
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Negishi cross-couplings in the synthesis of amino acids
William D. G.
Brittain
 * and
Steven L.
Cobb
*
* and
Steven L.
Cobb
*
Department of Chemistry, Durham University, South Road, Durham, DH1 3LE, UK. E-mail: steven.l.cobb@durham.ac.uk; william.d.brittain@durham.ac.uk
First published on 27th November 2017
Abstract
The Negishi cross-coupling is a powerful C–C bond-forming reaction widely utilised in many areas of organic synthesis. This review details the use of Negishi cross-couplings in the synthesis of unnatural amino acids. The application of this reaction in the preparation of aromatic, heteroaromatic, and, complex amino acid derivatives are reviewed and presented herein.
 William D. G. Brittain | William D. G. Brittain obtained an Msci degree from the University of Birmingham in 2013. He then went on to carry out PhD studies at the same institution on asymmetric copper catalysis under the supervision of Dr John S. Fossey (University of Birmingham) and Dr Benjamin R. Buckley (Loughborough University). During 2015/16 he spent time working with Professor Eric V. Anslyn (University of Texas at Austin) on boronic acid based assemblies. He is currently a postdoctoral research associate at Durham University in the Cobb group, where he is working in the areas of organic synthesis and peptide chemistry. |
 Steven L. Cobb | Steven L. Cobb received his PhD in Bioorganic Fluorine Chemistry from St. Andrews University, where he worked with Professor David O'Hagan on the biosynthesis of fluorinated natural products. He carried out postdoctoral studies on the development of peptide-based antibiotics with Professor John Vederas FRS at the University of Alberta. In 2008 he was awarded a Ramsay Memorial Fellowship at Durham University and he is now an Associate Professor of Chemical-Biology and Director of the Centre for Global Infectious Diseases. His research group looks to develop new peptide- and peptoid-based therapeutics for the treatment of antimicrobial infections. |
Introduction
Negishi cross-coupling, since its original development in 1977 by Negishi and co-workers, has become a widely-utilised reaction in the field of organic chemistry.1 Employing either palladium or nickel as a transition metal catalyst, the Negishi cross-coupling will form a new carbon–carbon bond between an organic halide/triflate and an organozinc reagent.2,3 As a versatile route to form carbon–carbon bonds the Negishi cross-coupling has seen widespread use in the synthesis of many novel structural motifs.4–6 In recent years, developments in Negishi cross-couplings by researchers, such as Fu, has allowed the transformation to be applied to more substrates than ever before.7–10One application of the Negishi cross-coupling reaction is in the synthesis of unnatural amino acids. The formation of such compounds is of interest to a multitude of chemical and biological research arenas. Introduction of unnatural amino acids into peptide sequences has been used to probe protein structure and function in both in vitro11,12 and in vivo systems.13,14 Antibiotic peptides have utilized unnatural amino acids to increase organism selectivity, potency and metabolic stability.15
In recent years asymmetric catalysis has employed unnatural amino acids as easily accessible chiral building blocks for the synthesis of new ligands.16–20 For example, chiral ligands such as PyBOX and BOX rely on the chirality afforded by natural or unnatural amino acids in their preparation and design.21–24
With wide-ranging applications, methods to form unnatural amino acids are without doubt important. This review gives an overview and update25 of the synthesis and derivatisation of amino acids through Negishi cross-couplings. Three major classes of derivatisation are discussed: aromatic-, heteroaromatic and more complex unnatural amino acids. Several mechanistic studies have been carried out on the Negishi cross-coupling of alkylamido zinc reagents, and these are also discussed.
Synthesis of aromatic amino acids
Aromatic amino acids have been shown to have a broad range of utility. Aromatic amino acids have been successfully employed in natural product synthesis,26 peptide chemistry27,28 and drug development.29 Therefore, facile synthesis of aromatic amino acids is important.The area of Negishi cross-coupling to form stereo-defined aromatic amino acids was pioneered in the seminal work by Jackson et al. in 1989.30 A range of novel single-enantiomer substituted phenylalanines 3a–d were reported from the cross-coupling between an iodozinc L-serine derivative 2 and a range of para-substituted iodobenzenes (Scheme 1). The transformation was found to be tolerant to Boc protecting groups and a range of electron-donating (Scheme 1, 3c) and withdrawing groups (Scheme 1, 3d) on the aryl iodide component. The novel phenylalanine derivatives were isolated in yields of between 35% and 65%.
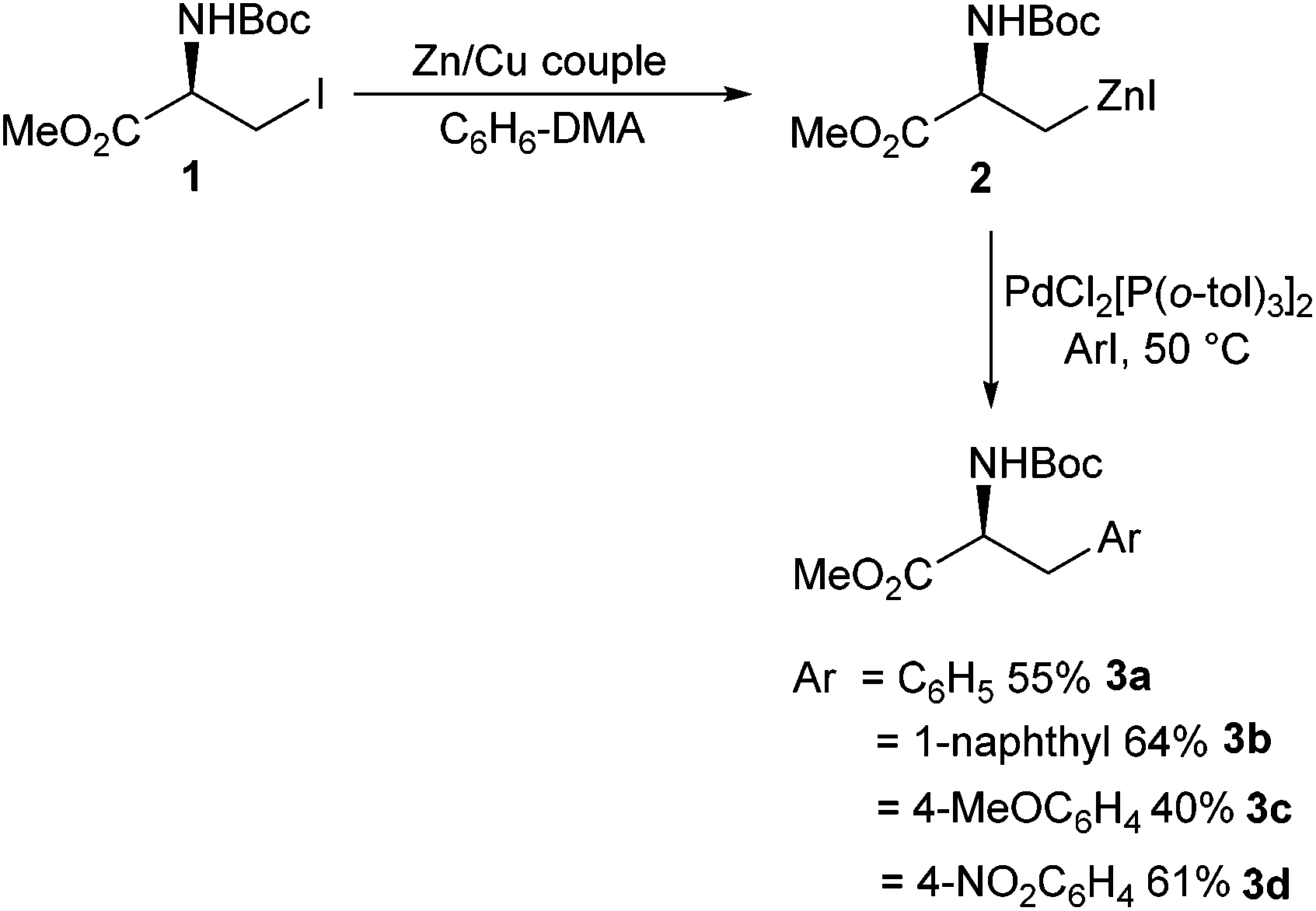 | ||
| Scheme 1 Synthesis of phenylalanine derivatives via Negishi cross coupling, Jackson and co-workers 1989.30 | ||
Jackson and Dexter would adapt their initial Negishi cross-coupling methods to also produce β and γ amino acids from aspartic and glutamic acid derivatives.31 Utilising activated zinc and Pd2(dba)3, a range of β and γ amino acids were furnished in yields between 33% and 89% (Scheme 2). The scope and reproducibility using this methodology was found to be superior to the conditions initially used to access α amino acids.30
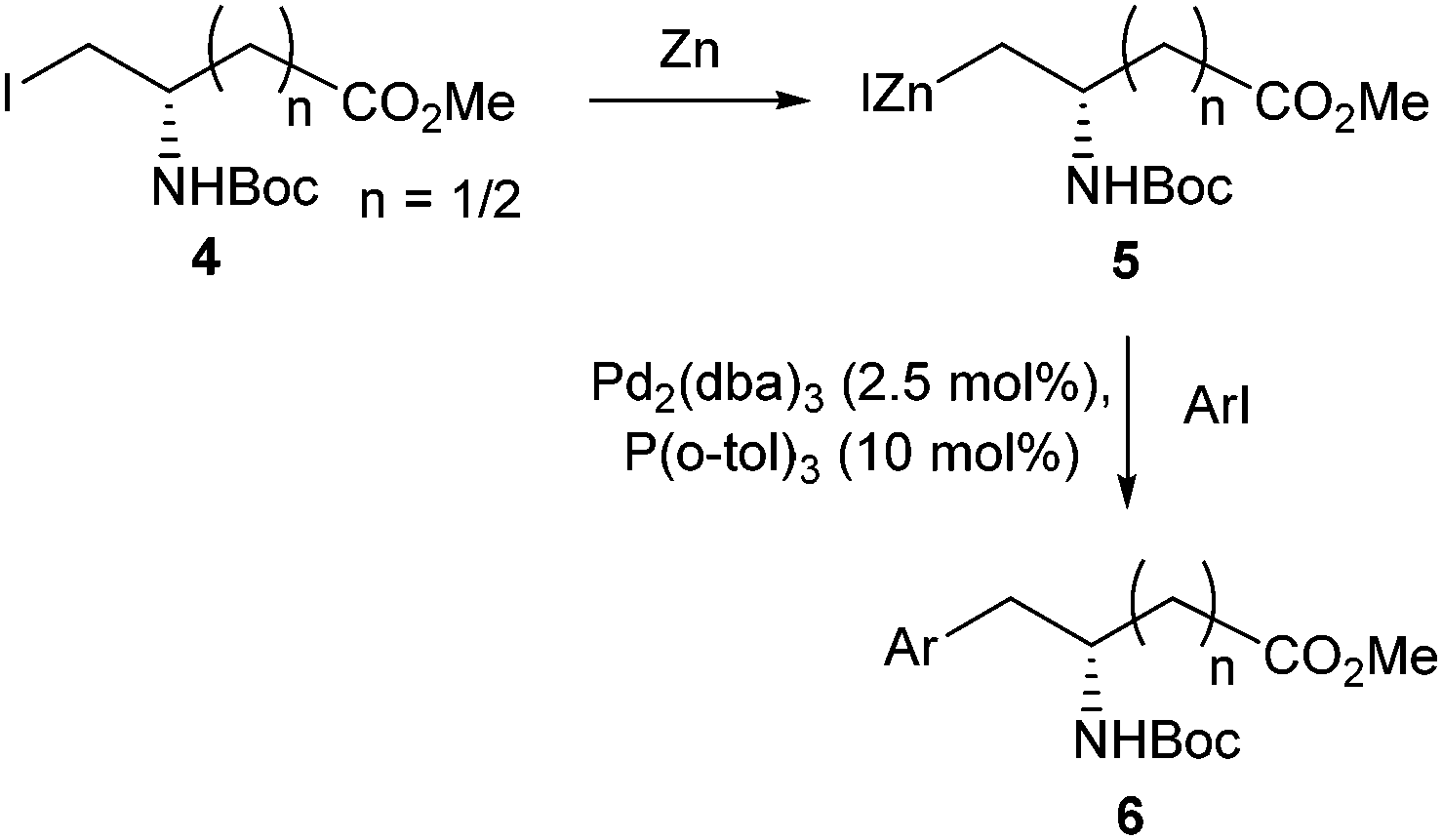 | ||
| Scheme 2 Synthesis of aspartic and glutamic acid derivatives utilising Negishi coupling, Jackson and Dexter, 1998.31 | ||
Modification of serine and phenylalanine via Negishi cross-coupling was reported by König and co-workers (Scheme 3).32 Using Rieke zinc they were able to successfully form organozinc species from amino acids, which could then be coupled with electron-rich iodoanilines and iodobenzyl amines. Using this approach, a series of metal-chelating side chain containing unnatural amino acids 9a–e were successfully synthesised in yields ranging between 65% and 86% (Scheme 3).
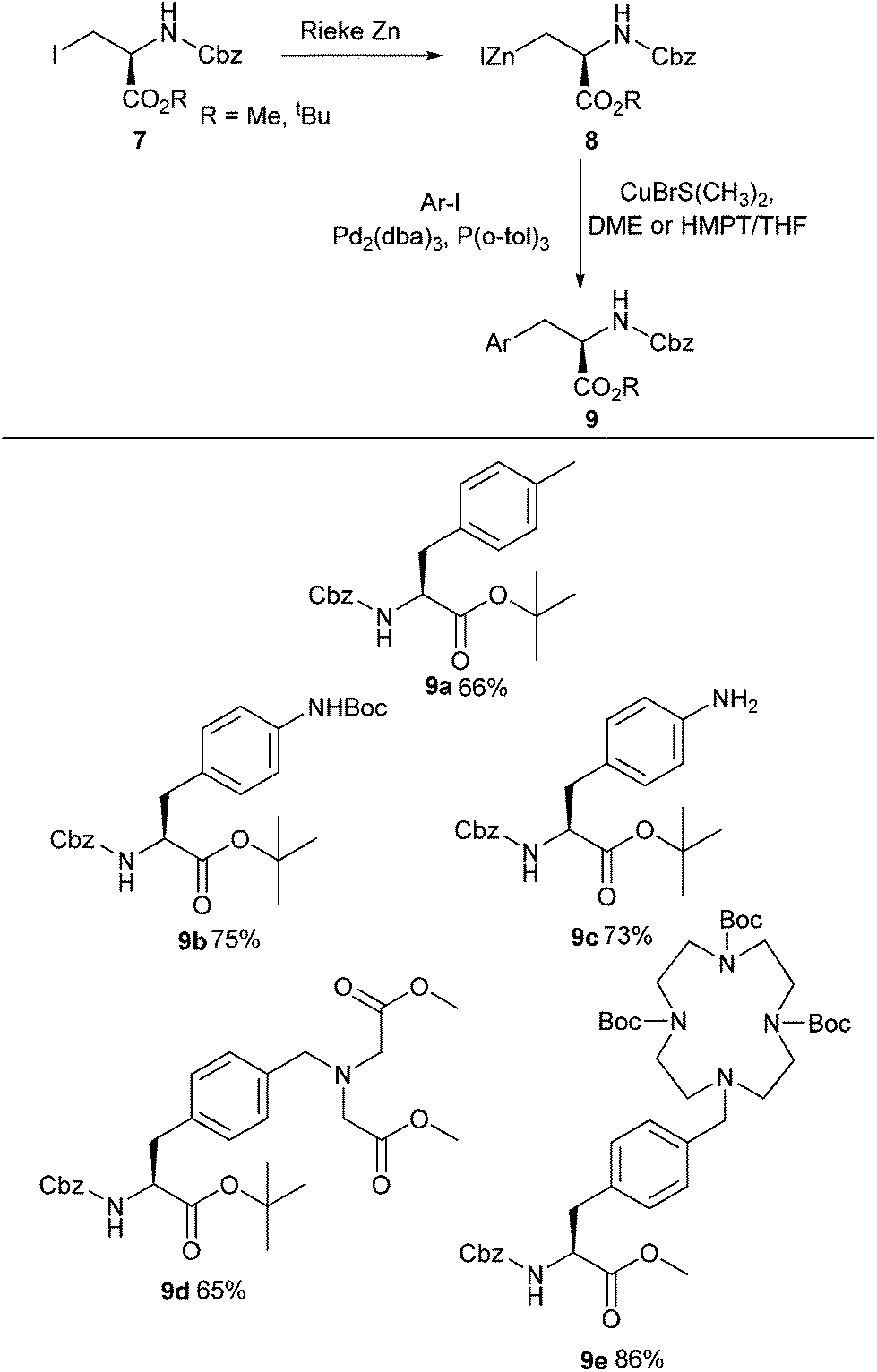 | ||
| Scheme 3 Synthesis of novel amino acids containing metal-coordinating side chains, König and co-workers.32 | ||
The synthesis of β2-homophenylalanine derivatives using Negishi cross-couplings were first reported in 2008 (Scheme 4).33 The synthesis of the corresponding β3-amino acids had already been reported in 1999,34,35 but the methodology had not been used to access β2-amino acids. Conjugate addition of benzylamine to compound 10, followed by Boc protection and alkyl iodide formation, led to the protected amino acid coupling partner 13 (Scheme 4). Cross-coupling was then achieved using a range of aryl iodides and bromides. In the case of nitro-substituted aryls lower yields were observed. This was attributed to a competing zinc catalysed reduction of the nitro groups. It was found that removal of excess zinc before Negishi cross-coupling gave greatly improved yields, with an increase from 24% to 45% in the case of 1-iodo-4-nitrobenzene (Scheme 4, 14d).
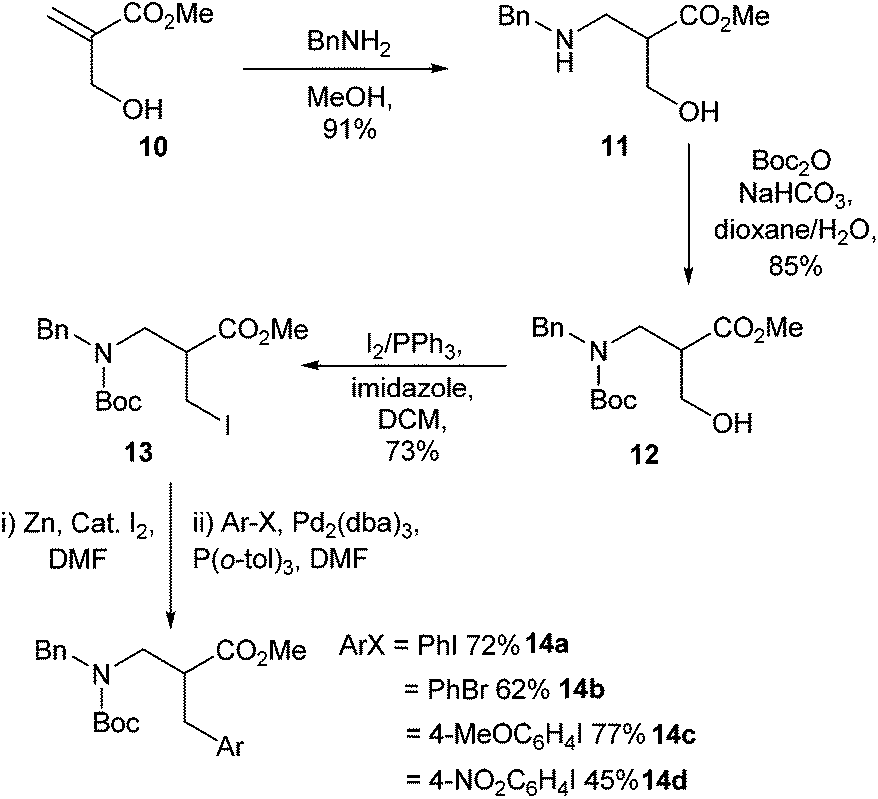 | ||
| Scheme 4 β2-Amino acids synthesised through Negishi cross couplings, Jackson and co-workers.33 | ||
In order to make the transformation as synthetically relevant as possible widening the scope of the haloaryl component of the Negishi cross-coupling was elaborated upon in 2008.36 Taking inspiration from the reported reaction of a serine derivative 1 and a bromo-substituted naphthalene by Szczepankiewicz et al. (Scheme 5)37 it was subsequently demonstrated that a wide range of bromides were applicable to the Negishi cross-coupling (Scheme 6).
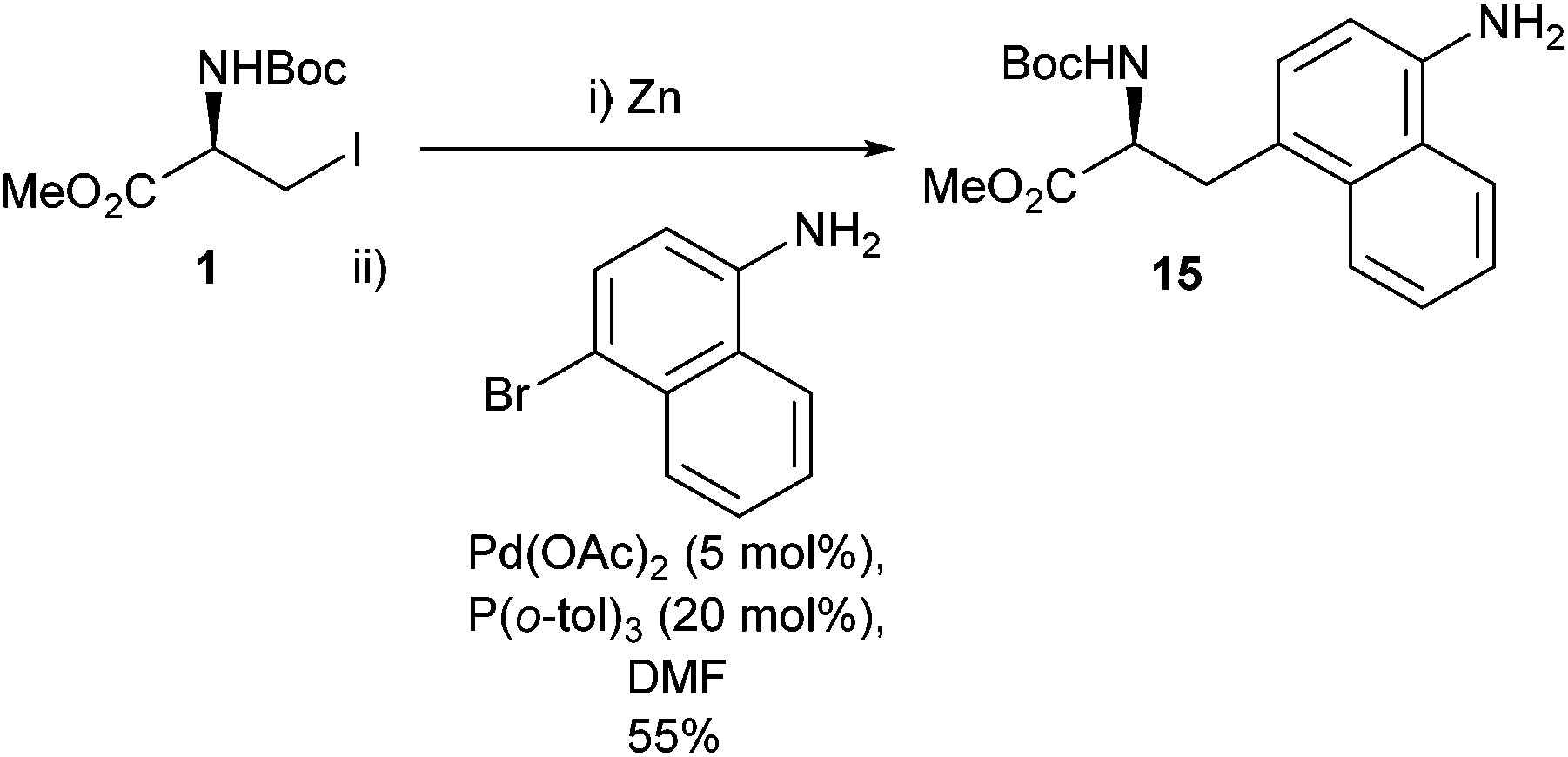 | ||
| Scheme 5 Synthesis of a naphthyl-appended amino acid precursor by Szczepankiewicz et al.37 | ||
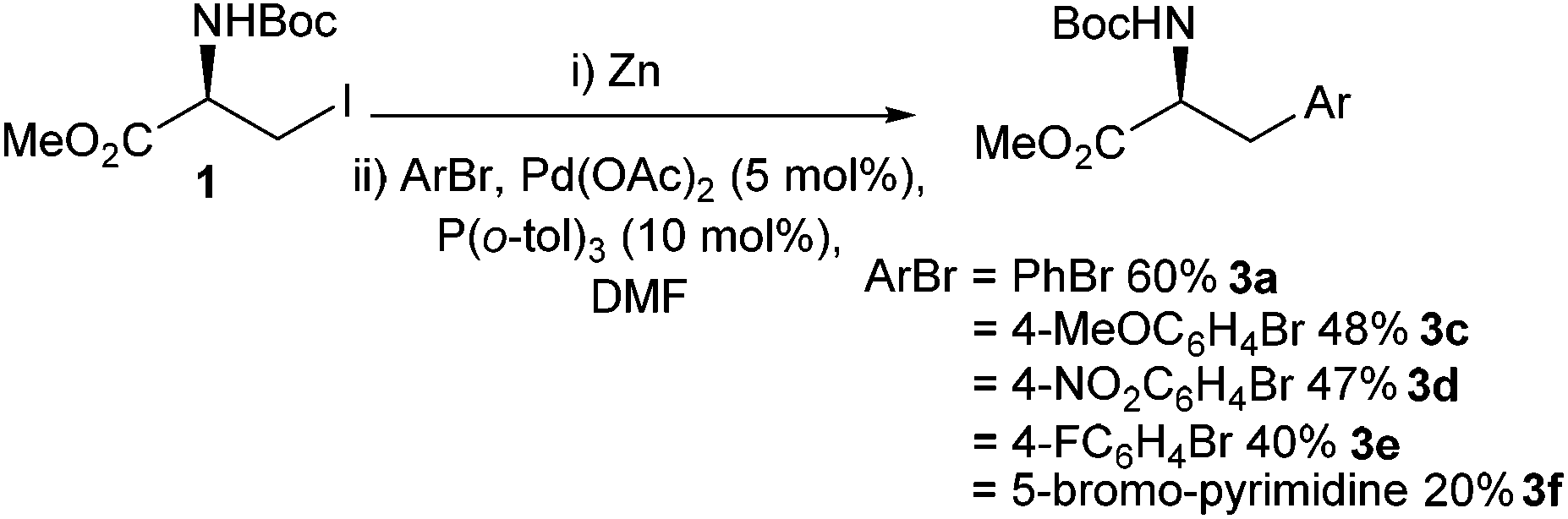 | ||
| Scheme 6 Negishi cross-couplings of aryl bromides with an iodo serine derivative. | ||
Yields for the cross-coupling reaction when an aryl bromide was employed were lower across all examples compared to their iodo equivalents.36 However, it should be noted that aryl bromides are often cheaper and more widely available than their corresponding iodides.
By exploiting the differences in cross-coupling reactivity between aromatic iodides and aromatic bromides it was demonstrated that asymmetrical bis-amino acids could be successfully synthesised. For example, the bis-amino acid 17 (Scheme 7) was synthesised through a double cross-coupling process in a 21% overall yield. This was an improvement over the previously reported palladium-catalysed Suzuki–Miyaura coupling approach to form asymmetrical biphenyl bis-amino acids which was found to return racemic products.38
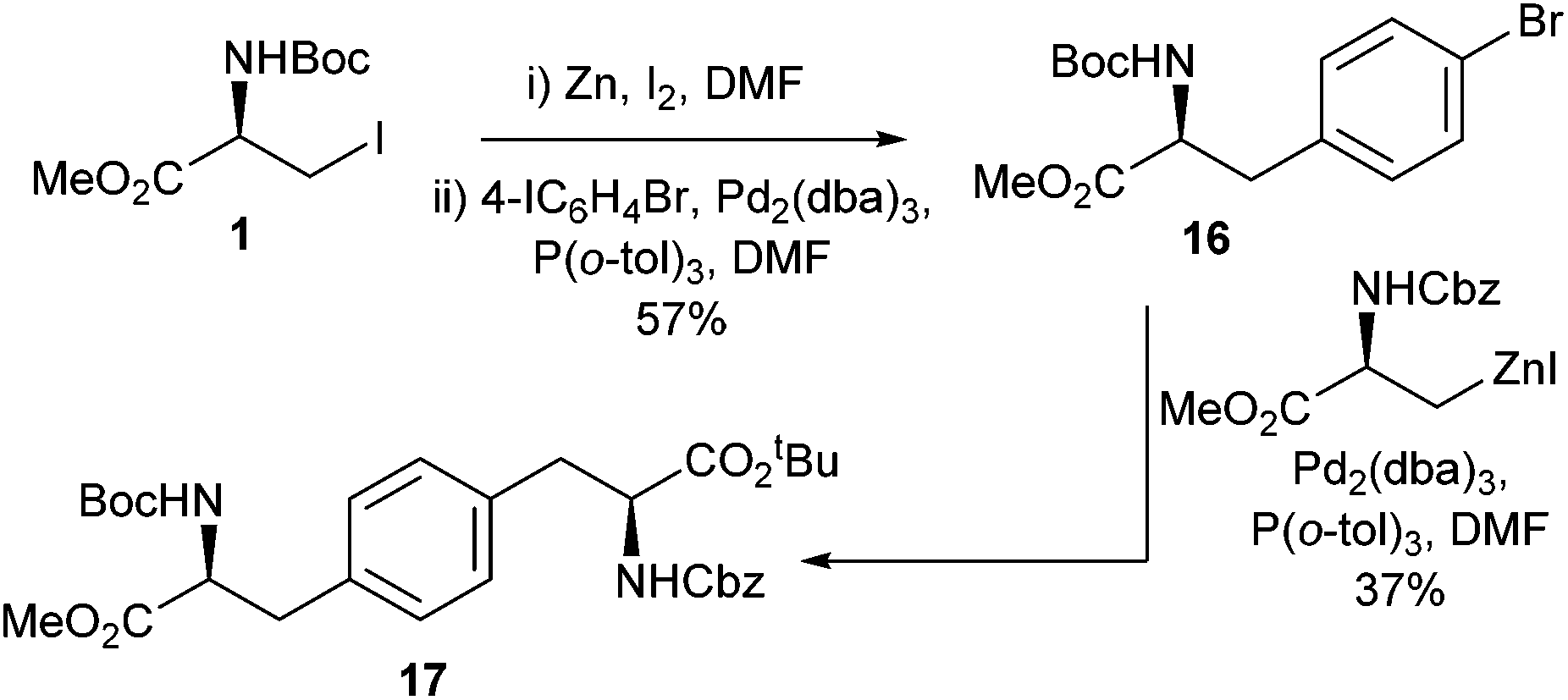 | ||
| Scheme 7 Synthesis of a bis(amino acid) derivative via a double Negishi cross coupling strategy. | ||
Biarylphosphine ligands such as SPhos are known to increase the rate and yield of palladium-catalysed reactions.39 These ligands are believed to stabilise monoligated Pd(0), and in addition, have the ability to stabilise Pd(II) centres.39,40 It was found that an active catalyst formed from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 SPhos to Pd2(dba)3 led to improved yields (compared to earlier reports) of Negishi cross-coupled phenylalanine derivatives 3a–d (Scheme 8).41
:1 SPhos to Pd2(dba)3 led to improved yields (compared to earlier reports) of Negishi cross-coupled phenylalanine derivatives 3a–d (Scheme 8).41
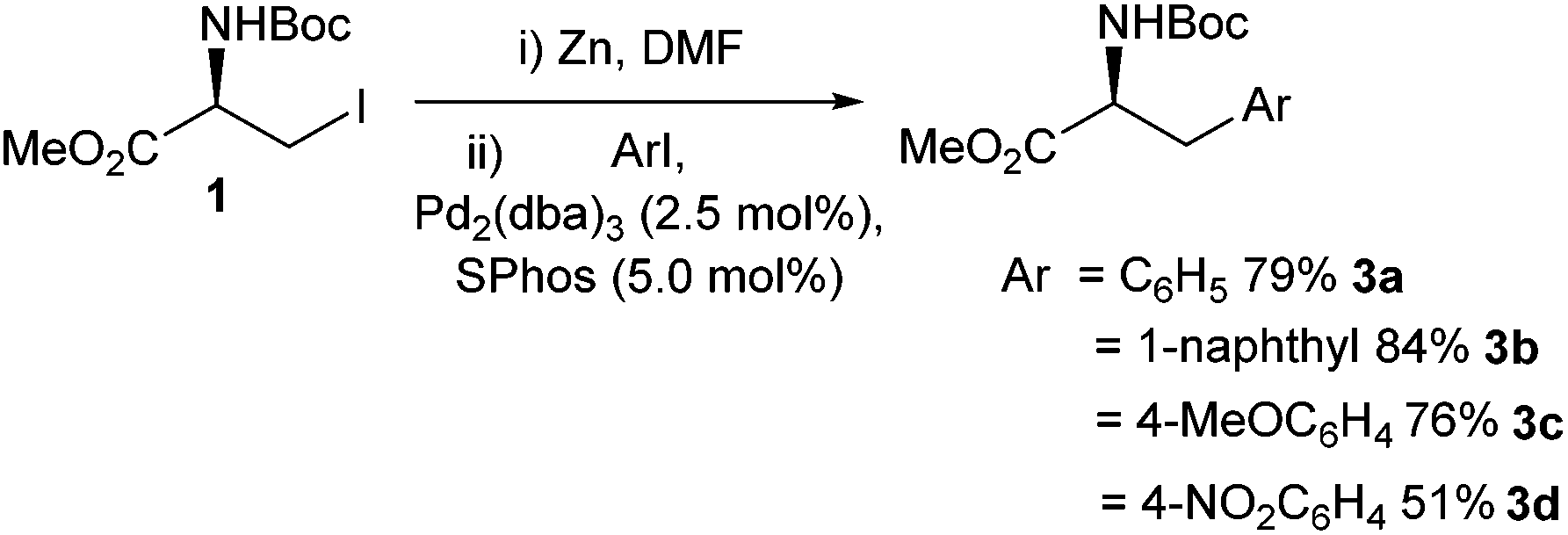 | ||
| Scheme 8 Improved cross-coupling conditions using SPhos reported by Jackson and co-workers.41 | ||
Interestingly it was found that when 2-iodoaniline was used in the presence of SPhos the expected amino acid derivative was not delivered and instead a lactam 18 was formed in good yield (85%) (Scheme 9).
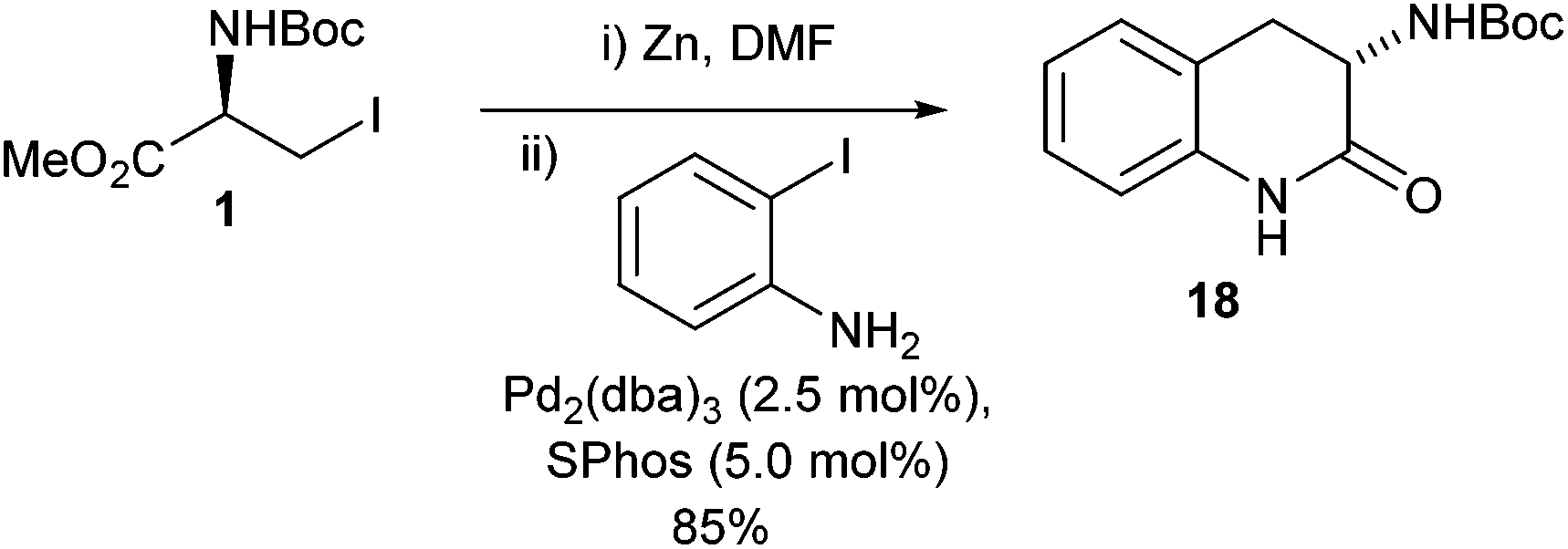 | ||
| Scheme 9 Unexpected lactam formation observed in the cross-coupling of an iodoaniline with an iodo serine derivative. | ||
Jackson's group built upon the observation that SPhos improved the yields of Negishi cross-couplings. By taking inspiration from their work on the kinetics of β-elimination, the Boc protecting group (see mechanistic studies)42 was replaced with a trifluoro-acyl (TFA) group (Scheme 10).43 The TFA protecting group had been shown previously to perturb the alkyl zinc halide from undergoing β-elimination, (see later section on mechanistic studies for further information) so it was hoped that the combination of SPhos and a TFA protecting group on the zinc halide would lead to improved reaction conditions for the synthesis of phenylethylamine derivatives. TFA-protected zinc iodides derived from phenylalanine 19 and valine 21 were shown to react smoothly with a range of aryl iodides, delivering good to excellent yields of aryl appended protected amines 20a–c/22a–c.
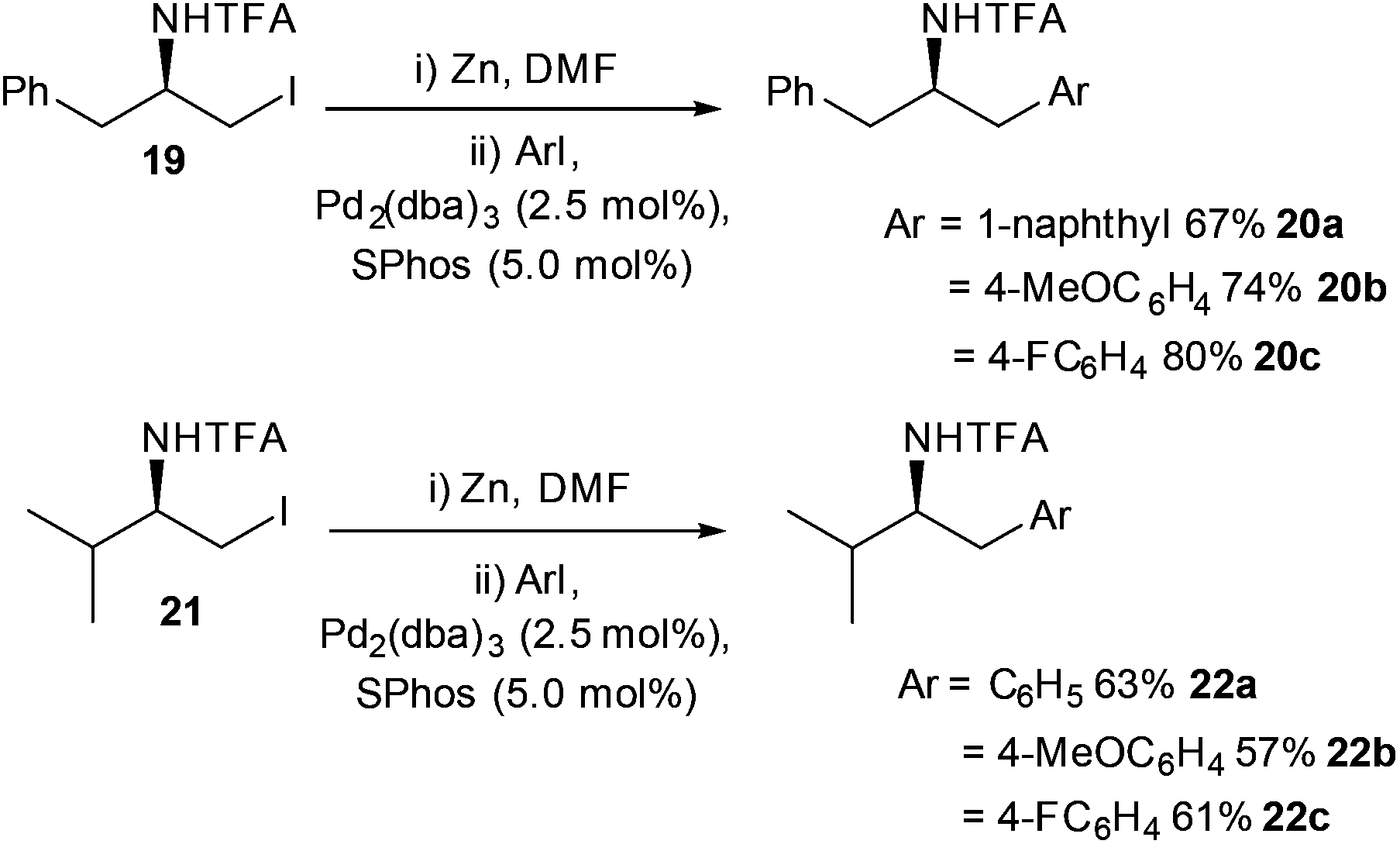 | ||
| Scheme 10 Amines prepared from amino acids by Negishi cross coupling, 19 and 21 prepared from phenylalanine and valine respectively in two steps. | ||
The synthesis of trifluoromethyl appended aromatic amino acids were reported by Featherston and Miller (Scheme 11).44 Cross-coupling was successfully carried out between Cbz-protected iodide 23 and a fluoro-aromatic to give 24 in a 54% yield. The aromatic amino acid 24 was then successfully transformed into the corresponding ketone through treatment with ozone. The peptides formed in the study were found to be catalytically active in the oxidation of substituted styrenes to their corresponding epoxides. In addition to their applications in catalysis, fluorinated amino acids have also been shown to be useful as biophysical probes and as structural components in peptide sequences.45
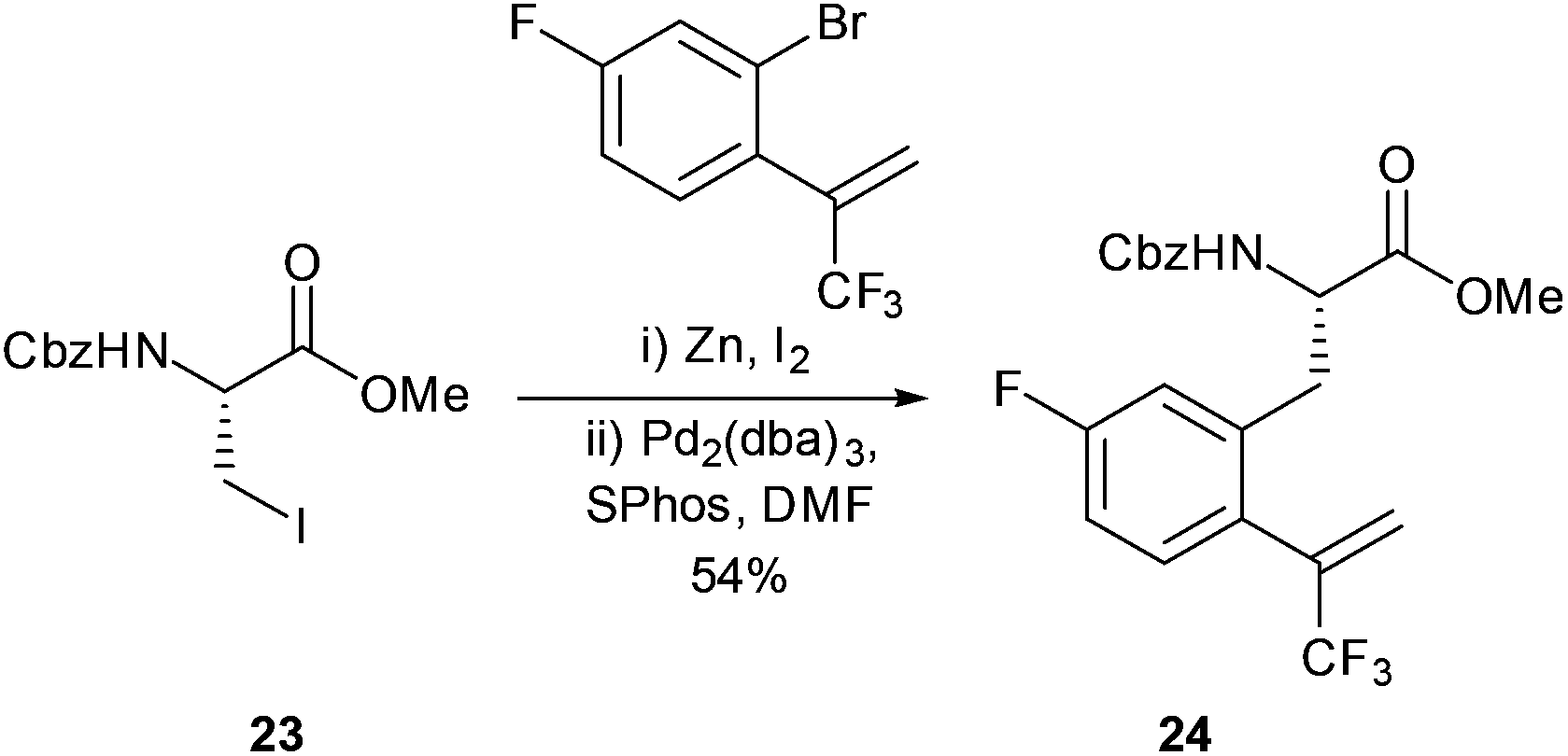 | ||
| Scheme 11 Synthesis of fluoroaromatic amino acid precursor for application in oxidation of substituted styrenes, Featherston and Miller.44 | ||
Synthesis of heteroaromatic amino acids
Drug development and discovery often utilise heteroaromatic cores to deliver potent biological activity,46,47 and elaboration of heterocyclic cores with other moieties is a cornerstone of fragment-based drug discovery (FBDD).48–50 Within this context heteroaromatic amino acids have been shown to be valuable building blocks for the synthesis of many biologically active compounds.51–53 The Negishi cross-coupling is one way in which heteroaromatic-appended amino acids can be synthesised in relatively few steps with great modularity.Synthesis of heteroaromatic amino acid derivatives formed through Negishi cross-coupling was reported by Göbel and co-workers.54 Negishi cross-coupling was successfully carried out using a range of aryl bromides and iodides with oxazolidine 27 (Scheme 12). Heteroaromatics such as quinoline 30e and pyrimidine 30f were found to be amenable to the methodology, and the inclusion of commonly used protecting groups (Boc and Fmoc) allowed the formed novel amino acids to be incorporated into longer peptide sequences by solid phase peptide synthesis (SPPS).
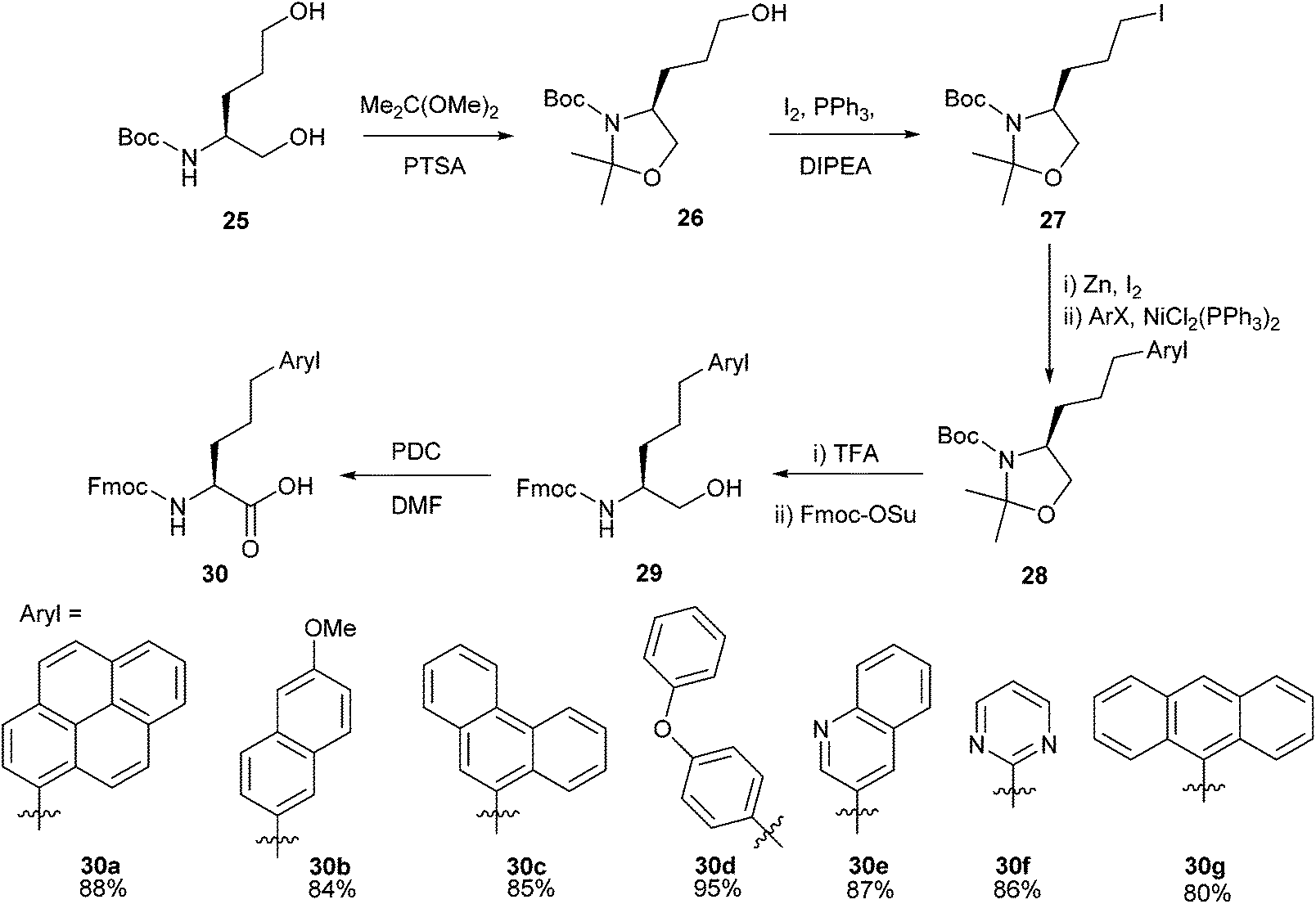 | ||
| Scheme 12 Synthesis of aromatic and heteroaromatic amino acids. | ||
A detailed study into the synthesis of ortho-substituted nitroaromatic containing amino acid derivatives was carried out by Tuttle et al. (Scheme 13).55 Reaction optimisation between 1-iodo-2-nitrobenzene and iodozinc compound 31 showed that Pd(OAc)2 in combination with XPhos in a 1:2 ratio gave a 92% yield of the desired cross-coupling product. In addition, it was found that 1-chloro-2-nitrobenzene was also amenable to cross-coupling in 87% yield. A range of ortho and multiply-substituted nitro-bromobenzenes and pyridines were used as cross-coupling partners with iodozinc compound 31. Yields for nitropyridines ranged between 22% and 29% whilst nitrobenzenes ranged between 41% and 77% yield.
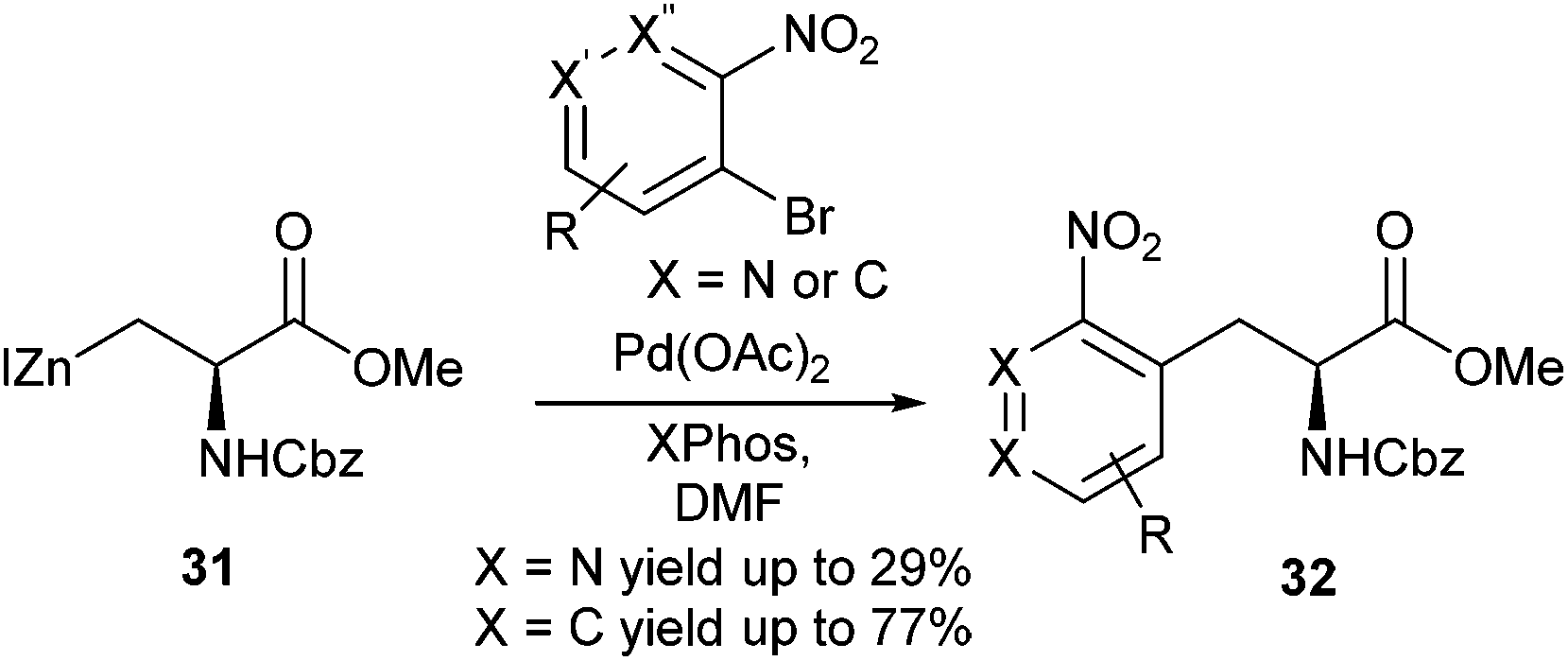 | ||
| Scheme 13 Nitro-aromatic and heteroaromatic protected amino acid derivatives, Tuttle et al.55 | ||
Using these novel protected amino acid derivatives, it was possible to deliver the intended hydroxamic acid targets 35 (Scheme 14) (similar to the species observed in Scheme 9). These heterocyclic motifs were of interest for their potential biological activity and the developed methodology allowed potential access to a library of these hydroxamic acids.
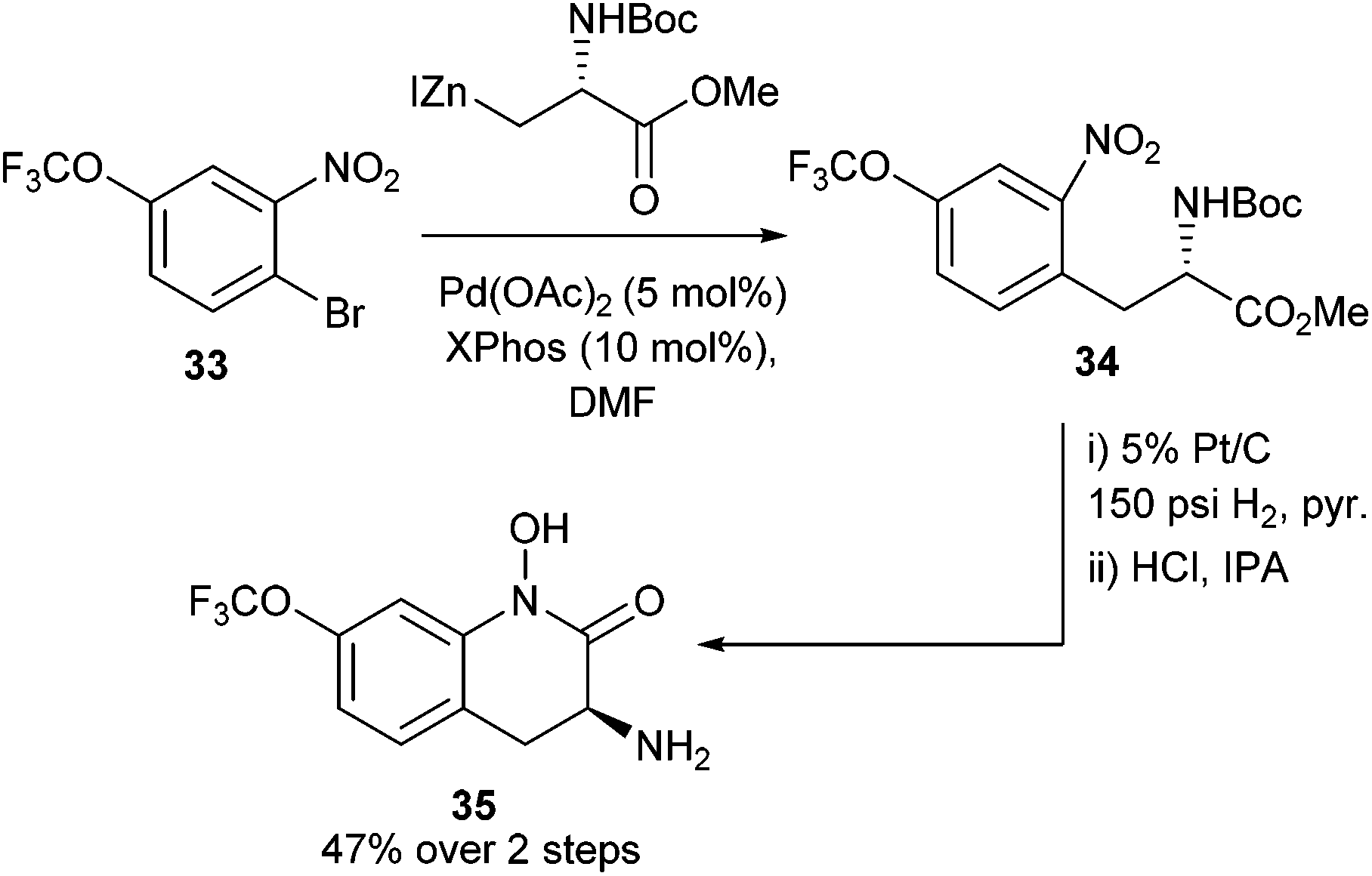 | ||
| Scheme 14 Synthesis of Boc-protected nitro-aromatic-appended amino acids and subsequent cyclisation to yield novel hydroxamic acids by Tuttle et al.55 | ||
In 2014 Usuki et al. reported the improved Negishi cross-coupling of halopyridines with an iodozinc species derived from aspartic acid (Scheme 15).56 Previous work such as that reported by Tuttle et al. (Scheme 13) had shown that yields for pyridine cross-coupling products significantly lagged behind their non-heteroaromatic equivalents.57 After extensive reaction optimisation, a catalytic system of Pd2(dba)3 in combination with a P(2-furyl)3 ligand was shown to be the best for the cross-coupling of halopyridines, with a 90% yield recorded in the reaction between 4-iodopyridine and 36. In moving to bromo-substituted pyridines lower yields were recorded for all substrates, with 2–3- and 4-bromopyridines giving 44%, 33% and 23% yields respectively. Chloropyridines, regardless of halide position on the ring, were found to be ineffectual in all cross-coupling reactions attempted. The Negishi cross-coupling of halopyridines would be utilised in the synthesis of complex pyridinium amino acids (see later section, Scheme 24).
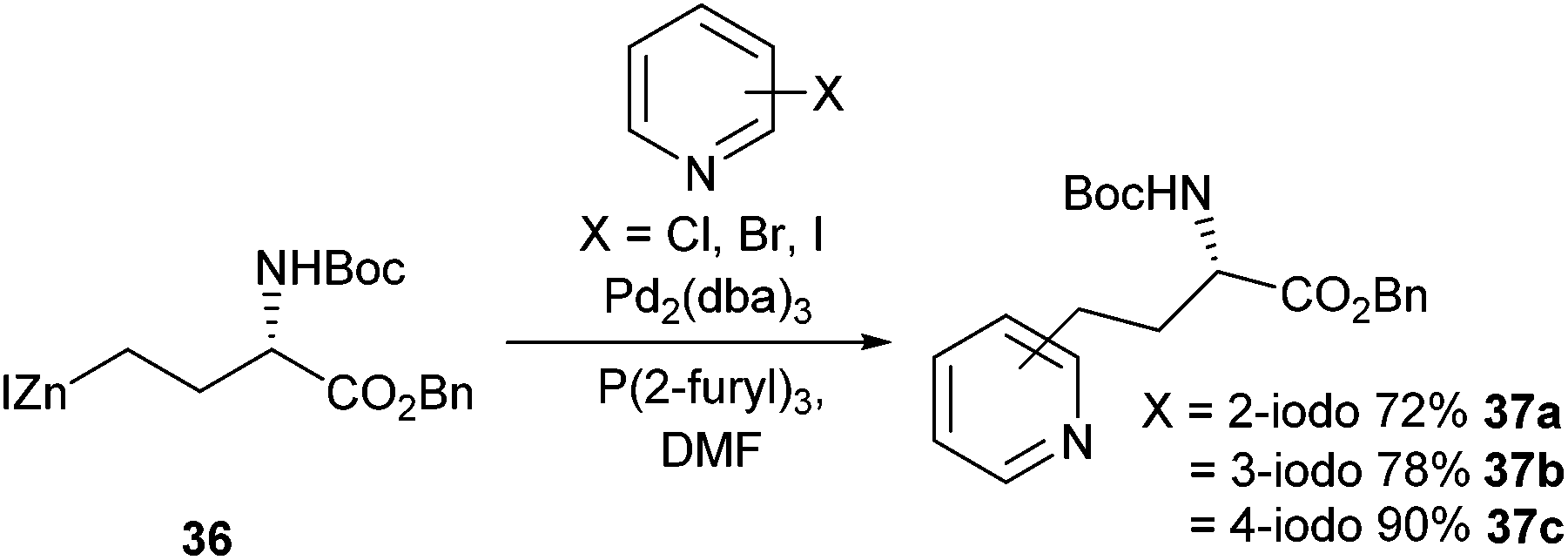 | ||
| Scheme 15 Synthesis of pyridine-containing amino acids via Negishi cross-couplings, Usuki et al.56 | ||
The ability to append furyl and thienyl functionalities onto amino acids was reported by Cobb and co-workers in 2015 (Scheme 16).58 The synthesis of 2-thienyl-alanine was achieved in a moderate 42% yield but this was an improvement over the previously reported, 10% yield.59 Both Boc and Fmoc protecting groups were shown to be tolerated by the cross-coupling conditions. A Boc-protected 2-furyl derivative was subsequently shown to be stable in a (Benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate (PyBOP) mediated peptide coupling.
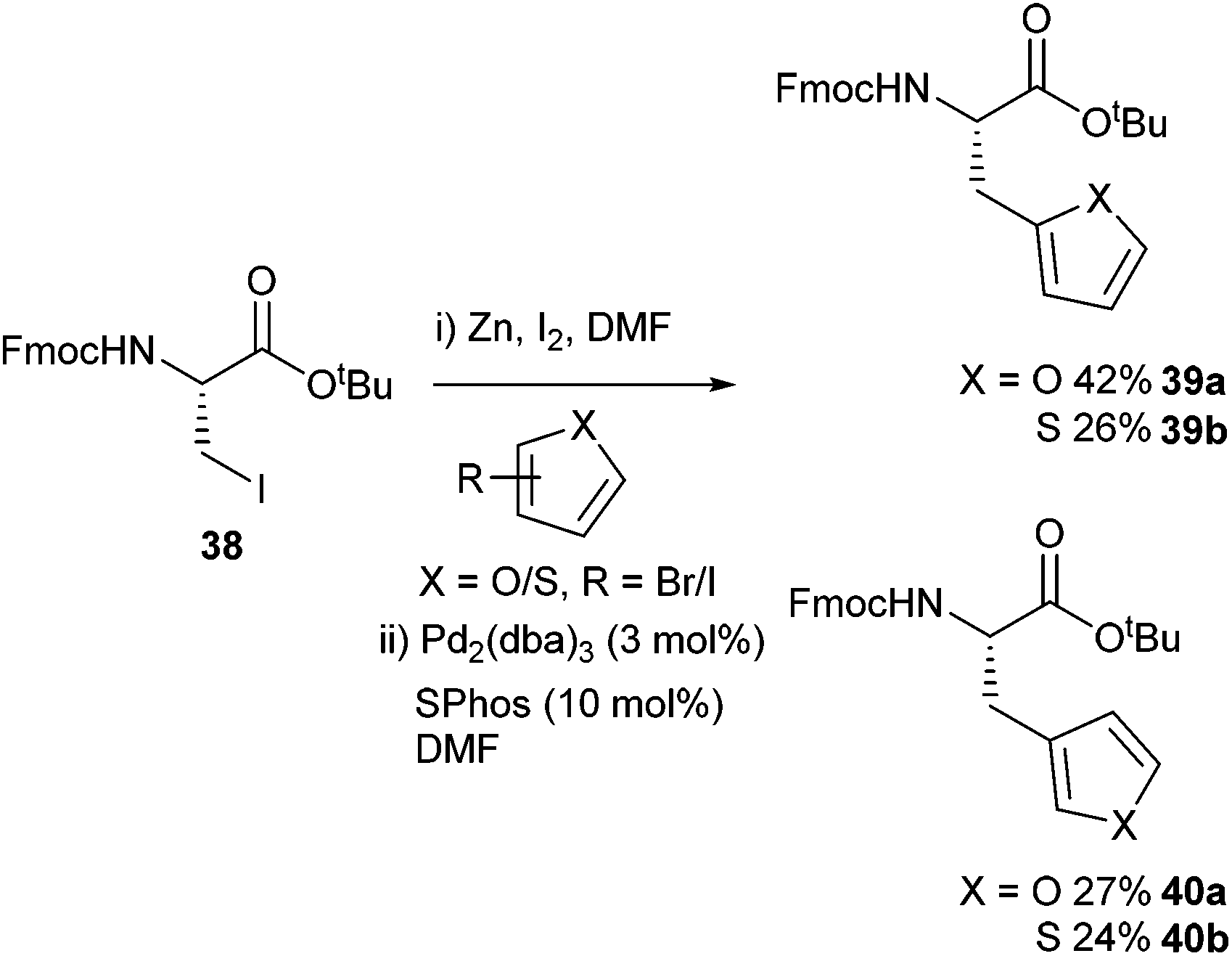 | ||
| Scheme 16 Furyl and thienyl-appended amino acids synthesised using Negishi cross-coupling, Cobb and co-workers.58 | ||
Synthesis of complex amino acids
Beyond aromatic and heteroaromatic examples, the Negishi cross-coupling reaction has been used to prepare a wide range of complex amino acids.The formation of biphenyl-linked amino acids and biphenyl amino acid derivatives can be easily achieved using a cross-coupling approach (Scheme 18).60 The synthesis of the macrocyclic tripeptide K1361 and a non-natural macrocyclic protease inhibitor used this approach.62 Building upon these studies the synthesis of biphenyl linked amino acids was reported where a strategy of coupling iodosubstituted bi-aryls with alkyl zinc reagents was adopted. Negishi cross-coupling was successfully carried out between iodozinc L-serine derivative 2 and a range of mono- and diiodo-substituted bi-phenyls.
The reaction was found to be applicable to both acyl and benzyl protecting groups on the bi-phenyl moiety. However, it was found that a large excess (4 equiv.) of the iodozinc species was required in order to achieve a satisfactory yield of 65% of the biaryl derivative 45 (Scheme 18).
Glycoproteins have been shown to be critical to cell adhesion,63,64 differentiation65 and growth, and they have also been used as new targets for therapeutics and diagnostics.66 Ousmer et al. utilised a Negishi cross-coupling to form a p-(C-glucopyranosyl)phenylalanine derivative (Scheme 17).67 An iodo-substituted phenylalanine derivative was successfully coupled to a glucal 41 in an excellent 90% yield. This was then further elaborated by stereo-selective hydroboration to yield the desired glucose modified phenylalanine 43 in a 37% overall yield.
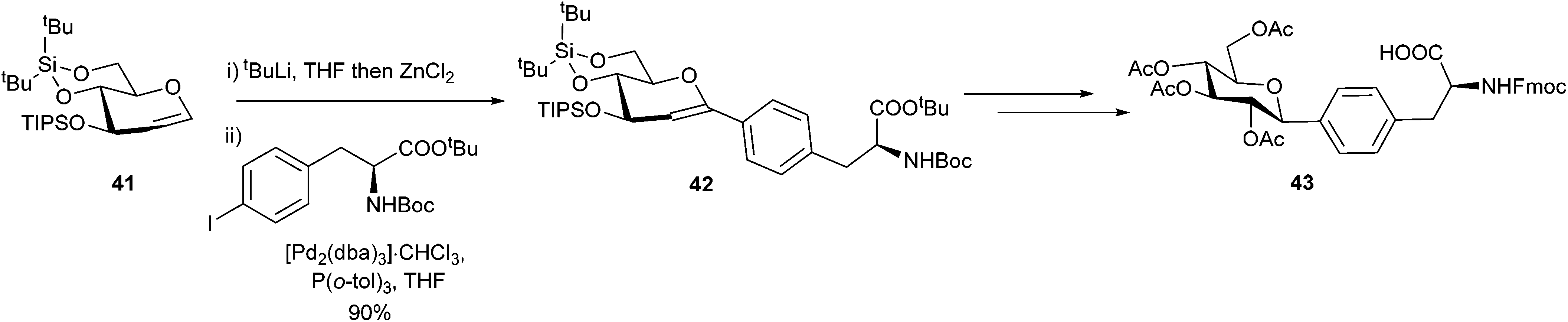 | ||
| Scheme 17 Synthesis of a p-(C-glucopyranosyl)phenylalanine derivative. | ||
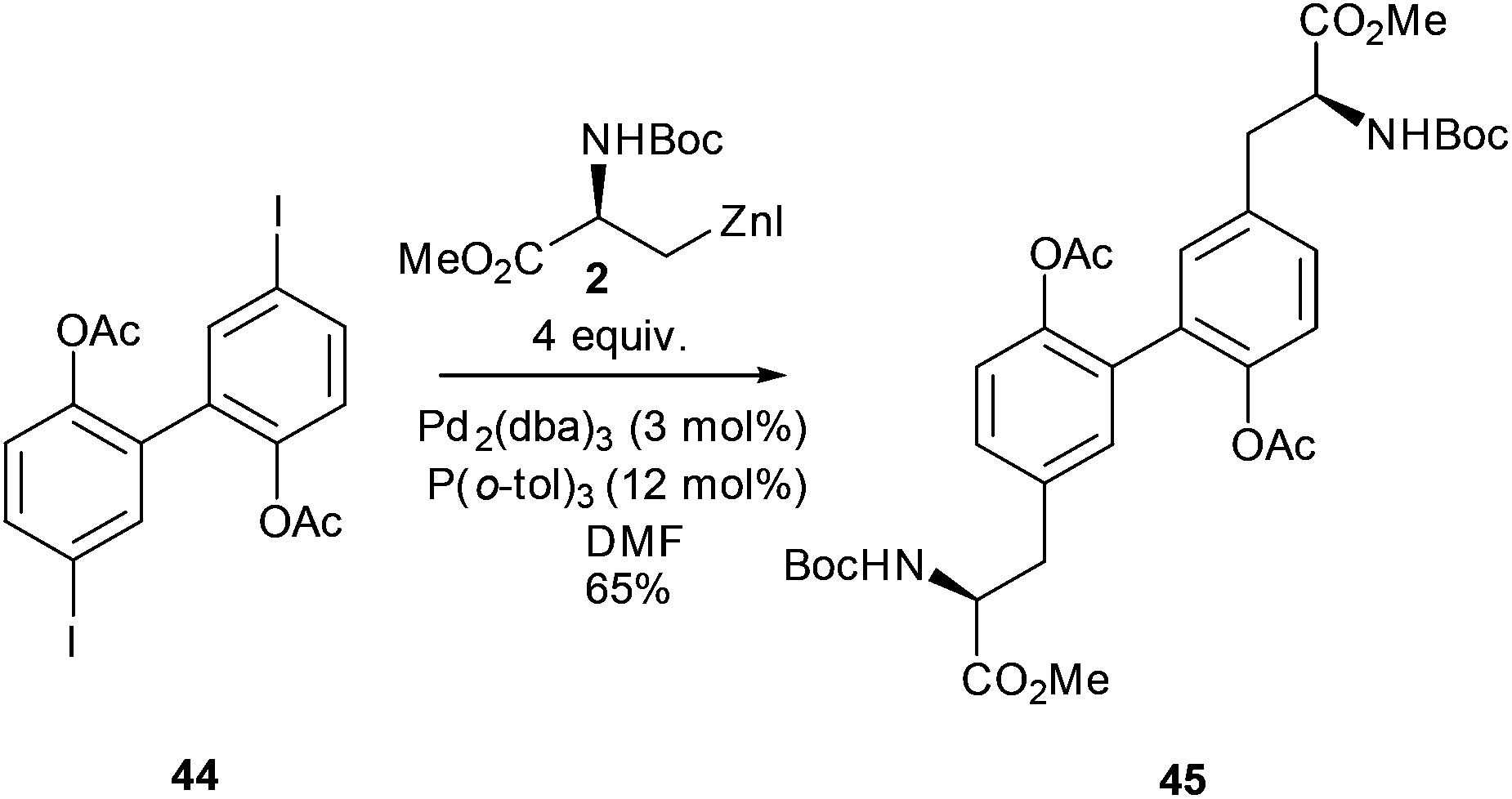 | ||
| Scheme 18 Synthesis of a bi-aryl, bis(amino acid), Jackson and co-workers.60 | ||
Negishi and Suzuki–Miyaura cross-couplings were shown to be amenable to the preparation of imidazoles derived from amino acids (Scheme 19).68 Imidazoles derived from (S)-Ala, (S)-Leu and (S)-Phe 46 protected with ethoxymethyl groups gave substrates 47 which were found to readily undergo Negishi cross-couplings to form corresponding bi-aryl systems. The bi-aryl protected amines 48a–c were successfully obtained in yields ranging from 34% to 88% (Scheme 19).
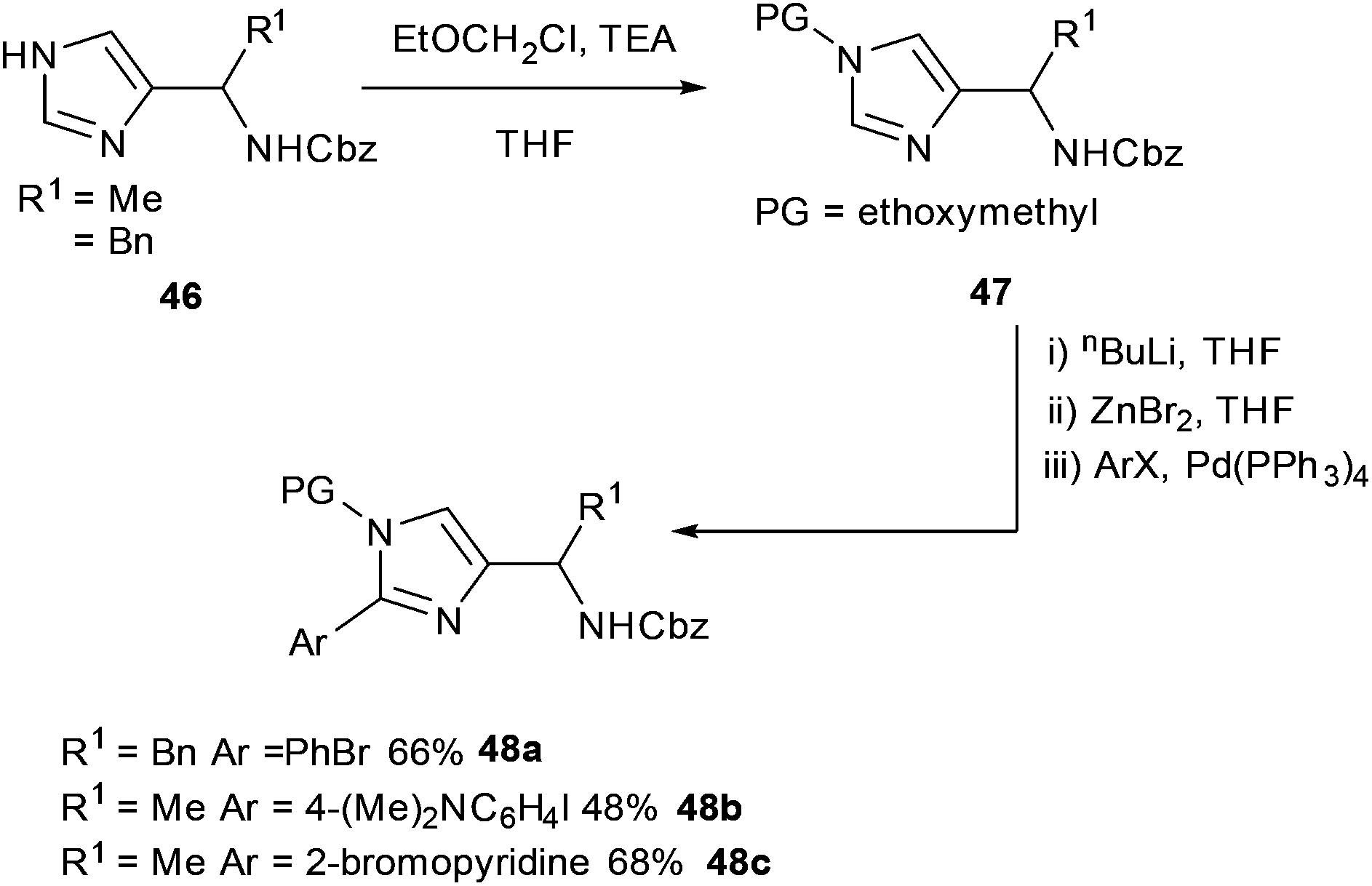 | ||
| Scheme 19 Derivitisation of imidazoles from amino acids by Negishi cross-coupling, Marek et al.68 | ||
Jackson and co-workers would revisit the synthesis of the macrocyclic peptide K1361 and expand their scope to the cyclic peptide OF4949-III 52 in 2009.69 Building on their earlier synthesis of K13 and their reported Negishi-based methodology for bi-aryl linked amino acids,60 Jackson et al. reported an intramolecular Negishi strategy to afford K13 and a Negishi cross-coupling key step synthesis of OF4949-III (Scheme 20). Utilising this approach OF4949-III 52 was synthesised in 12 steps with an overall yield of 20%.
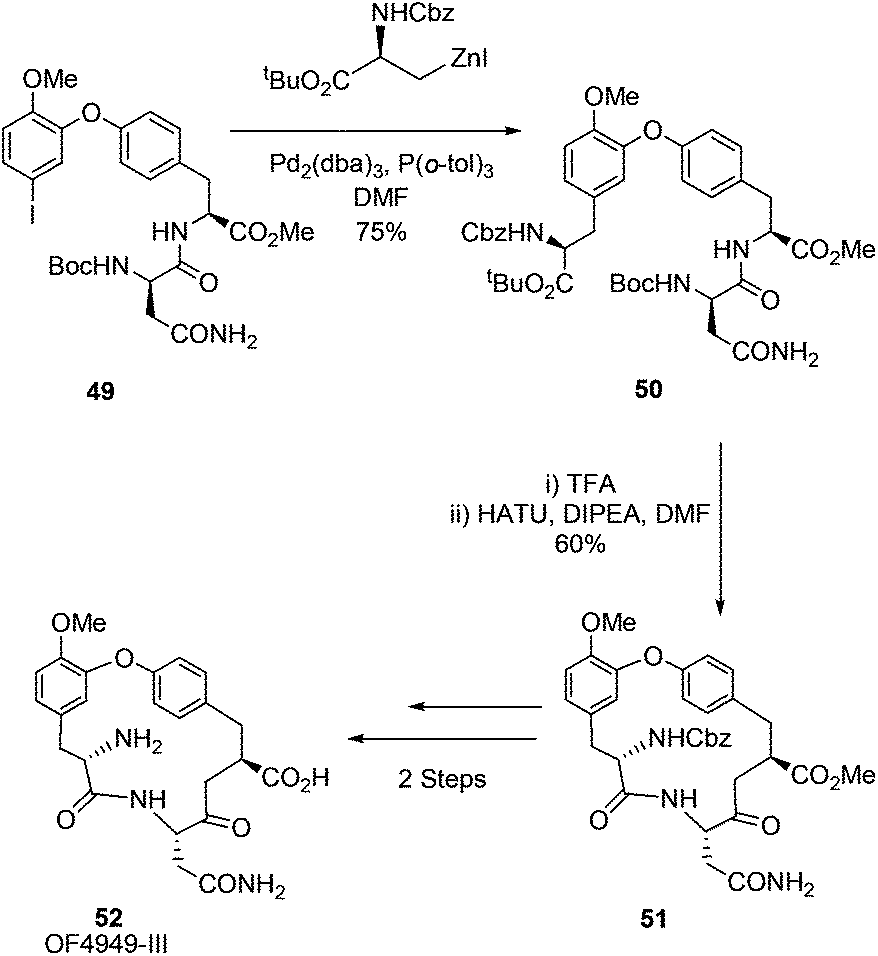 | ||
| Scheme 20 Synthesis of cyclic peptide OF4949-III, Jackson and co-workers.69 | ||
Other natural products have also been successfully synthesised using Negishi cross-couplings of amino acid building blocks as a key synthetic step. The Kapakahine family of natural products has shown promising preliminary biological activity against P388 murine leukemia cells and thus accessing these compounds was of significant interest.70 The synthesis of Kapakahine E and F, by Espejo and Rainier utilised a Negishi cross-coupling to form a key intermediate 54 which was subsequently taken through two end game synthetic routes to yield the two natural products (Scheme 21).70
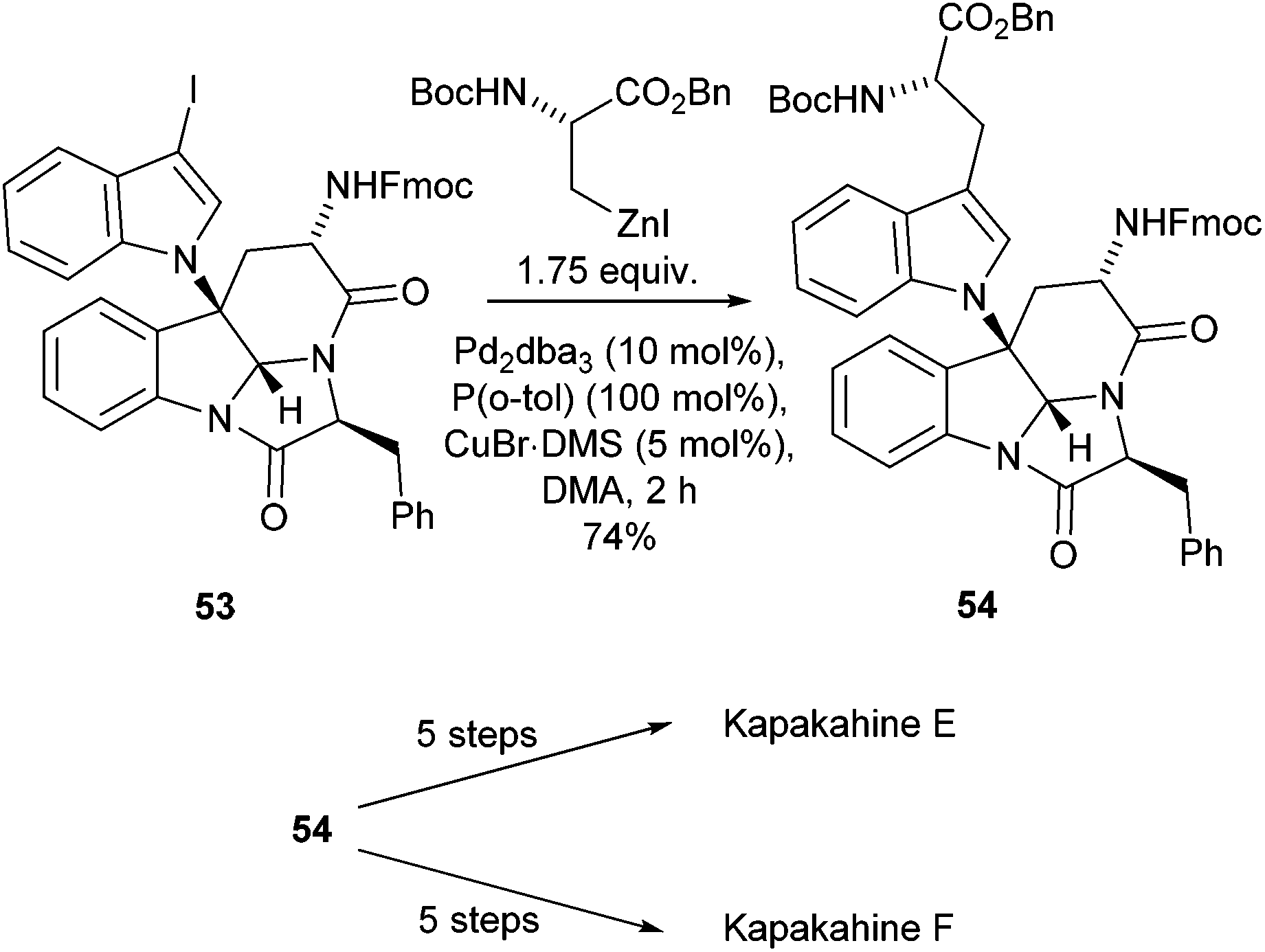 | ||
| Scheme 21 Synthesis of Kapakahine E and F, Espejo and Rainier.70 | ||
Negishi cross-couplings that can utilise triflates in the place of the halide component have been widely utilised in mainstream organic synthesis.71,72 Within the field of peptide chemistry it was reported that a PdCl2(PPh3)2 mediated cross-coupling between iodozinc reagents and cycloalkenyl triflates could be carried out, but the yields obtained were only moderate, ranging between 41% and 46%.73 The Negishi cross-coupling of iodozinc compounds with cycloalkenyl triflate derivatives was also reported by Jackson and co-workers in 2012 (Scheme 22).74 In this study, the addition of a phosphine ligand (SPhos), in combination with a Pd2(dba)3 catalyst, gives rise to an increase in the yield of the cross-coupling reaction product to 70% for the cyclopentenyl and cyclohexenyl derivatives 55a/b.
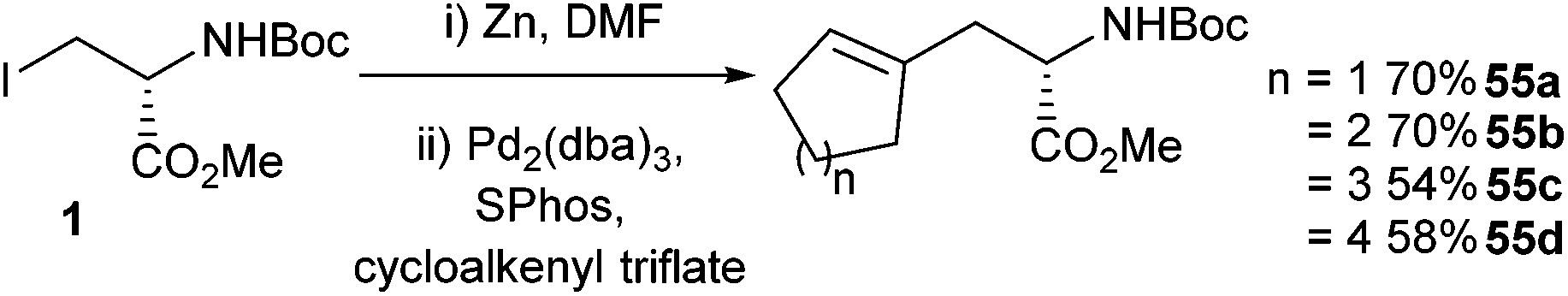 | ||
| Scheme 22 Cycloalkenyl-protected amino acid derivatives, Jackson and co-workers.74 | ||
Yields for cycloheptenyl and cyclooctenyl products 55c/d, however, were moderate at 54% and 58%, respectively. This led the authors to propose that the addition of lithium chloride could be advantageous. Lithium chloride had been shown previously to increase the yields in Stille reactions, and can increase the reactivity of organozinc halides.75 Indeed, the addition of 1.8 equivalents of LiCl to the Negishi cross-coupling reaction mixture led to increased yields for the cycloheptenyl and cyclooctenyl derivatives 55c/d, boosting yields to 62% and 63%, respectively.
In addition to the Negishi methodology developed it was shown that the cross-coupled product 55a could react with HF pyridine to furnish the fluoro compound 56 in 30% isolated yield (Scheme 23). This compound had been reported earlier as a building block for an enzyme inhibitor.76 The previous synthesis of compound 56 had been relatively low yielding and had required a chiral resolution step.77 The cross-coupling methodology therefore offered a simpler and more efficient synthesis of these fluorinated motifs.
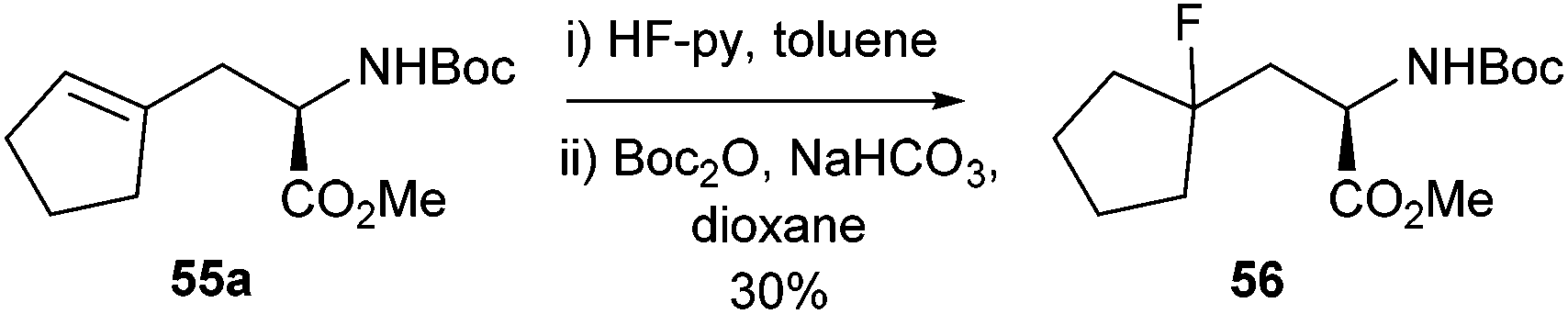 | ||
| Scheme 23 Fluorination of cycloalkenyl-protected amino acid derivatives. | ||
The Negishi cross-coupling of pyridines was extended to bis-halo pyridines by Usuki and co-workers (Scheme 24).78 Utilising a di-bromo-substituted pyridine 57 a double Negishi and Sonogashira cross-coupling was employed to garner key intermediate 58, which was further elaborated to yield desmosine 59. In the same report, a deuterated version of desmosine was also successfully synthesised following the same cross-coupling strategy. Desmosine is a biomarker for degradation of elastin-containing tissues, which is a symptom of diseases such as atherosclerosis, aortic aneurysm, cystic fibrosis and chronic obstructive pulmonary disease (COPD).79–81 The synthesis of desmosine therefore is of importance for drug discovery and the rapid diagnosis of diseases such as COPD.
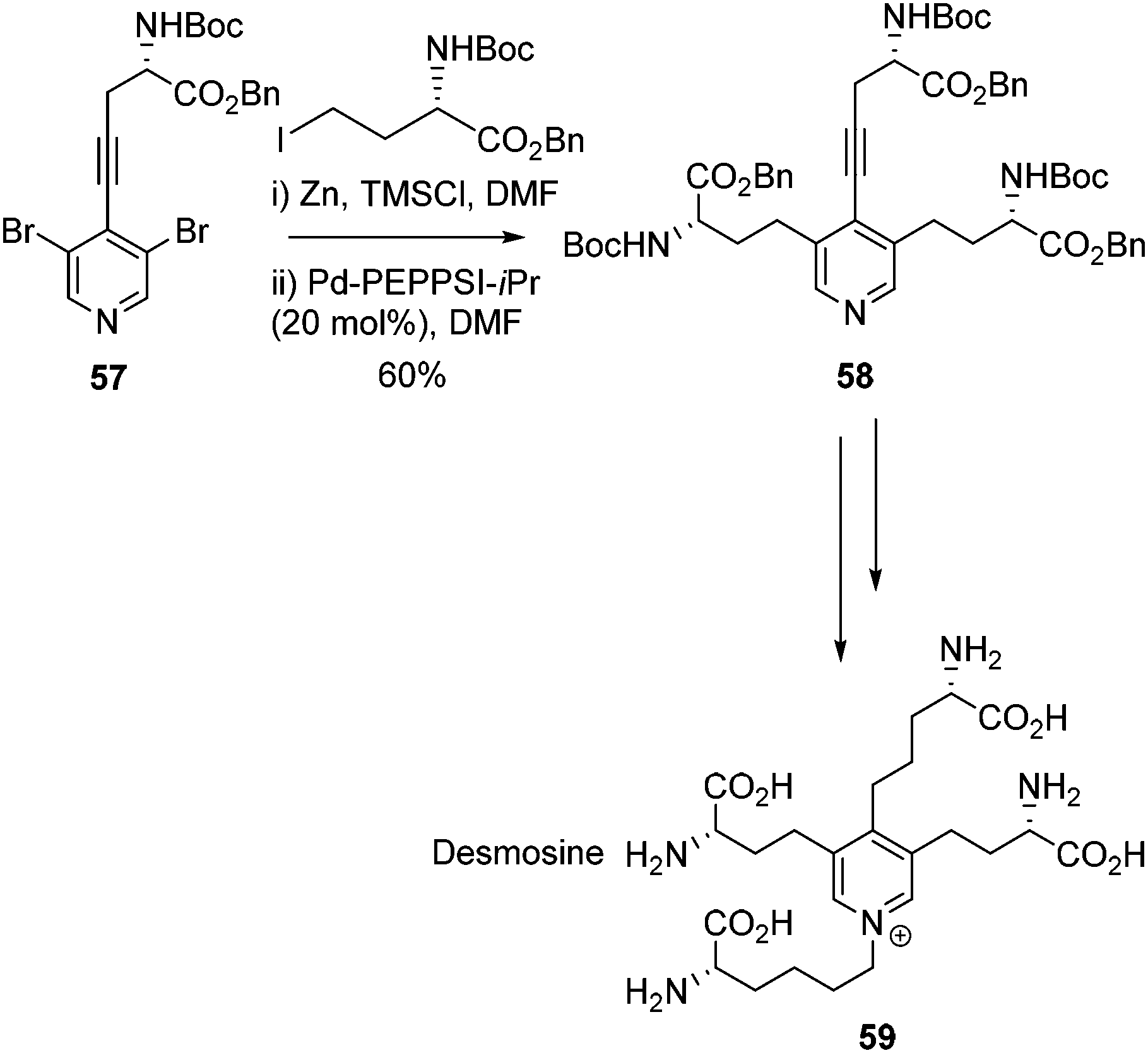 | ||
| Scheme 24 Synthesis of Desmosine via a Sonogashira, double Negishi cross coupling strategy. | ||
In 2017 Usuki and co-workers elaborted their synthesis of Desmosine to build it into a cyclic peptide (Scheme 25).82 This work was driven by the fact that the proposed stuctures of elastin suggest cross-linkage through Desmosine.83,84 Through the use of a Desmosine analogue the cross-linking motif within elastin was hoped to be probed. Usuki and co-workers utilised the same general strategy to build the Deomsine core as they had previously published.78
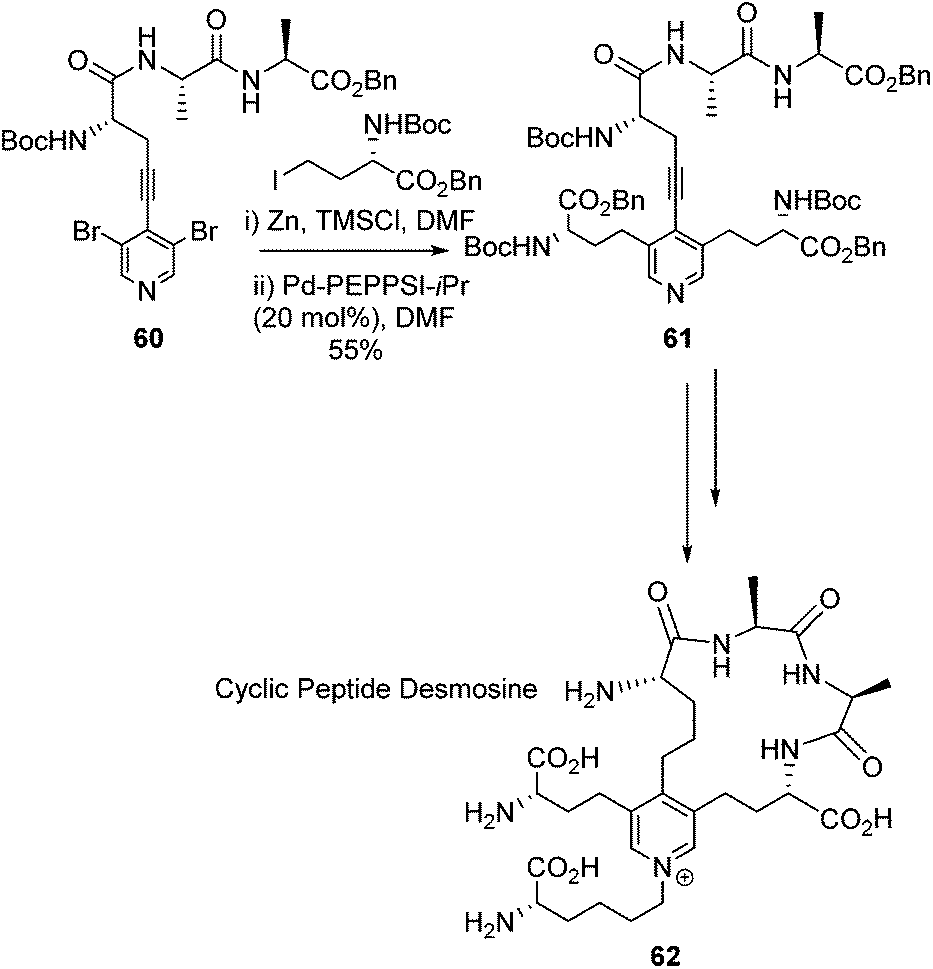 | ||
| Scheme 25 Synthesis of a cyclic peptide derivative of Desmosine, Usuki and co-workers.82 | ||
A Sonogashira cross-coupling afforded the intermediate 60 which was elaborated with a double Negishi coupling to afford intermediate 61. This intermediate was then successfully cyclised thtough an amide coupling reaction which led to the target compound 62. The overall synthesis gave the desired cyclic desmosine derivative in a 0.8% overall yield from L-alanine.
In 2017 the total synthesis of Nannocystin A 64 was reported by Liu et al.85 Within this report a Negishi cross-coupling of an iodo serine derviative was employed to build the key fragment 63 (Scheme 26). The final natural product was delivered with a longest linear sequence of 11 steps in a 5.3% overall yield. Nannocystin A 64 has been found to possess potent biological activity. It has been shown to be an inhibitor of elongation factor 1 and inhibits cancer cell growth.86 The Negishi cross-coupling reaction to form a novel amino acid derivative was vital to access this potent natural product synthetically.
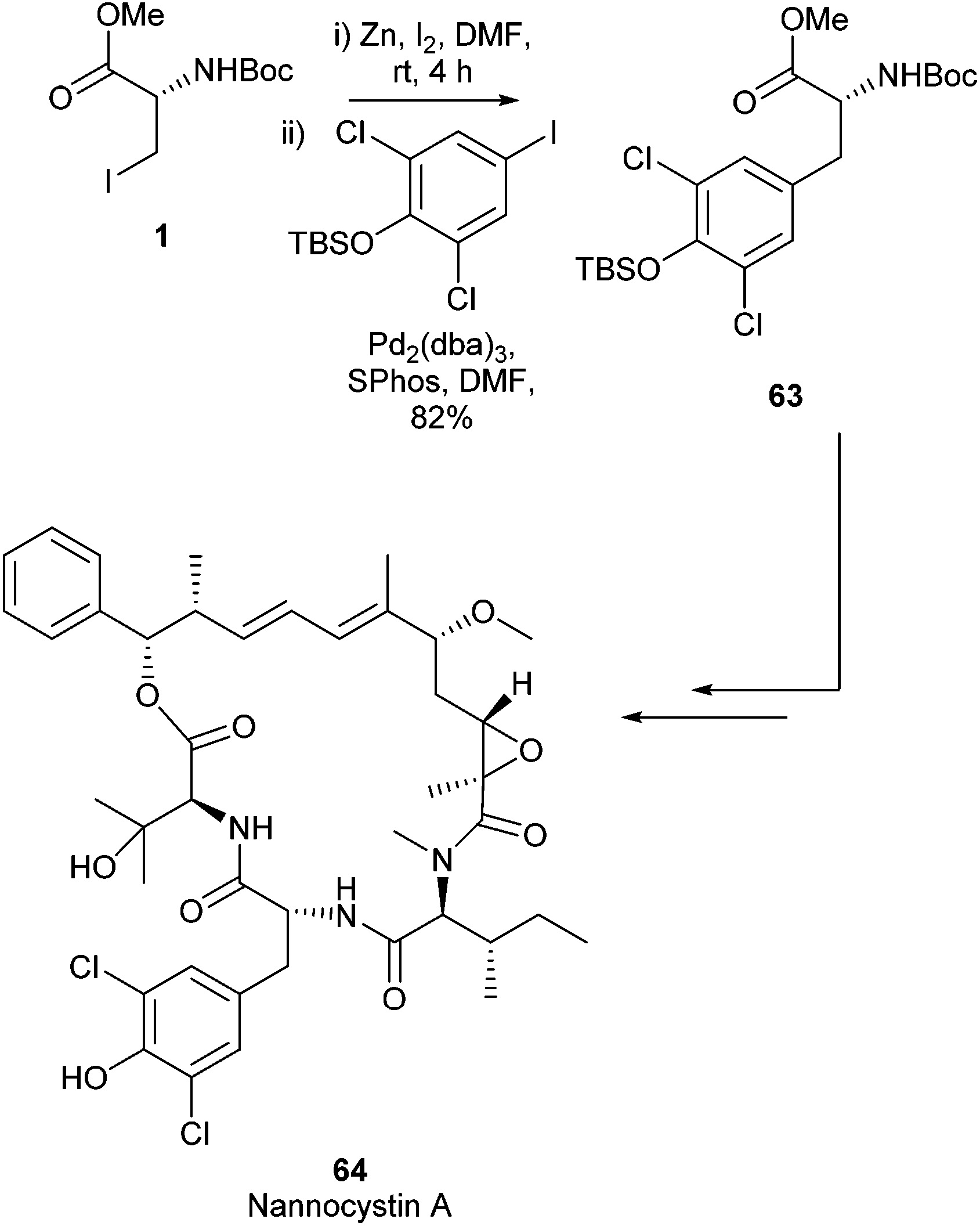 | ||
| Scheme 26 Total synthesis of nannocystin A, Liu et al.85 | ||
Mechanistic studies
Several mechanistic studies have been undertaken to try and gain fundamental knowledge of the reactivity of alkyl zinc amino acid derivatives. Using a range of analytical techniques, the species responsible for catalysis and mechanistic pathways have been probed. From these studies a greater understanding of the workings of Negishi cross-couplings of iodo amino acids has been established.One of the major problems in the synthesis of β-amino acids using Negishi cross-coupling is that the iodozinc cross-coupling partner (derived from species such as aspartic or glutamic acid) is intrinsically susceptible to β-elimination to form an alkene. In order to understand these problems, NMR spectroscopy was utilised to probe the decomposition of β-amidozinc reagents. NMR kinetic studies showed that the rates of β-elimination in both THF-d8 and DMF-d7 were first order and pointed towards a unimolecular rate-limiting step. In addition, it was observed that the rate constant in the case of DMF was four times smaller than that for THF. Therefore the elimination process is significantly perturbed in DMF. Calculation of the activation parameters led to the conclusion that the elimination process was syn in nature, invoking an intramolecular coordination between the carbamate and the zinc centre (Scheme 27). DMF therefore is believed to compete with the carbamate for zinc, thus slowing the rate of elimination.
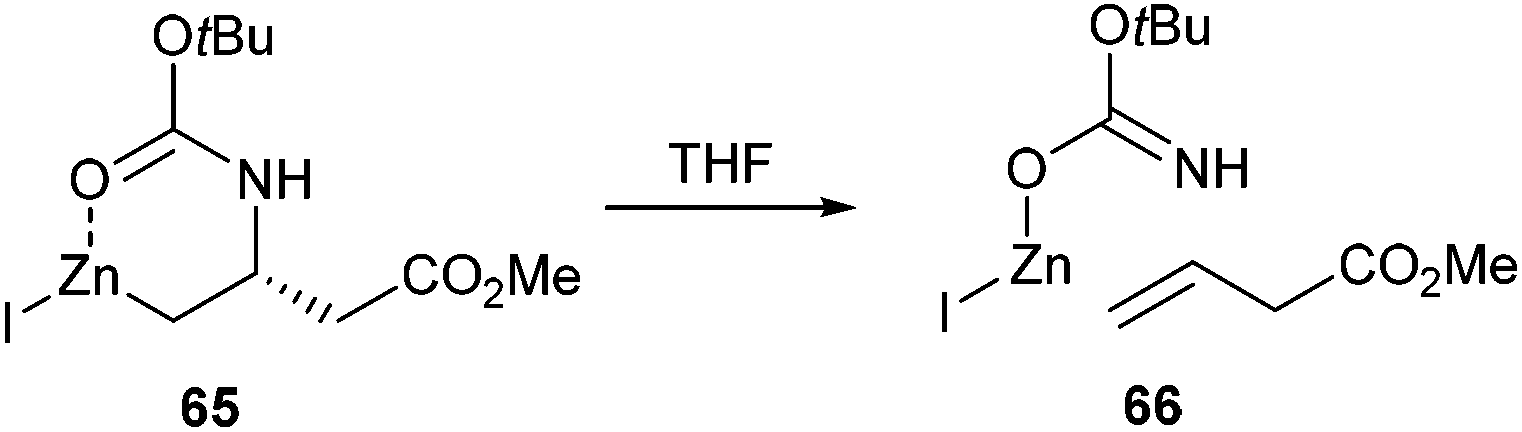 | ||
| Scheme 27 Syn elimination of Boc protected amidozinc reagents. | ||
In 2003 it was reported that a β-amidozinc reagent was more resistant to elimination when a better leaving group was employed.87 Jackson and Rilatt further studied this kinetic stability and reactivity of β-amino alkyl zinc iodides in 2008.88 They found that the N-protecting group on the zinc iodide was critical in determining the rate of β-elimination. Boc protecting groups were found to increase the rate of elimination, and from this it was theorised that the carbonyl group (within the carbamate) was competing with the ester for coordination with zinc, leading to increased elimination. Replacing the Boc group with a TFA group perturbed the rate of elimination. The lower Lewis basicity of TFA on its carbonyl group helps prevent coordination to the zinc, and thus elimination. On top of this, it was observed that upon changing the Boc group for TFA, the rate constant changed from being first order to second order, indicating that the TFA elimination pathway follows a different mechanism from the Boc elimination. In the same report the reactivity of the β-amino alkyl zinc iodides towards Negishi cross-coupling was also studied. It was found that the N-protecting group made little difference to the rate of cross-coupling with the main factor being the proximity of the ester from the Zn site. It was concluded that a TFA N-protecting group offers much greater stability toward β-elimination whilst not affecting the rate of Negishi cross-coupling.
Negishi cross-coupling was found to be applicable to aqueous environments. It was reported that the presence of tetramethylethylenediamine (TMEDA) allows the Negishi cross-coupling of phenylalanine derivatives to be successfully carried out in the presence of water (Scheme 28).89 Being able to carry out Negishi reactions in water is of significant interest to peptide chemists, as it allows the post-translational modification of peptides. It was found that the TMEDA helps prevent protonation of the alkylzinc iodides in water. It was proposed that the stabilisation occurs due to the TMEDA inducing ionisation in the zinc-iodine bond. This hypothesis was supported by evidence from tandum mass spectometery, infrared multiphoton dissociation and DFT calculations. Conditions for the successful synthesis of alkylzinc iodides and subsequent Negishi coupling in water were probed, and it was found that the use of nano zinc in combination with TMEDA and a PdCl2(AmPhos)2 gave good yields ranging between 28% and 73% of phenylalanine derivatives.
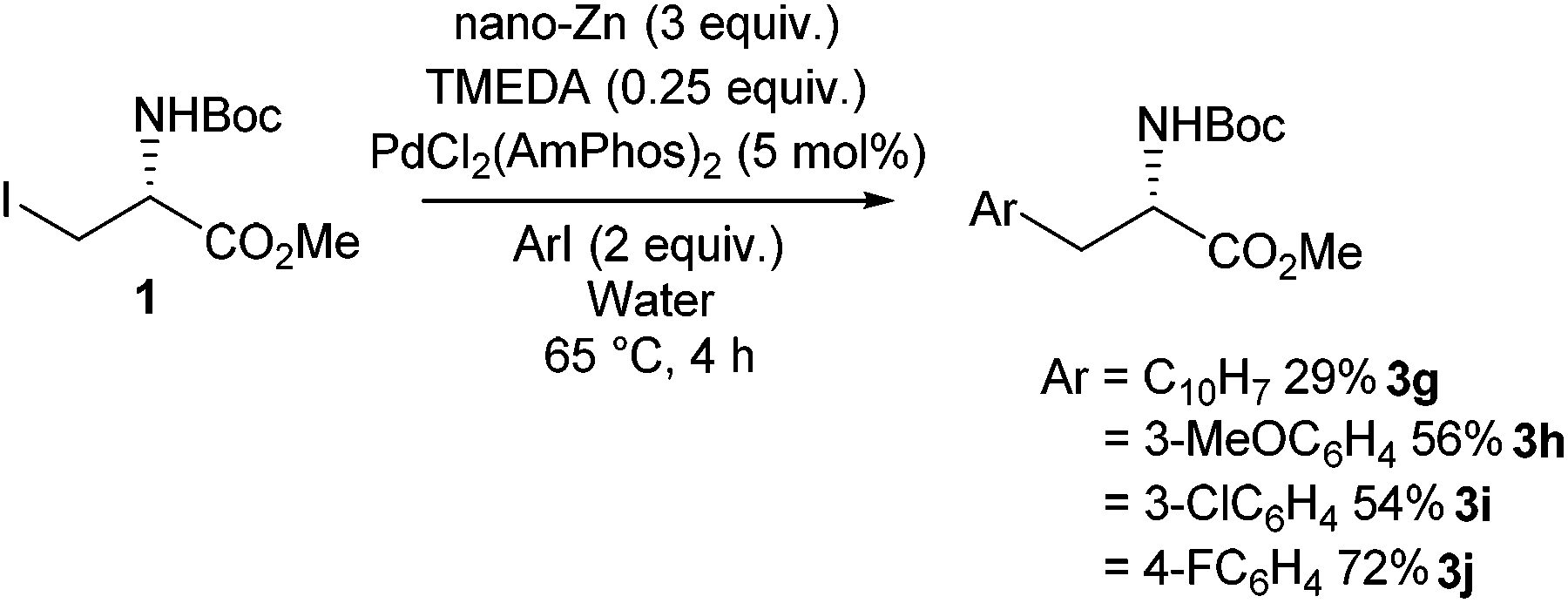 | ||
| Scheme 28 Negishi cross coupling in water of an iodo serine derivative, Jackson and co-workers.89 | ||
Conclusions
The Negishi cross-coupling reaction has been shown to be a highly versatile reaction allowing an expedient route to the synthesis of many unnatural amino acids. Since the pioneering work of Jackson and co-workers advances in available reagents (e.g. new ligands) has led to an increase in substrate tolerance and product yields. In addition mechanistic studies have helped researchers to understand the requirements for successful cross-couplings across a range of substrates and this has in turn allowed access to more novel unnatural amino acids than ever before. The ability to prepare novel aromatic, heteroaromatic and complex amino acids opens up a myriad of new potential applications for these motifs. For example, as highlighted in this review, unnatural amino acids accessed via Negishi cross-couplings have been exploited in the fields of natural product synthesis, drug discovery, materials/polymer chemistry and catalysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We wish to acknowledge the Biotechnology and Biological Sciences Research Council [BB/P003656/1] for financial support.Notes and references
- A. O. King, N. Okukado and E.-I. Negishi, J. Chem. Soc., Chem. Commun., 1977, 19, 683–684 RSC.
- D. Haas, J. M. Hammann, R. Greiner and P. Knochel, ACS Catal., 2016, 6, 1540–1552 CrossRef CAS.
- V. B. Phapale and D. J. Cardenas, Chem. Soc. Rev., 2009, 38, 1598–1607 RSC.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- M. C. Kozlowski, B. J. Morgan and E. C. Linton, Chem. Soc. Rev., 2009, 38, 3193–3207 RSC.
- E. Negishi, Acc. Chem. Res., 1982, 15, 340–348 CrossRef CAS.
- X. Mu, Y. Shibata, Y. Makida and G. C. Fu, Angew. Chem., Int. Ed., 2017, 56, 5821–5824 CrossRef CAS PubMed.
- Y. Liang and G. C. Fu, Angew. Chem., Int. Ed., 2015, 54, 9047–9051 CrossRef CAS PubMed.
- C. Dai and G. C. Fu, J. Am. Chem. Soc., 2001, 123, 2719–2724 CrossRef CAS PubMed.
- S. Son and G. C. Fu, J. Am. Chem. Soc., 2008, 130, 2756–2757 CrossRef CAS PubMed.
- G. F. Short, S. Y. Golovine and S. M. Hecht, Biochemistry, 1999, 38, 8808–8819 CrossRef CAS PubMed.
- S. Li, S. Millward and R. Roberts, J. Am. Chem. Soc., 2002, 124, 9972–9973 CrossRef CAS PubMed.
- S. E. Cellitti, D. H. Jones, L. Lagpacan, X. Hao, Q. Zhang, H. Hu, S. M. Brittain, A. Brinker, J. Caldwell, B. Bursulaya, G. Spraggon, A. Brock, Y. Ryu, T. Uno, P. G. Schultz and B. H. Geierstanger, J. Am. Chem. Soc., 2008, 130, 9268–9281 CrossRef CAS PubMed.
- W. Liu, A. Brock, S. Chen, S. Chen and P. G. Schultz, Nat. Methods, 2007, 4, 239–244 CrossRef CAS PubMed.
- R. P. Hicks, J. J. Abercrombie, R. K. Wong and K. P. Leung, Bioorg. Med. Chem., 2013, 21, 205–214 CrossRef CAS PubMed.
- H. Waldmann, J. Org. Chem., 1988, 53, 6133–6136 CrossRef CAS.
- C. Fu, M. Wenzel, E. Treutlein, K. Harms and E. Meggers, Inorg. Chem., 2012, 51, 10004–10011 CrossRef CAS PubMed.
- P. Singh, K. Samanta, S. K. Das and G. Panda, Org. Biomol. Chem., 2014, 12, 6297–6339 CAS.
- C. W. Phoon and C. Abell, Tetrahedron Lett., 1998, 39, 2655–2658 CrossRef CAS.
- Z. G. Hajos and D. R. Parrish, J. Org. Chem., 1974, 39, 1615–1621 CrossRef CAS.
- G. Desimoni, G. Faita and P. Quadrelli, Chem. Rev., 2003, 103, 3119–3154 CrossRef CAS PubMed.
- M. Nakamura, A. Hirai and E. Nakamura, J. Am. Chem. Soc., 1996, 118, 8489–8490 CrossRef CAS.
- A. Mori, H. Abet and S. Inoue, Appl. Organomet. Chem., 1995, 9, 189–197 CrossRef CAS.
- W. D. Brittain, B. R. Buckley and J. S. Fossey, Chem. Commun., 2015, 51, 17217–17220 RSC.
- I. Rilatt, L. Caggiano and R. F. W. Jackson, Synlett, 2005, 2701–2719 CAS.
- N. Colgin, T. Flinn and S. L. Cobb, Org. Biomol. Chem., 2011, 9, 1864–1870 CAS.
- S. Fleming and R. V. Ulijn, Chem. Soc. Rev., 2014, 43, 8150–8177 RSC.
- J. Li, X. Du, S. Hashim, A. Shy and B. Xu, J. Am. Chem. Soc., 2017, 139, 71–74 CrossRef CAS PubMed.
- P. Jenner, Nat. Rev. Neurosci., 2008, 9, 665–677 CrossRef CAS PubMed.
- R. F. W. Jackson, M. J. Wythes and A. Wood, Tetrahedron Lett., 1989, 30, 5941–5944 CrossRef CAS.
- C. S. Dexter and R. F. W. Jackson, Chem. Commun., 1998, 1, 75–76 RSC.
- M. Kruppa, G. Imperato and B. König, Tetrahedron, 2006, 62, 1360–1364 CrossRef CAS.
- H. E. Bartrum, H. Adams, L. Caggiano and R. F. W. Jackson, Tetrahedron, 2008, 64, 3701–3712 CrossRef CAS.
- C. S. Dexter, R. F. W. Jackson and J. Elliott, J. Org. Chem., 1999, 64, 7579–7585 CrossRef CAS.
- C. S. Dexter, C. Hunter, R. F. Jackson and J. Elliott, J. Org. Chem., 2000, 65, 7417–7421 CrossRef CAS PubMed.
- C. L. Oswald, T. Carrillo-Marquez, L. Caggiano and R. F. W. Jackson, Tetrahedron, 2008, 64, 681–687 CrossRef CAS.
- B. G. Szczepankiewicz, G. Liu, P. J. Hajduk, C. Abad-Zapatero, Z. Pei, Z. Xin, T. H. Lubben, J. M. Trevillyan, M. A. Stashko, S. J. Ballaron, H. Liang, F. Huang, C. W. Hutchins, S. W. Fesik and M. R. Jirousek, J. Am. Chem. Soc., 2003, 125, 4087–4096 CrossRef CAS PubMed.
- S. Kotha, V. R. Shah, S. Halder, R. Vinodkumar and K. Lahiri, Amino Acids, 2007, 32, 387–394 CrossRef CAS PubMed.
- T. E. Barder, S. D. Walker, J. R. Martinelli and S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 4685–4696 CrossRef CAS PubMed.
- R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461–1473 CrossRef CAS PubMed.
- A. J. Ross, H. L. Lang and R. F. Jackson, J. Org. Chem., 2010, 75, 245–248 CrossRef CAS PubMed.
- I. Rilatt and R. F. W. Jackson, J. Org. Chem., 2008, 73, 8694–8704 CrossRef CAS PubMed.
- C. M. L. Goddard, A. R. Massah and R. F. W. Jackson, Tetrahedron, 2010, 66, 9175–9181 CrossRef CAS.
- A. L. Featherston and S. J. Miller, Bioorg. Med. Chem., 2016, 24, 4871–4874 CrossRef PubMed.
- N. C. Yoder and K. Kumar, Chem. Soc. Rev., 2002, 31, 335–341 RSC.
- W. R. Pitt, D. M. Parry, B. G. Perry and C. R. Groom, Eur. J. Med. Chem., 2009, 52, 2952–2963 CrossRef CAS PubMed.
- T. J. Ritchie, S. J. F. Macdonald, S. Peace, S. D. Pickett and C. N. Luscombe, MedChemCommun, 2012, 3, 1062–1069 RSC.
- P. J. Hajduk and J. Greer, Nat. Rev. Drug Discovery, 2007, 6, 211–219 CrossRef CAS PubMed.
- D. C. Rees, M. Congreve, C. W. Murray and R. Carr, Nat. Rev. Drug Discovery, 2004, 3, 660–672 CrossRef CAS PubMed.
- M. Congreve, G. Chessari, D. Tisi and A. J. Woodhead, Eur. J. Med. Chem., 2008, 51, 3661–3680 CrossRef CAS PubMed.
- S. B. Kalindjian, I. M. Buck, J. M. R. Davies, D. J. Dunstone, M. L. Hudson, C. M. R. Low, I. M. McDonald, M. J. Pether, K. I. M. Steel, M. J. Tozer and J. G. Vinter, Eur. J. Med. Chem., 1996, 39, 1806–1815 CrossRef CAS PubMed.
- J. J. Nestor, B. L. Horner, T. L. Ho, G. H. Jones, G. I. McRae and B. H. Vickery, Eur. J. Med. Chem., 1984, 27, 320–325 CrossRef CAS.
- D. S. Agarwal, H. S. Anantaraju, D. Sriram, P. Yogeeswari, S. H. Nanjegowda, P. Mallu and R. Sakhuja, Steroids, 2016, 107, 87–97 CrossRef CAS PubMed.
- M. Suhartono, A. E. Schneider, G. Dürner and M. W. Göbel, Synthesis, 2010, 293–303 CAS.
- J. B. Tuttle, J. M. Azzarelli, B. M. Bechle, A. B. Dounay, E. Evrard, X. Gan, S. Ghosh, J. Henderson, J.-Y. Kim, V. D. Parikh and P. R. Verhoest, Tetrahedron Lett., 2011, 52, 5211–5213 CrossRef CAS.
- T. Usuki, H. Yanuma, T. Hayashi, H. Yamada, N. Suzuki and Y. Masuyama, J. Heterocycl. Chem., 2014, 51, 269–273 CrossRef CAS.
- J. B. Tuttle, J. M. Azzarelli, B. M. Bechle, A. B. Dounay, E. Evrard, X. M. Gan, S. Ghosh, J. Henderson, J. Y. Kim, V. D. Parikh and P. R. Verhoest, Tetrahedron Lett., 2011, 52, 5211–5213 CrossRef CAS.
- A. S. Hudson, L. Caron, N. Colgin and S. L. Cobb, Amino Acids, 2015, 47, 779–785 CrossRef CAS PubMed.
- R. F. W. Jackson, M. J. Wythes and A. Wood, Tetrahedron Lett., 1989, 30, 5941–5944 CrossRef CAS.
- E. Moreno, L. A. Nolasco, L. Caggiano and R. F. Jackson, Org. Biomol. Chem., 2006, 4, 3639–3647 CAS.
- M. Perez-Gonzalez and R. F. W. Jackson, Chem. Commun., 2000, 24, 2423–2424 RSC.
- E. Dumez, J. S. Snaith, R. F. W. Jackson, A. B. McElroy, J. Overington, M. J. Wythes, J. M. Withka and T. J. McLellan, J. Org. Chem., 2002, 67, 4882–4892 CrossRef CAS PubMed.
- J. Kruse, G. Keilhauer, A. Faissner, R. Timpl and M. Schachner, Nature, 1985, 316, 146–148 CrossRef CAS PubMed.
- P. J. Koch, M. J. Walsh, M. Schmelz, M. D. Goldschmidt, R. Zimbelmann and W. W. Franke, Eur. J. Cell Biol., 1990, 53, 1–12 CAS.
- M. M. Weiser, J. Biol. Chem., 1973, 248, 2536–2541 CAS.
- A. K. Shah, K.-A. L. Cao, E. Choi, D. Chen, B. Gautier, D. Nancarrow, D. C. Whiteman, N. A. Saunders, A. P. Barbour, V. Joshi and M. M. Hill, Mol. Cell. Proteomics, 2015, 14, 3023–3039 CAS.
- M. Ousmer, V. Boucard, N. Lubin-Germain, J. Uziel and J. Augé, Eur. J. Org. Chem., 2006, 2006, 1216–1221 CrossRef.
- A. Marek, J. Kulhánek and F. Bureš, Synthesis, 2009, 325–331 CAS.
- L. Nolasco, M. Perez Gonzalez, L. Caggiano and R. F. Jackson, J. Org. Chem., 2009, 74, 8280–8289 CrossRef CAS PubMed.
- V. R. Espejo and J. D. Rainier, Org. Lett., 2010, 12, 2154–2157 CrossRef CAS PubMed.
- S. Sase, M. Jaric, A. Metzger, V. Malakhov and P. Knochel, J. Org. Chem., 2008, 73, 7380–7382 CrossRef CAS PubMed.
- T. Zhang, X. Gao and H. B. Wood, Tetrahedron Lett., 2011, 52, 311–313 CrossRef CAS.
- T. Carrillo-Marquez, L. Caggiano, R. F. Jackson, U. Grabowska, A. Rae and M. J. Tozer, Org. Biomol. Chem., 2005, 3, 4117–4123 CAS.
- C. Wuttke, R. Ford, M. Lilley, U. Grabowska, D. Wiktelius and R. F. W. Jackson, Synlett, 2012, 243–246 CAS.
- A. M. Echavarren and J. K. Stille, J. Am. Chem. Soc., 1987, 109, 5478–5486 CrossRef CAS.
- T. Carrillo-Marquez, L. Caggiano, R. F. W. Jackson, U. Grabowska, A. Rae and M. J. Tozer, Org. Biomol. Chem., 2005, 3, 4117–4123 CAS.
- D. Hardick, M. Tozer, J. Canfield, M. Wilson, A. Rae, P. Fallon, B. Classon, C. Lindquist and S. Ayesa, WO PatentWO2006064286(A1), Medivir UK Ltd./UK; Peptimmune, Inc, 2006 Search PubMed.
- R. Suzuki, H. Yanuma, T. Hayashi, H. Yamada and T. Usuki, Tetrahedron, 2015, 71, 1851–1862 CrossRef CAS.
- M. Luisetti, S. Ma, P. Iadarola, P. J. Stone, S. Viglio, B. Casado, Y. Y. Lin, G. L. Snider and G. M. Turino, Eur. Respir. J., 2008, 32, 1146 CrossRef CAS PubMed.
- S. Ma, Y. Y. Lin and G. M. Turino, Chest, 2007, 131, 1363–1371 CrossRef CAS PubMed.
- S. Labib, S. Younes, E. Riad and A. Abdallah, Egypt. J. Chest Dis. Tuberc., 2014, 63, 569–573 CrossRef.
- K. Ogawa, T. Hayashi, Y. Y. Lin and T. Usuki, Tetrahedron, 2017, 73, 3838–3847 CrossRef CAS.
- R. P. Mecham and J. A. Foster, Biochem. J., 1978, 173, 617 CrossRef CAS PubMed.
- P. Brown-Augsburger, C. Tisdale, T. Broekelmann, C. Sloan and R. P. Mecham, J. Biol. Chem., 1995, 270, 17778–17783 CrossRef CAS PubMed.
- Q. Liu, P. Hu and Y. He, J. Org. Chem., 2017, 82, 9217–9222 CrossRef CAS PubMed.
- H. Hoffmann, H. Kogler, W. Heyse, H. Matter, M. Caspers, D. Schummer, C. Klemke-Jahn, A. Bauer, G. Penarier, L. Debussche and M. Brönstrup, Angew. Chem., Int. Ed., 2015, 54, 10145–10148 CrossRef CAS PubMed.
- R. F. W. Jackson, I. Rilatt and P. J. Murray, Chem. Commun., 2003, 11, 1242–1243 RSC.
- I. Rilatt and R. F. Jackson, J. Org. Chem., 2008, 73, 8694–8704 CrossRef CAS PubMed.
- A. J. Ross, F. Dreiocker, M. Schafer, J. Oomens, A. J. Meijer, B. T. Pickup and R. F. Jackson, J. Org. Chem., 2011, 76, 1727–1734 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |