
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and structural investigation of a series of mannose-containing oligosaccharides using mass spectrometry†
S.
Daikoku‡
a,
R.
Pendrill‡
b,
Y.
Kanie
 a,
Y.
Ito
c,
G.
Widmalm
*b and
O.
Kanie
*a
a,
Y.
Ito
c,
G.
Widmalm
*b and
O.
Kanie
*a
aDepartment of Applied Biochemistry, Tokai University, 4-1-1 Kitakaname, Hiratsuka, Kanagawa 259-1292, Japan. E-mail: kanie@u-tokai.ac.jp
bDepartment of Organic Chemistry, Arrhenius Laboratory, Stockholm University, S-106 91 Stockholm, Sweden
cSynthetic Cellular Chemistry Laboratory, RIKEN, 2-1 Hirosawa, Wako, Saitama 351-0198, Japan
First published on 5th December 2017
Abstract
A series of compounds associated with naturally occurring and biologically relevant glycans consisting of α-mannosides were prepared and analyzed using collision-induced dissociation (CID), energy-resolved mass spectrometry (ERMS), and 1H nuclear magnetic resonance spectroscopy. The CID experiments of sodiated species of disaccharides and ERMS experiments revealed that the order of stability of mannosyl linkages was as follows: 6-linked > 4-linked ≧ 2-linked > 3-linked mannosyl residues. Analysis of linear trisaccharides revealed that the order observed in disaccharides could be applied to higher glycans. A branched trisaccharide showed a distinct dissociation pattern with two constituting disaccharide ions. The estimation of the content of this ion mixture was possible using the disaccharide spectra. The hydrolysis of mannose linkages at 3- and 6-positions in the branched trisaccharide revealed that the 3-linkage was cleaved twice as fast as the 6-linkage. It was observed that the solution-phase hydrolysis and gas-phase dissociation have similar energetics.
Introduction
In order to elucidate the glycan structure of naturally occurring oligosaccharide samples, the use of a logically arranged array of compounds as a data source is considered advantageous.1–3 Although an array of compounds should provide a complete set of structural data, it is often very difficult to obtain such a library. A variety of manno-oligosaccharides exist in nature, some of which are shown in Fig. 1 with symbolic structures.4–7 A mammalian high mannose-type N-linked glycan is composed of nine mannoses with different anomeric configurations and linkage positions, and it plays an important role in the folding of glycoproteins in endoplasmic reticulum and is later reconstructed into complex N-glycans in Golgi apparatus.8–10 The hybrid type of N-linked glycan is associated with infectious diseases and immunity.11–15 Other forms of mannose-containing oligosaccharides i.e., glycosylphosphatidylinositol anchored glycoprotein,16 and cell wall polysaccharides of Saccharomyces cerevisiae17,18 and Mycobacterium smegmatis19 also exist. Surprisingly, when considering α-linked mannosyl residues, all the possible linkage isomers, namely mannose (Man) residues linked to 2-, 3-, 4-, and 6-positions of adjacent Man, exist in nature.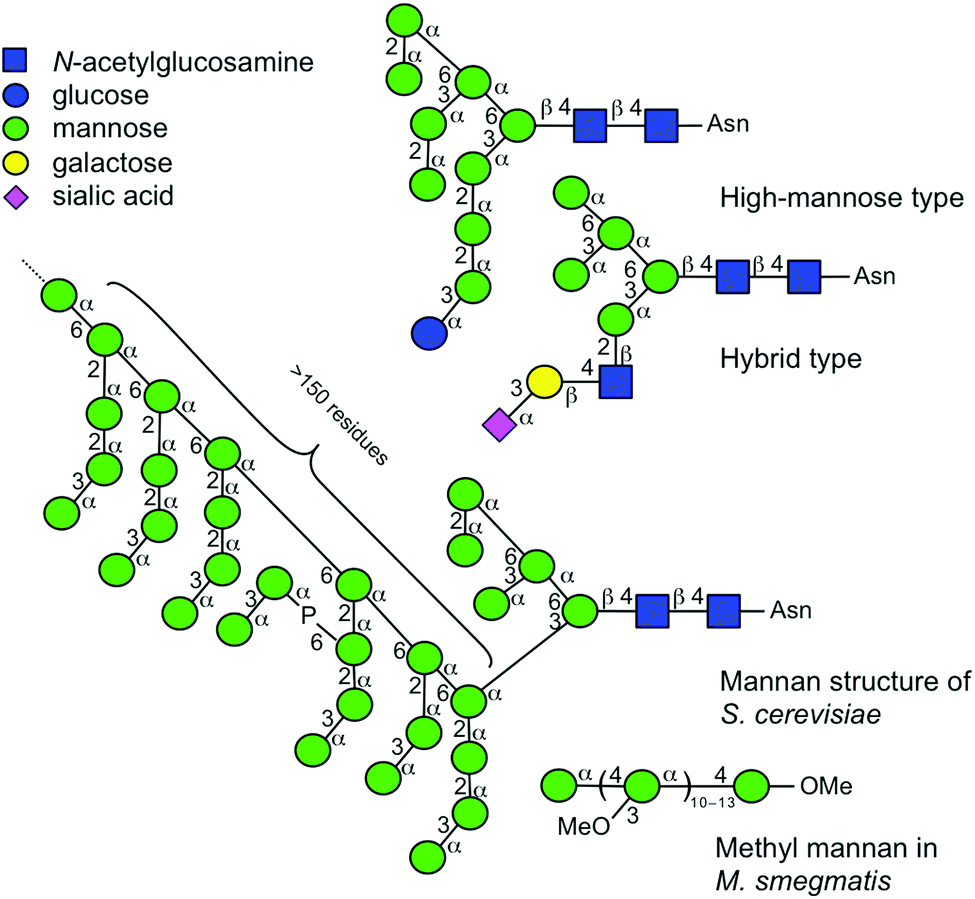 | ||
| Fig. 1 Symbolic representations of naturally occurring glycans consisting of multiple mannosyl residues. Symbols are used as suggested.4–7 Individual linkage isomers are shown with distinctive angle presentations.4 | ||
Therefore, it is important to discriminate individual structures. As the amounts of samples obtainable from natural sources are often limited, mass spectral analysis is an excellent technique for structural investigations. In particular, low-energy collision-induced dissociation (CID) and ion mobility mass spectrometry are suitable techniques for the differentiation of subtle structural differences.1,20,21
Herein, we perform the mass spectrometric analysis of synthetic compounds useful for investigating larger oligosaccharide structures. A series of disaccharides and trisaccharides, namely, αMan–(1 → 2)–αMan–OMe (1), αMan–(1 → 3)–αMan–OMe (2), αMan–(1 → 4)–αMan–OMe (3), αMan–(1 → 6)–αMan–OMe (4), αMan–(1 → 2)–αMan–(1 → 2)–αMan–OMe (5), αMan–(1 → 2)–αMan–(1 → 3)–αMan–OMe (6), αMan–(1 → 2)–αMan–(1 → 6)–αMan–OMe (7), and αMan–(1 → 3)–[αMan–(1 → 6)–]αMan–OMe (8) individually representing partial structures of biologically relevant glycan structures (Fig. 2) were analyzed. The results provide valuable information about the order of stability of mannosyl linkages, and the fragmentation analyses of individual ions with different linkages were carried out in detail. We also showed that the analysis of a mixture of ions obtained from a branched structure is possible. A comparison of the gas-phase CID reaction and acid hydrolysis reaction in solution was also carried out.
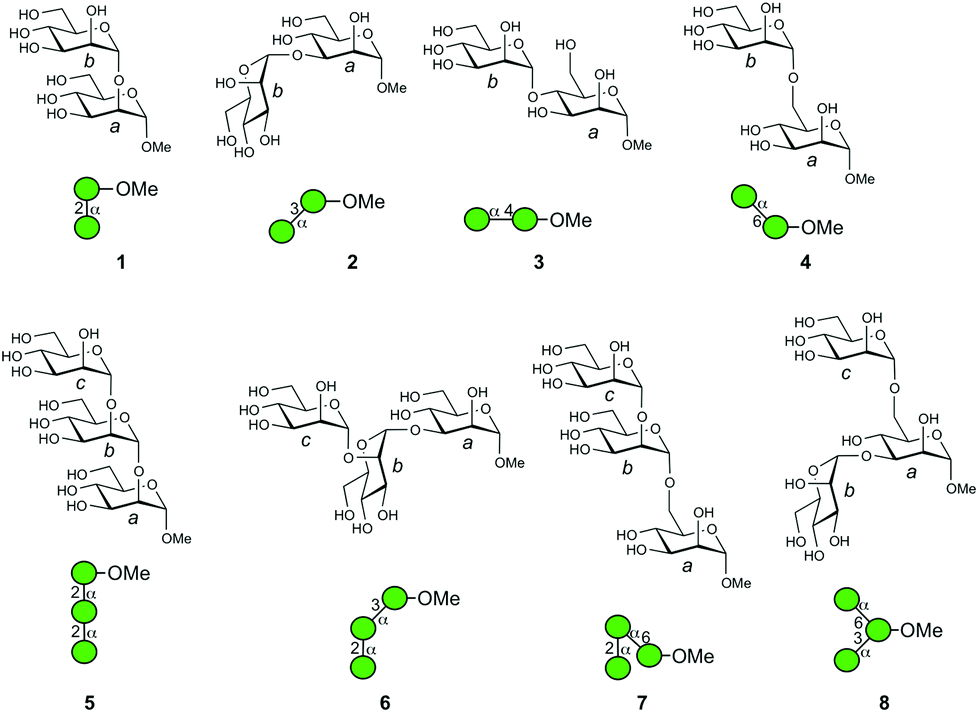 | ||
| Fig. 2 Structures of oligomannosides investigated. The individual mannose residues are designated as a, b, and c by reducing the end terminus for clarity in the molecular structures. Symbolic representations of the structures are also provided. | ||
Results and discussion
Oligosaccharide synthesis
The chemical synthesis of D-mannosyl-containing α-(1 → 4)-linked disaccharide 3 and αMan–(1 → 2)–αMan–(1 → 6)-linked trisaccharide 7 and its [6-13C]-isotopologue 7c was carried out via N-iodosuccinimide/silver triflate promoted glycosylation as the key reaction (Scheme 1). The coupling of donor 10 with acceptor 9 in the temperature interval of −50 °C to −30 °C proceeded smoothly to result in the anticipated α-(1 → 4)-linked disaccharide 11 in good yield (84%).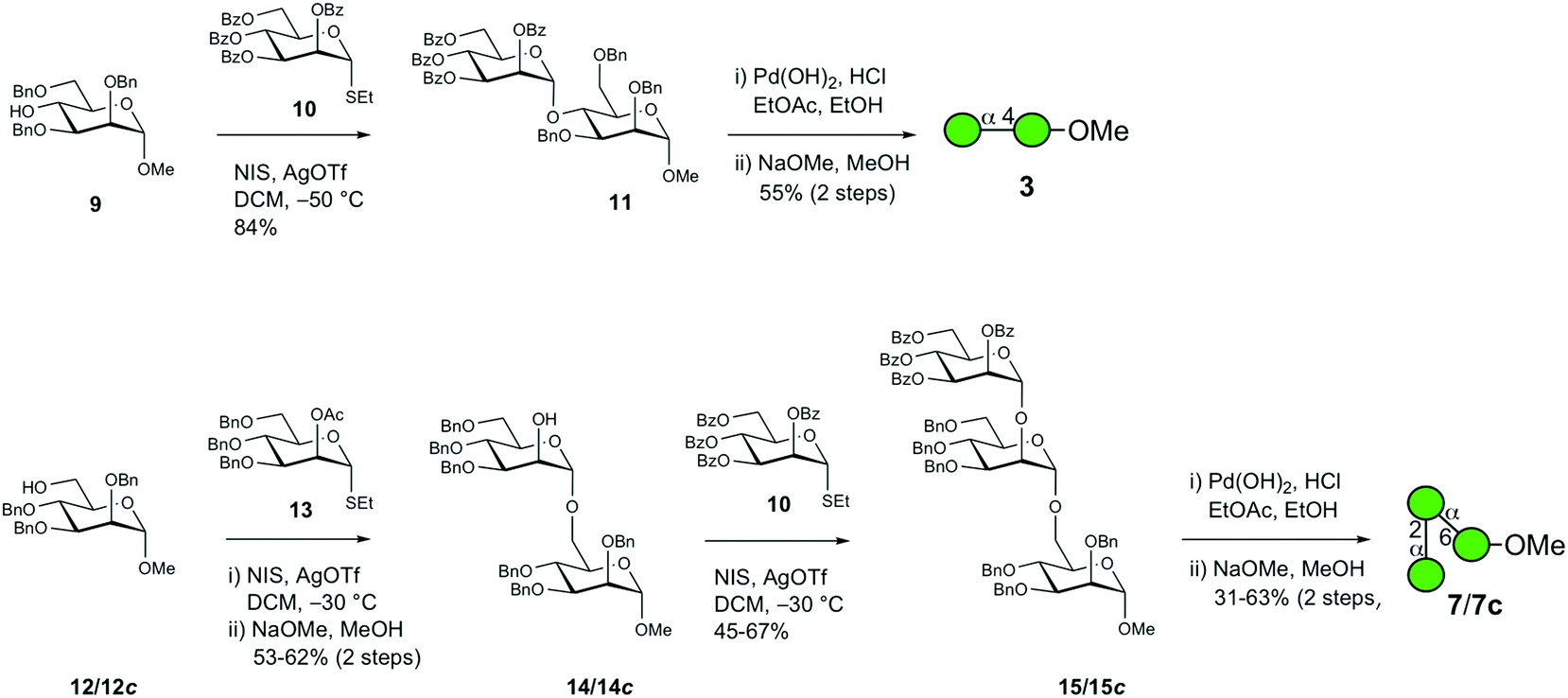 | ||
| Scheme 1 Synthesis of oligosaccharides 3, 7 and 7c. | ||
The subsequent two-step deprotection procedure employing hydrogenolysis first with palladium hydroxide on carbon as the catalyst followed by O-debenzoylation under Zemplén conditions resulted in target disaccharide 3 in 55% yield over two steps. In the synthesis of trisaccharide 7, the O-benzylated acceptor 12 was glycosylated by the suitably protected mannosyl donor 13 with an acetyl group installed at O2, thereby facilitating neighboring group participation ensuring that an α-linkage was formed in the disaccharide product; this was followed by the removal of the O-acetyl group using sodium methoxide in methanol, thereby unmasking the alcohol function to result in 14 in 53% yield over two steps. Disaccharide 14 was subsequently coupled with donor 10 under the same experimental conditions as in the previous glycosylation step, which subsequently resulted in trisaccharide 15 in 67% yield. Employing the same two-step deprotection procedure for the above disaccharide resulted in trisaccharide 7, albeit with 31% yield. Synthesis of the corresponding 4-aminophenyl glycoside of 7 was previously described by Khan et al.22 The 13C-isotopologue 7c, which has been investigated using nuclear magnetic resonance (NMR) relaxation experiments,23 was synthesized using the same protocol; the yields of the first glycosylation reaction and deprotection steps were, in this case, improved to 62% and 63%, respectively.
Structural details of disaccharides obtained from low-energy CID experiments
In order to understand the structural details of a series of oligosaccharides, the analysis of the fragmentation process of the corresponding precursor ions was carried out. Prior to the investigation, we decided to analyze sodium adducted molecules in the CID experiments, because they are frequently observed without any preparative manipulation and they provide data superior to those of proton adducts in energy-resolved mass spectrometry (ERMS).24,25 The common ions produced during the CID process of the sodiated precursor ions obtained for compounds 1–4 are ions with m/z 185 (B1) and 217 (Y1), each corresponding to the units b and a. (Fig. 3) (Assignment of ion species was done according to Domon and Costello.26) A pair of ions with m/z 185 and 217 correspond to the cleavage of a glycosidic bond at the non-reducing end of the mannosyl residue. As these ions are common in all disaccharides, the m/z values themselves cannot be used to diagnose the structures. However, the ratio of these cleavages can be used for the verification, called a “spectral matching technique.”27,28 The percentages of these glycosyl cleavages are shown in the table in Fig. 3. As evident from the table, these cleavage reactions are the major process in each case. A characteristic feature is the preferential methyl glycoside cleavage in sodiated compound 2. A clear explanation cannot be provided for this phenomenon but this can be a diagnostic event. Compounds 3 and 4 in their sodiated forms provided a similar fragmentation pattern with a preferential formation of the X-ion series. In these fragmentation reactions, the 3,5X1 ion (m/z = 319) was formed in the spectrum for 3, and 0,2X1 ion (m/z = 245) and 0,3X1 ion (m/z = 275) were characteristic of 4. We also investigated the fragmentation of sodiated isotopologue 4c [4c + Na+], in which C6 of the reducing terminus mannoside was replaced with 13C. A series of product ions produced during the CID of the sodiated compound was observed to be virtually identical to that of parent 4. This type of evidence might be useful in the investigation of pulse-chase experiments.29 Although cross-ring cleavages are useful in identifying linkage isomers, an ion with m/z = 319 which could be assigned to be the 2,4X1 ion might be used to identify 4-linked mannoside only.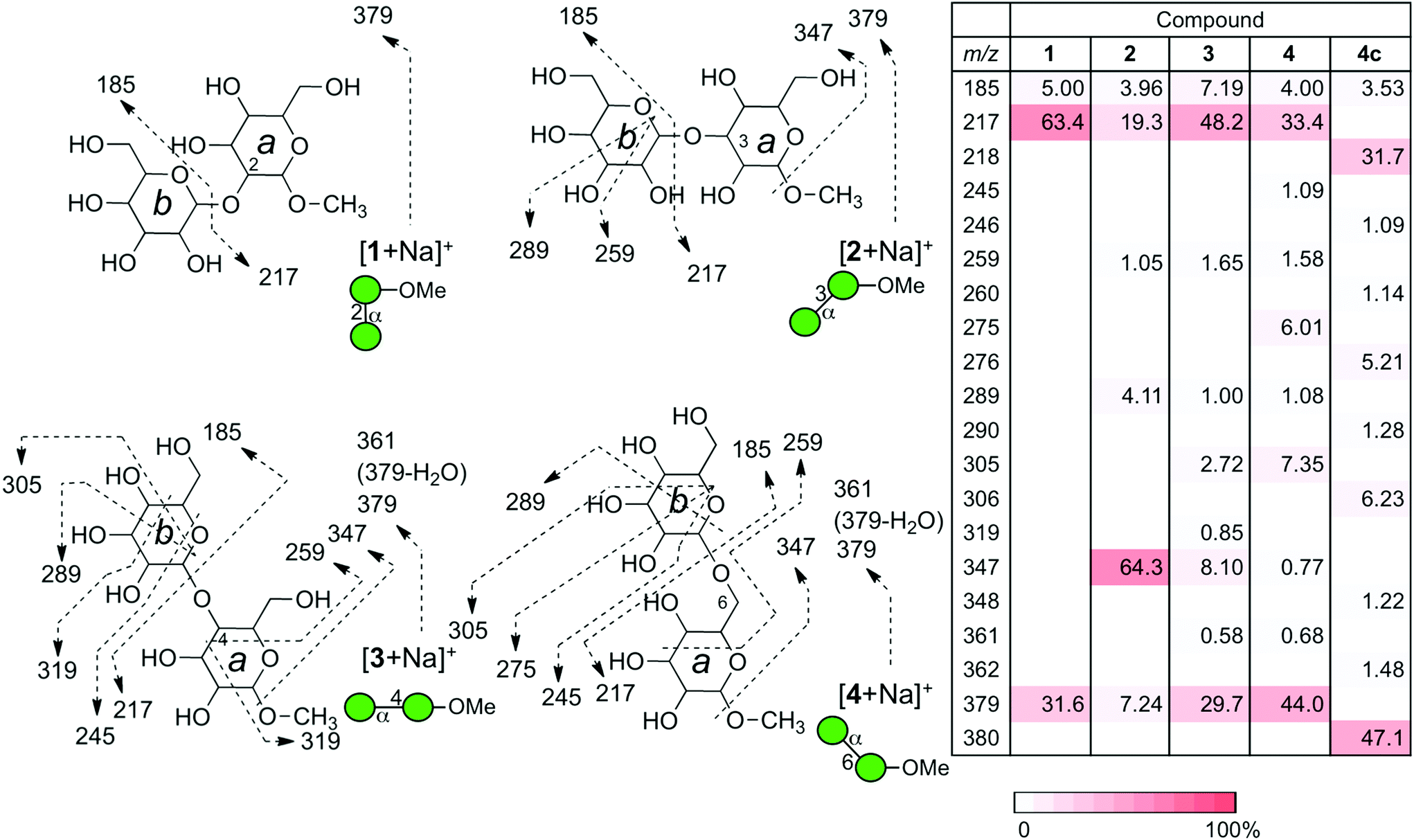 | ||
| Fig. 3 Fragmentation patterns of a series of sodiated disaccharides under CID conditions. The observed fragments are indicated in structures. The CID experiments were conducted at the end-cap radio frequency amplitude of 0.7 V. The table summarizes the intensities of individual product ions. The relative ion intensities are indicated by a red-colored scale. | ||
We subsequently analyzed the energy dependence of the fragmentation reactions of individual sodiated disaccharides. We focus on the decay process of precursor ions during ERMS (Fig. 4). The decay curves indicate the conversion process of individual precursor ions into product ions under CID conditions, which cannot be obtained via standard MS/MS experiments. The most labile disaccharide appeared to be the 3-linked compound 2 whereas 6-linked 4 was the most stable. The difference observed between sodiated 4 and its 13C-labeled counterpart 4c is believed to reflect the mass difference in the dissociation reaction. The decay curves of 2- (1) and 4-linked (3) disaccharides were in between these two disaccharides and showed similar reactivities under the given conditions. A visual inspection of the decay process of individual precursor ions in ERMS indicated the order of stability to be: 6-linked > 4-linked ≧ 2-linked > 3-linked disaccharide. It has been reported that the primary glycosidic linkage is stable, which can be explained by the rotational freedom around the glycosidic linkage compared to the secondary linkages.2
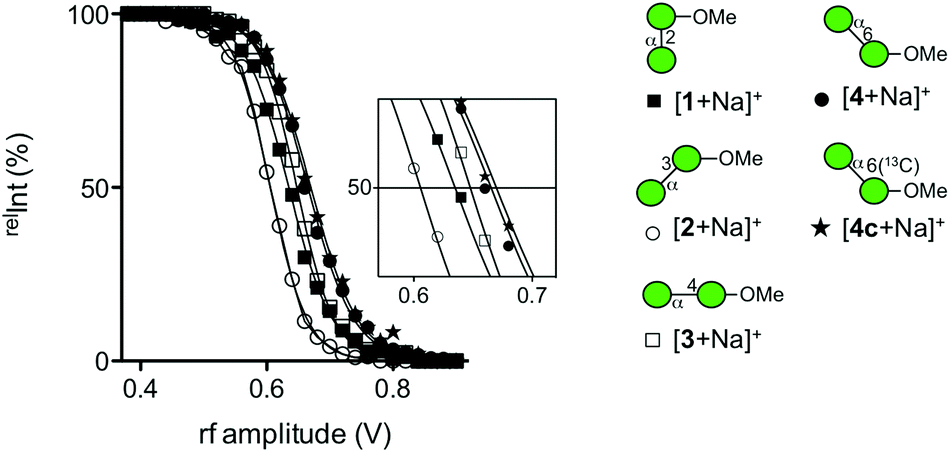 | ||
| Fig. 4 Decay curves of a series of sodiated disaccharides obtained via ERMS experiments. [1 + Na]+ (■), [2 + Na]+ (○), [3 + Na]+ (□), [4 + Na]+ (●), and [4c + Na]+ (★). | ||
Dissociation of a series of sodiated linear trisaccharides: c–b–a–OMe
A series of linear trisaccharides individually consisting of three distinct glycosidic linkages were subsequently analyzed. At a glance, dehydration ions with m/z = 523 ([M + Na-H2O]+) observed for 5 only, 3,5A3 ions with m/z = 451 for 6 and 1,5X2 ions with m/z = 467 for 7 are characteristic of the linkage isomers in the b–a units. Among a variety of product ions formed during the CID process, the major cleavage pathway in all the cases was the glycosyl rupture (Fig. 5). However, the intensities of signals corresponding to individual glycosyl cleavages were different.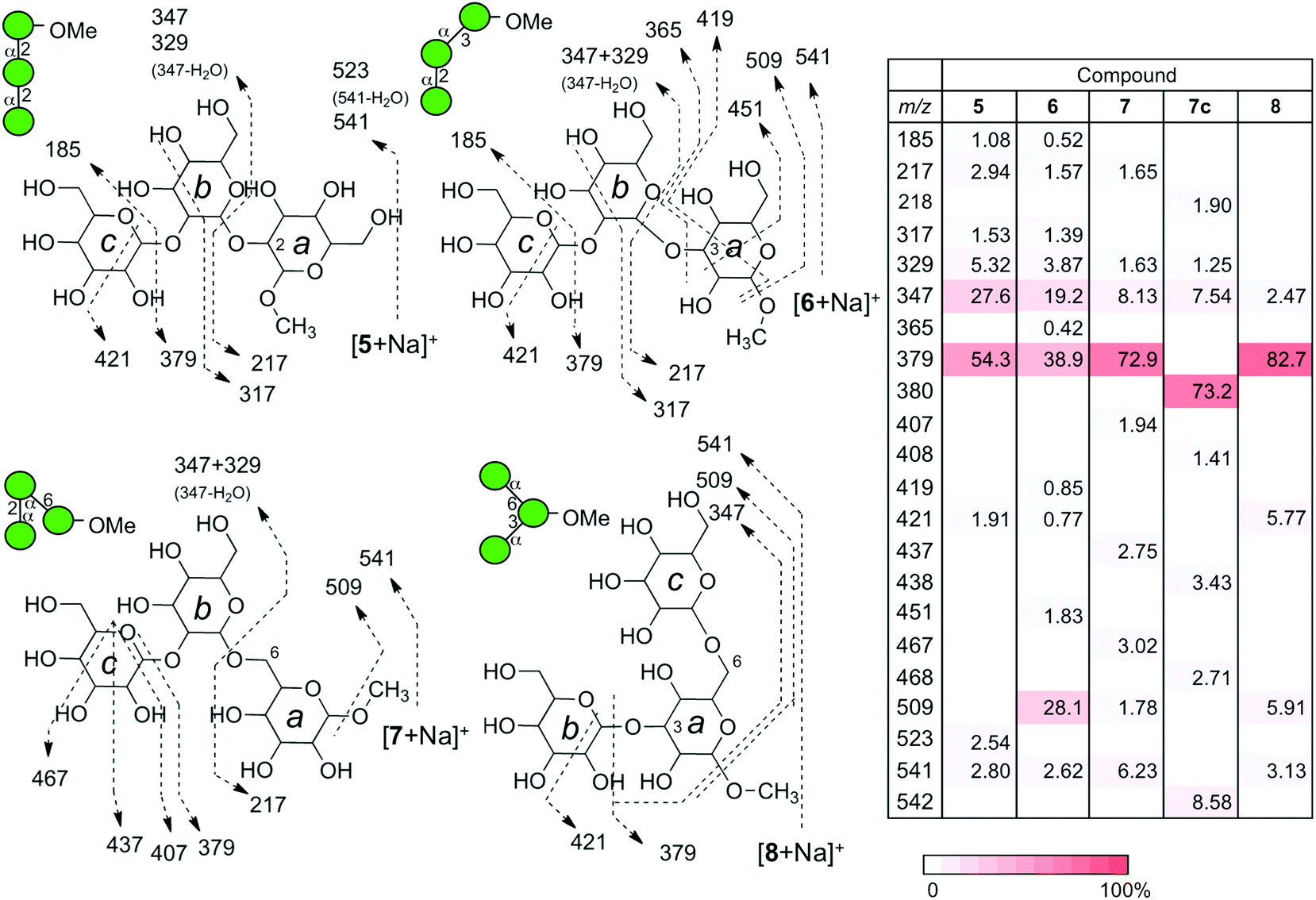 | ||
| Fig. 5 Fragmentation patterns of a series of sodiated trisaccharides under CID conditions. The CID experiments were conducted at the end-cap radio frequency amplitude of 0.7 V. The table summarizes the intensities of individual product ions. A designated red intensity scale is also shown. | ||
The sums of intensities for the glycosyl cleavages between units b and c (intensity (Int.) for [c + Na]+ + Int. [(b–a) + Na]+) were 55.4% for αMan–(1 → 2)–αMan–(1 → 2)–αMan–OMe (5), 39.4% for αMan–(1 → 2)–αMan–(1 → 3)–αMan–OMe (6), and 72.9% for αMan–(1 → 2)–αMan–(1 → 6)–αMan–OMe (7). The intensities of signals for the cleavage between units a and b (Int. m/z 217 + Int. m/z 347 + Int. m/z 329) were 35.9% (5), 24.6% (6), and 11.4% (7). The intensities associated with the dissociation of the methyl group were zero (not observed) (5), 28.1 (6), and 1.78% (7). Interestingly, the tendency of reducing the methyl glycoside cleavage in the ion of (6) can be observed in the CID data for one of the disaccharides, namely αMan–(1 → 3)–αMan–OMe (2), which shares the same 3-linked disaccharide structure.
Since the energy dependence of individual ions is quite complex and reflects various factors, we attempted to consider the internal energy as a reference point. The order of ion stability of a series of sodiated disaccharides observed under the CID conditions was as follows: 6-linked > 4-linked ≧ 2-linked > 3-linked, as described above. It is difficult to discuss the underlying mechanism using the data alone. However, as a series of linear trisaccharides contained the same disaccharide structure, we surmised that it would be possible to determine a correlation between the structure and the observed order of ion stability. Thus, the non-reducing mannosyl residue (unit c) is considered to be an internal energy reference because the position and anomeric configuration of the residue are the same and furthermore, the structural similarity makes it suitable to evaluate other inter-mannosyl linkages between units a and b. Although methyl glycoside was common to all the compounds, it was not considered to be appropriate as an internal energy reference because the residue was closely associated with the cleavage of linkage a–b.
Thus, we compared the sum of intensities of signals associated with individual glycosyl cleavages and obtained a ratio a–b/b–c to compare the relative strength of mannosyl linkages present in a structure. The obtained a–b/b–c values were 0.55 (5), 0.56 (6), and 0.10 (7). This indicates that the stability order is 6-linked > 2-linked ≥ 3-linked, which is consistent with the order observed for the stability of disaccharides. Moreover, the CID of an isotope labeled compound 7c, at C6 of reducing terminus mannoside, resulted in an almost identical fragmentation pattern with one atom unit shifts in the product ions carrying 13C instead of 12C.
Dissociation of the branched trisaccharide: b–(c–)a–OMe
Naturally occurring manno-oligosaccharides often contain a branching structure with α(1 → 3)- and α(1 → 6)-linkages (Fig. 1). Therefore, compound 8 would serve as a valuable information source. The CID analysis of sodiated αMan–(1 → 3)–[αMan–(1 → 6)–]αMan–OMe (8) was carried out (Fig. 5). The fragmentation pattern resembled that of sodiated 7 having a 6-linkage, rather than that of 6 having a 3-linkage, observed with an ion at m/z 379 corresponding to the Y-ion structure (b–a unit) that lost a non-reducing mannosyl residue (c unit) resulting in a prominent product ion. This indicates that the relative strengths of glycosidic linkages at the 3- and 6-positions are quite different, as clearly shown in the dissociation of disaccharides, where 6-linked mannose is more stable (Fig. 4). Therefore, the glycosyl cleavage at the 3-position will predominantly occur in sodiated 8.MS/MS results of a series of product ions with m/z 379 produced from sodiated trisaccharides
The CID experiments of sodiated disaccharide ions for unit b–a (m/z 379) produced from a series of sodiated trisaccharides were carried out at the MS3 stage to investigate whether the product ions are identical to the fragmentation patterns of those of the disaccharide library (Fig. 6). According to our previous results, the Y-ion species are treated as “pure” ions without any structurally isomeric ions, and thus can be used for the structural identification at different stages of MSn.25 Therefore, these types of ion species are suitable for structural identification. The fragmentation products and their intensities were strikingly similar to those of the corresponding disaccharides indicating that the structures of Y-ions produced from trisaccharides can be deduced from those of library disaccharides except for the branched compound. In the current experiment, using a library of disaccharides, the CID data were used to identify the partial structure of a trisaccharide. When we assume that the structure of a trisaccharide is not known, the partial structure [b–a + Na]+ is determined at the MS3 stage by comparing a series of data from a library.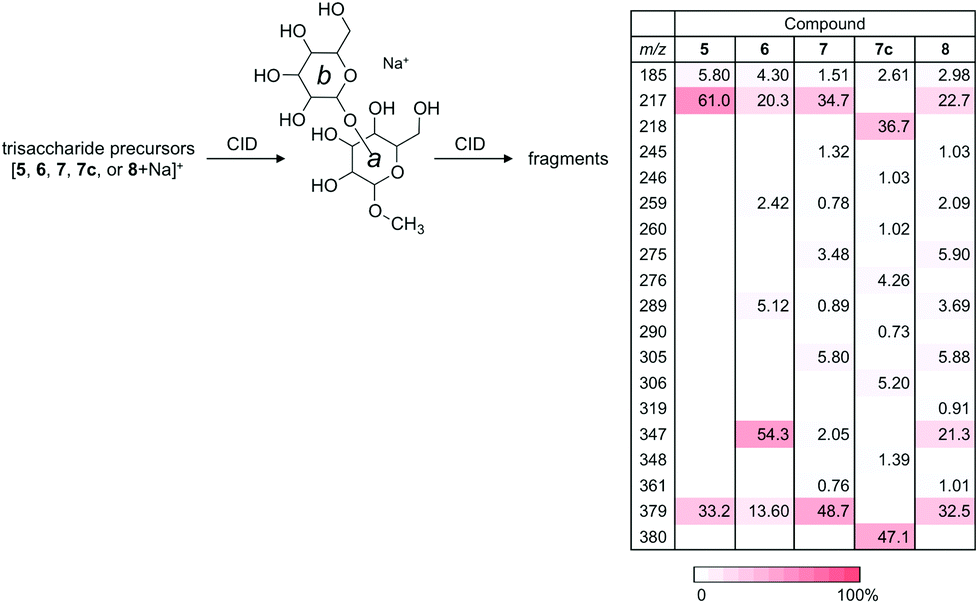 | ||
| Fig. 6 MS3 experiments of Y-ions produced from a series of sodiated trisaccharides under CID conditions. The series of product ions for individual precursors are virtually identical to those obtained from the corresponding disaccharide ions at MS2. | ||
Regarding the product ion obtained from the branched trisaccharide ion, the structure b–(c–)a-OMe (8) contains both 3- and 6-linked mannosyl residues. As mentioned above, the fragmentation pattern of the produced ion with m/z 379 was similar to that of [2 + Na]+ with some minor ions observed in the fragmentation of [4 + Na]+, indicating that the formed Y-ion may be a mixture of these two product ions.
We thus carried out an ERMS experiment on the produced Y-ion corresponding to the partial structure of trisaccharide 8, and compared it with those of authentic disaccharides, namely 2 and 4. As shown in Fig. 7, the decay curve obtained for the degradation of the Y-ion generated from [8 + Na]+ was observed in between those of individual decay curves of [2 + Na]+ and [4 + Na]+. This suggested that the Y-ion was a mixture of these two ions, which is consistent with the result of MS/MS experiments.
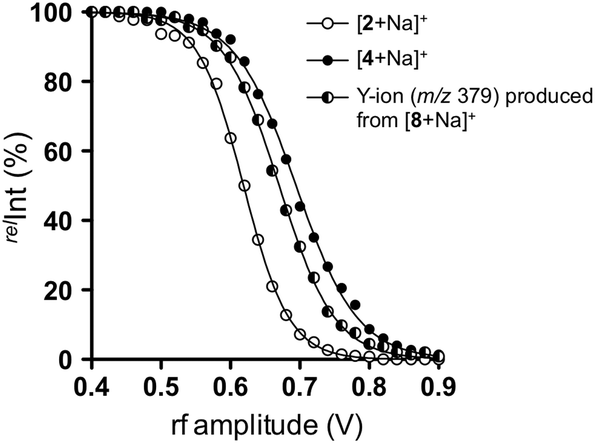 | ||
| Fig. 7 Comparison of decay curves of sodiated disaccharides (2 and 4) and Y-ion obtained from sodiated 8 in ERMS. [2 + Na]+ (○), [4 + Na]+ (●), and Y-ion (m/z 379) produced by CID of [8 + Na]+ (◐). | ||
We subsequently estimated the contents of [2 + Na]+ and [4 + Na]+ in the Y-ion mixture obtained from the branched trisaccharide [8 + Na]+. Accordingly, the ion intensities of individual product ions at the resonant frequency amplitude of 0.7 V were used. Consequently, it was estimated that the Y-ion produced was a mixture of [2 + Na]+ (37%) and [4 + Na]+ (63%) based on the assumption that there was no loss of ions during the CID (Fig. 8). Subtle differences could be observed in these spectra, which were attributed to the different populations of differently sodiated ion species (different coordination sites).
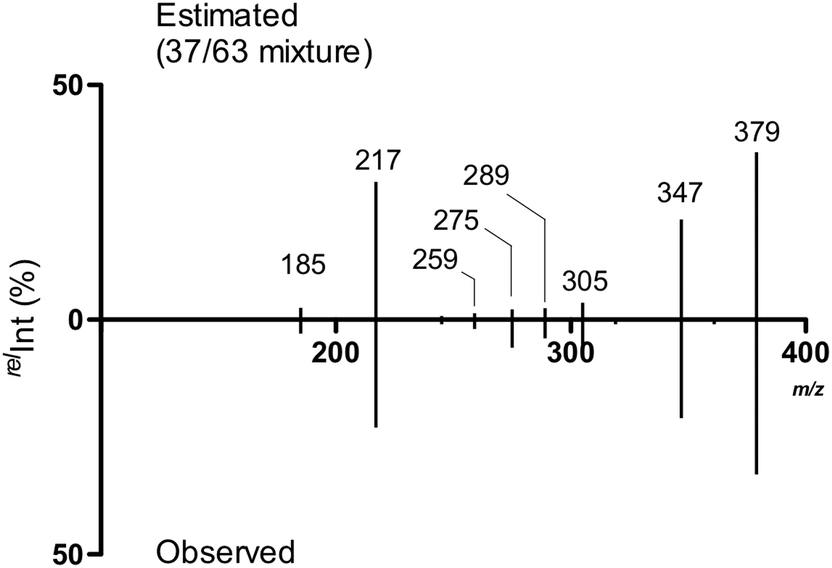 | ||
| Fig. 8 Estimated MS/MS spectrum of the ion mixture of constituting disaccharide ions and the observed spectrum. | ||
Oligosaccharide hydrolysis monitored using NMR
At this stage, we considered whether the gas phase CID reactions were related to the hydrolysis reactions. We performed acidic hydrolyses of compounds 2, 4, and 8 to examine the reactivities of 3- and 6-linked glycosidic bonds under 1H NMR monitoring (Fig. 9a). In trisaccharide 8, substitution by α-D-mannopyranosyl groups occurred at secondary and primary alcohol groups, i.e., at O3 and O6, respectively. In order to study the relative stability to hydrolysis at these glycosidic linkages, the trisaccharide was subjected to acid hydrolysis at an elevated temperature (1 M trifluoroacetic acid, D2O, 70 °C) and monitored using 1H NMR spectroscopy. As a reference, both the α(1 → 3)- and α(1 → 6)-linked disaccharides 2 and 4, respectively, were investigated under corresponding experimental conditions. The progress of the hydrolysis reaction was analyzed by monitoring the disappearance of the 1H resonances from anomeric protons of the terminal mannopyranosyl groups. While this is straightforward for disaccharides 2 and 4, notably, in trisaccharide 8, the anomeric 1H NMR chemical shifts at the α(1 → 3)-linkage are degenerate for 2 and 8 and this is also the case for the α(1 → 6)-linkages in compounds 4 and 8. This degeneracy indicates that in the trisaccharide, the hydrolysis of 2 and 8 is monitored at the α(1 → 3)-linkage and in the same way, the hydrolysis reaction for the α(1 → 6)-linkage is followed, but not differentiated, for compounds 4 and 8. In the disaccharides, the α-D-Manp group linked to O3 showed t1/2 = 5.3 h, whereas when linked to O6, t1/2 = 10.9 h. When these data were compared to the apparent hydrolysis rates in the trisaccharide, t1/2 = 5.4 h and t1/2 = 10.7 h, respectively, it became evident that the reactivity in the trisaccharide is similar to that of its constituent disaccharides. The experimentally determined half-lives were used to calculate the evolution of the composition of the trisaccharide and the two disaccharides over the course of the hydrolysis reaction of the trisaccharide (Fig. 9b). The hydrolysis of 8 (cyan line in Fig. 9b) is at its initial stage foremost owing to the cleavage of the α(1 → 3)-linkage resulting in the rapid formation of disaccharide 4 (red line in Fig. 9b). During the later course of the reaction, consequently, its α(1 → 6)-linkage is cleaved, which can be observed from the significantly higher concentration of 4 compared to that of 2 (red and black lines in Fig. 9b, respectively).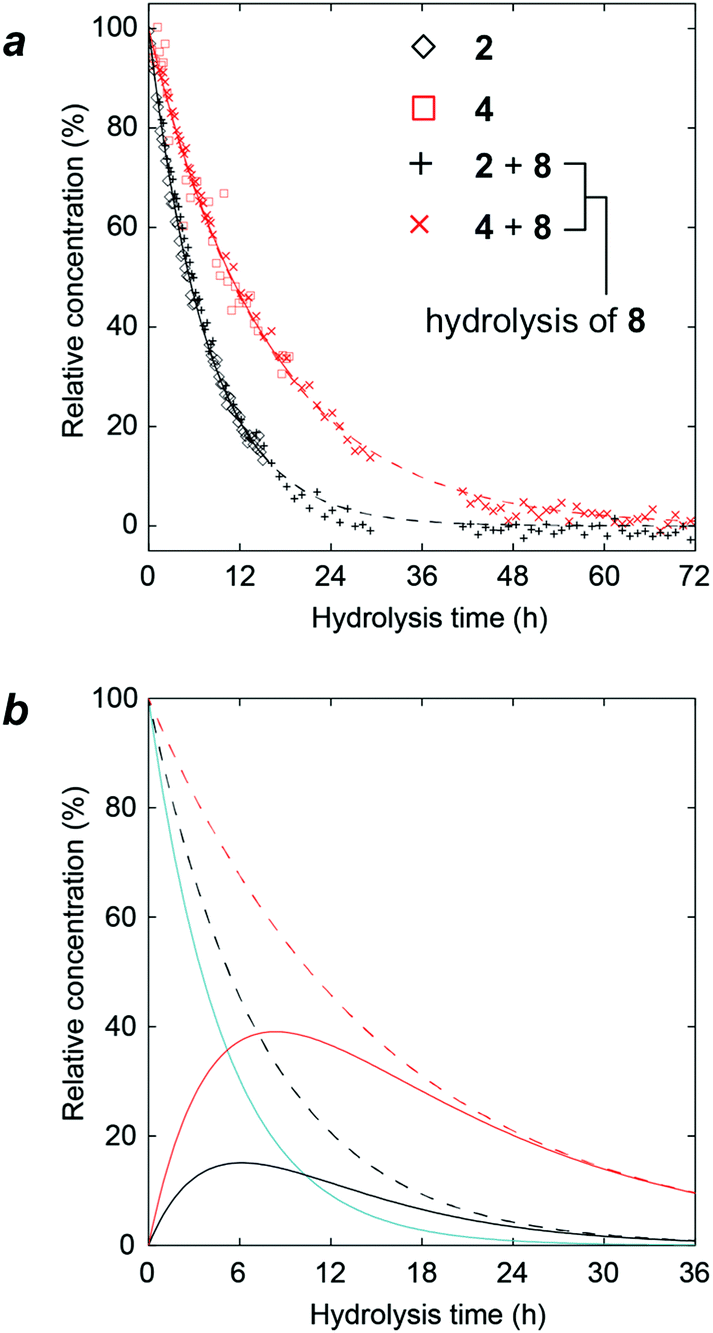 | ||
| Fig. 9 NMR analysis of hydrolysis reactions. (a) Time-course for acid hydrolysis (1 M TFA in D2O at 70 °C) of the 3,6-disubstituted trisaccharide 8, the constituent disaccharides 2 and 4, α-(1 → 3)- and α-(1 → 6)-linked, respectively, analyzed using 1H NMR spectroscopy with (◇) 2, (□) 4, (+) 2 and 8, (×) 4 and 8, whose fitted data are represented by solid black, solid red, dashed black, and dashed red lines, respectively; (b) calculated time-course for the acid hydrolysis of the 3,6-disubstituted trisaccharide 8 (cyan). The hydrolysis intermediates, i.e., disaccharides 2 and 4, are shown as black and red lines, respectively. The sum of 2 and 8 is shown by the dashed black line and the dashed red line corresponds to the sum of 4 and 8. | ||
The fact that hydrolysis at the α(1 → 3)-linkage occurred twice as fast as that at the α(1 → 6)-linkage for the mannose-containing oligosaccharides when using 1 M trifluoroacetic acid (TFA) at 70 °C is consistent with the previous results of hydrolysis rates for α(1 → 3)-linked nigerose, which was hydrolyzed 4.5 times faster in 0.1 N hydrochloric acid at 80 °C than α(1 → 6)-linked isomaltose; furthermore, at 99.5 °C, it was hydrolyzed 3 times faster.30 Notably, the hydrolysis rates at the α-D-Manp-linkages mirrored the stability of the mannose oligosaccharides when subjected to CID and the propensity to fragment as detected using MS. Although our result may be seen as partially contradictory to the hydrolysis results of S. cerevisiae mannan where the order of mannose release is not precisely controlled, the presence of branching mannoside such as mannosyl residues with the oligosaccharide attached to both 2- and 6-positions might be highly reactive due to steric factors.31 Thus the observed phenomena in this investigation cannot be directly applied to the hydrolysis of higher glycan structures.
Conclusions
A series of synthetic disaccharides and trisaccharides consisting of α-linked mannopyranoside were used to investigate gas-phase CID reactions and acid hydrolyses. Analyses data of all the possible linkage-isomers of α-mannosyl disaccharides were used in the analyses of trisaccharides. As the dissociation reactions of ions of trisaccharides produce their partial structures including disaccharide equivalent ions called Y-ions, it was confirmed that individual trisaccharides contained the corresponding disaccharides. This indicates that an array of low-level glycans is powerful in investigation higher glycan structures. The linkage isomers of these constituting disaccharides could be discriminated using low-energy CID experiments where the patterns of a series of produced product ions were different. Individual structures could also be differentiated based on ERMS. The analysis of linear trisaccharides revealed that the order of dissociation of the linkage isomers was the same as that of disaccharides when a common mannosyl cleavage was used as the internal energy reference. Furthermore, a 3- and 6-branched trisaccharide showed a distinctive dissociation pattern producing a lower number of product ions. As the branched compound produced two constituting disaccharide ions, the estimation of their population in the ion mixture was possible using the individual spectra of the disaccharides. Furthermore, the reaction progress of the hydrolytic glycosyl cleavages of 3- and 6-linked disaccharides and the branched trisaccharide was monitored using NMR, and compared with gas-phase dissociation reactions. The results indicated that the cleavage-reaction rates mirrored those observed in CID experiments. The results obtained herein may provide information for elucidating α-glycosidically linked mannosyl structures and may, in the future, be extended to other sugar residues with gluco- or galacto-configurations and β-linked oligosaccharides, some of which have been studied using the ERMS technique.32Experimental
General
The oligosaccharides methyl α-D-mannopyranosyl-(1 → 2)-α-D-mannopyranoside (1), methyl α-D-mannopyranosyl-(1 → 3)-α-D-mannopyranoside (2), methyl α-D-mannopyranosyl-(1 → 6)-α-D-mannopyranoside (4), methyl α-D-mannopyranosyl-(1 → 6)-α-D-[6-13C]mannopyranoside (4c), methyl α-D-mannopyranosyl-(1 → 2)-α-D-mannopyranosyl-(1 → 2)-α-D-mannopyranoside (5), methyl α-D-mannopyranosyl-(1 → 2)-α-D-mannopyranosyl-(1 → 3)-α-D-mannopyranoside (6) were available from previous studies33–35 and methyl α-D-mannopyranosyl-(1 → 3)[α-D-mannopyranosyl-(1 → 6)]-α-D-mannopyranoside (8) was purchased from Carbosynth, Compton, UK.Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 toluene:EtOAc) giving methyl 2-O-acetyl-3,4,6-tri-O-benzyl-α-D-mannopyranosyl-(1 → 6)-2,3,4-tri-O-benzyl-α-D-mannopyranoside in 58% yield (632 mg, 0.673 mmol). This compound (629 mg, 0.670 mmol) was then dissolved in MeOH and NaOMe was added until pH ≈ 9. After 21 hours, Dowex-50 (H+-form) was added prior to filtration and removal of the solvent under reduced pressure. After purification on silica gel (toluene/EtOAc: 7/3) product 1445 was obtained in 53% yield over two steps (547 mg, 0.610 mmol). 13C-NMR (CDCl3): δ = 54.74 (OMe), 66.4–80.2 (10 C2–C6, 6 PhCH2), 98.9 (C1, 1JC1,H1 = 169.8 Hz), 99.7 (C1′, 1JC1′,H1′ = 170.9 Hz), 127.6–128.5 (30 aromatic), 137.9–138.6 (6 i-Bn). 1H-NMR (CDCl3): δ = 2.32 (1H, d, J = 2.9 Hz, HO2′), 3.26 (1H, s, 3H, OMe), 3.57–4.93 (27H, H1, H2–H6, H2′–H6′, 6 PhCH2), 5.07 (1H, d, J = 1.8 Hz, H1′), 7.13–7.39 (30H, aromatic). ESI-MS: m/z [M + Na]+ calc. for C55H60NaO11 919.4028, found 919.4029.
:EtOH (1:1, 5 mL) and Pd(OH)2/C (spatula tip) as well as a drop of HCl were added (37% aq.). The mixture was stirred under H2 (110 psi) overnight before being filtered through Celite and the solvent being evaporated. The residue was dissolved in MeOH and NaOMe was added until pH ≈ 9 before stirring overnight. Dowex-50 (H+-form) was added and stirred for a few minutes before filtering and removing the solvent under reduced pressure. The crude material was applied to a C-18 reversed-phase cartridge eluting with H2O and lyophilized before being purified by gel-permeation chromatography on an ÄKTApurifier equipped with a SuperdexTM 30 column, prep. grade gel (GE Healthcare, Uppsala, Sweden), using water containing 1% n-butanol as the eluent. Compound 7 was obtained in 31% yield (12.7 mg, 24.4 μmol). 13C-NMR (D2O): δ = 55.46 (OMe), 61.60 (C6′), 61.79 (C6′′), 66.43 (C6), 67.19 (C4), 67.54 (C4′′), 67.59 (C4′), 70.55 (C2), 70.63 (C2′′), 70.91 (C3′), 70.99 (C3′′), 71.37 (C5), 71.42 (C3), 73.43 (C5′), 73.91 (C5′′), 79.36 (C2′), 98.65 (C1′, 1JC′,H′ = 172 Hz), 101.71 (1JC,H = 171 Hz, C1), 103.02 (1JC′′,H′′ = 171 Hz, C1′′). 1H-NMR (D2O): δ = 3.40 (3H, s, OMe), 3.62 (1H, dd, 3JH4′′,H5′′ = 10.0 Hz, H4′′), 3.69 (1H, ddd, 3JH5′,H6′pro-R = 5.7 Hz; 3JH5′,H6′pro-S = 2.0 Hz, H5′), 3.69 (1H, dd, 3JH4′,H5′ = 9.3 Hz, H4′), 3.72 (1H, dd, 3JH4,H5 = 9.9 Hz, H4), 3.73 (1H, dd, 2JH6′′pro-R,H6′′pro-S = –12.3 Hz, H6′′pro-R), 3.74 (1H, dd, 3JH3,H4 = 9.4 Hz, H3), 3.74 (1H, ddd, 3JH5,H6pro-R = 4.7 Hz; 3JH5,H6pro-S = 1.6 Hz, H5), 3.75 (1H, dd, 2JH6pro-R,H6pro-S = –11.7 Hz, H6pro-S), 3.76 (1H, dd, 2JH6′pro-R,H6′pro-S = –12.2 Hz, H6′pro-R), 3.77 (1H, ddd, 3JH5′′,H6′′pro-R = 6.6 Hz; 3JH5′′,H6′′pro-S = 2.2 Hz, H5′′), 3.84 (1H, dd, 3JH3′′,H4′′ = 9.7 Hz, H3′′), 3.89 (1H, dd, H6′′pro-S), 3.89 (1H, dd, H6′pro-S), 3.93 (1H, dd, 3JH2,H3 = 3.4 Hz, H2), 3.95 (1H, dd, 3JH3′,H4′ = 9.6 Hz, H3′), 3.96 (1H, dd, H6pro-R), 4.01 (1H, dd, 3JH2′,H3′ = 3.4 Hz, H2′), 4.07 (1H, dd, 3JH2′′,H3′′ = 3.4 Hz, H2′′), 4.74 (1H, d, 3JH1,H2 = 1.7 Hz, H1), 5.02 (1H, d, 3JH1′′,H2′′ = 1.8 Hz, H1′′), 5.14 (1H, d, 3JH1′,H2′ = 1.8 Hz, H1′). ESI-MS: m/z [M + Na]+ calc. for C19H34NaO16 541.1739, found 541.1731.
:1 toluene:EtOAc) giving methyl 2-O-acetyl-3,4,6-tri-O-benzyl-α-D-mannopyranosyl-(1 → 6)-2,3,4-tri-O-benzyl-α-D-mannopyranoside in 58% yield (632 mg, 0.673 mmol). This compound (629 mg, 0.670 mmol) was then dissolved in MeOH and NaOMe was added until pH ≈ 9. After 21 hours, Dowex-50 (H+-form) was added prior to filtration and removal of the solvent under reduced pressure. After purification on silica gel (toluene/EtOAc: 7/3) product 1445 was obtained in 53% yield over two steps (547 mg, 0.610 mmol). 13C-NMR (CDCl3): δ = 54.74 (OMe), 66.4–80.2 (10 C2–C6, 6 PhCH2), 98.9 (C1, 1JC1,H1 = 169.8 Hz), 99.7 (C1′, 1JC1′,H1′ = 170.9 Hz), 127.6–128.5 (30 aromatic), 137.9–138.6 (6 i-Bn). 1H-NMR (CDCl3): δ = 2.32 (1H, d, J = 2.9 Hz, HO2′), 3.26 (1H, s, 3H, OMe), 3.57–4.93 (27H, H1, H2–H6, H2′–H6′, 6 PhCH2), 5.07 (1H, d, J = 1.8 Hz, H1′), 7.13–7.39 (30H, aromatic). ESI-MS: m/z [M + Na]+ calc. for C55H60NaO11 919.4028, found 919.4029.
:EtOH (1:1, 5 mL) and Pd(OH)2/C (spatula tip) as well as a drop of HCl were added (37% aq.). The mixture was stirred under H2 (110 psi) overnight before being filtered through Celite and the solvent being evaporated. The residue was dissolved in MeOH and NaOMe was added until pH ≈ 9 before stirring overnight. Dowex-50 (H+-form) was added and stirred for a few minutes before filtering and removing the solvent under reduced pressure. The crude material was applied to a C-18 reversed-phase cartridge eluting with H2O and lyophilized before being purified by gel-permeation chromatography on an ÄKTApurifier equipped with a SuperdexTM 30 column, prep. grade gel (GE Healthcare, Uppsala, Sweden), using water containing 1% n-butanol as the eluent. Compound 7 was obtained in 31% yield (12.7 mg, 24.4 μmol). 13C-NMR (D2O): δ = 55.46 (OMe), 61.60 (C6′), 61.79 (C6′′), 66.43 (C6), 67.19 (C4), 67.54 (C4′′), 67.59 (C4′), 70.55 (C2), 70.63 (C2′′), 70.91 (C3′), 70.99 (C3′′), 71.37 (C5), 71.42 (C3), 73.43 (C5′), 73.91 (C5′′), 79.36 (C2′), 98.65 (C1′, 1JC′,H′ = 172 Hz), 101.71 (1JC,H = 171 Hz, C1), 103.02 (1JC′′,H′′ = 171 Hz, C1′′). 1H-NMR (D2O): δ = 3.40 (3H, s, OMe), 3.62 (1H, dd, 3JH4′′,H5′′ = 10.0 Hz, H4′′), 3.69 (1H, ddd, 3JH5′,H6′pro-R = 5.7 Hz; 3JH5′,H6′pro-S = 2.0 Hz, H5′), 3.69 (1H, dd, 3JH4′,H5′ = 9.3 Hz, H4′), 3.72 (1H, dd, 3JH4,H5 = 9.9 Hz, H4), 3.73 (1H, dd, 2JH6′′pro-R,H6′′pro-S = –12.3 Hz, H6′′pro-R), 3.74 (1H, dd, 3JH3,H4 = 9.4 Hz, H3), 3.74 (1H, ddd, 3JH5,H6pro-R = 4.7 Hz; 3JH5,H6pro-S = 1.6 Hz, H5), 3.75 (1H, dd, 2JH6pro-R,H6pro-S = –11.7 Hz, H6pro-S), 3.76 (1H, dd, 2JH6′pro-R,H6′pro-S = –12.2 Hz, H6′pro-R), 3.77 (1H, ddd, 3JH5′′,H6′′pro-R = 6.6 Hz; 3JH5′′,H6′′pro-S = 2.2 Hz, H5′′), 3.84 (1H, dd, 3JH3′′,H4′′ = 9.7 Hz, H3′′), 3.89 (1H, dd, H6′′pro-S), 3.89 (1H, dd, H6′pro-S), 3.93 (1H, dd, 3JH2,H3 = 3.4 Hz, H2), 3.95 (1H, dd, 3JH3′,H4′ = 9.6 Hz, H3′), 3.96 (1H, dd, H6pro-R), 4.01 (1H, dd, 3JH2′,H3′ = 3.4 Hz, H2′), 4.07 (1H, dd, 3JH2′′,H3′′ = 3.4 Hz, H2′′), 4.74 (1H, d, 3JH1,H2 = 1.7 Hz, H1), 5.02 (1H, d, 3JH1′′,H2′′ = 1.8 Hz, H1′′), 5.14 (1H, d, 3JH1′,H2′ = 1.8 Hz, H1′). ESI-MS: m/z [M + Na]+ calc. for C19H34NaO16 541.1739, found 541.1731.
Following the same procedure as that for the unlabeled compound, methyl 2,3,4,6-tetra-O-benzoyl-α-D-mannopyranosyl-(1 → 2)-3,4,6-tri-O-benzyl-α-D-mannopyranosyl-(1 → 6)-2,3,4-tri-O-benzyl-α-D-[6-13C]mannopyranoside (15c) (120.8 mg, 81.8 μmol) gave the title compound 7c in 63% yield (26.7 mg, 51.3 μmol). ESI-MS: m/z [M + Na]+ calc. for C1813CH34NaO16 542.1773, found 542.1771.
Mass spectrometric analysis of glycans
000, (7) maximum acquisition time: 200 ms, (8) average: 5 spectra, and (9) polarity, positive. In our MSn experiments, the end cap rf amplitude was raised by 0.02 V increments until the precursor ion could no longer be detected. Only the end cap rf amplitude was controlled during the CID experiment. The He pressure was 4.86 × 10−6 mbar and the CID time was 40 ms. Averages of 15 spectra were used for CID experiments. Isotopic peaks with [Ii + 1] and [Ii + 2], where In indicates a fragment ion, were included in the calculations. For the isolation of a product ion, m/z ± 2 (w = 2) were isolated and subjected to the CID experiments to include isotopes. Standard MS/MS spectra are the extracts of this ERMS at a designated amplitude.
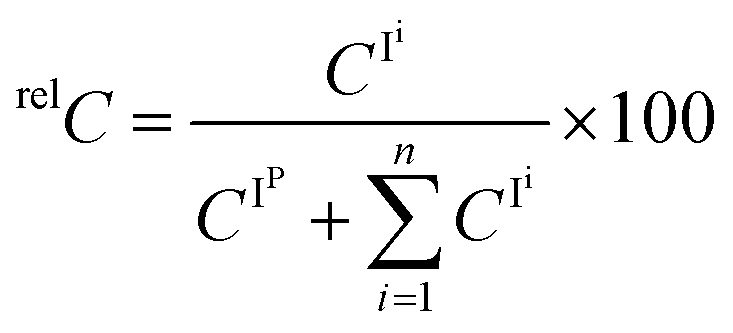 | (1) |
Hydrolysis of oligosaccharides
Acid hydrolysis reactions were carried out in 5 mm NMR tubes containing compound 2 (1.0 mg), 4 (1.0 mg) or 8 (1.5 mg) using 1 M TFA in D2O (0.5–0.6 mL) at 343.15 K. The reaction progress was monitored by one-dimensional 1H NMR spectroscopy at 500 MHz analyzing for the disappearance of pertinent anomeric proton resonances; the approximate degeneracy of the anomeric 1H chemical shifts in 8vs.2 or 4 was confirmed by NMR chemical shift predictions using the CASPER program.46Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge Professor K. F. Aoki-Kinoshita for her suggestions on symbolic nomenclature. This work was funded by grants from the Swedish Research Council (No. 621-2013-4859) and The Knut and Alice Wallenberg Foundation.Notes and references
- Y. Kanie and O. Kanie, Biol. Chem. Compos., 2017, 5, 3, DOI:10.7243/2052-9341-5-3
.
- S. Daikoku, T. Ako, R. Kato, I. Ohtsuka and O. Kanie, J. Am. Soc. Mass Spectrom., 2007, 18, 1873–1879 CrossRef CAS PubMed
- S. Daikoku, T. Ako, A. Kurimoto and O. Kanie, J. Mass Spectrom., 2007, 42, 714–723 CrossRef CAS PubMed
- D. J. Harvey, A. H. Merry, L. Royle, M. P. Campbell, R. A. Dwek and P. M. Rudd, Proteomics, 2009, 9, 3796–3801 CrossRef CAS PubMed
- A. Varki, R. D. Cummings, M. Aebi, N. H. Packer, P. H. Seeberger, J. D. Esko, P. Stanley, G. Hart, A. Darvill, T. Kinoshita, J. J. Prestegard, R. L. Schnaar, H. H. Freeze, J. D. Marth, C. R. Bertozzi, M. E. Etzler, M. Frank, J. F. Vliegenthart, T. Lütteke, S. Perez, E. Bolton, P. Rudd, J. Paulson, M. Kanehisa, P. Toukach, K. F. Aoki-Kinoshita, A. Dell, H. Narimatsu, W. York, N. Taniguchi and S. Kornfeld, Glycobiology, 2015, 25, 1323–1324 CrossRef CAS PubMed
-
Symbol Nomenclature for Glycans (SNFG), Essentials of Glycobiology, ed. A. Varki, R. D. Cummings, J. D. Esko, P. Stanley, G. Hart, M. Aebi, A. Darvill, T. Kinoshita, N. H. Packer, J. H. Prestegard, R. L. Schnaar and P. H. Seeberger, Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press, 3rd edn, 2015, Appendix 1B Search PubMed
- http://www.functionalglycomics.org/static/consortium/Nomenclature.shtml .
- A. Helenius and M. Aebi, Annu. Rev. Biochem., 2004, 73, 1019–1049 CrossRef CAS PubMed
- S. Dejgaard, J. Nicolay, M. Taheri, D. Y. Thomas and J. J. M. Bergeron, Curr. Issues Mol. Biol., 2004, 6, 29–42 CAS
- S.-H. Son, A. Seko, S. Daikoku, K. Fujikawa, K. Suzuki, Y. Ito and O. Kanie, ChemBioChem, 2016, 17, 630–639 CrossRef CAS PubMed
- K. J. Dooresa, C. Bonomelli, D. J. Harvey, S. Vasiljevic, R. A. Dwek, D. R. Burtona, M. Crispin and C. N. Scanlan, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13800–13805 CrossRef PubMed
- P. M. Rudd, M. R. Wormald and R. A. Dwek, Trends Biotechnol., 2004, 22, 524–530 CrossRef CAS PubMed
- S. J. Lee, S. Evers, D. Roeder, A. F. Parlow, J. Risteli, L. Risteli, Y. C. Lee, T. Feizi, H. Langen and M. C. Nussenzweig, Science, 2002, 295, 1898–1901 CrossRef CAS PubMed
- S. J. Van Dyken and R. M. Locksley, Immunol. Cell Biol., 2007, 85, 572–574 CrossRef CAS PubMed
- G. A. Rabinovich and D. O. Croci, Immunity, 2012, 36, 322–335 CrossRef CAS PubMed
- M. G. Paulick and C. R. Bertozzi, Biochemistry, 2008, 47, 6991–7000 CrossRef CAS PubMed
- P. Orlean, Cell Wall, Genetics, 2012, 192, 775–818 CrossRef CAS PubMed
- F. Cuskin, E. C. Lowe, M. J. Temple, Y. Zhu, E. A. Cameron, N. A. Pudlo, N. T. Porter, K. Urs, A. J. Thompson, A. Cartmell, A. Rogowski, B. S. Hamilton, R. Chen, T. J. Tolbert, K. Piens, D. Bracke, W. Vervecken, Z. Hakki, G. Speciale, J. L. Munōz-Munōz, A. Day, M. J. Pen, R. McLean, M. D. Suits, A. B. Boraston, T. Atherly, C. J. Ziemer, S. J. Williams, G. J. Davies, D. W. Abbott, E. C. Martens and H. J. Gilbert, Nature, 2015, 517, 165–169 CrossRef CAS PubMed
- S. K. Maitra and C. E. Ballou, J. Biol. Chem., 1977, 252, 2459–2469 CAS
- R. Lain, K. M. Pamidimukkala, A. D. French, R. W. Hall, S. Abbas, R. Jain and K. L. Matta, J. Am. Chem. Soc., 1988, 110, 6931–6939 CrossRef
- J. Hofmann, H. S. Hahm, P. H. Seeberger and K. Pagel, Nature, 2015, 526, 241–244 CrossRef CAS PubMed
- S. H. Khan, C. F. Piskorz and K. L. Matta, J. Carbohydr. Chem., 1994, 13, 1025–1035 CrossRef CAS
- D. Kotsyubynskyy, M. Zerbetto, M. Soltesova, O. Engström, R. Pendrill, J. Kowalewski, G. Widmalm and A. Polimeno, J. Phys. Chem. B, 2012, 116, 14541–14555 CrossRef CAS PubMed
- S. Daikoku, A. Kurimoto, S. Mutsuga, T. Ako, T. Kanemitsu, Y. Shioiri, A. Ohtake, R. Kato, C. Saotome, I. Ohtsuka, S. i. Koroghi, S. K. Sarkar, A. Tobe, S. Adachi, K. Suzuki and O. Kanie, Carbohydr. Res., 2009, 344, 384–394 CrossRef CAS PubMed
- A. Kurimoto, S. Daikoku, S. Mutsuga and O. Kanie, Anal. Chem., 2006, 78, 3461–3466 CrossRef CAS PubMed
- B. Domon and C. E. Costello, Glycoconjugate J., 1988, 5, 397–409 CrossRef CAS
- A. Kameyama, N. Kikuchi, S. Nakaya, H. Ito, T. Sato, T. Shikanai, Y. Takahashi, K. Takahashi and H. Narimatsu, Anal. Chem., 2005, 77, 4719–4725 CrossRef CAS PubMed
- H. Ito, Y. Takegawa, K. Deguchi, S. Nagai, H. Nakagawa, Y. Shinohara and S.-I. Nishimura, Rapid Commun. Mass Spectrom., 2006, 20, 3557–3565 CrossRef CAS PubMed
- R. Schoenheimer and D. Rittenberg, Science, 1938, 87, 221–226 CAS
- M. L. Wolfrom, A. Thompson and C. E. Timberlake, Cereal Chem., 1963, 40, 82–86 CAS
- K. Ogawa, J.-I. Nishikori, T. Ino and K. Matsuda, Biosci., Biotechnol., Biochem., 1994, 58, 560–562 CrossRef CAS PubMed
- S. Daikoku, G. Widmalm and O. Kanie, Rapid Commun. Mass Spectrom., 2009, 23, 3713–3719 CrossRef CAS PubMed
- P.-E. Jansson and G. Widmalm, J. Chem. Soc., Perkin Trans. 2, 1992, 1085–1090 RSC
- U. Olsson, E. Saewen, R. Stenutz and G. Widmalm, Chem. – Eur. J., 2009, 15, 8886–8894 CrossRef CAS PubMed
- C. Johannessen, R. Pendrill, G. Widmalm, L. Hecht and L. D. Barron, Angew. Chem., 2011, 123, 5461–5463 (
Angew. Chem. Int. Ed.
, 2011
, 50
, 5349–5351
) CrossRef
- R. Laatikainen, M. Niemitz, U. Weber, J. Sundelin, T. Hassinen and J. Vepsäläinen, J. Magn. Reson., Ser. A, 1996, 120, 1–10 CrossRef CAS
- C. Bernlind, S. Oscarson and G. Widmalm, Carbohydr. Res., 1994, 263, 173–180 CrossRef CAS
- S. D. Debenham and E. J. Toone, Tetrahedron: Asymmetry, 2000, 11, 385–387 CrossRef CAS
- P. J. Garegg, J.-L. Maloisel and S. Oscarson, Synthesis, 1995, 409–414 CrossRef CAS
- S. Sarbajna and N. Roy, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1998, 37, 252–256 Search PubMed
- D. Sail and P. Kovác, Carbohydr. Res., 2012, 357, 47–52 CrossRef CAS PubMed
- T. Peters, Liebigs Ann. Chem., 1991, 1991, 135–141 CrossRef
- R. K. Jain, X.-G. Liu, S. R. Oruganti, E. V. Chandrasekaran and K. L. Matta, Carbohydr. Res., 1995, 271, 185–196 CrossRef CAS PubMed
- F. Barresi and O. Hindsgaul, Can. J. Chem., 1994, 72, 1447–1465 CrossRef CAS
- V. K. Srivastava and C. Schurtch, J. Org. Chem., 1981, 46, 1121–1126 CrossRef CAS
- M. Lundborg and G. Widmalm, Anal. Chem., 2011, 83, 1514–1517 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ob02723k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |