
To a 10 mL oven-dried vial containing a magnetic stirring bar and 2 (0.2 mmol) in PhCF3 (0.5 mL) was added the solution of diazo compound 1 (0.4 mmol) in PhCF3 (1.5 mL) via a syringe pump in 5 hours at 105 °C under an argon atmosphere. The reaction mixture was stirred for an additional 7 h under these conditions. After the reaction was completed, the reaction mixture was directly purified by column chromatography on silica gel without any additional treatment (eluent: hexanes:EtOAc = 20:1 to 3:1) to give the pure products 3 in moderate to high yields.
Procedure for the synthesis of 6
To a 10 mL oven-dried vial containing a magnetic stirring bar and 3ga (77 mg, 0.2 mmol) in MeOH (2.0 mL) was added 85% hydrazine hydrate (64 μL, 1.0 mmol) dropwise over 2 min at 0 °C under stirring. The reaction mixture was stirred at 0 °C for an additional hour, and then warmed up to room temperature and reacted overnight. After the reaction completed, the solvents were removed in vacuo and the residue was dissolved in water (8.0 mL), and the pH of the solution was then adjusted to 1–2 by adding 1N HCl at 0 °C. The mixture was stirred for 1 h at room temperature, and then filtered. The filtrate was treated with solid Na2CO3 until the pH reached 11–12. The mixture was extracted with CH2Cl2 (5.0 mL × 3). The combined extracts were dried over Na2SO4, concentrated and dried in vacuo after filtration to provide a yellow oil 6 in 73% yield (28.4 mg). 1H NMR (400 MHz, CDCl3) (δ ppm) 7.26–7.22 (m, 2H), 7.03–6.98 (m, 2H), 6.21 (s, 1H), 3.84 (s, 3H), 3.83 (s, 3H); 13C NMR (100 MHz, CDCl3) (δ ppm) 165.2, 160.4, 137.4, 130.4, 121.4, 114.8, 55.5, and 52.5. HRMS (TOF MS CI+) calculated for C10H12NO3 [M + H]+: 194.0817, found 194.0818.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support for this research from the National Natural Science Foundation of China and the NSFC of Jiangsu (NSFC21602148 and BK20150315), the Priority Academic Program Development (PAPD) of Jiangsu Higher Education Institutions and the project of scientific and technologic infrastructure of Suzhou (SZS201708) are gratefully acknowledged.Notes and references
- For reviews: (a) Q.-Q. Cheng, Y.-M. Deng, M. Lankelma and M. P. Doyle, Chem. Soc. Rev., 2017, 46, 5425 RSC; (b) A. Padwa, Chem. Soc. Rev., 2009, 38, 3072 RSC; (c) H. M. L. Davies and D. Morton, Chem. Soc. Rev., 2011, 40, 1857 RSC; (d) D. Xing and W.-H. Hu, Tetrahedron Lett., 2014, 55, 777 CrossRef CAS; (e) X. Zhao, Y. Zhang and J.-B. Wang, Chem. Commun., 2012, 48, 10162 RSC; (f) S. Zhu and Q.-L. Zhou, Acc. Chem. Res., 2012, 45, 1365 CrossRef CAS PubMed.
- For reviews: (a) S.-B. Choi, S.-J. Ha and C.-M. Park, Chem. Commun., 2017, 53, 6054 RSC; (b) Y. Zhang and J.-B. Wang, Chem. Commun., 2009, 5350 RSC; (c) L. Liu and J. Zhang, Chem. Soc. Rev., 2016, 45, 506 RSC; (d) X. Xu and M. P. Doyle, Acc. Chem. Res., 2014, 47, 1396 CrossRef CAS PubMed; (e) M. P. Doyle, M. Ratnikov and Y. Liu, Org. Biomol. Chem., 2011, 9, 4007 RSC.
- For reviews: (a) T. Ye and M. A. McKervey, Chem. Rev., 1994, 94, 1091 CrossRef CAS; (b) N. R. Candeias, R. Paterna and P. M. P. Gois, Chem. Rev., 2016, 116, 2937 CrossRef CAS PubMed; (c) X. Guo and W. Hu, Acc. Chem. Res., 2013, 46, 2427 CrossRef CAS PubMed; (d) Q. Xiao, Y. Zhang and J. Wang, Acc. Chem. Res., 2013, 46, 236 CrossRef CAS PubMed.
- (a) M. Li, L. Zan, D. Prajapati and W.-H. Hu, Org. Biomol. Chem., 2012, 10, 8808 RSC; (b) H. Qiu, M. Li, L.-Q. Jiang, F.-P. Lv, L. Zan, C.-W. Zhai, M. P. Doyle and W.-H. Hu, Nat. Chem., 2012, 4, 733 CrossRef CAS PubMed; (c) D.-F. Chen, F. Zhao, Y. Hu and L.-Z. Gong, Angew. Chem., Int. Ed., 2014, 53, 10763 CrossRef CAS PubMed; (d) D. Zhang, J. Zhou, F. Xia, Z. Kang and W. Hu, Nat. Commun., 2015, 6, 5801 CrossRef CAS PubMed.
- Si–Si and Sn–Sn bond insertion (a) Z. Liu, H. Tan, T. Fu, Y. Xia, D. Qiu, Y. Zhang and J. Wang, J. Am. Chem. Soc., 2015, 137, 12800 CrossRef CAS PubMed. C–C bond insertion (b) Y. Xia, Z. Liu, Z. Liu, R. Ge, F. Ye, M. Hossain, Y. Zhang and J. Wang, J. Am. Chem. Soc., 2014, 136, 3013 CrossRef CAS PubMed.
- N–S Bond insertion (a) Z. Song, Y. Wu, T. Xin, C. Jin, X. Wen, H. Sun and Q.-L. Xu, Chem. Commun., 2016, 52, 6079 RSC. C–N bond insertion (b) G. Qin, L. Li, J. Li and H. Huang, J. Am. Chem. Soc., 2015, 137, 12490 CrossRef CAS PubMed. C–O bond insertion (c) J. Pospech, A. J. J. Lennox and M. Beller, Chem. Commun., 2015, 51, 14505 RSC. N–O bond insertion (d) X.-F. Xu, M. O. Ratnikov, P. Y. Zavalij and M. P. Doyle, Org. Lett., 2011, 13, 6122 CrossRef CAS PubMed. C–S bond insertion (e) M. Ma, L. Peng, C. Li, X. Zhang and J. Wang, J. Am. Chem. Soc., 2005, 127, 15016 CrossRef CAS PubMed; (f) Z. Zhang, Z. Sheng, W. Yu, G. Wu, R. Zhang, W. Chu, Y. Zhang and J. Wang, Nat. Chem., 2017, 9, 970 CrossRef CAS PubMed.
- (a) D. P. Hari and J. Waser, J. Am. Chem. Soc., 2016, 138, 2190 CrossRef CAS PubMed; (b) R. Panish, R. Selvaraj and J. M. Fox, Org. Lett., 2015, 17, 3978 CrossRef CAS PubMed.
- (a) W. Li, X. Liu, F. Tan, X. Hao, J. Zheng, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2013, 52, 10883 CrossRef CAS PubMed; (b) W.-J. Liu, B.-D. Lv and L.-Z. Gong, Angew. Chem., Int. Ed., 2009, 48, 6503 CrossRef CAS PubMed; (c) L. Gao, B. C. Kang and D. H. Ryu, J. Am. Chem. Soc., 2013, 135, 14556 CrossRef CAS PubMed.
- (a) H. Mao, Z. Tang, H. Hu, Y. Cheng, W.-H. Zheng and C. Zhu, Chem. Commun., 2014, 50, 9773 RSC; (b) G. Chen, J. Song, Y. Yu, X. Luo, C. Li and X. Huang, Chem. Sci., 2016, 7, 1786 RSC; (c) J. Tao, R. Tran and G. K. Murphy, J. Am. Chem. Soc., 2013, 135, 16312 CrossRef CAS PubMed; (d) H. Li, L. Wang, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2012, 51, 2943 CrossRef CAS PubMed; (e) K. E. Coffey and G. K. Murphy, Synlett, 2015, 1003 CAS; (f) C. Ma, D. Xing and W. Hu, Org. Lett., 2016, 18, 3134 CrossRef CAS PubMed.
- M. Hu, C. Ni, L. Li, Y. Han and J. Hu, J. Am. Chem. Soc., 2015, 137, 14496 CrossRef CAS PubMed.
- (a) S. R. Ovalles, J. H. Hansen and H. M. L. Davies, Org. Lett., 2011, 13, 4284 CrossRef CAS PubMed; (b) R. Barroso, A. Jimenez, M. C. Perez-Aguilar, M.-P. Cabal and C. Valdes, Chem. Commun., 2016, 52, 3677 RSC.
- (a) S. R. Hansen, J. E. Spangler, J. H. Hansen and H. M. L. Davies, Org. Lett., 2012, 14, 4626 CrossRef CAS PubMed; (b) J. Barluenga, M. Tomás-Gamasa, F. Aznar and C. Valdés, Angew. Chem., Int. Ed., 2010, 49, 4993 CrossRef CAS PubMed; (c) C. Tortoreto, D. Rackl and H. M. L. Davies, Org. Lett., 2017, 19, 770 CrossRef CAS PubMed.
- (a) Y. Zheng, R.-J. Bian, X.-L. Zhang, R.-W. Yao, L.-H. Qiu, X.-G. Bao and X.-F. Xu, Eur. J. Org. Chem., 2016, 3872 CrossRef CAS; (b) D. Arunprasath and G. Sekar, Adv. Synth. Catal., 2017, 359, 698 CrossRef CAS.
- (a) M. Kischkewitz, C.-G. Daniliuc and A. Studer, Org. Lett., 2016, 18, 1206 CrossRef CAS PubMed; (b) Z.-X. Liu, H.-C. Tan, L. Wang, T.-R. Fu, Y. Xia, Y. Zhang and J.-B. Wang, Angew. Chem., Int. Ed., 2015, 54, 3056 CrossRef CAS PubMed.
- (a) C. Zhang, J.-J. Huang, L.-H. Qiu and X.-F. Xu, Org. Lett., 2016, 18, 6208 CrossRef CAS PubMed; (b) C. Zhang, S.-L. Chang, L.-H. Qiu and X.-F. Xu, Chem. Commun., 2016, 52, 12470 RSC; (c) Y. Zheng, M. Bao, L.-H. Qiu and X.-F. Xu, Tetrahedron Lett., 2017, 58, 3390 CrossRef CAS; (d) K. Dong, B. Yan, S. Chang, Y. Chi, L.-H. Qiu and X.-F. Xu, J. Org. Chem., 2016, 81, 6887 CrossRef CAS PubMed.
- (a) M. P. Doyle, M. Yan, H.-M. Gau and E. C. Blossey, Org. Lett., 2003, 5, 561 CrossRef CAS PubMed; (b) S. Cui, Y. Zhang, D. Wang and Q. Wu, Chem. Sci., 2013, 4, 3912 RSC; (c) N. R. O'Connor, P. Bolgar and B. M. Stoltz, Tetrahedron Lett., 2016, 57, 849 CrossRef.
- (a) D. F. Taber, R. B. Sheth and P. V. Joshi, J. Org. Chem., 2005, 70, 2851 CrossRef CAS PubMed; (b) S. Bachmann, D. Fielenbach and K. A. Jørgensen, Org. Biomol. Chem., 2004, 2, 3044 RSC; (c) G. G. Cox, D. Haigh, R. M. Hindley, D. J. Miller and C. J. Moody, Tetrahedron Lett., 1994, 35, 3199 CrossRef.
- See ESI† for details.
- J. Shen, S.-F. Zhu, Y. Cai, H. Xu, X. Xie and Q.-L. Zhou, Angew. Chem., Int. Ed., 2014, 53, 13188 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, characterization of all compounds, copies of 1H and 13C NMR spectra for all target compounds. See DOI: 10.1039/c7ob02488f |
| This journal is © The Royal Society of Chemistry 2018 |