
Synthesis of 2-methylbenzoxazoles directly from N-phenylacetamides catalyzed by palladium acetate†
Biying
Wang
,
Chengfei
Jiang
,
Jiasheng
Qian
,
Shuwei
Zhang
 *,
Xiaodong
Jia
and
Yu
Yuan
*
*,
Xiaodong
Jia
and
Yu
Yuan
*
School of Chemistry and Chemical Engineering, Yangzhou University, 225002 Yangzhou, People's Republic of China. E-mail: shuweiz@yzu.edu.cn; yyuan@yzu.edu.cn
First published on 22nd November 2017
Abstract
A method to synthesize 2-methylbenzoxazoles directly from N-phenylacetamides catalyzed by Pd(OAc)2 in the presence of K2S2O8 and TfOH has been developed. The desired products were obtained in moderate to excellent yields. This approach provides a facile procedure to prepare benzoxazoles with available substrates. It is found that TfOH is the key factor for this cyclization reaction. A plausible mechanism of the reaction is proposed according to the control reactions and the literature.
1. Introduction
The construction of functionalized benzoxazoles has been receiving considerable attention, since the benzoxazole moiety as one of the important subunits is widely present in natural products and pharmaceutical compounds, as well as in some optical materials.1 A number of methods for the preparation of benzoxazoles have been developed in the past few decades. The classical approaches are the oxidative cyclization of the compounds condensed from 2-aminophenols and aldehydes or carboxylic acids.2 Some transition-metal-catalyzed C–N or C–O coupling reactions have also been reported for the synthesis of benzoxazoles from intramolecular cyclization of halogenated arene precursors.3 Although these methods provide available access to benzoxazoles, there are still some limitations such as substrate scope, harsh reaction conditions, and low atom-economy. To overcome these drawbacks, considerable efforts have been devoted to developing new approaches for the construction of C–O bonds via C–H functionalization processes. For example, C–H functionalization followed by C–O bond formation has been explored for the synthesis of benzoxazoles by using palladium-, ruthenium-, iron-, copper-based catalytic, or organocatalytic systems.4 Among these research studies, 2-aryl substituted benzoxazole derivatives were synthesized with the corresponding aryl substrates in most cases. Unfortunately, few examples were reported for the preparation of 2-alkyl substituted benzoxazole derivatives from N-phenylacetamides (Scheme 1).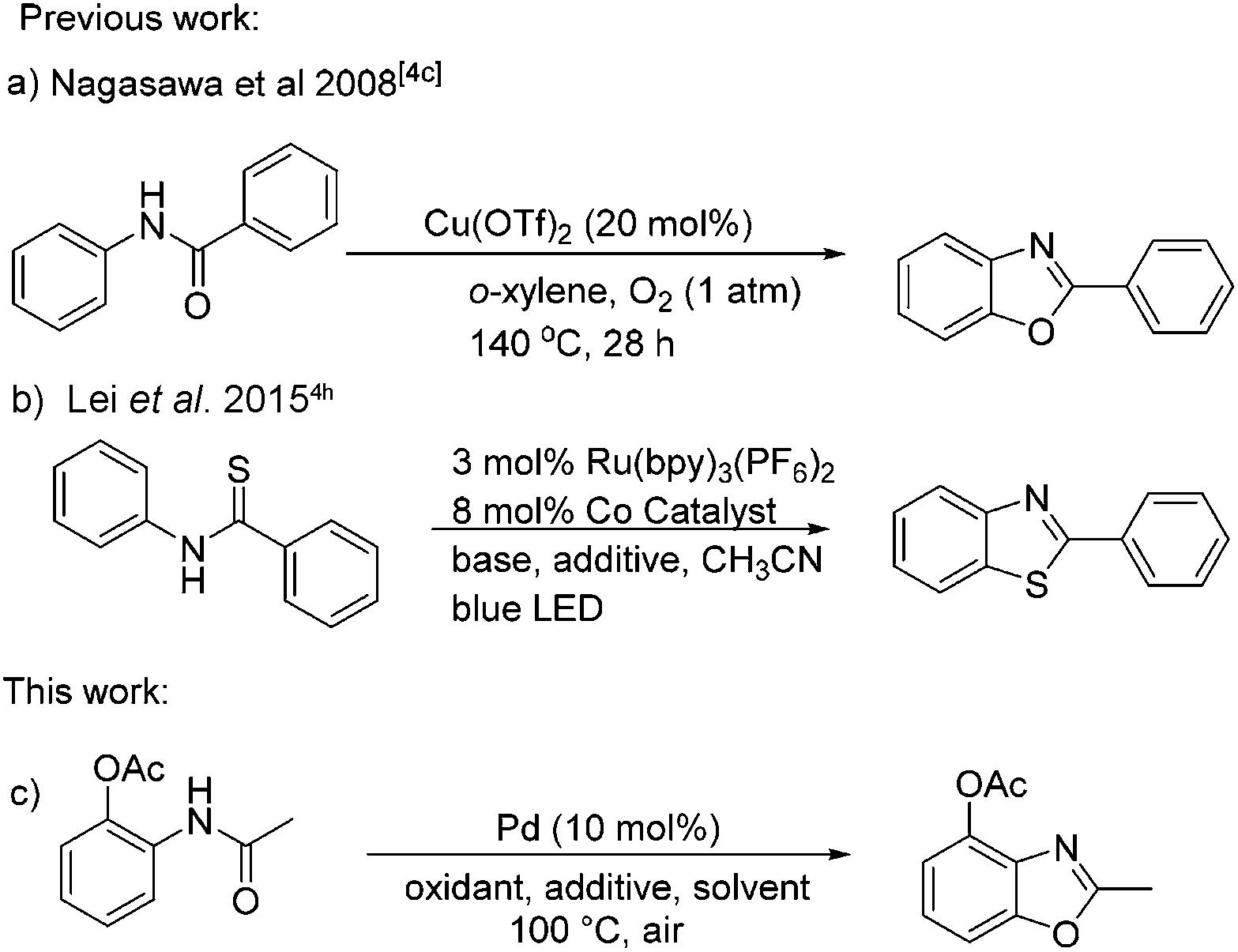 | ||
| Scheme 1 Different pathways for the synthesis of 2-substituted arene derivatives. | ||
Recently, our group focused on the oxidative C–H bond coupling reactions of N-phenylacetamides and benzothiazoles.5 Herein, we report a direct method to prepare 2-methyl benzoxazoles by palladium-catalyzed C–H functionalization and intramolecular oxidative C–O coupling reaction with readily available N-phenylacetamides.
2. Results and discussion
According to our previous work,5a the combination of an oxidant, an acid, and palladium could affect the coordination of N-phenylacetamide. Firstly, we used 2-acetamidophenyl acetate (1a) as the substrate to test the cyclization reaction in the presence of K2S2O8 and TsOH in mixed solvents (AcOH/DMF) by using Pd(OAc)2 as the catalyst to obtain 42% 2-methylbenzo[d]oxazol-4-yl acetate (2a) (Table 1, entry 1). The yield improved to 64% with the use of TfOH as the additive (Table 1, entry 2), whereas 2a was not formed when TFA was in the system (Table 1, entry 3). After adjusting the amount of TfOH, the results indicated that 1.0 equiv. of TfOH was optimal for the reaction (Table 1, entries 2 and 4–6). Other oxidants (TBHP, TBAI, and O2) were also used to investigate the influence on the reactions, but all failed. It was shown that the best oxidant is 1.5 equiv. potassium peroxodisulfate (Table 1, entries 4, 7 and 8).|
|
|||||
|---|---|---|---|---|---|
| Entry | Pd (10 mol%) | Additive (equiv.) | Oxidants (equiv.) | Solvent | Yieldsb (%) |
| a Unless otherwise specified, the reaction was carried out with 1a (0.2 mmol), catalysts, additives and oxidants at 100 °C in AcOH/DMF (8/1) for 24 h. b Yields after purification. c AcOH/DMF (4/1). d AcOH/DMF (10/1). | |||||
| 1 | Pd(OAc)2 | p-TsOH (0.5) | K2S2O8 (2) | AcOH/DMF | 42 |
| 2 | Pd(OAc)2 | TfOH (0.5) | K2S2O8 (2) | AcOH/DMF | 64 |
| 3 | Pd(OAc)2 | TFA | K2S2O8 (2) | AcOH/DMF | NR |
| 4 | Pd(OAc)2 | TfOH (1) | K2S2O8 (2) | AcOH/DMF | 66 |
| 5 | Pd(OAc)2 | TfOH (1.5) | K2S2O8 (2) | AcOH/DMF | 55 |
| 6 | Pd(OAc)2 | TfOH (2) | K2S2O8 (2) | AcOH/DMF | 48 |
| 7 | Pd(OAc)2 | TfOH (1) | K2S2O8 (1) | AcOH/DMF | 43 |
| 8 | Pd(OAc)2 | TfOH (1) | K2S2O8 (1.5) | AcOH/DMF | 68 |
| 9 | Pd(TFA)2 | TfOH (0.5) | K2S2O8 (2) | AcOH/DMF | 47 |
| 10 | Pd(PPh3)4 | TfOH (0.5) | K2S2O8 (2) | AcOH/DMF | 23 |
| 11 | Pd(OAc)2 | TfOH (1) | K2S2O8 (1.5) | AcOH | Trace |
| 12 | Pd(OAc)2 | TfOH (1) | K2S2O8 (1.5) | DMF | Trace |
| 13 | Pd(OAc)2 | TfOH (1) | K2S2O8 (1.5) | AcOH/DMFc | 55 |
| 14 | Pd(OAc)2 | TfOH (1) | K2S2O8 (1.5) | AcOH/DMFd | 47 |
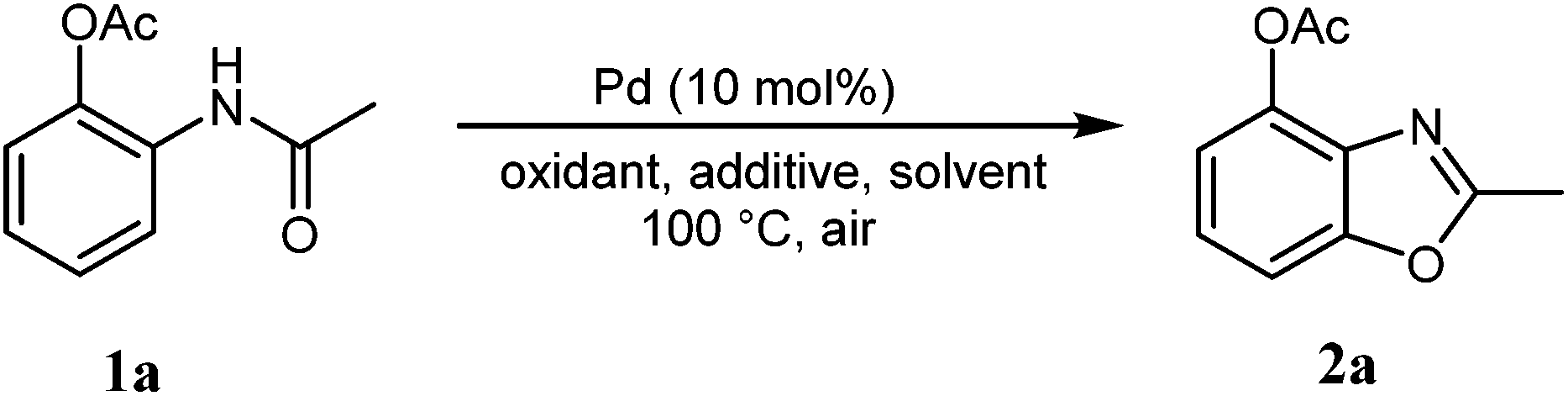
The catalysts Pd(TFA)2 and Pd(PPh3)4 were also tested under the conditions (Table 1, entries 9 and 10). A little lower yield of 2a (47%) was obtained by employing Pd(TFA)2. When the sole solvent AcOH or DMF (Table 1, entries 11 and 12) was applied into the reaction system, only a few products were detected. It is evident that the mixed solvents are necessary for the reaction. The ratio of the mixed solvents also affected the reactions, and AcOH/DMF (8/1) was suitable for the reactions (Table 1, entries 13 and 14). As a result, the best result was achieved under these conditions including 10 mol% of Pd(OAc)2 as the catalyst, 1.0 equiv. TfOH as the additive, 1.5 equiv. of K2S2O8 as the oxidant, and AcOH/DMF (8/1) as the solvent at 100 °C.
Under the optimal reaction conditions, we further investigated the scope and functional group compatibility of the palladium-catalyzed intramolecular oxidative C–O coupling reaction. A variety of o-substituted N-phenylacetamides were employed as substrates to verify the scope of the reactions. The results indicated that the reactions were mild and tolerated many functional groups resulting in good to mild yields (Table 2). When N-phenylacetamide (1b) was reacted under the optimal conditions, 2a was isolated rather than benzoxazole. We proposed that the reaction should first form 2-acetamidophenyl acetate,6 and then cyclize via intramolecular oxidative C–O coupling reaction to obtain 2a. For either mono- or di-halogen substrates, the reactions worked well (2b–2d, 2r–2s). It is found that electron-donating moieties on the o-substituted compounds resulted in lower yields (2g–2h). With stronger electron-withdrawing substituents on the aromatic ring, better yields were obtained, especially for methyl 2-acetamidobenzoate whose yield reached 89% (2f). There is no obvious influence on the yields when multiple substituents are on the ring, and even when containing a methyl group (2i–2p). The best yield could reach 93% with methyl and ester groups (2p). However, when the methyl group connected to the carbonyl group was changed to an ethyl or phenyl group, the desired products were hardly detected.
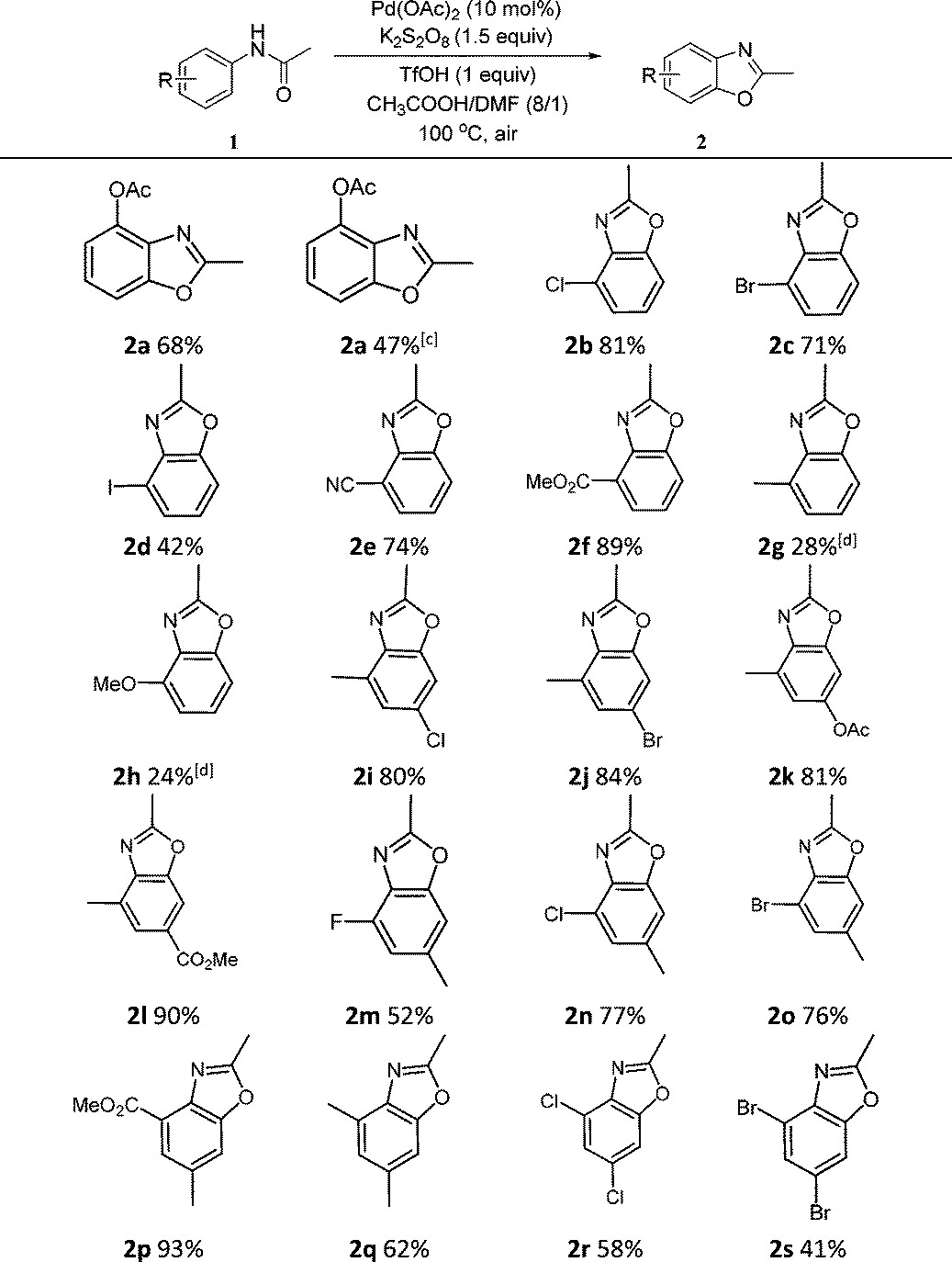
In Table 2, the same product 2a could be obtained using 1a and N-phenylacetamide, respectively. In order to investigate the reaction mechanism, some control reactions were carried out (Scheme 2). Firstly, 2-methylbenzo[d]oxazole was employed as the substrate, where no reaction occurred under the optimal conditions. Then, N-phenylacetamide was used, and the process of this reaction was detected by HPLC at 3 hour intervals. The results are shown in Fig. 1. It was found that compound 1a was formed, and the maximum concentration of 1a appeared at the 6th hour. Meanwhile, in the first 3 h, no desired product was formed in the system. Interestingly, the product was formed while 1a reached the high concentration (6 h). The substrate and 1a almost disappeared after 9 h and 21 h, respectively. In the whole reaction, the doubly acetoxylated N-phenylacetamide was never generated. It was suggested that N-phenylacetamide first underwent an ortho-acetoxylation and then formed the benzoxazole.
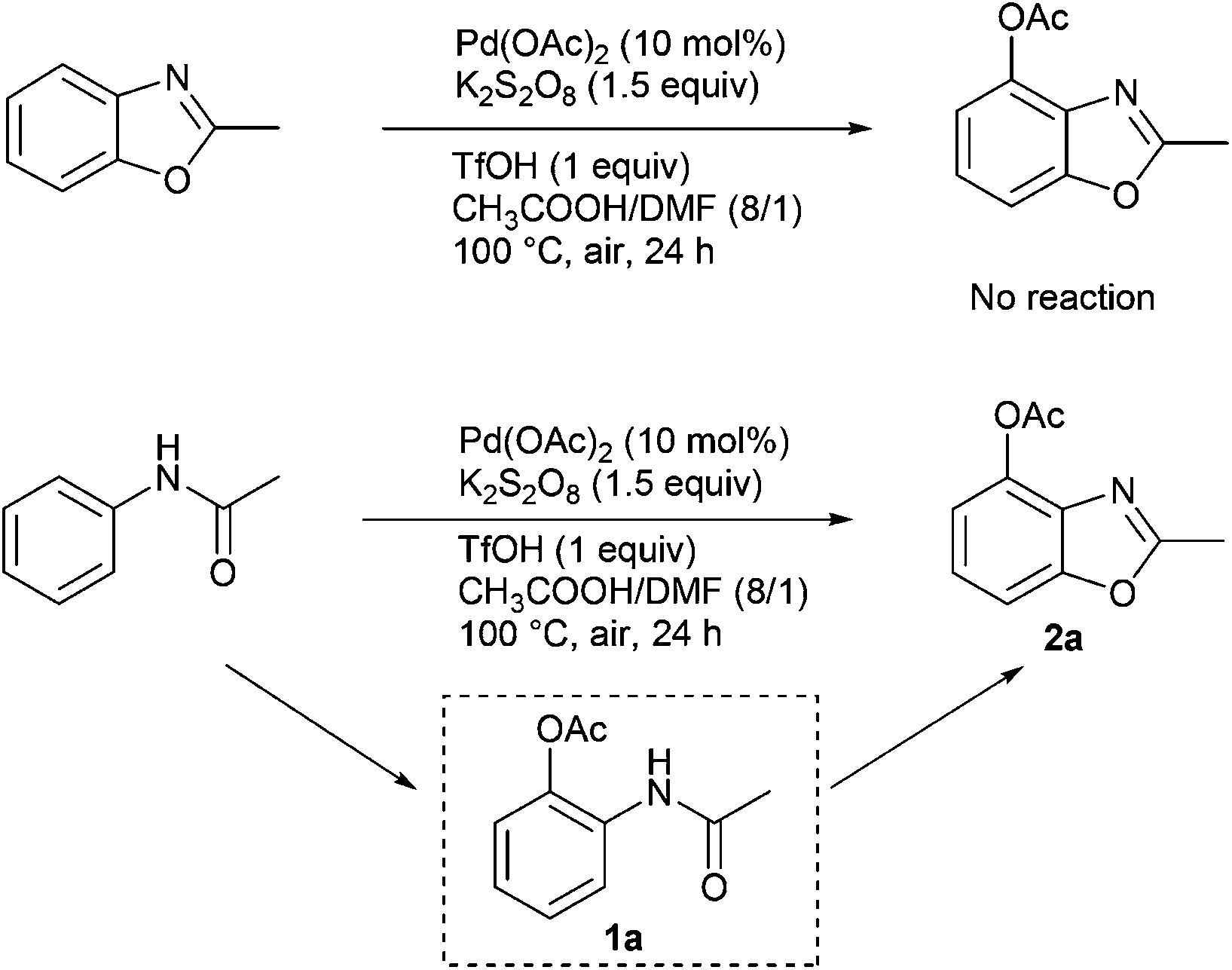 | ||
| Scheme 2 Control experiments. | ||
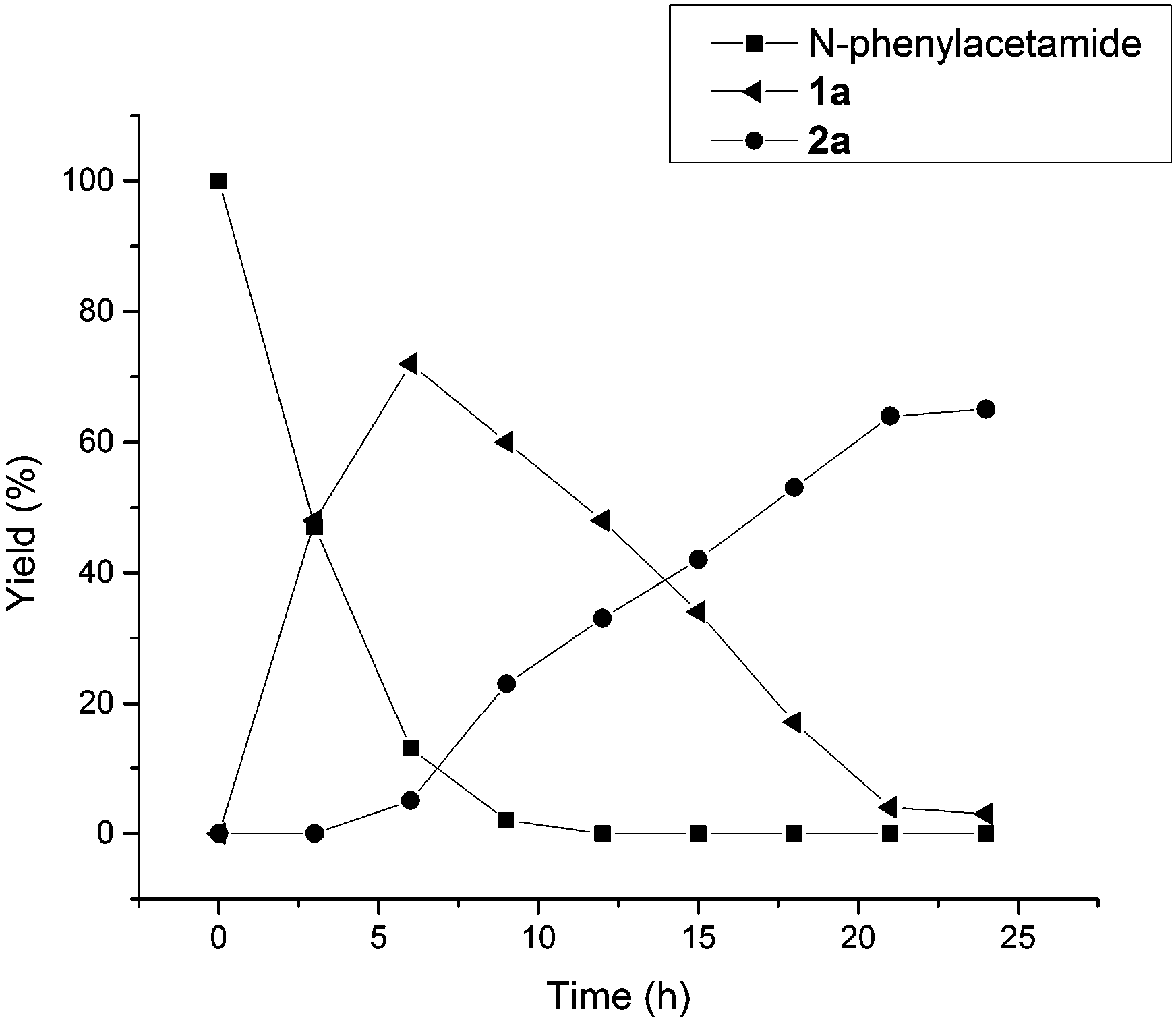 | ||
| Fig. 1 The time history plot of the reaction of N-phenylacetamide. | ||
Based on our results and reported studies, a plausible mechanism is proposed in Scheme 3. The reaction was initiated by electrophilic attack of a Pd(II) center to the aromatic ring with the assistance of the acetamino group to form a bimetallic palladium complex.7 The Pd(II) intermediate would undergo the oxidative addition to form Pd(IV) in the presence of K2S2O8.8 The anion species linked with Pd(IV) could be the TfO− anion that enhanced the catalytic reactivity, since AcO− coordinated with palladium could be replaced by TfO− in the system.5a,9 The acetyl group connected with an amine was transferred to enol formation since TfO− promoted the coordination of palladium with an oxygen atom. After reductive elimination the desired compound was produced, and Pd(II) was regenerated for the cycle.
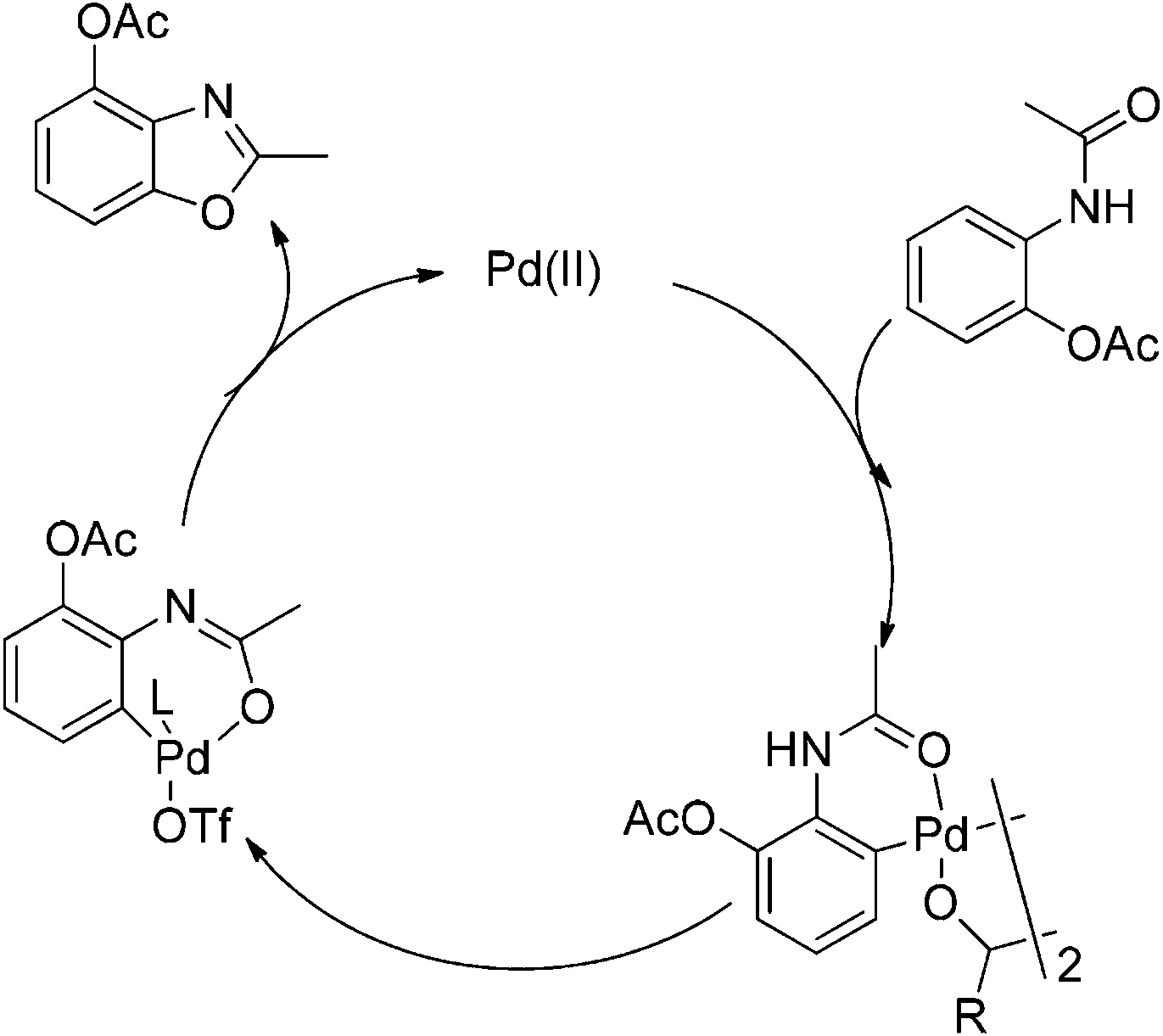 | ||
| Scheme 3 Plausible mechanism for palladium catalyzed intramolecular oxidative C–O coupling reaction. | ||
3. Conclusions
In conclusion, we have developed an efficient and clean method to synthesize 2-methyl benzoxazoles from N-phenylacetamides by C–H functionalization and intramolecular oxidative C–O coupling reaction with a catalytic amount of palladium(II) acetate. The reaction reveals high efficiency in atom-economy, and could be carried out with a wide scope of substrates which are readily available.4. Experimental section
4.1. General information
1H NMR and 13C NMR spectra were obtained with an Agilent Technologies 400 spectrometer in CDCl3 with TMS as an internal standard. Chemical shifts (δ) are reported in ppm, and coupling constants (J) are in hertz (Hz). The following abbreviations were used to explain the multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, and br = broad. Mass spectra were obtained on a Bruker Dalton maXis instrument. All reactions were monitored by TLC. Flash column chromatography was carried out using 300–400 mesh silica gel at medium pressure.4.2. Preparation of starting materials
In a 50 mL round bottom flask, aniline (5.0 mmol, 1.0 equiv.) was dissolved in 20 mL dichloromethane. Then, acetic anhydride (6.0 mmol, 1.2 equiv.) was added and the reaction mixture was stirred at room temperature until the complete consumption of aniline. After completion of the reaction, the mixture was washed with a saturated solution of sodium carbonate, the organic layer was dried with Na2SO4 and the solvent was removed under reduced pressure to obtain the desired N-phenylacetamide in quantitative yield. In most cases, analytically pure N-phenylacetamides can be obtained after extraction however if necessary purification by flash chromatography with ethyl acetate/hexane was used.All other commercially available reagents were purchased from commercial sources or prepared according to the literature and purified by standard techniques without special instructions. Solvents were freshly dried and degassed according to the purification handbook Purification of Laboratory Chemicals before using.
4.3. General procedure for substrate scope
Pd(OAc)2 (4.5 mg, 0.02 mmol), 2-acetamidophenyl acetate 1a (38.6 mg, 0.2 mmol), glacial acetic acid (0.8 mL), N,N-dimethylformamide (0.1 mL), potassium peroxodisulfate (100.0 mg, 0.3 mmol), and TfOH (30 mg, 0.2 mmol) were placed into a 15 mL sealed tube, and heated in an oil bath at 100 °C. The reaction mixture was stirred for 24 h. After cooling to room temperature, the reaction mixture was filtered through Celite, and the filtrate was concentrated under vacuum to afford an oily residue. The residue was purified by column chromatography on silica gel. The fraction was collected and concentrated to obtain 26.2 mg product 2a.Yield: 68%. White solid; mp: 68–71 °C; 1H NMR (400 MHz, CDCl3) δ 7.35 (d, J = 8.2 Hz, 1H), 7.29–7.23 (m, 1H), 7.04 (d, J = 8.0 Hz, 1H), 2.62 (s, 3H), 2.40 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 168.92, 164.23, 152.36, 141.08, 134.16, 125.67, 124.59, 108.16, 20.93, 14.54; IR (KBr): ν = 2925, 2855, 1769, 1620, 1493, 1438, 1373, 1253, 1193, 1044, 866, 803, 743, 582 cm−1; HRMS: calculated for C10H9NO3: 192.0660, found: 192.0653.
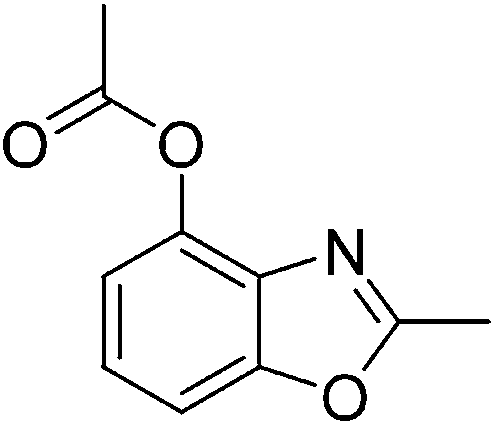
Yield: 68%. White solid; mp: 68–71 °C; 1H NMR (400 MHz, CDCl3) δ 7.35 (d, J = 8.2 Hz, 1H), 7.29–7.23 (m, 1H), 7.04 (d, J = 8.0 Hz, 1H), 2.62 (s, 3H), 2.40 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 168.92, 164.23, 152.36, 141.08, 134.16, 125.67, 124.59, 108.16, 20.93, 14.54; IR (KBr): ν = 2925, 2855, 1769, 1620, 1493, 1438, 1373, 1253, 1193, 1044, 866, 803, 743, 582 cm−1; HRMS: calculated for C10H9NO3: 192.0660, found: 192.0653.
Yield: 81%. White solid; mp: 52–55 °C; 1H NMR (400 MHz, CDCl3) δ 7.34 (d, J = 8.0 Hz, 1H), 7.27 (d, J = 7.9 Hz, 1H), 7.18 (t, J = 8.0 Hz, 1H), 2.63 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 164.63, 151.47, 139.20, 124.95, 124.31, 123.87, 108.86, 14.56; IR (KBr): ν = 3079, 2926, 2855, 1677, 1611, 1471, 1425, 1254, 1159, 1092, 1031, 946, 866, 800, 146, 520 cm−1; HRMS: calculated for C8H6BrNO: 211.9711, found: 211.9705.
Yield: 71%. White solid; mp: 74–77 °C; 1H NMR (600 MHz, CDCl3) δ 7.46 (d, J = 8.0 Hz, 1H), 7.41 (d, J = 8.1 Hz, 1H), 7.16 (t, J = 8.0 Hz, 1H), 2.66 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 164.49, 151.08, 140.95, 127.48, 125.68, 111.96, 109.43, 14.45; IR (KBr): ν = 3556, 3069, 2922, 2852, 1605, 1560, 1422, 1248, 1127, 1038, 916, 860, 790 cm−1; HRMS: calculated for C8H6BrNO: 211.9711, found: 211.9705.
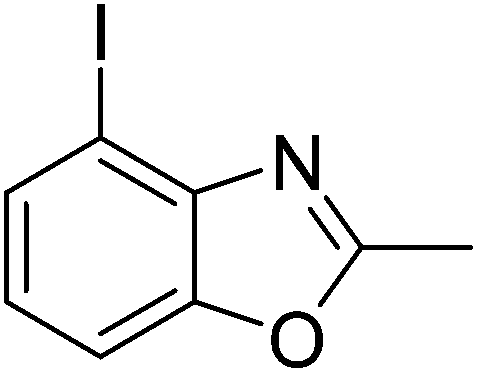
Yield: 42%. White solid; mp: 71–74 °C; 1H NMR (600 MHz, CDCl3) δ 7.68 (d, J = 7.8 Hz, 1H), 7.43 (d, J = 8.1 Hz, 1H), 7.06 (t, J = 8.0 Hz, 1H), 2.67 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 162.98, 148.46, 143.30, 132.32, 124.98, 109.22, 83.24, 13.59; IR (KBr): ν = 3357, 3190, 3064, 2921, 2854, 1599, 1417, 1332, 1254, 1028, 904, 799, 697, 505 cm−1; HRMS: calculated for C8H6INO: 259.9572, found: 259.9574.
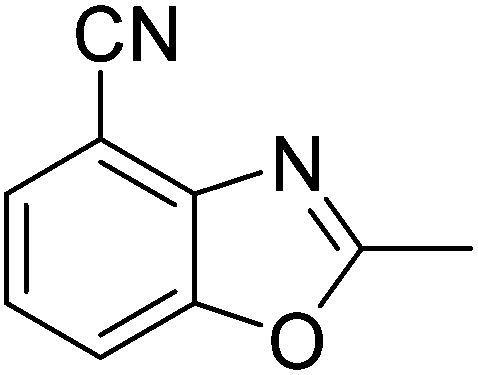
Yield: 74%. White solid; mp: 110–113 °C; 1H NMR (600 MHz, CDCl3) δ 7.46 (d, J = 8.0 Hz, 1H), 7.41 (d, J = 8.1 Hz, 1H), 7.16 (t, J = 8.0 Hz, 1H), 2.66 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 164.28, 153.03, 149.37, 134.74, 126.98, 126.38, 126.21, 120.22, 22.27; IR (KBr): ν = 3172, 3040, 2914, 2019, 1684, 1619, 1462, 1259, 1026, 885, 780, 667, 595 cm−1; HRMS: calculated for C9H6N2O: 159.0558, found: 159.0560.
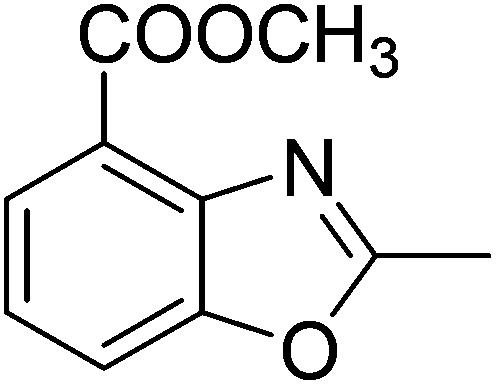
Yield: 89%. White solid; mp: 80–83 °C; 1H NMR (400 MHz, CDCl3) δ 7.95 (d, J = 7.8 Hz, 1H), 7.64 (d, J = 8.1 Hz, 1H), 7.32 (t, J = 7.9 Hz, 1H), 4.00 (s, 3H), 2.69 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 165.89, 165.72, 151.49, 140.94, 126.60, 123.87, 121.52, 114.56, 52.48, 14.77; IR (KBr): ν = 3360, 2933, 2852, 1719, 1573, 1430, 1294, 1144, 1023, 926, 865, 807, 758 cm−1; HRMS: calculated for C10H9NO3: 192.0660, found: 192.0652.
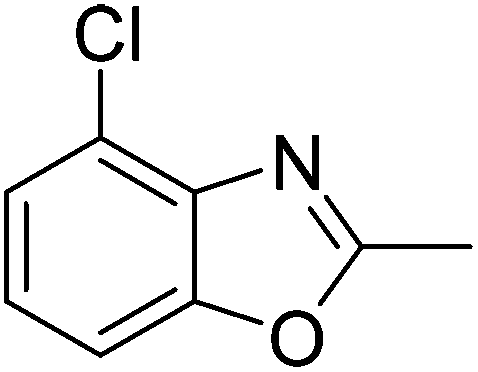
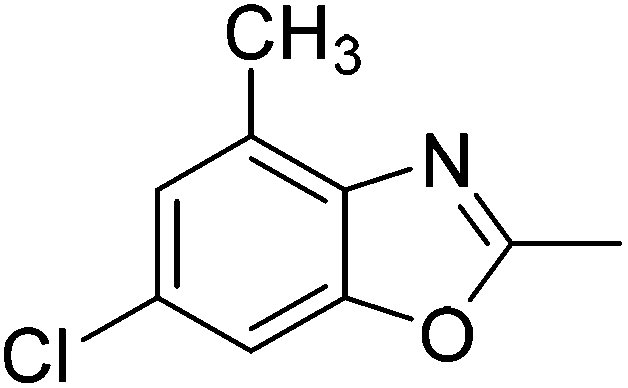
Yield: 80%. White solid; mp: 82–85 °C; 1H NMR (400 MHz, CDCl3) δ 7.30 (s, 1H), 7.09 (s, 1H), 2.62 (s, 3H), 2.55 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 163.55, 150.71, 139.41, 130.67, 129.64, 125.19, 108.19, 16.27, 14.44; IR (KBr): ν = 3426, 2926, 2858, 1732, 1617, 1459, 1254, 1160, 925, 823 cm−1; HRMS: calculated for C9H8ClNO: 182.0373, found: 182.0365.
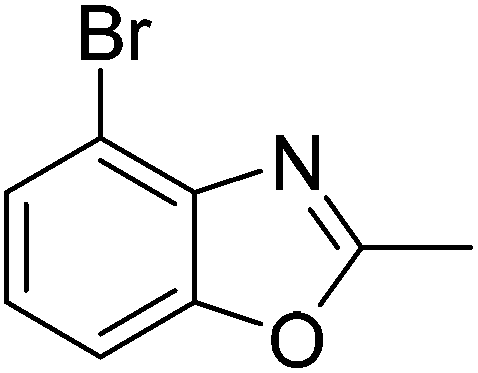
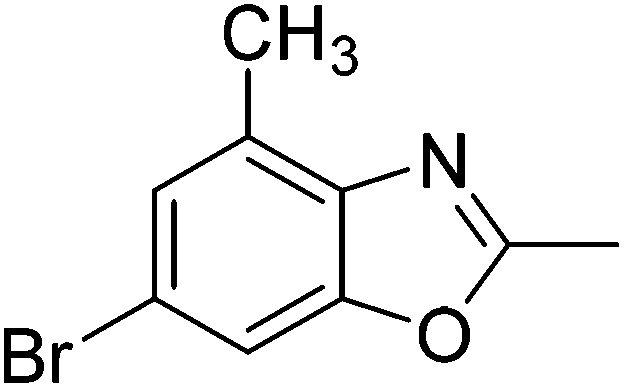
Yield: 84%. White solid; mp: 101–104 °C; 1H NMR (600 MHz, CDCl3) δ 7.44 (s, 1H), 7.23 (s, 1H), 2.61 (s, 3H), 2.54 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 163.48, 151.04, 139.92, 131.21, 127.96, 117.03, 111.10, 16.24, 14.46; IR (KBr): ν = 3359, 3177, 3101, 3038, 2921, 2853, 1695, 1610, 1451, 1391, 1256, 1202, 1050, 921, 803, 684, 591 cm−1; HRMS: calculated for C9H8BrNO: 225.9868, found: 225.9857.
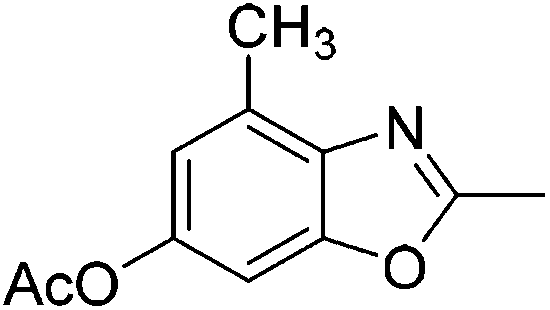
Yield: 81%. White solid; mp: 101–104 °C; 1H NMR (400 MHz, CDCl3) δ 7.78 (s, 1H), 7.45 (s, 1H), 4.00 (s, 3H), 2.67 (s, 3H), 2.48 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 165.88, 165.24, 151.85, 138.78, 134.33, 127.55, 120.78, 114.85, 52.45, 21.41, 14.70; IR (KBr): ν = 3424, 2928, 1765, 1624, 1434, 1374, 1208, 1118, 1064, 1020, 903, 600 cm−1; HRMS: calculated for C11H11NO3: 206.0817, found: 206.0790.
Yield: 90%. White solid; mp: 105–108 °C; 1H NMR (400 MHz, CDCl3) δ 7.95 (s, 1H), 7.82 (s, 1H), 3.92 (s, 3H), 2.65 (s, 3H), 2.59 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 166.90, 165.78, 150.27, 144.55, 129.48, 126.34, 126.17, 109.32, 52.21, 16.37, 14.68; IR (KBr): ν = 3712, 3434, 2934, 1712, 1625, 1449, 1050, 889, 796, 731, 659, 540 cm−1; HRMS: calculated for C11H11NO3: 206.0817, found: 206.0790.
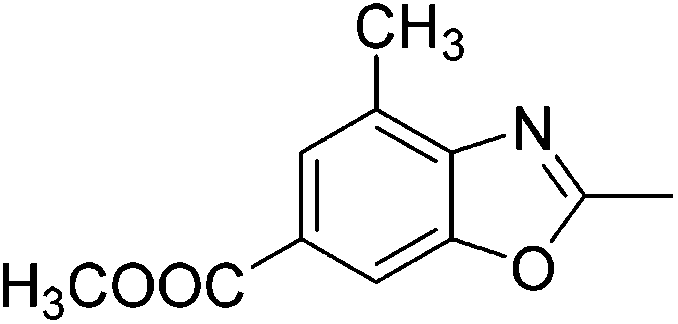
Yield: 52%. White solid; mp: 55–58 °C; 1H NMR (600 MHz, CDCl3) δ 7.07 (s, 1H), 6.84 (d, J = 10.6 Hz, 1H), 2.62 (s, 3H), 2.45 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 163.42, 153.53, 151.01, 135.79, 127.73, 111.56, 106.53, 21.68, 14.39; IR (KBr): ν = 3364, 2924, 2826, 1624, 1424, 1259, 1089, 1027, 805, 697, 503 cm−1; HRMS: calculated for C9H8FNO: 166.0668, found: 166.0665.
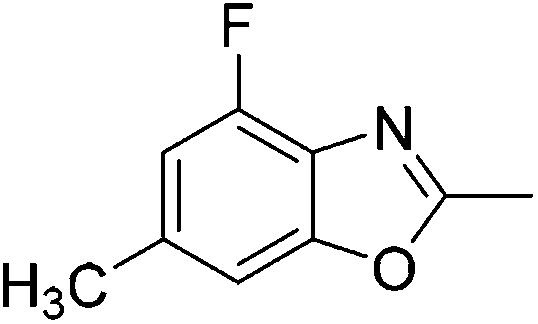
Yield: 77%. White solid; mp: 72–75 °C; 1H NMR (600 MHz, CDCl3) δ 7.09 (s, 1H), 7.05 (s, 1H), 2.56 (s, 3H), 2.36 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 163.03, 150.73, 136.05, 134.71, 124.33, 122.14, 108.16, 20.47, 13.45; IR (KBr): ν = 3492, 2924, 2858, 1696, 1613, 1468, 1256, 1188, 1099, 1025, 923, 811, 698, 594, 467 cm−1; HRMS: calculated for C9H8ClNO: 182.0373, found: 182.0372.
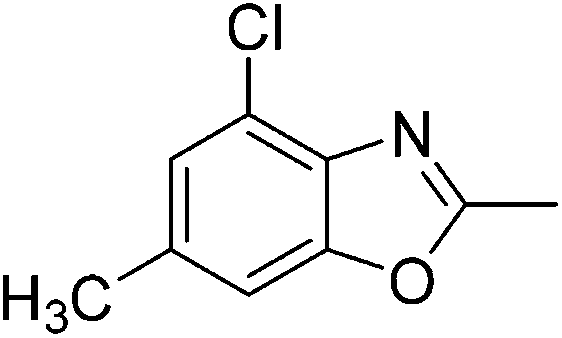
Yield: 76%. White solid; mp: 96–99 °C; 1H NMR (400 MHz, CDCl3) δ 7.29 (s, 1H), 7.20 (s, 1H), 2.63 (s, 3H), 2.43 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 163.90, 151.11, 138.71, 136.12, 128.23, 111.14, 109.72, 21.38, 14.49; IR (KBr): ν = 3430, 2926, 2858, 1748, 1611, 1458, 1249, 1117, 834 cm−1; HRMS: calculated for C9H8BrNO: 225.9868, found: 225.9850.
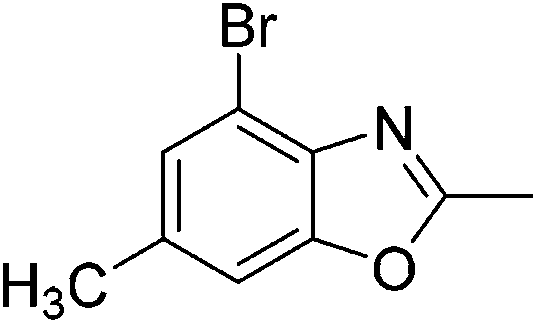
Yield: 93%. White solid; mp: 119–122 °C; 1H NMR (400 MHz, CDCl3) δ 7.06 (s, 1H), 6.82 (s, 1H), 2.61 (s, 3H), 2.55 (s, 3H), 2.30 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 169.66, 163.64, 150.38, 147.27, 138.48, 130.15, 118.53, 101.73, 21.03, 16.42, 14.45; IR (KBr): ν = 2932, 2580, 1714, 1523, 1439, 1323, 1245, 1197, 1036, 859, 789, 594 cm−1; HRMS: calculated for C11H11NO3: 206.0817, found: 206.0790.
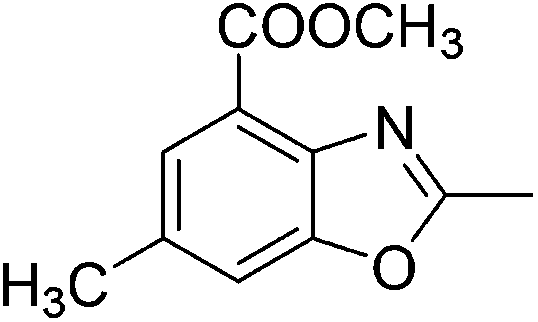
Yield: 62%. White solid; mp: 66–69 °C; 1H NMR (400 MHz, CDCl3) δ 7.09 (s, 1H), 6.92 (s, 1H), 2.61 (s, 3H), 2.54 (s, 3H), 2.42 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 162.42, 150.95, 138.36, 134.29, 128.90, 125.94, 107.72, 21.59, 16.38, 14.50; IR (KBr): ν = 3721, 3362, 2923, 2855, 1644, 1458, 1261, 1093, 1029, 805, 695 cm−1; HRMS: calculated for C10H11NO: 162.0919, found: 162.0915.
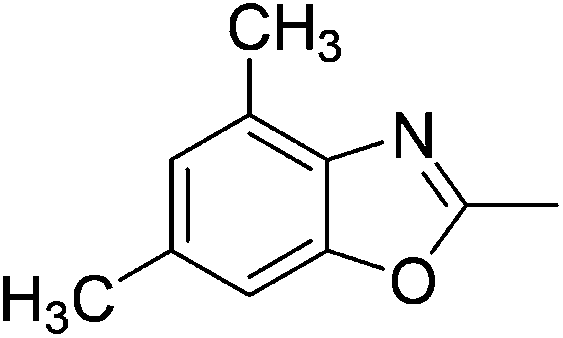
Yield: 58%. White solid; mp: 62–65 °C; 1H NMR (600 MHz, CDCl3) δ 7.40 (s, 1H), 7.33 (s, 1H), 2.66 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 165.26, 151.33, 138.20, 130.33, 124.83, 124.31, 109.73, 14.52; IR (KBr): ν = 3360, 2924, 2856, 1624, 1424, 1259, 1089, 1027, 805, 697, 503 cm−1; HRMS: calculated for C8H5Cl2NO: 201.9826, found: 201.9815.
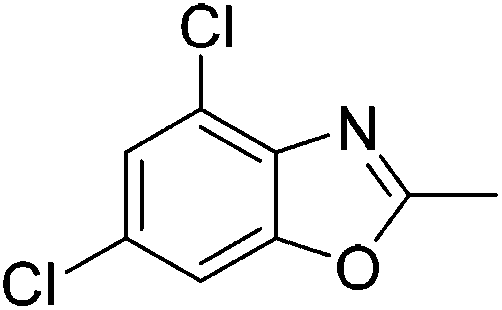
Yield: 41%. White solid; mp: 148–151 °C; 1H NMR (400 MHz, CDCl3) δ 7.63 (s, 1H), 7.59 (s, 1H), 2.65 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 165.26, 151.33, 138.20, 130.33, 124.83, 124.31, 109.73, 14.52; IR (KBr): ν = 3717, 3424, 3087, 2926, 2858, 1725, 1608, 1448, 1388, 1258, 1158, 1051, 939, 848, 745 cm−1; HRMS: calculated for C8H5Br2NO: 289.8816, found: 289.8798.
4.4. The control reaction using N-phenylacetamide as the substrate
Pd(OAc)2 (22.5 mg, 0.10 mmol), N-phenylacetamide (135.0 mg, 1.0 mmol), glacial acetic acid (4.0 mL), N,N-dimethylformamide (0.5 mL), potassium peroxodisulfate (500.0 mg, 1.5 mmol), and TfOH (150.0 mg, 1.0 mmol) were placed into a 15 mL sealed tube, and heated in an oil bath at 100 °C for 24 h. The reaction mixture (0.2 mL) was taken out from the tube at 3 hour intervals. After work up, the oily residue was detected by HPLC on a C18 reversed phase column, methanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water = 50: 50, flow rate = 1.0 mL min−1. The yields were determined with 1,3,5-trimethoxybenzene as an internal standard.
:water = 50: 50, flow rate = 1.0 mL min−1. The yields were determined with 1,3,5-trimethoxybenzene as an internal standard.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Jiangsu Provincial Nature Science Foundation (SBK2016021885), the Top-notch Academic Programs Project of Jiangsu Higher Education Institutions, and the Priority Academic Program Development of Jiangsu Higher Education Institutions.Notes and references
- (a) A. A. Nagel, D. R. Liston, S. Jung, M. Mahar, L. A. Vincent, D. Chapin, Y. L. Chen, S. Hubbard, J. L. Ives, S. B. Jones, J. A. Nielsen, A. Ramirez, I. A. Shalaby, A. Villalobos and W. F. White, J. Med. Chem., 1995, 38, 1084 CrossRef CAS PubMed; (b) M. Taki, J. L. Wolford and T. V. O'Halloran, J. Am. Chem. Soc., 2004, 126, 712 CrossRef CAS PubMed; (c) D. N. Rana, M. T. Chhabria, N. K. Shah and P. S. Brahmkshatriya, Med. Chem. Res., 2014, 23, 2218 CrossRef CAS; (d) D. Wee, S. Yoo, Y. H. Kang, Y. H. Kim, J. W. Ka, S. Y. Cho, C. Lee, J. Ryu, M. H. Yi and K. S. Jang, J. Mater. Chem. C, 2014, 2, 6395 RSC; (e) P. C. Xue, P. Chen, J. H. Jia, Q. X. Xu, J. B. Sun, B. Q. Yao, Z. Q. Zhang and R. Lu, Chem. Commun., 2014, 50, 2569 RSC; (f) Z. Zhang, Z. Y. Li, B. Fu and Z. H. Zhang, Chem. Commun., 2015, 51, 16312 RSC; (g) Y. Leng, F. Yang, W. Zhu, Y. Wu and X. Li, Org. Biomol. Chem., 2011, 9, 5288 RSC; (h) K. Seth, M. Nautiyal, P. Purohit, N. Parikh and A. K. Chakraborti, Chem. Commun., 2015, 51, 191 RSC.
- (a) C. L. Galatis, J. Am. Chem. Soc., 1948, 70, 1967 CrossRef PubMed; (b) J. Chang, K. Zhao and S. Pan, Tetrahedron Lett., 2002, 43, 951 CrossRef CAS; (c) F. Z. Su, S. C. Mathew, L. Möhlmann, M. Antonietti, X. C. Wang and S. Blechert, Angew. Chem., Int. Ed., 2011, 50, 657 CrossRef CAS PubMed; (d) T. B. Nguyen, L. Ermolenko, W. A. Dean and A. Al-Mourabit, Org. Lett., 2012, 14, 5948 CrossRef CAS PubMed; (e) W. C. Li, C. C. Zeng, L. M. Hu, H. Y. Tian and R. D. Little, Adv. Synth. Catal., 2013, 355, 2884 CrossRef CAS; (f) L. J. Gu, C. Jin, J. M. Guo, L. Z. Zhang and W. Wang, Chem. Commun., 2013, 49, 10968 RSC; (g) X. Shi, J. Guo, J. Liu, M. Ye and Q. Xu, Chem. – Eur. J., 2015, 21, 9988 CrossRef CAS PubMed; (h) J. Jiang, H. X. Zou, Q. Z. Dong, R. J. Wang, L. H. Lu, Y. G. Zhu and W. M. He, J. Org. Chem., 2016, 81, 51 CrossRef CAS PubMed.
- (a) V. N. Bochatay, P. J. Boissarie, J. A. Murphy, C. J. Suckling and S. Lang, J. Org. Chem., 2013, 78, 1471 CrossRef CAS PubMed; (b) L. Tang, X. F. Guo, Y. Yang, Z. G. Zha and Z. Y. Wang, Chem. Commun., 2014, 50, 6145 RSC; (c) K. R. Babu, N. B. Zhu and H. L. Bao, Org. Lett., 2017, 19, 46 CrossRef CAS PubMed; (d) G. Altenhoff and F. Glorius, Adv. Synth. Catal., 2004, 346, 1661 CrossRef CAS; (e) V. Kavala, D. Janreddy, M. J. Raihan, C. W. Kuo, C. Ramesh and C. F. Yao, Adv. Synth. Catal., 2012, 354, 2229 CrossRef CAS.
- (a) M. M. Guru, M. A. Ali and T. Punniyamurthy, J. Org. Chem., 2011, 76, 5295 CrossRef CAS PubMed; (b) X. X. Guo, D. W. Gu, Z. X. Wu and W. B. Zhang, Chem. Rev., 2015, 115, 1622 CrossRef CAS PubMed; (c) S. Ueda and H. Nagasawa, Angew. Chem., Int. Ed., 2008, 47, 6411 CrossRef CAS PubMed; (d) Z. Wang, K. Z. Li, D. B. Zhao, J. B. Lan and J. S. You, Angew. Chem., Int. Ed., 2011, 50, 5365 CrossRef CAS PubMed; (e) L. J. Gu, C. Jin, J. M. Guo, L. Z. Zhang and W. Wang, Chem. Commun., 2013, 49, 10968 RSC; (f) K. V. N. Esguerra and J. P. Lumb, ACS Catal., 2017, 7, 3477 CrossRef CAS; (g) H. Wang, L. Wang, J. Shang, X. Li, H. Wang, J. Gui and A. Lei, Chem. Commun., 2012, 48, 76 RSC; (h) G. T. Zhang, C. Liu, H. Yi, Q. Y. Meng, C. L. Bian, H. Chen, J. X. Jian, L. Z. Wu and A. W. Lei, J. Am. Chem. Soc., 2015, 137, 9273 CrossRef CAS PubMed.
- (a) Y. Yuan, D. T. Chen and X. W. Wang, Adv. Synth. Catal., 2011, 353, 3373 CrossRef CAS; (b) H. Liu, H. Xu and Y. Yuan, Tetrahedron, 2014, 70, 6474 CrossRef CAS; (c) X. W. Wang, Y. J. Li and Y. Yuan, Synthesis, 2013, 45, 1247 CrossRef CAS.
- G. W. Wang, T. T. Yuan and X. L. Wu, J. Org. Chem., 2008, 73, 4717 CrossRef CAS PubMed.
- (a) R. Das and M. Kapur, J. Org. Chem., 2017, 82, 1114 CrossRef CAS PubMed; (b) J. Jiang, W. M. Zhang, J. J. Dai, J. Xu and H. J. Xu, J. Org. Chem., 2017, 82, 3622 CrossRef CAS PubMed; (c) O. Tischler, Z. Bokányi and Z. Novák, Organometallics, 2016, 35, 741 CrossRef CAS; (d) S. J. Lou, Y. J. Mao, D. Q. Xu, J. Q. He, Q. Chen and Z. Y. Xu, ACS Catal., 2016, 6, 3890 CrossRef CAS; (e) Y. Wu, P. Y. Choy, F. Mao and F. Y. Kwong, Chem. Commun., 2013, 49, 689 RSC; (f) S. J. Tremont and H. Ur Rahman, J. Am. Chem. Soc., 1984, 106, 5759 CrossRef CAS; (g) X. B. Wan, Z. X. Ma, B. J. Li, K. Y. Zhang, S. K. Cao, S. W. Zhang and Z. J. Shi, J. Am. Chem. Soc., 2006, 128, 7416 CrossRef CAS PubMed; (h) M. Tobisu, Y. Ano and N. Chatani, Org. Lett., 2009, 11, 3250 CrossRef CAS PubMed; (i) D. C. Powers, D. Benitez, E. Tkatchouk, W. A. Goddard III and T. Ritter, J. Am. Chem. Soc., 2010, 132, 14092 CrossRef CAS PubMed; (j) F. Szabo, J. Daru, D. Simko, T. Z. Nagy, A. Stirling and Z. Novak, Adv. Synth. Catal., 2013, 355, 685 CrossRef CAS; (k) F. H. Luo, J. Yang, Z. K. Li, H. F. Xiang and X. G. Zhou, Eur. J. Org. Chem., 2015, 2463 CrossRef CAS.
- (a) H. Xu, M. Shang, H. X. Dai and J. Q. Yu, Org. Lett., 2015, 17, 3830 CrossRef CAS PubMed; (b) K. Seth, M. Nautiyal, P. Purohit, N. Parikh and A. K. Chakraborti, Chem. Commun., 2015, 51, 191 RSC; (c) A. R. Dick, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2004, 126, 2300 CrossRef CAS PubMed; (d) T. S. Jiang and G. W. Wang, J. Org. Chem., 2012, 77, 9504 CrossRef CAS PubMed; (e) Z. W. Yin and P. P. Sun, J. Org. Chem., 2012, 77, 11339 CrossRef CAS PubMed; (f) G. W. Wang and T. T. Yuan, J. Org. Chem., 2010, 75, 476 CrossRef CAS PubMed; (g) B. S. Kim, C. Jang, D. J. Lee and S. W. Youn, Chem. – Asian J., 2010, 5, 2336 CrossRef CAS PubMed; (h) J. Q. Weng, Z. Q. Yu, X. H. Liu and G. F. Zhang, Tetrahedron Lett., 2013, 54, 1205 CrossRef CAS.
- (a) R. Giri, J. K. Lam and J. Q. Yu, J. Am. Chem. Soc., 2010, 132, 686 CrossRef CAS PubMed; (b) D. K. Li, N. Xu, Y. C. Zhang and L. Wang, Chem. Commun., 2014, 50, 14862 RSC; (c) Y. Wu, C. Jiang, D. Wu, Q. Gu, Z. Y. Luo and H. B. Luo, Chem. Commun., 2016, 52, 1286 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ob02566a |
| This journal is © The Royal Society of Chemistry 2018 |