
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiomimetic antimicrobial polymers: recent advances in molecular design
Cansu
Ergene
a,
Kazuma
Yasuhara
b and
Edmund F.
Palermo
 *a
*a
aMaterials Science and Engineering, Rensselaer Polytechnic Institute, Troy, NY 12180, USA. E-mail: palere@rpi.edu
bGraduate School of Materials Science, Nara Institute for Science and Technology, 8916-5 Takayama-cho, Ikoma, Nara, Japan
First published on 23rd April 2018
Abstract
The increasing prevalence of antibiotic-resistant bacterial infections, coupled with the decline in the number of new antibiotic drug approvals, has created a therapeutic gap that portends an emergent public health crisis. Since the 1980s, host defense peptides (HDPs) have been recognized as antibacterial compounds that do not induce resistance, but are hampered by their high cost and lack of synthetic scalability. Starting in the early 2000s, synthetic (co)polymers have been designed to mimic the salient physiochemical features of HDPs. These polymers have shown broad-spectrum antimicrobial activity, rapid bactericidal kinetics, and a very low propensity to induce resistance. Systematic optimization of the (co)polymer composition, chain length, hydrophobicity, and cationic charge has generated select examples that are also highly biocompatible (non-hemolytic and non-cytotoxic in vitro). These polymers are derived from inexpensive feedstocks and are produced using cost-effective, scalable processes. Accordingly, such polymers may be viewed as early stage pre-clinical candidates for potential use in pharmaceutical or therapeutic applications. In this review, we focus on the key macromolecular design principles that have been gleaned from more than a decade of structure–activity relationship (SAR) studies, as well as some key mechanistic investigations, across this multidisciplinary field. A fundamental understanding of these functional (co)polymers has arisen from a convergence of ideas in polymer chemistry, microbiology, and biophysics. In this context, we emphasize the recent advances from the past few years and emerging opportunities surrounding the rapidly growing field of HDP-mimetic antimicrobial polymers.
1. Motivation and background
1.1. Host defense peptides
The rise of antibiotic drug resistance in infectious pathogens presents one of the most daunting challenges facing modern medicine.1 Tens of millions of lives are saved annually by the routine use of broad-spectrum antibiotics. Without these indispensable drugs, society would abruptly return to the status quo ante of the 1930s, an unimaginably bleak prospect by modern standards. For example, procedures such as caesarean section birth carried a disturbingly high risk of mortality prior to the advent of antibiotics.Host defense peptides (HDPs) are components of innate immunity expressed by all multicellular organisms2–4 (representative structural examples are shown in Fig. 1). The putative function of an HDP, at least in part, is to kill bacteria without harming host cells and without inducing resistance. The ability to exert broad-spectrum activity without causing resistance is related to their mechanism of action, which is thought to involve membrane permeabilization.5–7 Instead of a precise “lock and key” mechanism, the membrane disruption pathway is much less specific and thus more challenging for bacteria to circumvent. Cationic residues (Lys and Arg) in the HDPs experience electrostatic attraction to anionic components of the bacterial cell membrane whereas their abundant hydrophobic residues readily insert in the non-polar membrane core. Such peptides have been shown to permeabilize model liposome vesicles8 and to induce leakage of potassium ions and enzymes from bacterial cells, presumably leading to concomitant cell death. Electron microscopy,9 confocal microscopy, and even high-speed AFM10 of bacterial cells upon exposure to HDPs and related compounds also support the notion of membrane permeabilization. It is important to note that the antibacterial activity of HDPs is in fact more specific than originally thought; there is convincing evidence that defensins, for example, sequester microbe-specific lipid receptors (lipid II), inhibiting cell wall biosynthesis.11 In this review, however, we focus on the HDP-mimetic design and optimization of synthetic polymers that aim to capture the essence of a non-specific membrane-disruption mechanism.
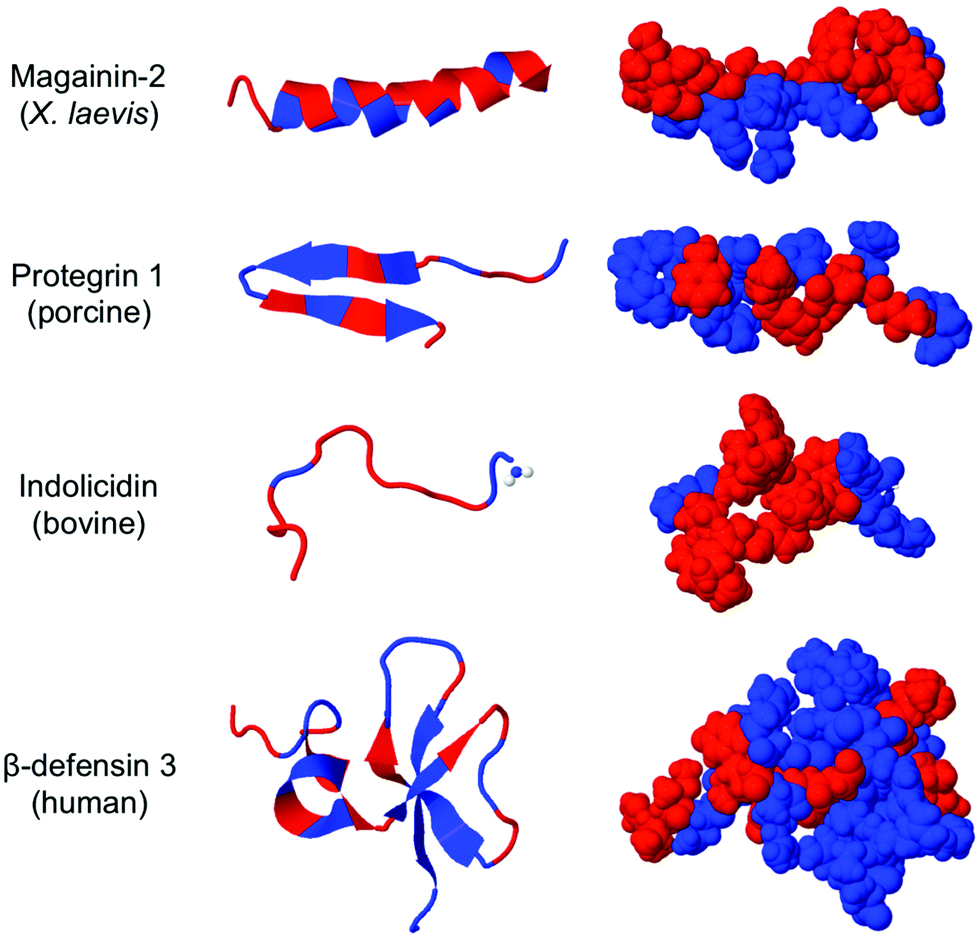 | ||
| Fig. 1 Molecular models of four HDPs. The hydrophobic residues are red and the hydrophilic residues are blue. There is a lack of conserved secondary structure but hydrophobic residues appear to cluster into distinct domains. The images were generated using the RCSB Protein Databank (http://www.rcsb.org/pdb/home/home.do). | ||
One of the earliest discoveries of an HDP was Magainin-2, isolated from the skin of the African clawed frog Xenopus laevis, by M. Zasloff in 1987.12 Since that time, thousands of other antimicrobial peptides have been discovered and it is now understood that they exist in all multicellular organisms. Wang and Wang maintain a detailed online database of all known antimicrobial peptides (AMPs), with 2903 current entries.13–15 Among this class, there is a remarkable lack of conserved sequence or preferred secondary structure. Rather, the commonalities seem to be physiochemical in nature: AMPs (1) possess cationic charge at neutral pH, (2) contain a substantial fraction of hydrophobic residues, and (3) are relatively short chain peptides (Fig. 2). The hydrophobic groups are key to their interaction with phospholipid bilayers and the cationic charge is considered crucial for their electrostatic attraction to anionic components of the bacterial cell surface. Non-natural analogs of HDPs, such as all-D peptides,16,17 β-peptides,18–20 and peptoids,21,22 successfully reproduced the antibacterial activity and low toxicity to human cells that are the hallmarks of HDP efficacy, simply by designing sequences that possess the requisite cationic amphiphilicity. These abiotic compounds are referred to as antimicrobial peptides “AMPs”, but they are not HDPs.
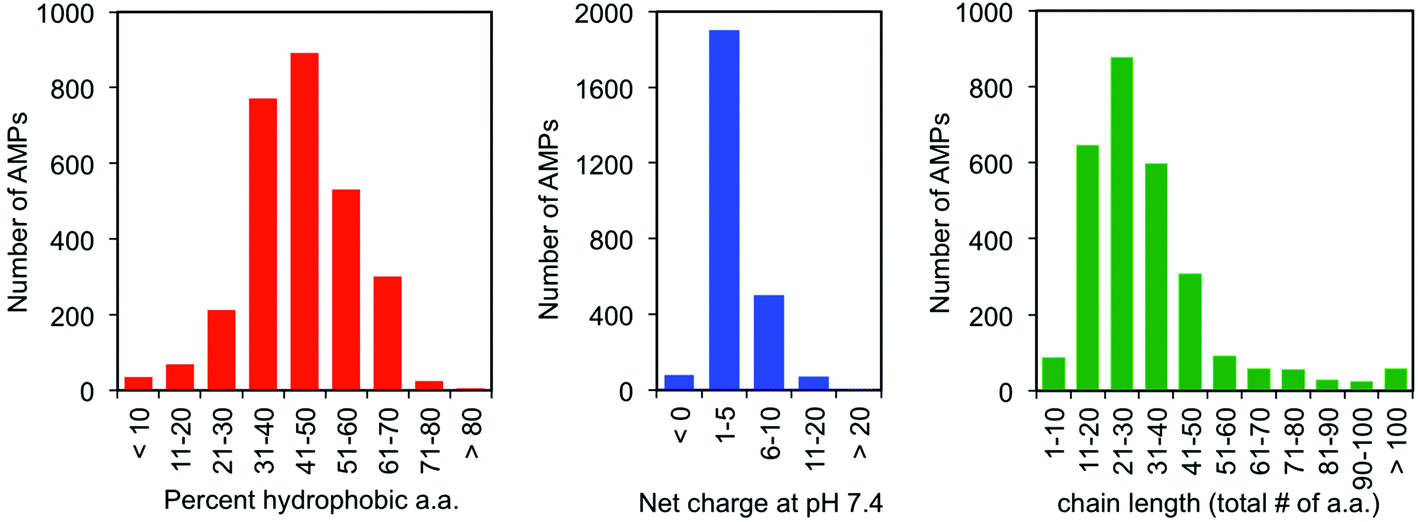 | ||
| Fig. 2 Despite their lack of conserved sequence or secondary structure, there are three common physiochemical characteristics of antimicrobial peptides: (a) hydrophobicity, (b) cationic charge, and (c) short chain length. The data were collected from The Antimicrobial Peptide Database (http://aps.unmc.edu/AP/structure.php). | ||
As an interesting side note, a small fraction of AMPs appear to bear a net negative charge at neutral pH. One such example is the surfactant-associated anionic peptide (SAAP, charge −7) from Ovis aries, which only exerts activity when complexed with Zn2+ ions.23 Another anionic example is microplusin (charge −8), which requires binding to Cu2+ as a cofactor to exert activity against bacteria.24 Although the number of such anionic AMPs represents less than 3% of the total, it is possible that they possess some unique characteristics worthy of biomimicry.
The conspicuous lack of any conserved sequence or defined secondary structure led DeGrado's group to the hypothesis that synthetic (co)polymers – with all their structural imperfections and heterogeneities – could perhaps be designed to capture the essential physiochemical features of HDPs that are key to their antimicrobial activity. Indeed, this strategy has been widely employed by numerous research groups over the past decade with a great deal of promise. In this review, we will focus on the key structural determinants of antibacterial activity, and toxicity to human cells, in this class of peptide-mimetic macromolecules. We begin with summarizing the classical design approaches to cationic amphiphilicity and then expand on this idea to include the most recent advances. We also summarize the relatively limited, but rapidly growing, number of studies on in vivo activity, and finally we review the current understanding of their mechanism(s) of bactericidal action. This article is by no means intended to represent an exhaustive summary of all the valuable contributions to the field, and the keen reader is encouraged to reference a large number of other excellent reviews.25–35 Here, we place substantial emphasis on new work published in the past two years, which are not covered by numerous prior reviews, in addition to a summary of the major achievements of the past 10 years that have laid the foundation for future development.
1.2. Convergent trends: HDPs and benzalkonium chlorides
Concurrently with the discovery of HDPs, conceptually and structurally related polymeric disinfectants were being developed in the 1980s,36 although there appears to have been little connection made between these two areas initially. Common commercial biocides containing the so-called “benzalkonium chlorides” (BACs) are cationic surfactants composed of a benzylic quaternary ammonium salt (QAS) and a long alkyl chain (C8H17 to C18H37). The monomeric surfactants indiscriminately lyse biomembranes at high concentrations, regardless of cell type (i.e. they are “biocidal”). Thus, BACs are bactericidal but also toxic to mammals.In 1984, Ikeda and co-workers reported the polymerization of a styrene derivative that contained a pendant BAC surfactant in the side chain (Fig. 3).37 Not surprisingly, both the styrenic monomer and the polymer thereof exerted antibacterial activity, albeit in the mg mL−1 concentration range. The toxicity data were not shown, but the intensely hydrophobic nature of the polymer side chains, as well as the high concentrations of the polymer employed, suggests that these materials would almost certainly exert surfactant-like membrane lysis regardless of the cell type.
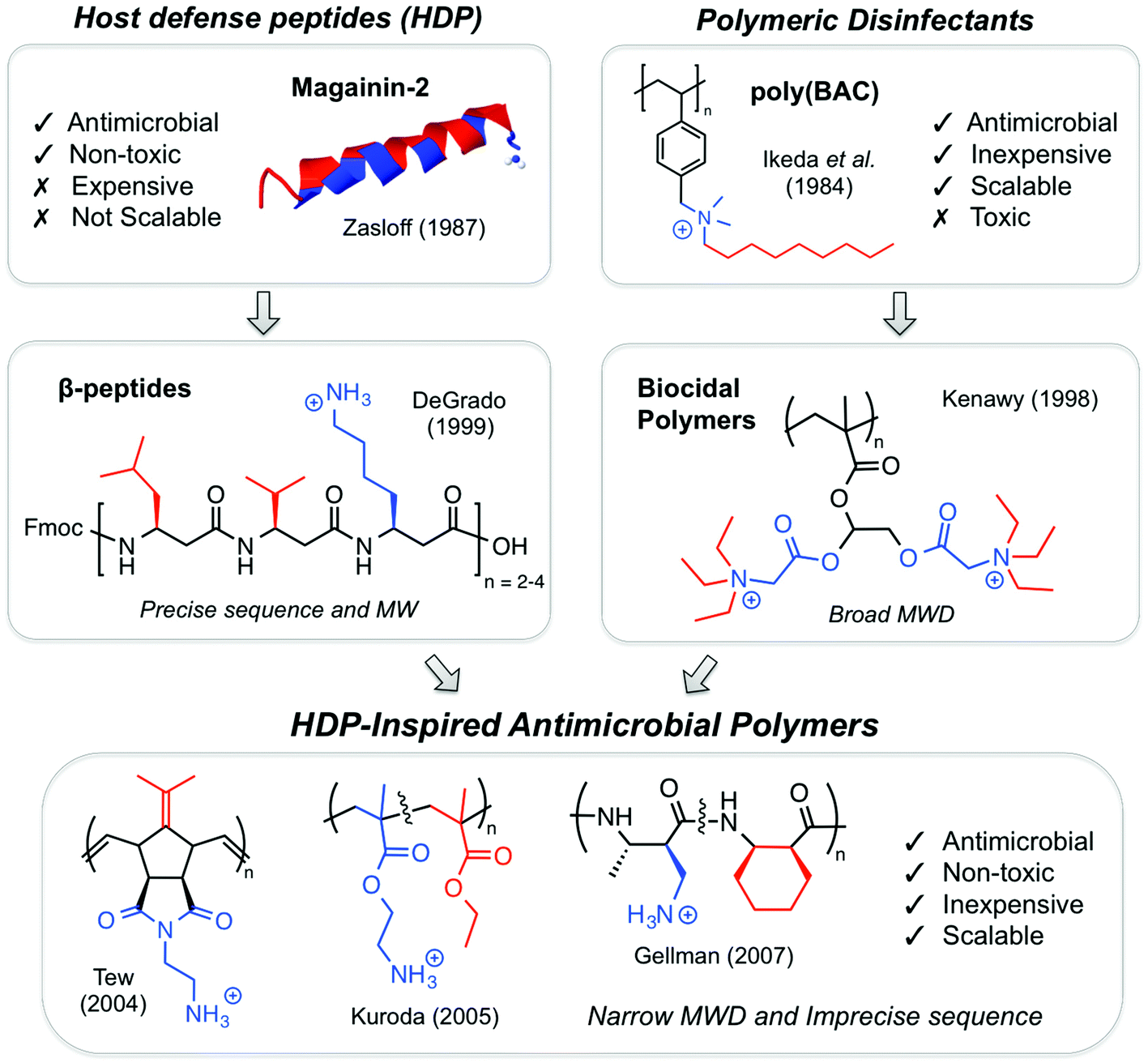 | ||
| Fig. 3 The evolution of antimicrobial polymers. Molecular design principles were gleaned from a convergence of ideas on antimicrobial peptides and synthetic polymer disinfectants. HDP-inspired polymers are intended to combine the advantages of peptide activity with the cost-effectiveness and scalability of synthetic polymer chemistry, without the need for precise sequence, unimolecular chain length, or defined secondary structure. | ||
During the late 1990s, HDPs inspired various peptidomimetics such as β-peptides,20,38 α/β-peptides39 and peptoids40 with antimicrobial activity. In parallel, poly(BAC)s inspired biocidal polymers with a variety of cationic and hydrophobic substituents. By the mid-2000s, the two fields effectively converged into HDP-mimetic polymers, and the field began to experience rapid growth that continues today (Fig. 4). The convergence of trends is a prime example of how multidisciplinary science can vertically advance technologies at the interface of conventionally unrelated fields.
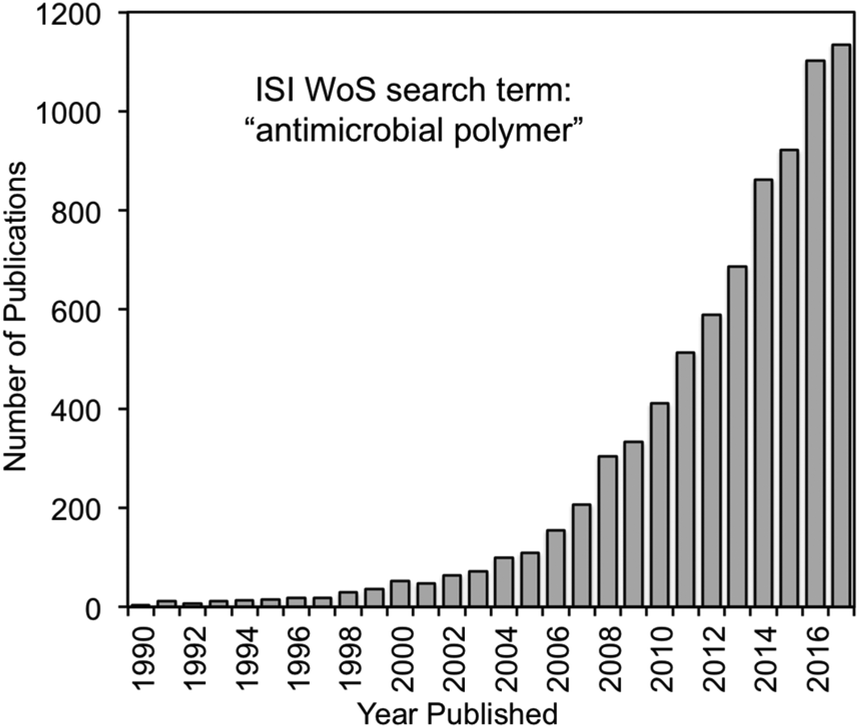 | ||
| Fig. 4 Number of publications as a function of time that contain the phrase “antimicrobial polymer” via ISI Web of Science. These data include the HDP-mimetic polymer literature (the scope of this review), as well as biocidal polymers, biopolymers, polymer/drug conjugates, and composites of polymers with antibacterial inorganics. | ||
In this comparative context, it is instructive to consider the bee venom toxin melittin.41 This peptide is 26 a.a. residues in length, contains 46% hydrophobic residues, and bears a +6 cationic charge at neutral pH. Viewing these data alone, the structure may appear to have more in common with HDPs than with biocidal polymers. However, the activity profile of melittin is more similar to that of a QAS-containing disinfectant: it completely lyses both bacterial cells and human red blood cells alike at approximately the same concentrations. Such a comparison invites one to ponder: What are the key design principles that distinguish a cell-selective antibacterial macromolecule from a biocidal one? Can biocidal polymers be re-optimized to confer cell-type selectivity?
1.3. Terminology
Before a discussion of structure–activity relationships (SAR), the technical jargon of this field deserves an explanation for the non-expert (Table 1). The MIC, a widely used metric, is the lowest concentration of a polymer that prevents the proliferation of bacteria. The MIC is the bacteriostatic concentration at which bacteria do not grow (but may or may not be dead). In contrast, the MBC reflects the bactericidal concentration at which most or all of the bacterial cells are confirmed dead. The MBC represents at least a 99.9% (or 99.99%) reduction in colony-forming units (CFU) per mL. The MBC test is more labor intensive because it requires colony counting on nutrient agar plates, but is more informative. Frequently for antibacterial polymers, the MIC and MBC values are identical or very similar, but it must always be the case that MIC ≥ MBC for a given set of test conditions (it is not possible to kill a cell but also fail to inhibit its growth, whereas the converse is indeed possible).| Term | Definition | Comments |
|---|---|---|
| Minimum inhibitory concentration (MIC) | The lowest concentration of a polymer that completely inhibits the growth of a microorganism in nutrient media. | • Turbidity-based, high-throughput assay |
| • Lower MIC = better activity | ||
| • Bacteriostatic activity | ||
| Minimum bactericidal concentration (MBC) | The lowest concentration of a polymer that reduces bacterial cell viability by at least 99.9% of the initial inoculum. | • Colony counting, low-throughput assay |
| • Lower MBC = better activity | ||
| • Bactericidal activity | ||
| Hemolytic concentration (HC50) | The concentration that induces 50% release of hemoglobin from red blood cells in buffer. | • Colorimetric, high-throughput assay |
| • Lower HC50 = worse toxicity | ||
| Lethal concentration (LC50) | The concentration that reduces mammalian cell viability by 50% in culture media. | • Colorimetric, high-throughput assay |
| • Lower LC50 = worse toxicity |
The most commonly used measure of toxicity to human cells is the hemolysis assay. At the HC50 concentration, a compound induces 50% release of hemoglobin from a suspension of RBCs. This colorimetric assay is amenable to high throughput, and provides a quantification of the membrane-lytic activity against mammalian cell membranes. Cytotoxicity against mammalian cell cultures in vitro is less frequently reported but arguably more informative. The concentration of the polymer that causes a 50% reduction in cell viability is termed the LC50. Various cell lines (HeLa, HEp-2, Cos7, Jurkat, etc.) along with various high-throughput colorimetric assay kits (LDH, MTT, XTT, etc.) are routinely employed to that end.
It is important to recognize that all the benchmark activity values are highly assay-dependent.42 The same polymer can show different MIC values, for example, based on the type of assay media, the initial concentration of bacteria in the inoculum (typically ∼106 CFU mL−1), and – surprisingly – even the material of which the microplate is made. Melittin gives a 4× higher MIC value when tested on “tissue-culture treated” polystyrene microplates compared to polypropylene microplates, all else being equal. Similarly, the HC50 value can vary significantly depending on the pH and the concentration of the RBCs in the buffer (typically 107 cells per mL, with higher RBC concentrations resulting in higher HC50). For these reasons, it is important to view MIC and HC50 literature values relative to a standard of known activity (such as Magainin-2 or Melittin) tested under strictly identical assay conditions.
The terms “antibacterial” and “antimicrobial” are distinct: the former is a compound that inhibits (or kills) bacterial cells, whereas the latter is one that inhibits (or kills) all microorganisms, including bacteria, yeast and viruses. Although sometimes used interchangeably, “antibacterial” is the more accurate term for a polymer that has not been tested against yeast or viruses, strictly speaking. The terms “host defense peptide” (HDP) and “antimicrobial peptide” (AMP) are also subtly distinct: the former refers to the naturally-occurring components of innate immunity in host organisms, whereas the latter is any peptide with antimicrobial activity, whether it be a natural HDP or a synthetic peptide. Finally, the term “biocidal” refers to a compound that indiscriminately kills both prokaryotic and eukaryotic cells.
In this article, we denote the concentration range of 1–10 μg mL−1 as “very potent” activity, whereas the range of 10–100 μg mL−1 is called “moderate” activity, 100–1000 μg mL−1 is “weak” and >1000 μg mL−1 is “inactive”. These ranges are only intended as a rough guide to facilitate our discussion and are by no means intended as strict rules.
2. Structure–activity relationships (SAR)
2.1. Fundamental design principles
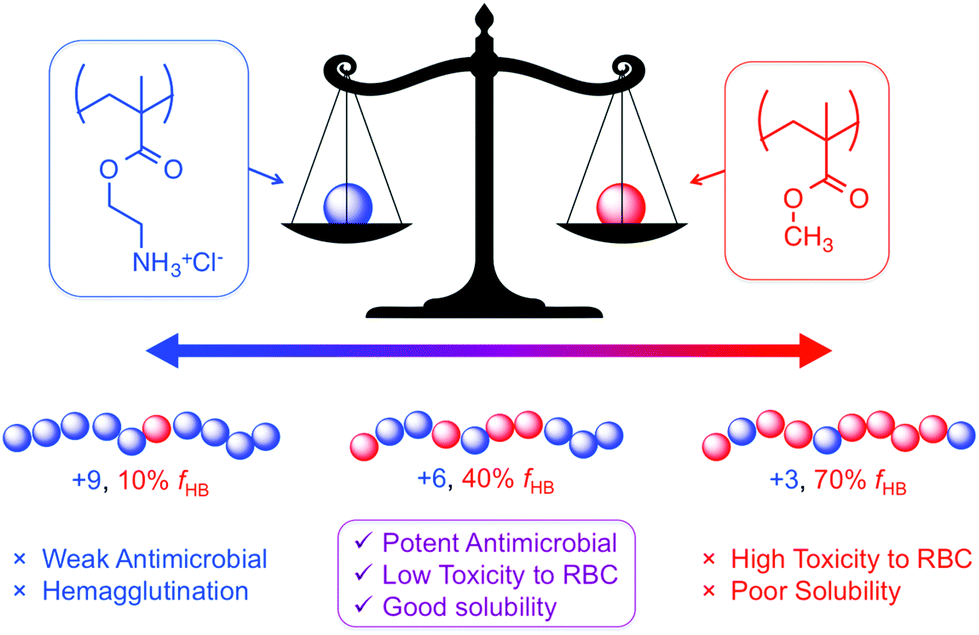 | ||
| Fig. 5 Amphiphilic balance is a widely employed concept for the optimization of copolymer composition in antibacterial polymers. In random copolymers of methacrylates, the ratio of cationic to hydrophobic comonomer dictates the antibacterial and hemolytic activities. | ||
It should be acknowledged that the concept of “amphiphilic balance” in fact predates HDP-mimetic polymers; it was extensively discussed for biocidal polymers since the 1990s.36 Benzalkonium chlorides, and their polymeric derivatives, contain very long alkyl chains (up to C18) attached to their cationic QAS groups and are toxic to human cells. In stark contrast, the most hydrophobic amino acid is isoleucine (with a C4 side chain). Thus, the use of modestly hydrophobic side chain groups (C1–C4) in HDP-mimetic polymers is a crucial factor determining their cell-type selectivity. It has been shown that the overall hydrophobicity of the polymer will dictate the hemolytic toxicity.43 The fundamental design principle is therefore quite simple: incorporate the minimum amount of hydrophobic content needed to confer antibacterial activity. Fig. 6 shows the diversity of structural modifications that have been carried out on the polymethacrylate platform in order to optimize the balance of hydrophobic and cationic character.
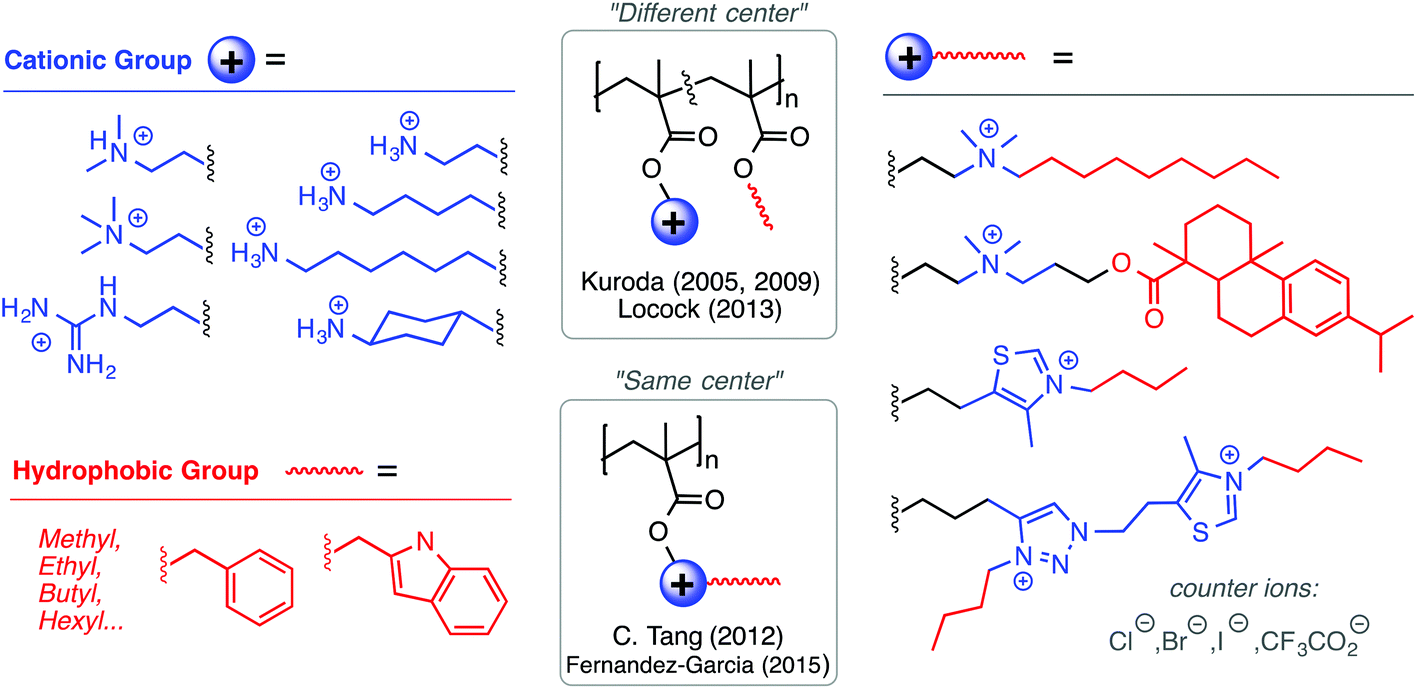 | ||
| Fig. 6 The broad diversity of cationic and hydrophobic group structures, and their relative spatial arrangements, have been extensively tailored in the poly(methacrylate) design platform towards the optimization of biological activity. | ||
Among the polynorbornene derivatives studied by the Tew group (Fig. 7), a variety of different cationic structures have been utilized. Primary ammonium cations were used first52 and most frequently,53–55 but pyridinium56 and guanidinium groups54 have also been utilized. In one study for example, Tew et al. showed that increasing the density of amine groups on each monomer unit enhances the efficacy of the polymers by decreasing hemolytic toxicity substantially.57 Charge density was also examined by the Yan group in the context of geminized polyacrylates containing multiple same-centered QAS amphiphiles in each repeat unit. They found that the increased charge density per monomer unit enhanced antibacterial activity.58
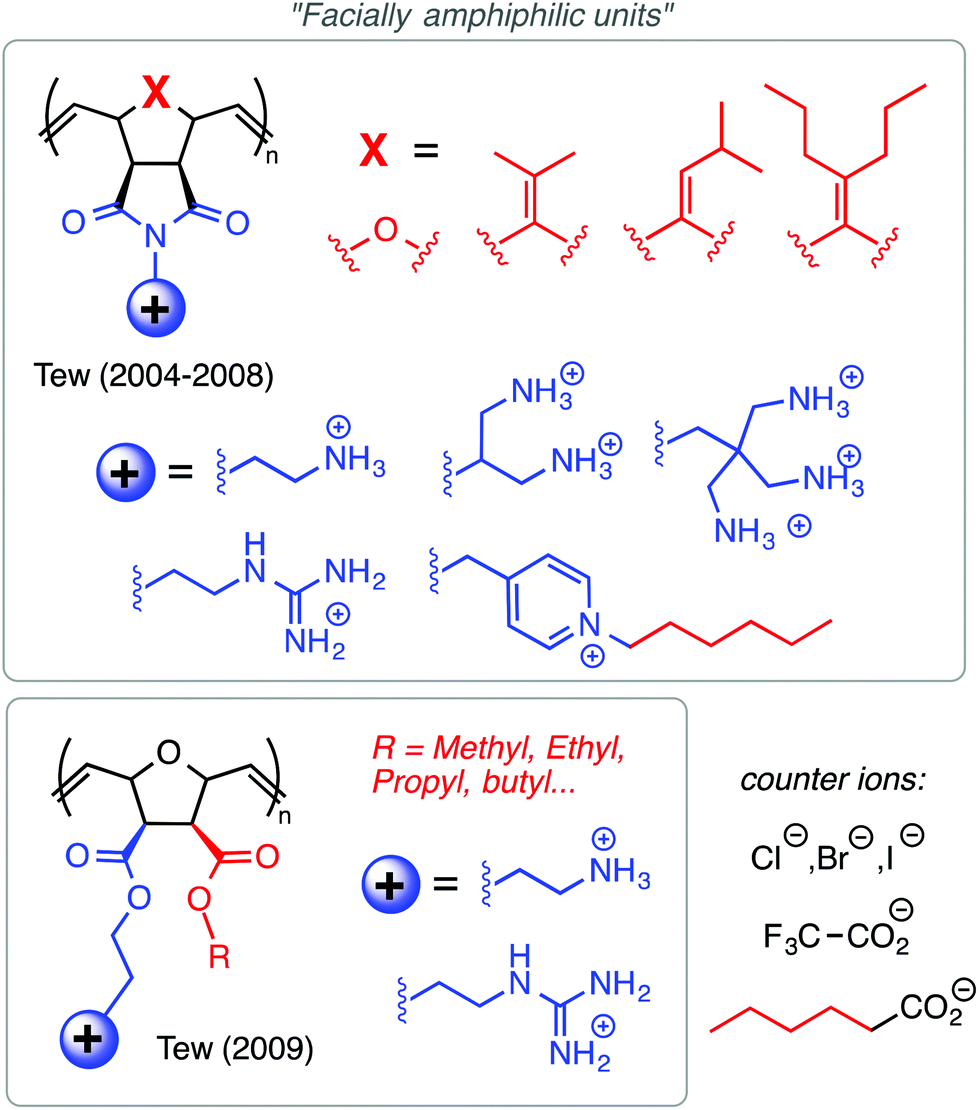 | ||
| Fig. 7 Examples from the polynorbornene design platform of Tew and co-workers. The identity and density of cationic charges, as well as the identity, relative amount, and spatial arrangement of the hydrophobic groups, have been extensively optimized. | ||
Hedrick, Yang, and co-workers have extensively studied antibacterial polycarbonates (some example structures are given in Fig. 8) with a variety of different QAS groups, including pyridinium and imidazolium.59,60 These materials showed remarkable selectivity for S. aureus relative to human RBCs (HC50/MIC > 250) but were less effective against Gram-negatives and C. albicans. The J. Cai group studied amphiphilic polycarbonates (block and random) with primary ammonium functionality in the side chains, as compared to the QAS groups, and found potent selectivity against multidrug-resistant Gram-positives but not Gram-negatives.61
 | ||
| Fig. 8 Polycarbonates prepared by organo-catalyzed ROP have been structurally tuned to optimize the hydrophobic and cationic properties. | ||
Besides high Lys content, AMPs are also enriched in Arg as a source of cationic charge. The guanidinium group in Arg is known to complex anionic phospholipids by a combination of Coloumbic attraction and precisely orientated bidentate hydrogen-bonding interactions. Because anionic phospholipids are present on the outer leaflet of bacterial cell membranes, several groups have reported cationic amphiphilic polymers containing the guanidinium functional group as the source of cationic charge, instead of primary ammonium cations. Locock's group has shown that polymethacrylates with guanidinium cations were more active against S. epidermidis and C. albicans relative to primary ammonium-containing analogues.62 Morgan and co-workers observed similar effects in guanidinium-functionalized poly(methacrylamide)s.63 Recently, the Tew group has pioneered guanidinium-rich polymers as mimics of cell-penetrating peptides,64,65 which is very interesting but outside the scope of this review.
Fernández-García and co-workers developed polymethacrylates bearing thiazolium and triazolium cations, which gave excellent antibacterial activity (4–8 μg mL−1) against P. aeruginosa and S. aureus with very high HC50 values (>5000 μg mL−1).66 Tang and co-workers have pioneered the use of natural products in antimicrobial formulations. In one example,67 they used tertiary amine groups quaternized with a derivative of the natural product rosin, which showed moderate antibacterial activity against E. coli and S. aureus. The same group has shown that cationic polymethacrylates bearing cationic metallocene groups (cobaltocenium) kill multidrug-resistant bacteria and can also complex with small molecule antibiotic drugs in a synergistic formulation, paving the way for polymers to reinvigorate antibiotics that are presently considered obsolete.68
Tew's group has employed a strategy that is distinct from both the “same center” and “different center” motifs: in their polynorbornene derivatives, the cationic group and the hydrophobic group reside within the same repeat unit but are not directly attached to one another (Fig. 7). Rather, norbornenes with a hydrophobic group on one side and a cationic group on the other side are sometimes referred to as “facially amphiphilic” monomer units. The polymer chain composed of such units possesses conformational freedom, but is expected to adopt an overall facially amphiphilic conformation upon binding to biomembranes. It is reasonable to speculate that the monomer-level facial amphiphilicity may facilitate the formation of facially amphiphilic conformations at biointerfaces.
The Tew group presented a direct comparison of the “facially amphiphilic” monomer approach versus the “different center” approach, in polynorbornenes, and they showed a profound difference in activity and membrane-disruption ability in model membranes.75 The Gellman group has argued that flexible random copolymers will naturally adopt “globally” amphiphilic conformations upon binding to biomembranes, without the need (or even the desire) for pre-programmed facial amphiphilicity at the monomer level or in terms of defined secondary structure.46 Kuroda recently advocated a holistic view of antimicrobial polymer structures, emphasizing that these polymers should be designed to mimic the globally amphiphilic properties of the whole HDP molecule, instead of focusing on side chain identity.76
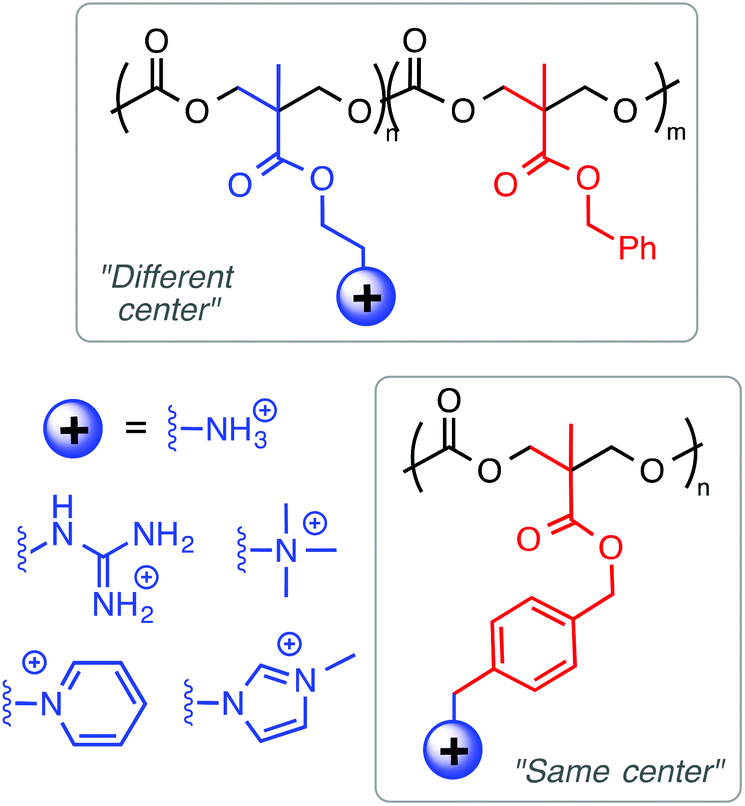
Considering the combined results from the “same center”, “different center” and “facially amphiphilic” approaches (Fig. 9), it is clear that each one requires independent optimization to yield antibacterial potency and low toxicity to RBCs. Overall, it is not clear whether any one strategy is fundamentally “better” than either of the other two: all three have yielded select examples with excellent activity profiles in terms of MIC (low μM) and HC50 (>1000 μM) in vitro. More studies that show a direct comparison between these approaches in terms of cytotoxicity and in vivo activity would perhaps clarify this issue further.
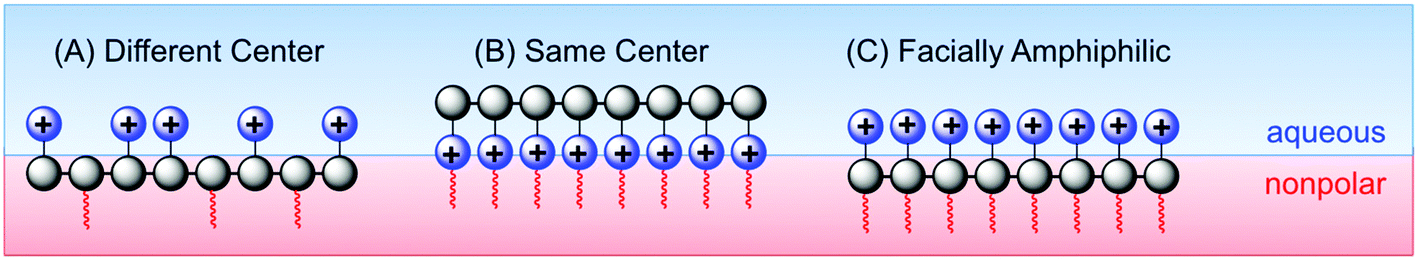 | ||
| Fig. 9 The relative spatial arrangements of cationic and hydrophobic groups in an antibacterial polymer generally fall into three categories: (A) “different-centered”, (B) “same-centered” and (C) “facially” amphiphilic structures. The approach in part (A) is evocative of the HDP primary structure, whereas (C) more closely mimics the typical secondary structure of a membrane-bound HDP. The approach in (B) appears similar to that of poly(BAC) disinfectants, but can exert activity similar to that of HDPs in some cases. | ||
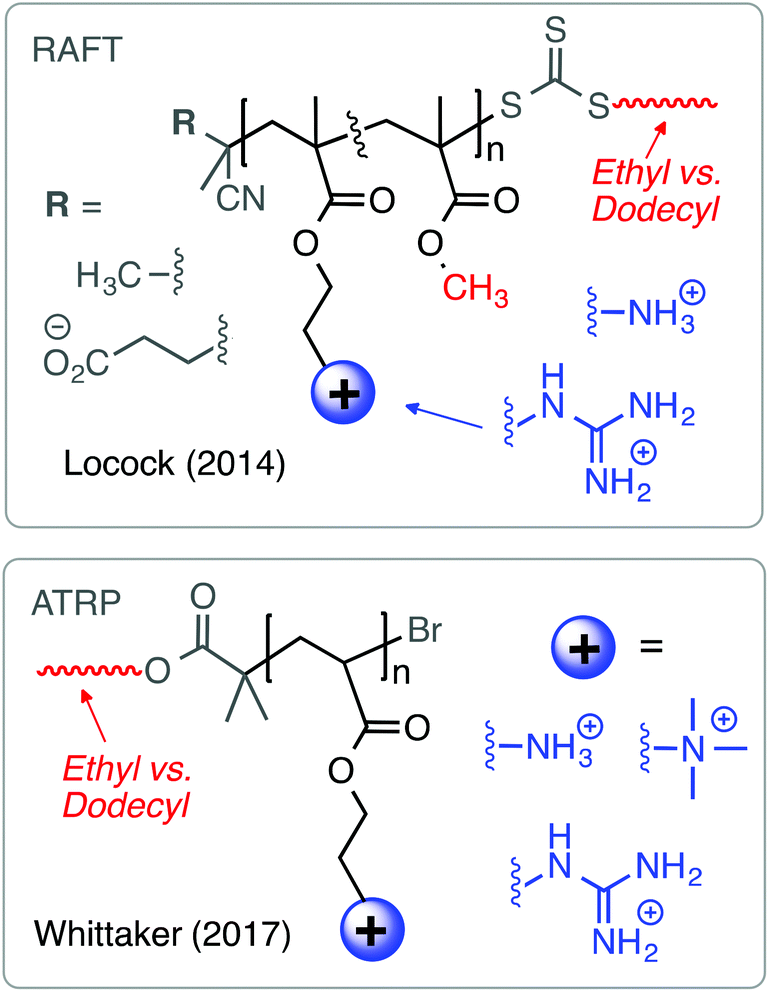 | ||
| Fig. 10 End group affects the antibacterial and hemolytic activity in polymethacrylates prepared by RAFT and polyacrylates prepared by ATRP. | ||
2.2. Emerging SAR trends
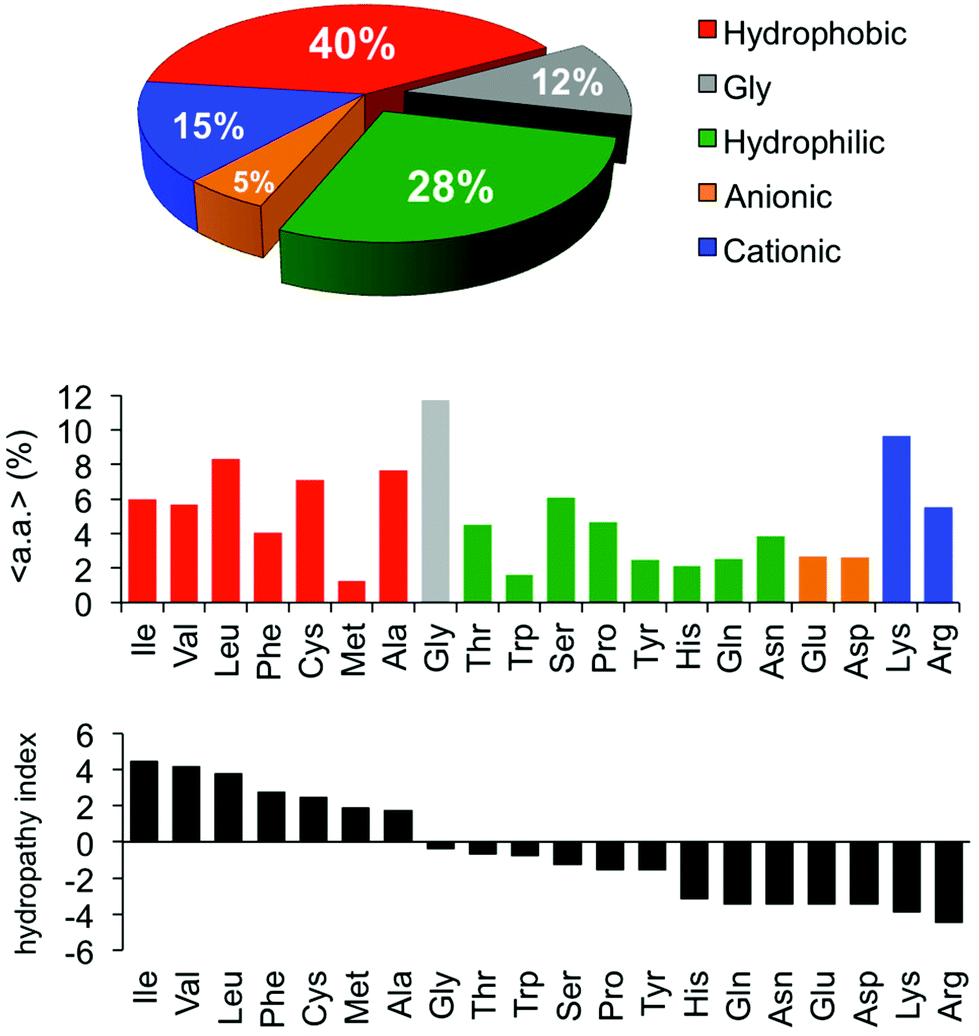 | ||
| Fig. 11 Averaged composition of all 2903 AMPs in the database, percent incidence of each a.a. and their hydropathy indices. A substantial fraction (28%, or 40% including Gly) of the residues are neutral and hydrophilic. This portion of the AMP composition, previously neglected in binary systems, is mimicked effectively in ternary (co)polymer systems. | ||
One of the earliest examples of a ternary antimicrobial polymer with neutral, hydrophilic groups, in addition to cationic amphiphilicity, was reported by Youngblood and co-workers in 2007 (Fig. 12).83 They copolymerized 4-vinyl pyridine (4VP) with PEG methacrylate and then quaternized the pyridine groups with alkyl halides. It was previously known that quaternized poly(4VP) is inherently antibacterial but also highly hemolytic. Incorporating the PEGylated comonomer reduced the hemolytic toxicity and did not abrogate the antibacterial properties. Thus, by including a third component in the design, these authors effectively demonstrated that a nominally biocidal polymer can be modified to confer cell-type selectivity.
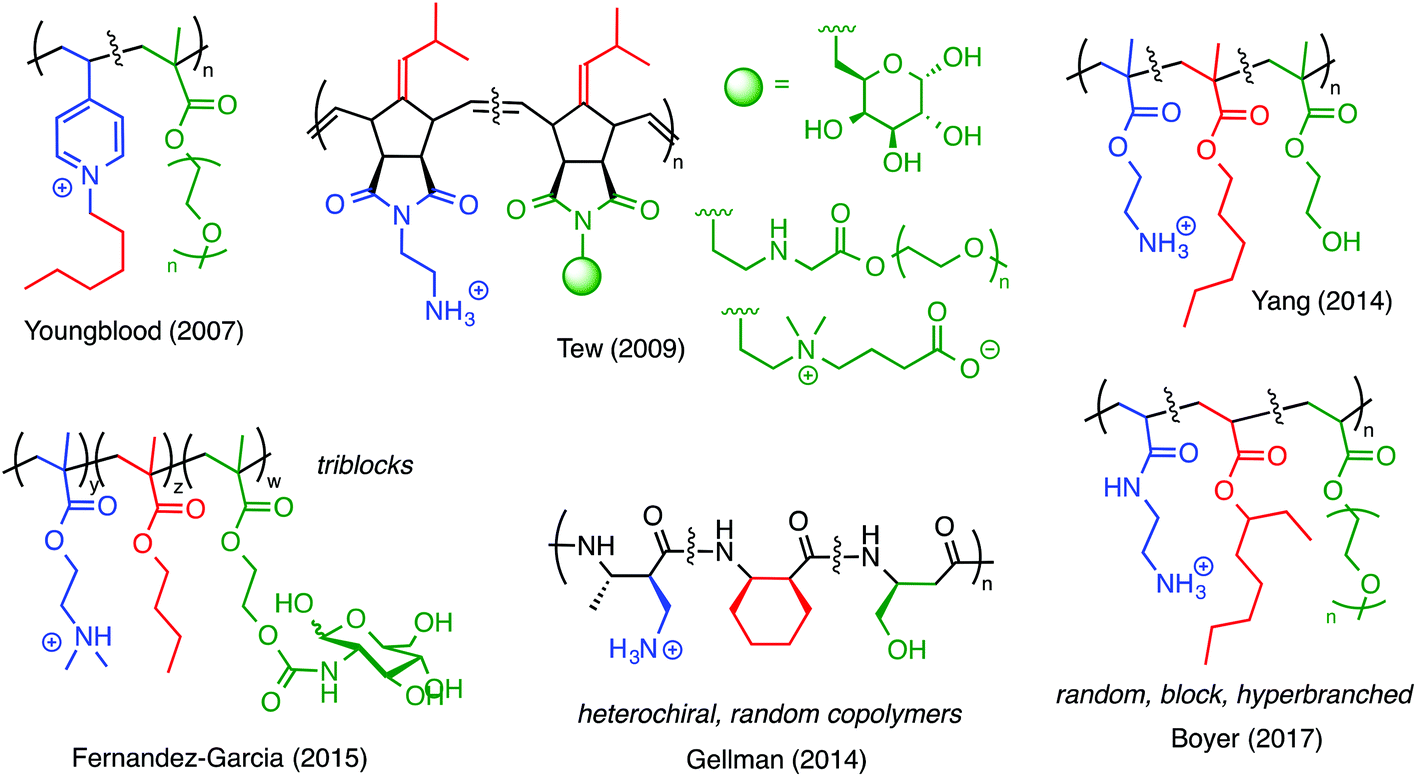 | ||
| Fig. 12 Ternary amphiphilic polymers containing hydrophobic, cationic, and neutral/hydrophilic groups (hydroxyl, sugar, PEG, and zwitterionic). The inclusion of the third component reduces the hemolytic toxicity without sacrificing antibacterial potency. | ||
Numerous other examples (Fig. 12) have since shown that the same design principle can be applied to polymers of norbornenes,84 methacrylates,85 and nylon-3 type materials.86 PEG has frequently been employed for such purposes, but hydroxyl substituents (which mimic Ser residues),87,88 sugars,89,90 and zwitterionic moieties84 also give comparable results. In our opinion, these ternary systems are likely promising candidates for continued study.
G. G. Qiao and co-workers used PAMAM dendrimers to initiate the ring-opening polymerization of N-carboxyanhydrides to yield particles decorated with random copolymers of Lys and Val (Fig. 13).93 These nanoparticles exhibited potent activity against Gram-negative bacteria, were non-toxic to human cells, and did not induce drug resistance. It is critically important for in vivo applications that serum proteins do not abrogate the activity of the polymers, although some interaction between a polycation and a protein in the media is likely unavoidable. The dendrimer-based nanoparticles were remarkably active even in the presence of simulated body fluid and blood serum, which mimic the in vivo environment. Interestingly, the presence of added divalent salt slowed the kinetics of E. coli outer membrane permeabilization, which suggests that ion exchange may play an important role in their mechanism of action.
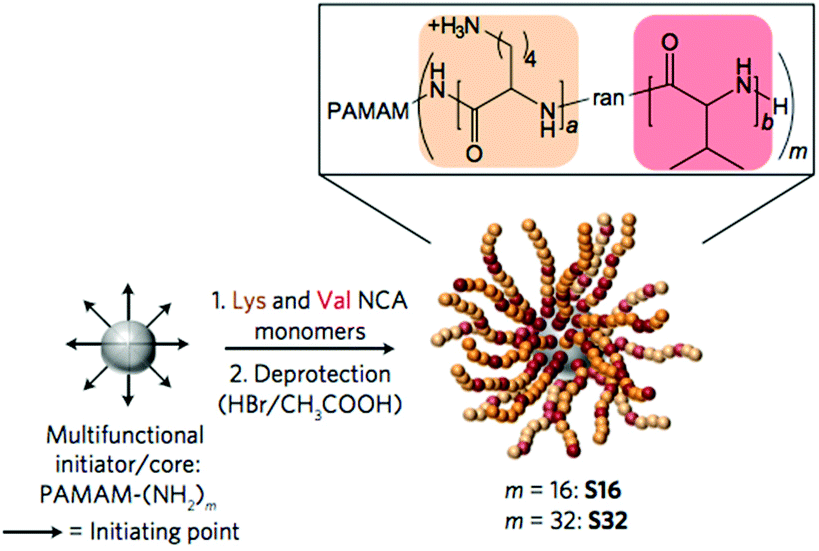 | ||
| Fig. 13 Antibacterial nanoparticles by ring-opening polymerization initiated by a PAMAM dendrimer core. This unique chain architecture influences the mechanism of activity relative to linear chains. Reproduced with permission from ref. 93, copyright 2016, Springer Nature. | ||
Antibacterial nanostructures composed of spherical and rod-shaped graft copolymers that display quaternized poly(vinyl pyridine)s showed a strong dependence of activity on chain architecture.94 Interestingly, the smaller spherical-shaped nanostructures exhibited stronger antibacterial activity as compared to rod-like graft copolymers and linear polymer chains.
Combined, the results from these studies strongly support the notion that polymer chain microstructure, chain architecture, and nanostructures are important determinants of biological activity. Efforts to examine the role of sequence, tacticity, regioregularity, and chain architecture appear worthwhile and ought to be the focus of more attention in the future.
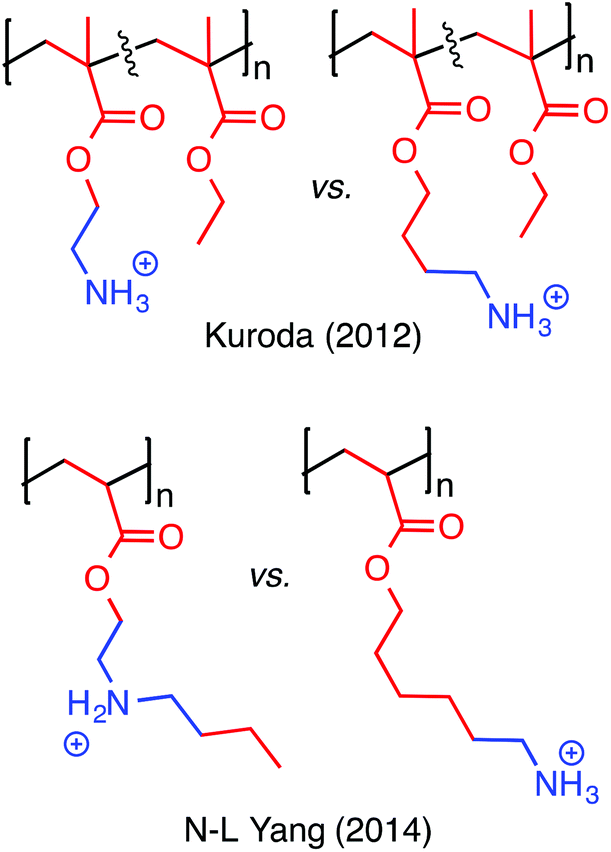 | ||
| Fig. 14 The length of the spacer arm that connects a cation to the polymer backbone is a key design parameter. | ||
Ergene and Palermo developed the first generation of self-immolative antibacterial polymers122 (Fig. 15). They synthesized poly(benzyl ether)s with pendant allyl side chains and silyl ether end-caps (responsive to fluoride ions),123 following the route reported by Phillips and co-workers. They further modified side chains with cysteamine HCl via photo-initiated thiol–ene chemistry. Cationic poly(benzyl ether)s bearing primary ammonium groups exerted potent, rapid and broad-spectrum antibacterial activity, but were also highly hemolytic. These antibacterial polycations displayed the ability to unzip into their small molecules when activated by a specific external stimulus. Upon triggered depolymerization, bactericidal activity was mostly maintained, while the hemolytic activity was remarkably decreased.
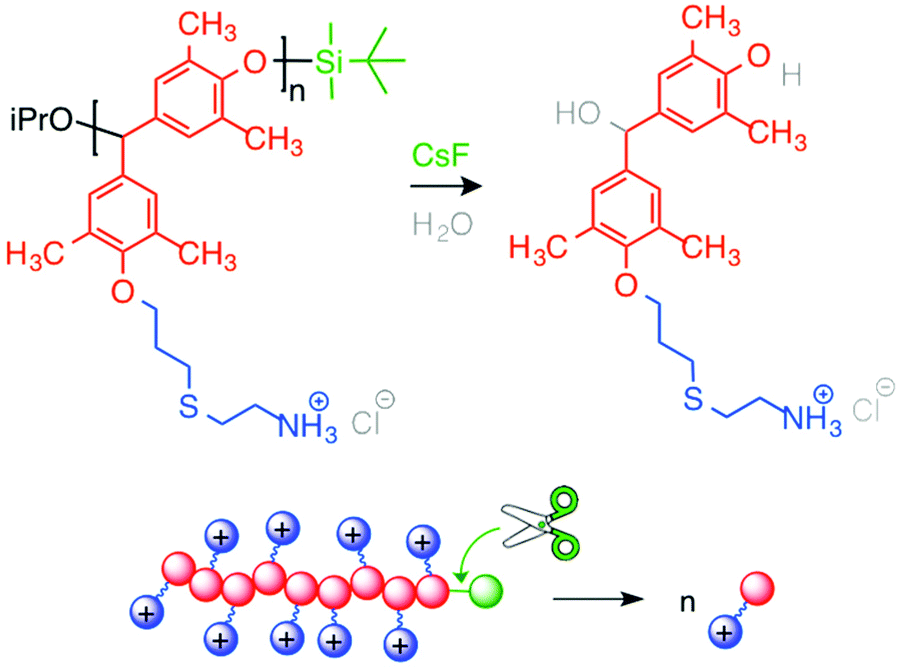 | ||
| Fig. 15 Self-immolative antibacterial polymers spontaneously unzip into small molecules in response to a specific chemical stimulus. | ||
2.3. Remaining questions
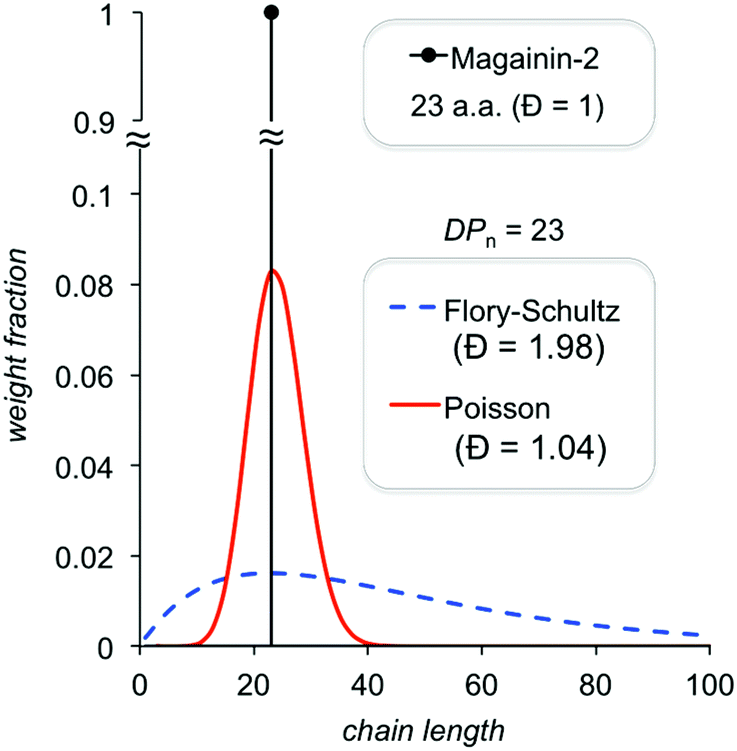 | ||
| Fig. 16 Comparison of theoretical chain length distributions for controlled and uncontrolled polymerizations. | ||
There is also dispersity in terms of copolymer composition, which is difficult to quantify, whenever a “different center” approach is employed (compositional dispersity is not an issue for the “same center” approach). For example, a population with an average of 50% hydrophobic repeat units in a random copolymer chain likely contains a non-negligible fraction of individual chains with ∼25% and ∼75% hydrophobes. If the former subpopulation is inactive and the latter subpopulation is highly hemolytic, such compositional dispersity may fundamentally limit efforts to improve on cell-type selectivity. To our knowledge, no study has quantified this effect, perhaps because it is challenging (though not impossible with well-resolved MALDI data) to quantify the compositional distribution. Mowery et al. discussed the possibility that sub-populations within heterogeneous mixtures of their nylon-3 copolymers may exert different mechanisms of antibacterial activity as early as 2009.45 But to date, the extent to which a truly unimolecular compound confers any advantage relative to a narrow distribution (we would hypothesize that it does not) remains to be demonstrated in a clear side-by-side comparison.
Alternating copolymers are particularly interesting as antimicrobials because they display very high degrees of sequence fidelity and very low (or nil) compositional dispersity. Perhaps due to the limited monomer scope of comonomer pairs with high propensity for alternation in chain growth polymerization, only a few reports on alternating antibacterial copolymers exist in the literature. It is straightforward to prepare alternating copolymers by step-growth, but these materials will necessarily have quite broad chain length dispersity. An outstanding example by the Sampson group127 showed that the ROMP of 1-substituted cyclobutenes and cyclohexenes affords alternating copolymers with precise spacing between functional groups (Fig. 17). They found that alternating sequences of cationic and hydrophobic groups outperformed random copolymer analogues in terms of potent antibacterial activity and minimal hemolytic toxicity, with better than 100-fold selectivity for the best examples. Interestingly, increasing the hydrophobicity of the groups between the cations did not dramatically aggravate the hemolytic toxicity, as typically seen with other copolymer mimics of HDPs. Related advances have been made by the Haldar group using alternating copolymers of maleimide and isobutylene.128 These findings suggest that alternating sequence is a prudent and perhaps under-appreciated design strategy.
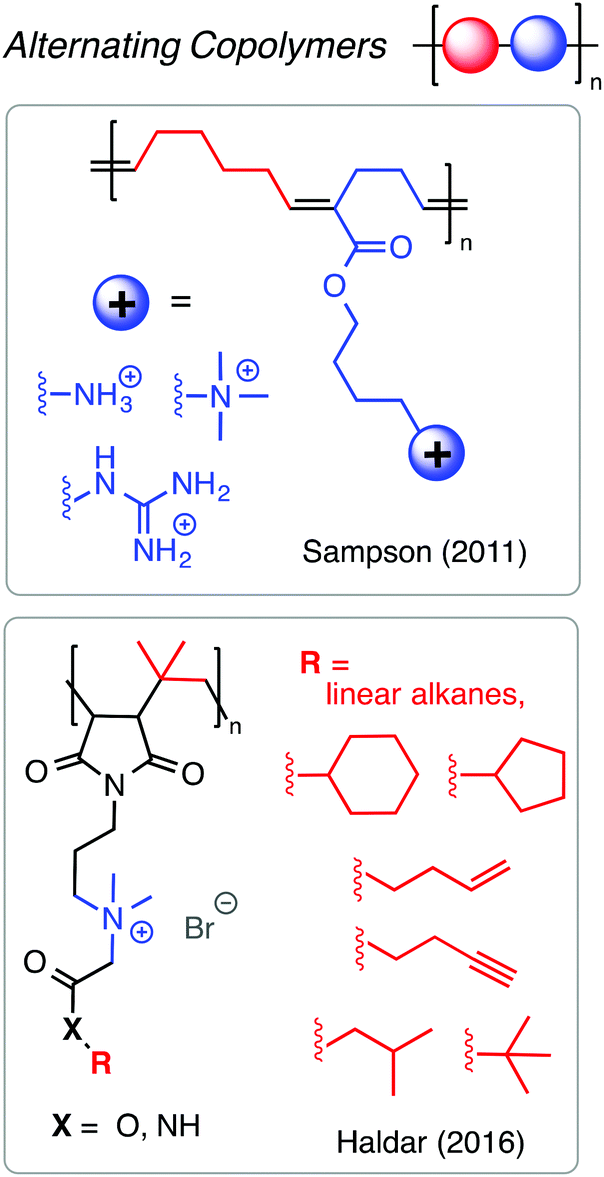 | ||
| Fig. 17 Alternating copolymers as platforms for antibacterial polymers. | ||
Very recently, Perrier and co-workers undertook a detailed study of sequence control in antimicrobial poly(acrylamide)s bearing hydrophobic isopropyl and cationic aminoethyl side chains.129 They prepared statistical, diblock, and multiblock linear copolymers. The antibacterial and hemolytic activities are significantly impacted by the sequence distribution; the MIC against P. aeruginosa decreased from 1000 μg mL−1 for the statistical copolymer to 32 μg mL−1 for the diblock and 8 μg mL−1 for the heptablock whereas all three samples were non-hemolytic. Thus, one may conclude that the clustering of hydrophobic side chains into small domains along the chain, as in a multiblock architecture, is a prudent strategy to enhance the antibacterial activity without affecting the toxicity to human cells.
2.4. The real deal: activity in vivo
The field of antibacterial polymers initially focused on the optimization of structures to minimize MIC and maximize HC50 values in vitro. Having identified many promising examples from a large pool of candidates, the next logical step is to quantify the activity in vivo using various animal models. This is a non-trivial advance because polymers that perform well in vitro may be inactivated in vivo by serum proteins, platelet adhesion, or other non-specific interactions. Nevertheless, a growing number of recent studies have shown encouraging results, which collectively represent a tremendous vertical advance of the field.Thoma et al. showed that a topical formulation based on cationic homopolymers of aminoethyl methacrylate eradicated S. aureus infection in a mouse nasal infection model (Fig. 18).130 These low MW polymers (3–10 kDa) are bactericidal to S. aureus at low micromolar concentrations even in the presence of serum in vitro and are non-toxic to mammalian cells (hRBCs, HEp-2 and COS-7 cell lines). They also demonstrated eradication efficacy in vivo that was significantly better than the small molecule antibiotic drug mupirocin. Further, it was confirmed that the polymers do not induce resistance after 15 sub-inhibitory exposures. These results all strongly suggest that such polymers are good candidates for topical applications to cure S. aureus infections.
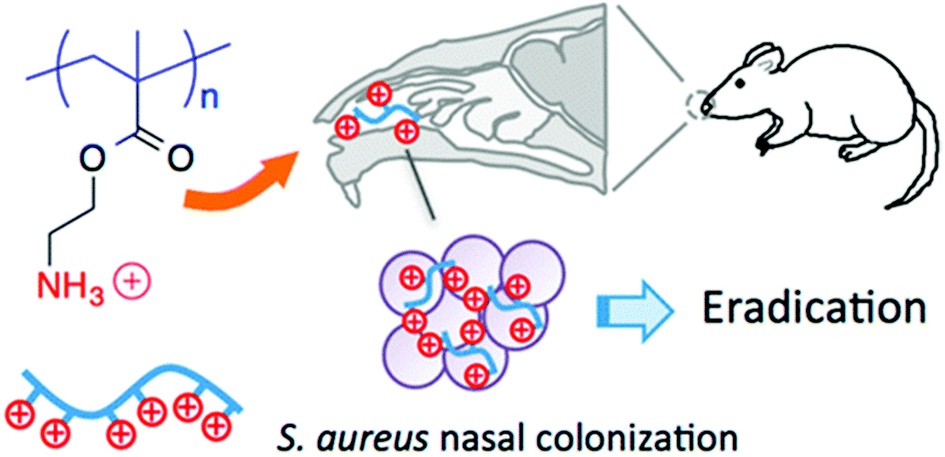 | ||
| Fig. 18 Cationic polymethacrylates eradicated S. aureus in a mouse nasal infection model. Reproduced from ref. 130, ACS AuthorChoice (Open Access). | ||
Haldar and co-workers have reported the in vivo efficacy of various HDP-mimetic copolymers131 and small molecules.132 Alternating copolymers of maleimide and isobutylene bearing a same-centered QAS amphiphile in the side chains were effective against A. baumannii in a burn infection mouse model via topical application.131 The concentration of viable bacterial cells in the burn was reduced from ∼1010 to 107 CFU mL−1 upon treatment with 50 mg kg−1 of polymer, which is comparable to the efficacy of the antibiotic minocycline at the same dosage. When both the polymer and minocycline were used in combination, the A. baumannii concentration was further reduced to ∼104 CFU mL−1. The same group showed that cationic and amphiphilic small molecules composed of a single fatty acid chain conjugated to two equivalents of lysine (+4 charge at pH 7.4), which could be viewed as a minimalist design to mimic the HDP structure, successfully eradicated MRSA biofilms from infected mouse skin.132
P. X. Ma and co-workers reported the in vivo activity of antimicrobial bottle-brush polymethacrylates grafted to PDMS in a subcutaneous rat infection model.133 Random copolymers of lysine and phenylalanine prepared by ROP were conjugated to heterobifunctional PEG methacrylate and then subjected to surface-initiated/UV-triggered polymerization to yield the bottle-brush grafted surface. These coated PDMS samples, alongside bare PDMS, were inoculated with 107 CFU of E. coli and then implanted into rats subcutaneously. After 5 days, the excised implants coated with the polymer contained only 0–10 CFU (a 6- or 7-log reduction) whereas the bare PDMS surface was still host to ∼105 CFU. Visually, the bare PDMS surface side of the wound showed extensive bacterial infection whereas the coated side appeared to be normal tissue with no sign of infection. Thus, these polymethacrylate-graft-(PEG-poly(Lys-block-Phe)) materials substantially reduced the implant-associated infection under rat skin, representing a major advance for implant materials.
The G. G. Qiao group reported in vivo efficacy of their antimicrobial polymer nanoparticles in a mouse peritonitis model. They found that the concentration of A. baumannii in the peritoneal wash was dramatically reduced (a 5-log reduction) upon dosing with antibacterial dendrimers, which was similar to the efficacy of the antibiotic drug imipenem.93
The cationic and amphiphilic polycarbonates of Hedrick and Yang showed high in vivo efficacy against MRSA by systemic application in immunosuppressed mice (Fig. 19).134 The mice were injected with 4 × 108 CFU mL−1 of MRSA, which was determined to be a lethal dose. Amazingly, the polymer out-performed the antibiotic vancomycin in terms of reduction in MRSA concentration (to 103 CFU mL−1) over a time period of 4 and 8 hours post-injection. The effective dose of the polymer was ∼2 mg L−1 against MRSA whereas the lethal dose against the mice was ∼20 mg L−1, suggesting a 10-fold selectivity. After 7 days post-infection, 14 out of 15 mice that received the polymer treatment survived whereas none of the control mice (injected with MRSA in PBS buffer) survived. Hence, this breakthrough study provides very strong evidence that cationic, amphiphilic polymers can cure infection in mice when administered systemically.
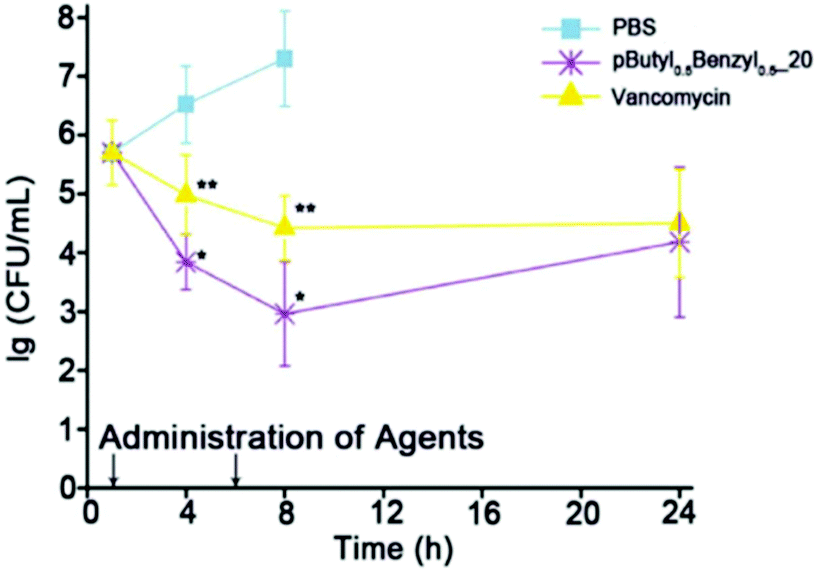 | ||
| Fig. 19 Rapid reduction in the concentration of MRSA cells in the bloodstream of a mouse was achieved with antibacterial poly(carbonate)s compared to the antibiotic drug vancomycin. Reproduced with permission from ref. 134, copyright 2015, Wiley-VCH. | ||
Overall, these early-stage in vivo studies collectively demonstrate that HDP-mimetic polymers possess great promise as therapeutic agents to combat drug-resistant bacterial infections. Still, the relative paucity of survival studies at this early stage indicates a continued need for further progress.
3. Mechanism of action
3.1. HDP mechanism
Since the 1980s, strongly cationic peptides have been known to disrupt the integrity of bacterial membranes.12,135 At the simplest level, the peptide–membrane interaction is understood in terms of electrostatic attraction between the anionic bacterial cell envelope and cationic residues in the peptide, as well as partitioning of hydrophobic residues into the nonpolar membrane core. The putative mechanism of HDP action is often described in molecular detail by the Shai–Matsuzaki–Huang (SMH) model.7,136,137 As a classical example, Magainin-2 adopts a facially amphiphilic α-helix at the membrane interface upon binding, and then cooperatively forms transient pentameric pores across the bilayer.138 An abundance of evidence supports some aspects of the SMH model for a wide variety of HDPs.4,139–142 Nonetheless, HDPs may exert mechanisms other than (or in addition to) membrane disruption, including immunomodulatory effects, metabolic inhibition, and programmed cell death.5,143,144 Here, we review mechanistic studies that probe HDP-mimetic polymers, with a sole focus on the membrane disruption aspects. Because the polymers possess the critical features of HDPs, and show similar antibacterial activity, it is widely hypothesized that they act by related mechanisms of action, despite their molecular heterogeneity and lack of defined secondary structure.3.2. Liposomes as model membranes
Liposomes are frequently employed as simplified model membrane systems to probe the interaction of polymers with phospholipid bilayers using a wide range of physicochemical methods such as spectroscopy, microscopy and thermal analysis.A classical experiment to evaluate membrane permeabilization induced by polymers is to monitor the release of a fluorescent dye entrapped within a liposome upon mixing with a dilute polymer solution (Fig. 20). A water-soluble fluorescent dye, such as carboxyfluorescein, calcein or sulforhodamine-B, is entrapped in large unilamellar vesicles (LUVs) above the self-quenching concentration (typically 50 mM). Upon the disruption of the bilayer barrier function, the dye is released into the dilute solution and fluorescence intensity is markedly recovered.43,56,145 Phospholipid composition is chosen to mimic various cell types. For instance, the mixture of anionic phosphatidylglycerol (PG) and zwitterionic phosphatidylethanolamine (PE) is evocative of E. coli membranes whereas phosphatidylcholine (PC) and cholesterol are used as a proxy for mammalian cell membranes.146 Despite its simplicity, dye leakage provides a useful demonstration of the inherent membrane-disputing ability of a polymer, which may contribute to the mechanism of antibacterial and hemolytic action (at least in part).147,148 This technique has been employed for polynorbornenes,56 polymethacrylates,43 and nylon-3 copolymers.147 The fundamental basis for cell-type selectivity can be observed by comparing dye leakage from liposomes that mimic anionic bacterial membranes (e.g. POPE/POPG) and those that mimic zwitterionic mammalian cell membranes (POPC/chol). Membrane permeabilization by HDPs depends on the lipid charge as well as intrinsic curvature.149 Gellman et al. showed that their HDP-mimetic nylon-3 polymer selectively disrupts lipid membranes depending on the lipid composition; the polymer efficiently disrupts liposomes of DOPG/DOPG (mimicking E. coli membranes), but not those of POPC (mimicking mammalian cell membranes).147 These data suggest that optimized formulations of HDP-mimetic polymers are inherently more active against membranes that possess anionic charge and negative curvature. Fluorescence lifetime assays can distinguish between all-or-none leakage (a fraction of the vesicles released all of their contents) versus graded leakage (all vesicles released a portion of their contents). The type of mechanism depends on polymer hydrophobicity: a nylon-3 derivative consisting solely of cationic units induces all-or-none leakage, whereas a cationic/hydrophobic copolymer exhibits graded leakage.147
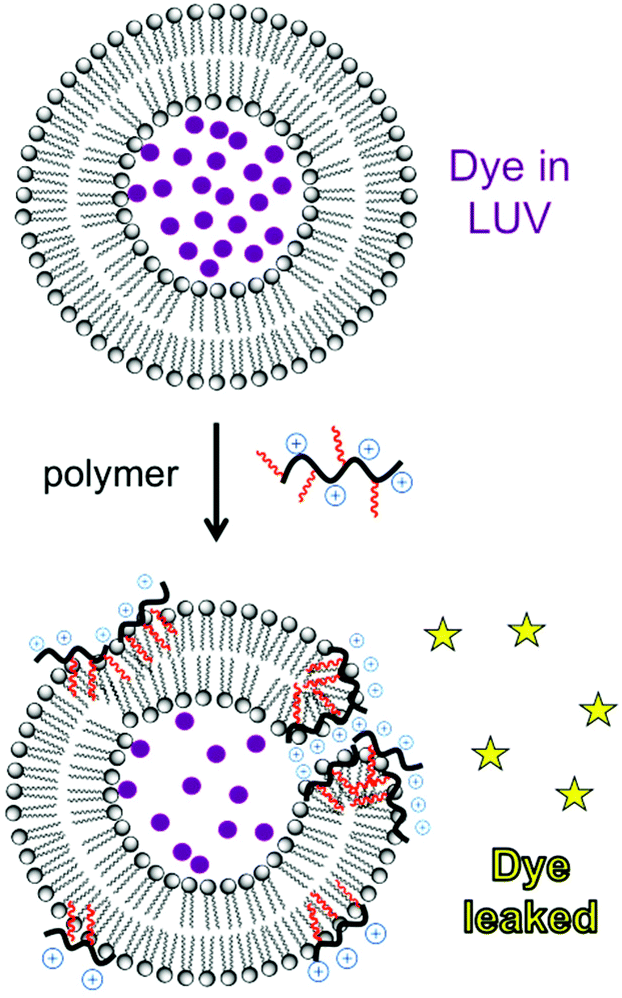 | ||
| Fig. 20 Schematic of the liposome dye leakage induced by a cationic amphiphilic polymer. | ||
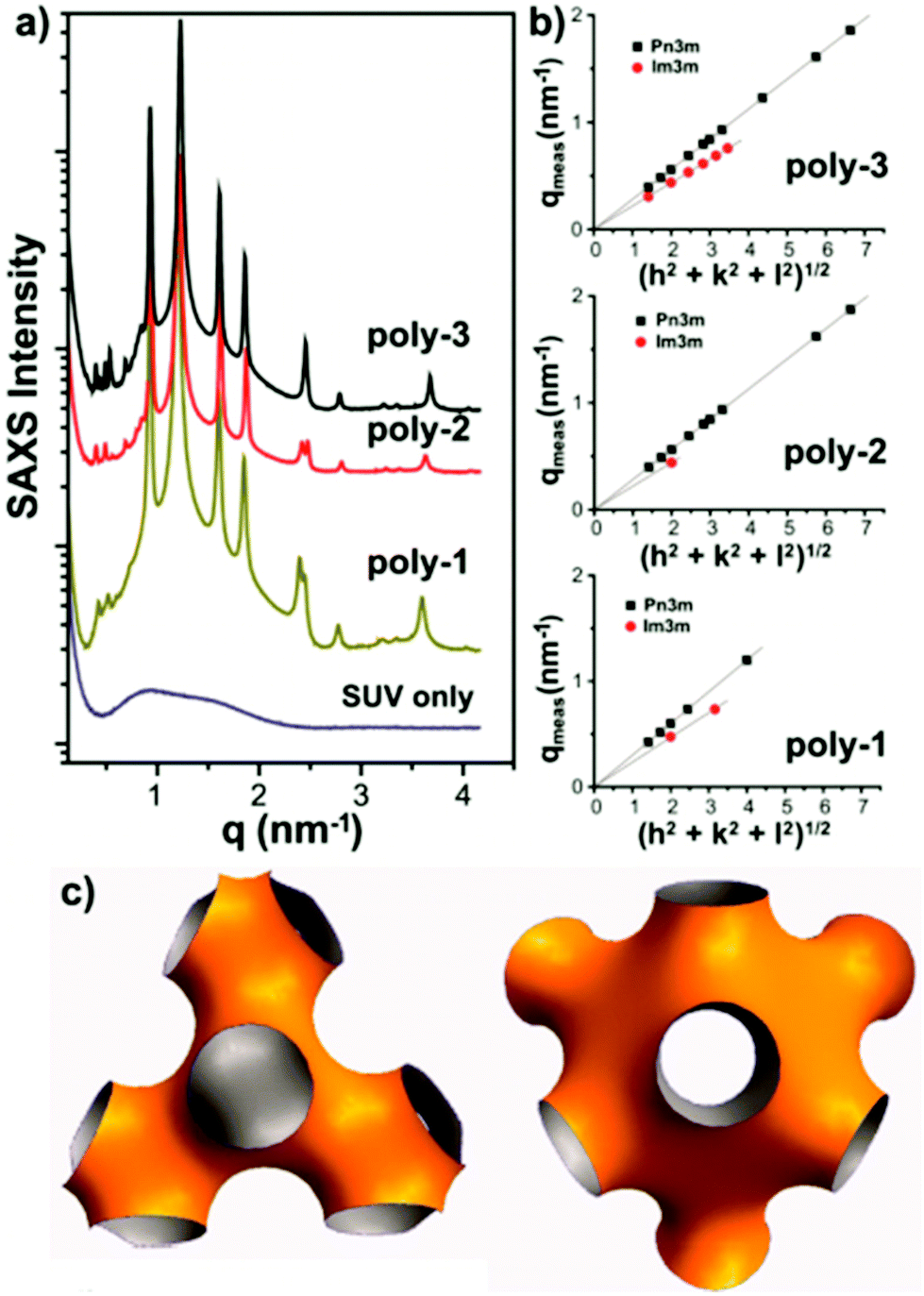 | ||
| Fig. 21 SAXS data (a,b) for a lipid bilayer containing antimicrobial polymethacrylates and corresponding real space model (c). Reproduced with permission from ref. 157, copyright 2013, American Chemical Society. | ||
3.3. Molecular dynamics simulation
All-atom molecular dynamics simulations are rapidly emerging as a very powerful tool to probe polymer–lipid interactions in precise detail at the molecular level (Fig. 22). The MD simulation of an antimicrobial polymethacrylate binding to a phospholipid bilayer revealed that primary amines in the side chains associate with phosphate head groups in the lipids, whereas the insertion of the polymer into the membrane is mediated predominately by the hydrophobic effect, as expected.167 Most interestingly, it was found that the comonomer sequence plays a pivotal role in dictating the overall shape of the chain molecule, which implies that sequence control may indeed be an important molecular design consideration that remains underexplored to date.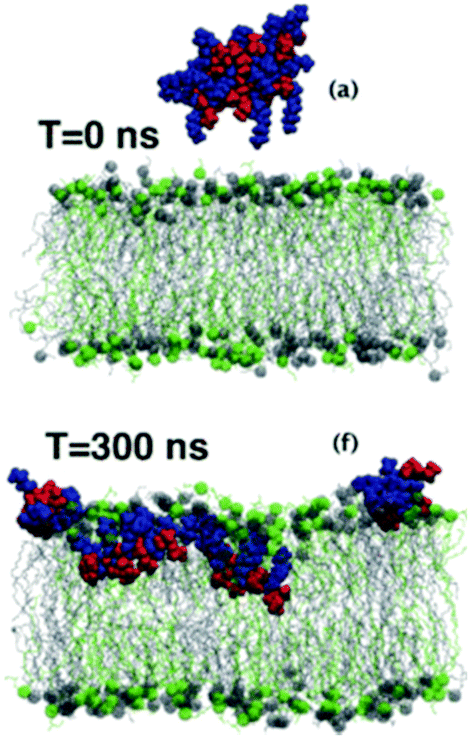 | ||
| Fig. 22 Snapshots from an MD simulation of a polymethacrylate on a lipid bilayer. Reproduced with permission from ref. 170, copyright 2017, RSC Publishing. | ||
Also in the context of antimicrobial polymethacrylates, MD was employed to clarify the role of the cationic spacer group (i.e., the length of the carbon chain connecting the cationic ammonium group to the backbone). Interestingly, the elongation of the spacer group promotes the deeper insertion of the polymer backbone into the membrane core, in analogy to the “snorkel effect” in transmembrane peptides.95,168,169 The polymers with intermediate spacer length (C4) adopt a facially-amphiphilic structure upon binding to the membrane, in which cationic and hydrophobic sidechains are segregated to opposite faces.97,170 This is a remarkable observation reminiscent of the secondary structure in membrane-bound Magainin-2, despite the complete lack of structural perfection in random copolymethacrylates. Related MD simulations also showed the clustering of negatively-charged lipids into distinct domains upon polymer binding, which resulted in lipid coarsening and bimodal membrane thickness.171 Such “charge clustering” in the membrane is known to produce a packing defect, wherein the permeability barrier is compromised, in corroboration with the experimental DSC data from Epand.159
3.4. Seeing is believing
Direct observation of bacterial cell membranes in the presence of an antimicrobial polymer provides the most convincing evidence that their mechanism of action involves the membrane as a target, albeit with less quantitative molecular-level understanding. Such visualization has been done extensively by TEM, which can reveal detailed morphological changes in the membrane structure upon exposure to antimicrobial polymers (Fig. 23). For example, Yang and Hedrick reported biodegradable nanostructures by the self-assembly of cationic and hydrophobic triblock copolycarbonates.134 Comparative TEM images of methicillin-resistant S. aureus (MRSA) in the presence of a polymer (slightly above the MIC) showed significant damage to the cell membrane accompanied by the leakage of cell contents, whereas the control images show smooth, healthy cell surfaces.98 It is apparent from the images that cell lysis occurs by an all-or-none mechanism. The authors hypothesize that micelles of these triblock copolymers, which are attracted to the anionic cell surface by electrostatic interactions, may inhibit cell wall synthesis due to their steric bulk, leading to membrane disruption. Alternatively, the micelles might permeabilize the cytoplasmic membrane as a result of electroporation or the sinking raft model, which could also lead to cell death.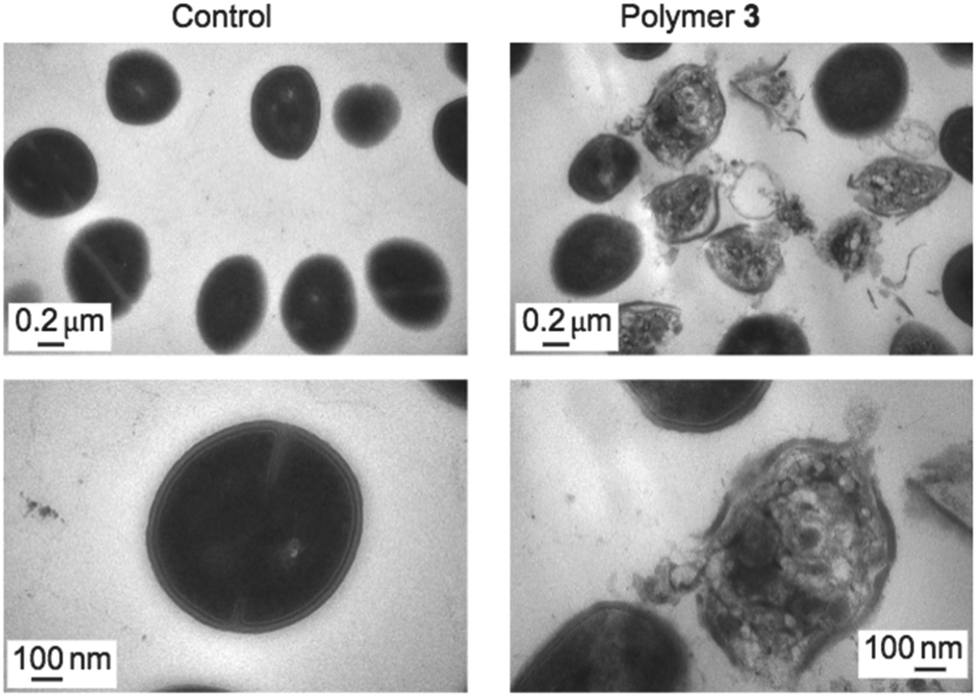 | ||
| Fig. 23 TEM images of intact MRSA cells (left) and damaged cells upon exposure to an inhibitory dose of antibacterial polycarbonate (right), at two magnifications. Reproduced with permission from ref. 98, copyright 2011, Springer Nature. | ||
In TEM images, the shape of the bacterial cells can be visualized, but the polymers themselves are essentially invisible. In order to directly track the fate of the polymers in vitro, Reynolds and Qiao prepared dendrimers of an antimicrobial polymer tagged with AlexaFlour-488 (green) and employed super-resolution fluorescence 3D structured illumination microscopy (3D-SIM) for visualization (Fig. 24).93E. coli cells labeled with an optically orthogonal membrane dye (red) were mixed with an AF488-tagged polymer, above and below the lethal dose, to investigate the polymer particle localization in relation to the bacterial membrane.
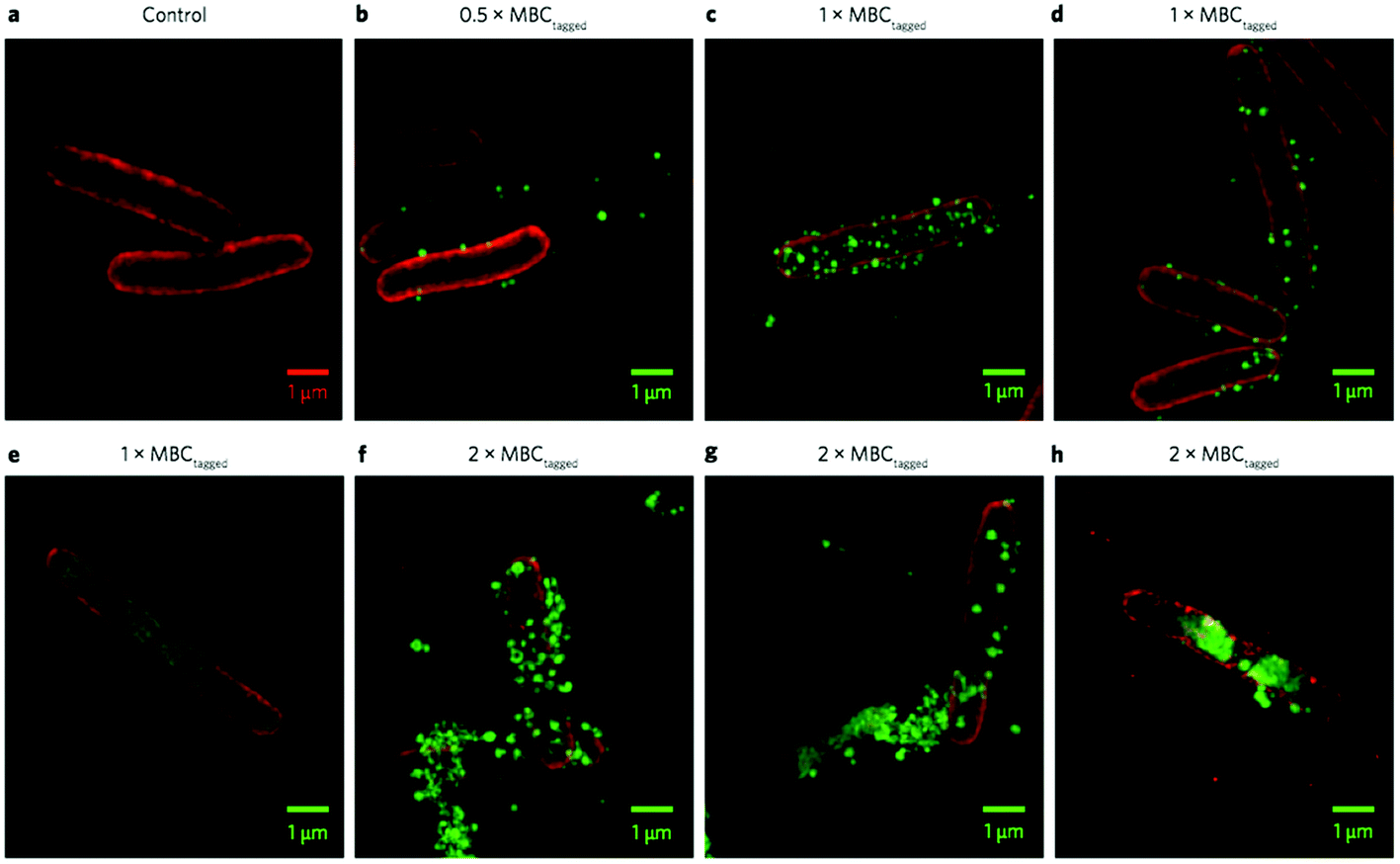 | ||
| Fig. 24 3D-SIM superresolution fluorescence images of an antibacterial polymer particle (PAMAM dendrimer with surface-initiated random copolymers of Lys and Val) with E. coli cells. The particles are tagged with AF488 (green) and the E. coli cell membrane is dyed with FM4-64FX (red). Control E. coli (a), polymer at half the MBC (b), at the MBC (c–e), and at twice the MBC (f–h). Reproduced with permission from ref. 93, copyright 2016, Springer Nature. | ||
Whereas various microscopy techniques have been employed to study polymer–membrane interactions previously,172,173 this elegant study was the one of the first that used super-resolution optics to unambiguously confirm the co-localization of an antimicrobial particle on a bacterial cell membrane at high resolution.93 Below the lethal dose (1/2 MBC), these particles sparsely populate the outer surface of the E. coli cells, whereas the surface is nearly saturated with particles at the MBC. At double the MBC, particles aggregate on and penetrate into the cells, leading to a clustering of lipids in the membrane and clear loss of membrane integrity. It is clearly seen in the red channel that the membrane lipids are clustered into dense domains, leaving behind patchy dark regions (Fig. 24, panels e–h). Also, aggregates of red lipid can be seen in the surrounding media, suggesting perhaps that micelles or vesicles of lipid have been shed from the cell surface. Based on these observations, the authors speculated that the polymers initially adhere to the outer membrane (OM) due to electrostatic attraction, which might destabilize the OM and permit translocation to the cytoplasmic membrane (CM). In support of this view, they demonstrated that the polymers avidly bind LPS, a major component of the OM. The polymer particles did not induce pore formation in model membranes, but did cause unregulated ion movement and dissipation of membrane potential. Finally, the polymers induced a programmed cell death (PCD)144 pathway that is triggered under stressful conditions such as membrane perturbation, although the polymer retains the antibacterial activity in the presence of an inhibitor that blocks this PCD pathway. Hence, the authors conclude that PCD is not a requisite mechanism for antibacterial activity but that it may occur as a supplemental, though not required, alternative mechanism of bacterial lethality.93
It is important to note that nanoparticles composed of antibacterial polymers may exert subtly different mechanism(s) of action as compared to the corresponding individually solvated polymer chains. These nanoparticles contain multiple chains that are co-localized in space and are thus inherently more locally concentrated upon membrane binding. By comparison, individual chains must cooperatively bind membranes in order to achieve local concentrations comparable to that of the nanoparticles. In addition, non-specific binding to serum proteins, kinetics of degradation, details of membrane permeabilization and/or translocation and the intracellular fate of the nanoparticles may all differ from those of their constituent polymeric chains. While the bulk of prior mechanistic work has been done on individual polymer chains, many of the most cutting-edge materials currently being explored for in vivo application are increasing focused on nanoparticles and nano-assemblies of antibacterial polymers. Thus, mechanistic studies on polymer nanoparticles, both in vitro and in vivo, are still urgently needed at the forefront of this field. In summary, the literature is replete with evidence, ranging from the molecular level to the nano- and micro-scales, that HDP-mimetic antibacterial polymers indeed act, at least in part, by a mechanism involving the disruption of the bacterial cell membrane barrier function.
4. Conclusions
The fascinating discovery of HDPs and the development of polymeric disinfectants in the 1980s set the stage for molecular level biomimicry using synthetic polymers. Since the ground-breaking studies of DeGrado and others in the early 2000s, the field of peptide-mimetic antimicrobial polymers has continuously expanded. By focusing on the key physiochemical structural determinants of activity in host defense peptides, some well-established design rules are now in place: (1) a finely tuned balance of hydrophobic and cationic moieties, (2) protonated primary amine (or guanidine) groups as the source of cationic charge, and (3) relatively low MW, which tend to confer the desired bioactivity profiles. Further fine-tuning of the activity profiles has been achieved by incorporating neutral/hydrophilic components, and by exploring the roles of chain sequence and architecture. PEGylation in particular is a proven method to alleviate hemolytic toxicity without sacrificing antibacterial potency. Molecular platforms for optimization over the past ten years have included the polynorbornene class of Tew and co-workers, the polymethacrylates of Kuroda's and several other groups, and the nylon-3 class spearheaded by Gellman's group.Ever more diverse and creative variants continue to appear in the literature. Hyperbranched polymers, dendrimers, and single chain nanoparticles have shown excellent promise as antibacterials with low toxicity. Other contributions have included the demonstration of in vivo efficacy, optimization of biodegradable platforms, the self-immolative examples, and the use of renewable feedstocks, as just a few examples. These recent efforts, and many others, continue to invigorate the rapidly expanding multidisciplinary subject. Still, much work remains to be done. Precise control structural features including comonomer sequence, tacticity, chain length, and compositional dispersity have not been exhaustively correlated with activity. While many excellent studies on the interaction between polymers and model lipid bilayers provided a valuable foundation of knowledge, a complete understanding of the mechanism of action exerted in vivo by this increasingly diverse class of biomacromolecules is still urgently needed.
In terms of practical applications, antimicrobial polymers have found use in the solid state as coatings/surfaces on consumer products and medical devices, food-packaging materials, and textiles.174 Numerous HDP-inspired antimicrobial peptides have completed or are currently in clinical trials,175 but thus far no synthetic polymer HDP-mimic has reached such a benchmark, to our knowledge. Potential pitfalls abound in the development of any new drug and antimicrobial polymers are certainly no exception: pharmacokinetics, long-term toxicity, biodistribution, possible septicemia caused by bacterial cell lysis (if that is the mechanism of action), and potential immunogenicity arising from a polymer's degradation by-products (if it is degradable) are all challenges that will likely require sustained efforts to surmount. Although synthetic polymers possess heterogeneity in chain length, composition, and stereochemistry, we can see no fundamental reason why these features inherently prohibit synthetic polymers from exploration in the clinical setting. Moreover, continued advances in the precision control of polymer synthesis may soon circumvent such concerns altogether. In closing, we look forward to continued growth and future developments in this exciting interdisciplinary field of polymer science.
Conflicts of interest
There are no conflicts to declare.References
- H. W. Boucher, G. H. Talbot, J. S. Bradley, J. E. Edwards, D. Gilbert, L. B. Rice, M. Scheld, B. Spellberg and J. Bartlett, Clin. Infect. Dis, 2009, 48, 1–12 CrossRef PubMed
.
- R. E. W. Hancock and H. G. Sahl, Nat. Biotechnol., 2006, 24, 1551–1557 CrossRef CAS PubMed
- R. I. Lehrer and T. Ganz, Curr. Opin. Immunol., 1999, 11, 23–27 CrossRef CAS PubMed
- M. Zasloff, Nature, 2002, 415, 389–395 CrossRef CAS PubMed
- K. A. Brogden, Nat. Rev. Microbiol., 2005, 3, 238–250 CrossRef CAS PubMed
- K. A. Brogden, M. Ackermann, P. B. McCray and B. F. Tack, Int. J. Antimicrob. Agents, 2003, 22, 465–478 CrossRef CAS PubMed
- Y. Shai, Biochim. Biophys. Acta, Biomembr., 1999, 1462, 55–70 CrossRef CAS
- K. Matsuzaki, Biochim. Biophys. Acta, Rev. Biomembr., 1998, 1376, 391–400 CrossRef CAS
- M. L. Juba, D. K. Porter, E. H. Williams, C. A. Rodriguez, S. M. Barksdale and B. M. Bishop, Biochim. Biophys. Acta, Biomembr., 2015, 1848, 1081–1091 CrossRef CAS PubMed
- G. E. Fantner, R. J. Barbero, D. S. Gray and A. M. Belcher, Nat. Nanotechnol., 2010, 5, 280–285 CrossRef CAS PubMed
- M. Wilmes, B. P. A. Cammue, H. G. Sahl and K. Thevissen, Nat. Prod. Rep., 2011, 28, 1350–1358 RSC
- M. Zasloff, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 5449–5453 CrossRef CAS
- G. S. Wang, X. Li and Z. Wang, Nucleic Acids Res., 2016, 44, D1087–D1093 CrossRef CAS PubMed
- G. S. Wang, X. Li and Z. Wang, Nucleic Acids Res., 2009, 37, D933–D937 CrossRef CAS PubMed
- Z. Wang and G. S. Wang, Nucleic Acids Res., 2004, 32, D590–D592 CrossRef CAS PubMed
- Y. Lan, J. T. Lam, G. K. H. Siu, W. C. Yam, A. J. Mason and J. K. W. Lam, Tuberculosis, 2014, 94, 678–689 CrossRef CAS PubMed
- A. Won, M. Khan, S. Gustin, A. Akpawu, D. Seebun, T. J. Avis, B. O. Leung, A. P. Hitchcock and A. Ianoul, Biochim. Biophys. Acta, Biomembr., 2011, 1808, 1592–1600 CrossRef CAS PubMed
- K. Hall, E. Porter, N. Umezawa, S. H. Gellman and M. Aguilar, J. Pept. Sci., 2004, 10, 175–175 Search PubMed
- T. L. Raguse, E. A. Porter, B. Weisblum and S. H. Gellman, J. Am. Chem. Soc., 2002, 124, 12774–12785 CrossRef CAS PubMed
- E. A. Porter, B. Weisblum and S. H. Gellman, J. Am. Chem. Soc., 2002, 124, 7324–7330 CrossRef CAS PubMed
- W. Huang, J. Seo, S. B. Willingham, A. M. Czyzewski, M. L. Gonzalgo, I. L. Weissman and A. E. Barron, PLoS One, 2014, 9, e90397 Search PubMed
- S. A. Fowler and H. E. Blackwell, Org. Biomol. Chem., 2009, 7, 1508–1524 CAS
- K. A. Brogden, A. J. DeLucca, J. Bland and S. Elliott, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 412–416 CrossRef CAS
- F. D. Silva, C. A. Rezende, D. C. P. Rossi, E. Esteves, F. H. Dyszy, S. Schreier, F. Gueiros-Filho, C. B. Campos, J. R. Pires and S. Daffre, J. Biol. Chem., 2009, 284, 34735–34746 CrossRef CAS PubMed
- G. N. Tew, R. W. Scott, M. L. Klein and W. F. Degrado, Acc. Chem. Res., 2010, 43, 30–39 CrossRef CAS PubMed
- M. S. Ganewatta and C. B. Tang, Polymer, 2015, 63, A1–A29 CrossRef CAS
- W. Ren, W. R. Cheng, G. Wang and Y. Liu, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 632–639 CrossRef CAS
- L. Timofeeva and N. Kleshcheva, Appl. Microbiol. Biotechnol., 2011, 89, 475–492 CrossRef CAS PubMed
- A. Munoz-Bonilla and M. Fernandez-Garcia, Prog. Polym. Sci., 2012, 37, 281–339 CrossRef CAS
- H. Takahashi, E. F. Palermo, K. Yasuhara, G. A. Caputo and K. Kuroda, Macromol. Biosci., 2013, 13, 1285–1299 CrossRef CAS PubMed
- E. F. Palermo and K. Kuroda, Appl. Microbiol. Biotechnol., 2010, 87, 1605–1615 CrossRef CAS PubMed
- M. Hartlieb, E. G. L. Williams, A. Kuroki, S. Perrier and K. E. S. Locock, Curr. Med. Chem., 2017, 24, 2115–2140 CrossRef CAS PubMed
-
J. Rodríguez-Hernández, in Polymers against Microorganisms: On the Race to Efficient Antimicrobial Materials, Springer International Publishing, Cham, 2017, pp. 39–69, DOI:10.1007/978-3-319-47961-3_3
- A. C. Engler, N. Wiradharma, Z. Y. Ong, D. J. Coady, J. L. Hedrick and Y. Y. Yang, Nano Today, 2012, 7, 201–222 CrossRef CAS
- M. R. E. Santos, A. C. Fonseca, P. V. Mendona, R. Branco, A. C. Serra, P. V. Morais and J. F. J. Coelho, Materials, 2016, 9, 599 CrossRef PubMed
- E.-R. Kenawy, S. D. Worley and R. Broughton, Biomacromolecules, 2007, 8, 1359–1384 CrossRef CAS PubMed
- T. Ikeda, S. Tazuke and Y. Suzuki, Makromol. Chem., 1984, 185, 869–876 CrossRef CAS
- D. H. Liu and W. F. DeGrado, J. Am. Chem. Soc., 2001, 123, 7553–7559 CrossRef CAS PubMed
- M. A. Schmitt, B. Weisblum and S. H. Gellman, J. Am. Chem. Soc., 2004, 126, 6848–6849 CrossRef CAS PubMed
- Y. M. Song, Y. Park, S. S. Lim, S. T. Yang, E. R. Woo, I. S. Park, J. S. Lee, J. I. Kim, K. S. Hahm, Y. Kim and S. Y. Shin, Biochemistry, 2005, 44, 12094–12106 CrossRef CAS PubMed
- K. Matsuzaki, S. Yoneyama and K. Miyajima, Biophys. J., 1997, 73, 831–838 CrossRef CAS PubMed
-
L. Otvos and M. Cudic, in Peptide Characterization and Application Protocols, ed. G. B. Fields, Humana Press, Totowa, NJ, 2007, pp. 309–320, DOI:10.1007/978-1-59745-430-8_12
- K. Kuroda, G. A. Caputo and W. F. DeGrado, Chem. – Eur. J., 2009, 15, 1123–1133 CrossRef CAS PubMed
- M. F. Ilker, K. Nusslein, G. N. Tew and E. B. Coughlin, J. Am. Chem. Soc., 2004, 126, 15870–15875 CrossRef CAS PubMed
- B. P. Mowery, A. H. Lindner, B. Weisblum, S. S. Stahl and S. H. Gellman, J. Am. Chem. Soc., 2009, 131, 9735–9745 CrossRef CAS PubMed
- B. P. Mowery, S. E. Lee, D. A. Kissounko, R. F. Epand, R. M. Epand, B. Weisblum, S. S. Stahl and S. H. Gellman, J. Am. Chem. Soc., 2007, 129, 15474–15476 CrossRef CAS PubMed
- E. F. Palermo and K. Kuroda, Biomacromolecules, 2009, 10, 1416–1428 CrossRef CAS PubMed
- E. F. Palermo, D.-K. Lee, A. Ramamoorthy and K. Kuroda, J. Phys. Chem. B, 2011, 115, 366–375 CrossRef CAS PubMed
- M. A. Gelman, B. Weisblum, D. M. Lynn and S. H. Gellman, Org. Lett., 2004, 6, 557–560 CrossRef CAS PubMed
- A. Punia, E. He, K. Lee, P. Banerjee and N. L. Yang, Chem. Commun., 2014, 50, 7071–7074 RSC
- A. Punia, P. R. Debata, P. Banerjee and N. L. Yang, RSC Adv., 2015, 5, 95300–95306 RSC
-
M. F. Ilker, G. N. Tew and E. B. Coughlin, Antiterrorism and Homeland Defense: Polymers and Materials, 2007, vol. 980, pp. 175–197 Search PubMed
-
K. Lienkamp, A. E. Madkour and G. N. Tew, Polymer Composites - Polyolefin Fractionation - Polymeric Peptidomimetics - Collagens, 2013, vol. 251, pp. 141–172 Search PubMed
- K. Lienkamp and G. N. Tew, Chem. – Eur. J., 2009, 15, 11784–11800 CrossRef CAS PubMed
- K. Lienkamp, A. E. Madkour, A. Musante, C. F. Nelson, K. Nuesslein and G. N. Tew, J. Am. Chem. Soc., 2008, 130, 9836–9843 CrossRef CAS PubMed
- T. Eren, A. Som, J. R. Rennie, C. F. Nelson, Y. Urgina, K. Nusslein, E. B. Coughlin and G. N. Tew, Macromol. Chem. Phys., 2008, 209, 516–524 CrossRef CAS
- Z. M. Al-Badri, A. Som, S. Lyon, C. F. Nelson, K. Nusslein and G. N. Tew, Biomacromolecules, 2008, 9, 2805–2810 CrossRef CAS PubMed
- H. Wang, X. F. Shi, D. F. Yu, J. Zhang, G. Yang, Y. X. Cui, K. J. Sun, J. B. Wang and H. K. Yan, Langmuir, 2015, 31, 13469–13477 CrossRef CAS PubMed
- V. W. L. Ng, J. P. K. Tan, J. Y. Leong, Z. X. Voo, J. L. Hedrick and Y. Y. Yang, Macromolecules, 2014, 47, 1285–1291 CrossRef CAS
- W. Chin, C. A. Yang, V. W. L. Ng, Y. Huang, J. C. Cheng, Y. W. Tong, D. J. Coady, W. M. Fan, J. L. Hedrick and Y. Y. Yang, Macromolecules, 2013, 46, 8797–8807 CrossRef CAS
- A. Nimmagadda, X. Liu, P. Teng, M. Su, Y. Q. Li, Q. Qiao, N. K. Khadka, X. T. Sung, J. J. Pan, H. Xu, Q. Li and J. F. Cai, Biomacromolecules, 2017, 18, 87–95 CrossRef CAS PubMed
- K. E. S. Locock, T. D. Michl, J. D. P. Valentin, K. Vasilev, J. D. Hayball, Y. Qu, A. Traven, H. J. Griesser, L. Meagher and M. Haeussler, Biomacromolecules, 2013, 14, 4021–4031 CrossRef CAS PubMed
- S. E. Exley, L. C. Paslay, G. S. Sahukhal, B. A. Abel, T. D. Brown, C. L. McCormick, S. Heinhorst, V. Koul, V. Choudhary, M. O. Elasri and S. E. Morgan, Biomacromolecules, 2015, 16, 3845–3852 CrossRef CAS PubMed
- A. Som, A. O. Tezgel, G. J. Gabriel and G. N. Tew, Angew. Chem., Int. Ed., 2011, 50, 6147–6150 CrossRef CAS PubMed
- B. M. de Ronde, A. Birke and G. N. Tew, Chem. – Eur. J., 2015, 21, 3013–3019 CrossRef CAS PubMed
- R. Tejero, D. Lopez, F. Lopez-Fabal, J. L. Gomez-Garces and M. Fernández-García, Biomacromolecules, 2015, 16, 1844–1854 CrossRef CAS PubMed
- Y. Chen, P. A. Wilbon, Y. P. Chen, J. H. Zhou, M. Nagarkatti, C. P. Wang, F. X. Chu, A. W. Decho and C. B. Tang, RSC Adv., 2012, 2, 10275–10282 RSC
- J. Y. Zhang, Y. P. Chen, K. P. Miller, M. S. Ganewatta, M. Bam, Y. Yan, M. Nagarkatti, A. W. Decho and C. B. Tang, J. Am. Chem. Soc., 2014, 136, 4873–4876 CrossRef CAS PubMed
- V. Sambhy, B. R. Peterson and A. Sen, Angew. Chem., Int. Ed., 2008, 47, 1250–1254 CrossRef CAS PubMed
- R. W. Williams, M. Zasloff and D. Covell, Biophys. J., 1988, 53, A631–A631 Search PubMed
- M. Pillong, J. A. Hiss, P. Schneider, Y. C. Lin, G. Posselt, B. Pfeiffer, M. Blatter, A. T. Muller, S. Bachler, C. S. Neuhaus, P. S. Dittrich, K. H. Altmann, S. Wessler and G. Schneider, Small, 2017, 13, 1701316 CrossRef PubMed
- A. Giangaspero, L. Sandri and A. Tossi, Eur. J. Biochem., 2001, 268, 5589–5600 CrossRef CAS PubMed
- A. Tossi, C. Tarantino and D. Romeo, Eur. J. Biochem., 1997, 250, 549–558 CAS
- A. Som, S. Vemparala, I. Ivanov and G. N. Tew, Biopolymers, 2008, 90, 83–93 CrossRef CAS PubMed
- G. J. Gabriel, J. A. Maegerlein, C. E. Nelson, J. M. Dabkowski, T. Eren, K. Nusslein and G. N. Tew, Chem. – Eur. J., 2009, 15, 433–439 CrossRef CAS PubMed
- H. Takahashi, G. A. Caputo, S. Vemparala and K. Kuroda, Bioconjugate Chem., 2017, 28, 1340–1350 CrossRef CAS PubMed
- K. Lienkamp, K. N. Kumar, A. Som, K. Nusslein and G. N. Tew, Chem. – Eur. J., 2009, 15, 11710–11714 CrossRef CAS PubMed
- T. D. Michl, K. E. S. Locock, N. E. Stevens, J. D. Hayball, K. Vasilev, A. Postma, Y. Qu, A. Traven, M. Haeussler, L. Meagher and H. J. Griesser, Polym. Chem, 2014, 5, 5813–5822 RSC
- J. L. Grace, J. X. Huang, S. E. Cheah, N. P. Truong, M. A. Cooper, J. Li, T. P. Davis, J. F. Quinn, T. Velkov and M. R. Whittaker, RSC Adv., 2016, 6, 15469–15477 RSC
- J. L. Grace, A. G. Elliott, J. X. Huang, E. K. Schneider, N. P. Truong, M. A. Cooper, J. Li, T. P. Davis, J. F. Quinn, T. Velkov and M. R. Whittaker, J. Mater. Chem. B, 2017, 5, 531–536 RSC
- D. J. Coady, Z. Y. Ong, P. S. Lee, S. Venkataraman, W. Chin, A. C. Engler, Y. Y. Yang and J. L. Hedrick, Adv. Healthcare Mater., 2014, 3, 882–889 CrossRef CAS PubMed
- K. Hu, N. W. Schmidt, R. Zhu, Y. Jiang, G. H. Lai, G. Wei, E. F. Palermo, K. Kuroda, G. C. L. Wong and L. Yang, Macromolecules, 2013, 46, 1908–1915 CrossRef CAS PubMed
- B. C. Allison, B. M. Applegate and J. P. Youngblood, Biomacromolecules, 2007, 8, 2995–2999 CrossRef CAS PubMed
- S. Colak, C. F. Nelson, K. Nusslein and G. N. Tew, Biomacromolecules, 2009, 10, 353–359 CrossRef CAS PubMed
- A. Punia, A. Mancuso, P. Banerjee and N. L. Yang, ACS Macro Lett., 2015, 4, 426–430 CrossRef CAS
- S. Chakraborty, R. H. Liu, Z. Hayouka, X. Y. Chen, J. Ehrhardt, Q. Lu, E. Burke, Y. Q. Yan, B. Weisblum, G. C. L. Wong, K. S. Masters and S. H. Gellman, J. Am. Chem. Soc., 2014, 136, 14530–14535 CrossRef CAS PubMed
- X. Yang, K. Hu, G. T. Hu, D. Y. Shi, Y. J. Jiang, L. W. Hui, R. Zhu, Y. T. Xie and L. H. Yang, Biomacromolecules, 2014, 15, 3267–3277 CrossRef CAS PubMed
- A. Punia, K. Lee, E. He, S. Mukherjee, A. Mancuso, P. Banerjee and N. L. Yang, Int. J. Mol. Sci., 2015, 16, 23867–23880 CrossRef CAS PubMed
- M. Alvarez-Paino, A. Munoz-Bonilla, F. Lopez-Fabal, J. L. Gomez-Garces, J. P. A. Heuts and M. Fernandez-Garcia, Biomacromolecules, 2015, 16, 295–303 CrossRef CAS PubMed
- E. H. H. Wong, M. M. Khin, V. Ravikumar, Z. Y. Si, S. A. Rice and M. B. Chan-Park, Biomacromolecules, 2016, 17, 1170–1178 CrossRef CAS PubMed
- T. K. Nguyen, S. J. Lam, K. K. K. Ho, N. Kumar, G. G. Qiao, S. Egan, C. Boyer and E. H. H. Wong, ACS Infect. Dis., 2017, 3, 237–248 CrossRef CAS PubMed
- R. Namivandi-Zangeneh, R. J. Kwan, T.-K. Nguyen, J. Yeow, F. L. Byrne, S. H. Oehlers, E. H. H. Wong and C. Boyer, Polym. Chem., 2018 10.1039/C7PY01069A
- S. J. Lam, N. M. O'Brien-Simpson, N. Pantarat, A. Sulistio, E. H. H. Wong, Y. Y. Chen, J. C. Lenzo, J. A. Holden, A. Blencowe, E. C. Reynolds and G. G. Qiao, Nat. Microbiol., 2016, 1, 16162 CrossRef CAS PubMed
- Y. J. Jiang, W. Zheng, L. J. Kuang, H. R. Ma and H. J. Liang, ACS Infect. Dis., 2017, 3, 676–687 CrossRef CAS PubMed
- E. Strandberg, S. Morein, P. C. A. van der Wel, R. E. Koeppe and J. A. Killian, Biophys. J., 2001, 80, 541a–542a Search PubMed
- E. Strandberg and J. A. Killian, FEBS Lett., 2003, 544, 69–73 CrossRef CAS PubMed
- E. F. Palermo, S. Vemparala and K. Kuroda, Biomacromolecules, 2012, 13, 1632–1641 CrossRef CAS PubMed
- F. Nederberg, Y. Zhang, J. P. K. Tan, K. J. Xu, H. Y. Wang, C. Yang, S. J. Gao, X. D. Guo, K. Fukushima, L. J. Li, J. L. Hedrick and Y. Y. Yang, Nat. Chem., 2011, 3, 409–414 CrossRef CAS PubMed
- M. Mizutani, E. F. Palermo, L. M. Thoma, K. Satoh, M. Kamigaito and K. Kuroda, Biomacromolecules, 2012, 13, 1554–1563 CrossRef CAS PubMed
- E. A. Chamsaz, S. Mankoci, H. A. Barton and A. Joy, ACS Appl. Mater. Interfaces, 2017, 9, 6704–6711 CAS
- J. A. Grapski and S. L. Cooper, Biomaterials, 2001, 22, 2239–2246 CrossRef CAS PubMed
- S. Mankoci, R. L. Kaiser, N. Sahai, H. A. Barton and A. Joy, ACS Biomater. Sci. Eng., 2017, 3, 2588–2597 CrossRef CAS
- D. N. Amato, D. V. Amato, O. V. Mavrodi, W. B. Martin, S. N. Swilley, K. H. Parsons, D. V. Mavrodi and D. L. Patton, ACS Macro Lett., 2017, 6, 171–175 CrossRef CAS
- M. Vert, Biomacromolecules, 2005, 6, 538–546 CrossRef CAS PubMed
- M. A. Dewit and E. R. Gillies, J. Am. Chem. Soc., 2009, 131, 18327–18334 CrossRef CAS PubMed
- A. Sagi, R. Weinstain, N. Karton and D. Shabat, J. Am. Chem. Soc., 2008, 130, 5434–5435 CrossRef CAS PubMed
- A. M. DiLauro, J. S. Robbins and S. T. Phillips, Macromolecules, 2013, 46, 2963–2968 CrossRef CAS
- S. T. Phillips and A. M. DiLauro, ACS Macro Lett., 2014, 3, 298–304 CrossRef CAS
- C. E. Diesendruck, G. I. Peterson, H. J. Kulik, J. A. Kaitz, B. D. Mar, P. A. May, S. R. White, T. J. Martinez, A. J. Boydston and J. S. Moore, Nat. Chem., 2014, 6, 624–629 CrossRef PubMed
- G. I. Peterson, M. B. Larsen and A. J. Boydston, Macromolecules, 2012, 45, 7317–7328 CrossRef CAS
- M. A. Dewit, A. Beaton and E. R. Gillies, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3977–3985 CrossRef CAS
- B. Fan, J. F. Trant, A. D. Wong and E. R. Gillies, J. Am. Chem. Soc., 2014, 136, 10116–10123 CrossRef CAS PubMed
- A. D. Wong, M. A. DeWit and E. R. Gillies, Adv. Drug Delivery Rev., 2012, 64, 1031–1045 CrossRef CAS PubMed
- K. Yeung, H. Kim, H. Mohapatra and S. T. Phillips, J. Am. Chem. Soc., 2015, 137, 5324–5327 CrossRef CAS PubMed
- A. P. Esser-Kahn, N. R. Sottos, S. R. White and J. S. Moore, J. Am. Chem. Soc., 2010, 132, 10266–10268 CrossRef CAS PubMed
- M. Shamis, H. N. Lode and D. Shabat, J. Am. Chem. Soc., 2004, 126, 1726–1731 CrossRef CAS PubMed
- H. Wang, Q. Huang, H. Chang, J. R. Xiao and Y. Y. Cheng, Biomater. Sci., 2016, 4, 375–390 RSC
- Y. Xie, T. Murray-Stewart, Y. Z. Wang, F. Yua, J. Li, L. J. Marton, R. A. Casero and D. Oupicky, J. Controlled Release, 2017, 246, 110–119 CrossRef CAS PubMed
- A. M. DiLauro, G. G. Lewis and S. T. Phillips, Angew. Chem., Int. Ed., 2015, 54, 6200–6205 CrossRef CAS PubMed
- H. Zhang, K. Yeung, J. S. Robbins, R. A. Pavlick, M. Wu, R. Liu, A. Sen and S. T. Phillips, Angew. Chem., Int. Ed., 2012, 51, 2400–2404 CrossRef CAS PubMed
- A. P. Esser-Kahn, S. A. Odom, N. R. Sottos, S. R. White and J. S. Moore, Macromolecules, 2011, 44, 5539–5553 CrossRef CAS
- C. Ergene and E. F. Palermo, Biomacromolecules, 2017, 18, 3400–3409 CrossRef CAS PubMed
- M. G. Olah, J. S. Robbins, M. S. Baker and S. T. Phillips, Macromolecules, 2013, 46, 5924–5928 CrossRef CAS
- J. F. Lutz, Polym. Chem., 2010, 1, 55–62 RSC
- K. Hilpert, M. R. Elliott, R. Volkmer-Engert, P. Henklein, O. Donini, Q. Zhou, D. F. H. Winkler and R. E. W. Hancock, Chem. Biol., 2006, 13, 1101–1107 CrossRef CAS PubMed
- Y. Oda, S. Kanaoka, T. Sato, S. Aoshima and K. Kuroda, Biomacromolecules, 2011, 12, 3581–3591 CrossRef CAS PubMed
- A. R. Song, S. G. Walker, K. A. Parker and N. S. Sampson, ACS Chem. Biol., 2011, 6, 590–599 CrossRef CAS PubMed
- D. S. S. M. Uppu, M. Bhowmik, S. Samaddar and J. Haldar, Chem. Commun., 2016, 52, 4644–4647 RSC
- A. Kuroki, P. Sangwan, Y. Qu, R. Peltier, C. Sanchez-Cano, J. Moat, C. G. Dowson, E. G. L. Williams, K. E. S. Locock, M. Hartlieb and S. Perrier, ACS Appl. Mater. Interfaces, 2017, 9(46), 40117–40126 CAS
- L. M. Thoma, B. R. Boles and K. Kuroda, Biomacromolecules, 2014, 15, 2933–2943 CrossRef CAS PubMed
- D. S. S. M. Uppu, S. Samaddar, J. Hoque, M. M. Konai, P. Krishnamoorthy, B. R. Shome and J. Haldar, Biomacromolecules, 2016, 17, 3094–3102 CrossRef CAS PubMed
- M. M. Konai and J. Haldar, Bioconjugate Chem., 2017, 28, 1194–1204 CrossRef CAS PubMed
- Q. Gao, M. Yu, Y. J. Su, M. H. Xie, X. Zhao, P. Li and P. X. Ma, Acta Biomater., 2017, 51, 112–124 CrossRef CAS PubMed
- J. C. Cheng, W. Chin, H. H. Dong, L. Xu, G. S. Zhong, Y. Huang, L. J. Li, K. J. Xu, M. Wu, J. L. Hedrick, Y. Y. Yang and W. M. Fan, Adv. Healthcare Mater., 2015, 4, 2128–2136 CrossRef CAS PubMed
- H. G. Sahl, Microbiol. Sci., 1985, 2, 212–217 CAS
- K. Matsuzaki, Biochim. Biophys. Acta, Biomembr., 1999, 1462, 1–10 CrossRef CAS
- L. Yang, T. M. Weiss, R. I. Lehrer and H. W. Huang, Biophys. J., 2000, 79, 2002–2009 CrossRef CAS PubMed
- K. Matsuzaki, O. Murase and K. Miyajima, Biochemistry, 1995, 34, 12553–12559 CrossRef CAS PubMed
- G. Bocchinfuso, A. Palleschi, B. Orioni, G. Grande, F. Formaggio, C. Toniolo, Y. Park, K. S. Hahm and L. Stella, J. Pept. Sci., 2009, 15, 550–558 CrossRef CAS PubMed
- Y. Shai, Biochim. Biophys. Acta, Biomembr., 2002, 66, 236–248 CAS
- D. Sengupta, H. Leontiadou, A. E. Mark and S. J. Marrink, Biochim. Biophys. Acta, Biomembr., 2008, 1778, 2308–2317 CrossRef CAS PubMed
- E. Gazit, I. R. Miller, P. C. Biggin, M. S. P. Sansom and Y. Shai, J. Mol. Biol., 1996, 258, 860–870 CrossRef CAS PubMed
- A. J. Krauson, J. He and W. C. Wimley, Biochim. Biophys. Acta, Biomembr., 2012, 1818, 1625–1632 CrossRef CAS PubMed
- K. W. Bayles, Nat. Rev. Microbiol., 2014, 12, 63–69 CrossRef CAS PubMed
- L. P. Ding, E. Y. Chi, S. Chemburu, E. Ji, K. S. Schanze, G. P. Lopez and D. G. Whitten, Langmuir, 2009, 25, 13742–13751 CrossRef CAS PubMed
- K. Matsuzaki, K. Sugishita, N. Fujii and K. Miyajima, Biochemistry, 1995, 34, 3423–3429 CrossRef CAS PubMed
- S. G. Hovakeemian, R. H. Liu, S. H. Gellman and H. Heerklotz, Soft Matter, 2015, 11, 6840–6851 RSC
- J. R. Lai, R. F. Epand, B. Weisblum, R. M. Epand and S. H. Gellman, Biochemistry, 2006, 45, 15718–15730 CrossRef CAS PubMed
- K. Matsuzaki, K. Sugishita, N. Ishibe, M. Ueha, S. Nakata, K. Miyajima and R. M. Epand, Biochemistry, 1998, 37, 11856–11863 CrossRef CAS PubMed
- Y. H. Li, L. M. Chan, L. Tyer, R. T. Moody, C. M. Himel and D. M. Hercules, J. Am. Chem. Soc., 1975, 97, 3118–3126 CrossRef CAS
- E. F. Palermo, D. K. Lee, A. Ramamoorthy and K. Kuroda, J. Phys. Chem. B, 2011, 115, 366–375 CrossRef CAS PubMed
- Y. Tamba and M. Yamazaki, Biochemistry, 2005, 44, 15823–15833 CrossRef CAS PubMed
- J. M. Alam, T. Kobayashi and M. Yamazaki, Biochemistry, 2012, 51, 5160–5172 CrossRef CAS PubMed
- E. E. Ambroggio, F. Separovic, J. H. Bowie, G. D. Fidelio and L. A. Bagatolli, Biophys. J., 2005, 89, 1874–1881 CrossRef CAS PubMed
- T. M. Domingues, K. A. Riske and A. Miranda, Langmuir, 2010, 26, 11077–11084 CrossRef CAS PubMed
- L. H. Yang, V. D. Gordon, A. Mishra, A. Sorn, K. R. Purdy, M. A. Davis, G. N. Tew and G. C. L. Wong, J. Am. Chem. Soc., 2007, 129, 12141–12147 CrossRef CAS PubMed
- K. Hu, N. W. Schmidt, R. Zhu, Y. J. Jiang, G. H. Lai, G. Wei, E. F. Palermo, K. Kuroda, G. C. L. Wong and L. H. Yang, Macromolecules, 2013, 46, 1908–1915 CrossRef CAS PubMed
- R. F. Epand, G. S. Wang, B. Berno and R. M. Epand, Antimicrob. Agents Chemother., 2009, 53, 3705–3714 CrossRef CAS PubMed
- R. F. Epand, W. L. Maloy, A. Ramamoorthy and R. M. Epand, Biochemistry, 2010, 49, 4076–4084 CrossRef CAS PubMed
- R. M. Epand, S. Rotem, A. Mor, B. Berno and R. F. Epand, J. Am. Chem. Soc., 2008, 130, 14346–14352 CrossRef CAS PubMed
- R. F. Epand, B. P. Mowery, S. E. Lee, S. S. Stahl, R. I. Lehrer, S. H. Gellman and R. M. Epand, J. Mol. Biol., 2008, 379, 38–50 CrossRef CAS PubMed
- C. W. Avery, A. Som, Y. J. Xu, G. N. Tew and Z. Chen, Anal. Chem., 2009, 81, 8365–8372 CrossRef CAS PubMed
- C. W. Avery, E. F. Palermo, A. McLaughin, K. Kuroda and Z. Chen, Anal. Chem., 2011, 83, 1342–1349 CrossRef CAS PubMed
- X. Y. Chen, J. Wang, A. P. Boughton, C. B. Kristalyn and Z. Chen, J. Am. Chem. Soc., 2007, 129, 1420–1427 CrossRef CAS PubMed
- A. Ramamoorthy, Solid State Nucl. Magn. Reson., 2009, 35, 201–207 CrossRef CAS PubMed
- Y. C. Su, W. F. DeGrado and M. Hong, J. Am. Chem. Soc., 2010, 132, 9197–9205 CrossRef CAS PubMed
- I. Ivanov, S. Vemparala, V. Pophristic, K. Kuroda, W. F. DeGrado, J. A. McCammon and M. L. Klein, J. Am. Chem. Soc., 2006, 128, 1778–1779 CrossRef CAS PubMed
- E. Strandberg, S. Morein, D. T. S. Rijkers, R. M. J. Liskamp, P. C. A. van der Wel and J. A. Killian, Biochemistry, 2002, 41, 7190–7198 CrossRef CAS PubMed
- V. K. Mishra, M. N. Palgunachari, J. P. Segrest and G. M. Anantharamaiah, J. Biol. Chem., 1994, 269, 7185–7191 CAS
- U. Baul and S. Vemparala, Soft Matter, 2017, 13, 7665–7676 RSC
- U. Baul, K. Kuroda and S. Vemparala, J. Chem. Phys., 2014, 141, 084902 CrossRef PubMed
- M. Benincasa, S. Pacor, R. Gennaro and M. Scocchi, Antimicrob. Agents Chemother., 2009, 53, 3501–3504 CrossRef CAS PubMed
- C. S. Alves, M. N. Melo, H. G. Franquelim, R. Ferre, M. Planas, L. Feliu, E. Bardaji, W. Kowalczyk, D. Andreu, N. C. Santos, M. X. Fernandes and M. A. R. B. Castanho, J. Biol. Chem., 2010, 285, 27536–27544 CrossRef CAS PubMed
- K. S. Huang, C. H. Yang, S. L. Huang, C. Y. Chen, Y. Y. Lu and Y. S. Lin, Int. J. Mol. Sci., 2016, 17 Search PubMed
- C. D. Fjell, J. A. Hiss, R. E. W. Hancock and G. Schneider, Nat. Rev. Drug Discovery, 2012, 11, 37–51 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2018 |