DOI:
10.1039/C8PY00062J
(Paper)
Polym. Chem., 2018,
9, 2313-2327
SET-LRP in biphasic mixtures of fluorinated alcohols with water
Received
13th January 2018
, Accepted 16th March 2018
First published on 20th March 2018
Abstract
Biphasic-binary mixtures of 2,2,2-trifluoroethanol (TFE) or 2,2,3,3-tetrafluoropropanol (TFP) with water were used as reaction media to synthesize well-defined poly(methyl acrylate) (PMA) with chain end functionality close to 100% by SET-LRP. Non-activated Cu(0) wire was used as a catalyst, taking advantage of the Cu(0)-activation property that these fluorinated alcohols possess. Biphasic-binary mixtures of water, containing a ligand and Cu(II)Br2 either generated by disproportionation of Cu(I)Br or externally added, and an organic solvent, containing a monomer and a polymer, were studied. Two N-ligands were investigated: the classic tris(2-dimethylaminoethyl)amine (Me6-TREN) and tris(2-aminoethyl)amine (TREN), as a more economically attractive alternative for technological purposes. The results reported here support the replacement of Me6-TREN by TREN, taking into account the fact that the latter requires small loadings of an externally added Cu(II)Br2 deactivator and a ligand in the water phase to mediate a living radical polymerization process. Both catalytic systems ensure efficient SET-LRP processes with first order kinetics to high conversion, linear dependence of experimental Mn on conversion, narrow molecular weight distribution, and near-quantitative chain end functionality.
Introduction
Single-Electron Transfer Living Radical Polymerization (SET-LRP) is sustained by the self-regulated generation of the Cu(0) activator and Cu(II)X2/ligand deactivator via the solvent–ligand mediated disproportionation of Cu(I)X species.1–8 This fundamental step is known to occur not only in water but also in many polar organic solvents in the presence of N-donor ligands that preferentially bind Cu(II)X2 rather than Cu(I)X. Hence, water,9–13 dimethyl sulfoxide (DMSO),14–17 dimethyl formamide (DMF), dimethyl acetamide (DMAC), ethylene carbonate, alcohols,18–25 and some of their binary mixtures12,26,27 are excellent reaction media to practice SET-LRP. The list of preferred ligands that mediate disproportionation includes tris(2-dimethylaminoethyl)amine (Me6-TREN) as well as other less expensive alternatives such as tris(2-aminoethyl)amine (TREN) and branched poly(ethyleneimine) (PEI).2,5,28–31 According to the required disproportionation of Cu(I)X, the use of solvents that do not mediate sufficient disproportionation (for e.g. acetone and CH3CN) and/or others in which Cu(II)X2 is not sufficiently soluble like in the case of toluene, anisole and other non-polar solvents poses a threat to the livingness of the SET-LRP methodology.16,32–34
Our laboratory recently developed a library of “programmed” multiphasic SET-LRP systems based on mixtures of organic solvents with water.35–41 Previous experiments that inspired this concept will be discussed in a later subchapter. This approach relies on the unexpected immiscibility between a solution of water containing Cu(II)Br2 and a ligand with organic solvents, including water miscible organic solvents, containing a monomer and a polymer. Of particular interest are mixtures of organic solvents based on combinations of polar and/or non-polar non-disproportionating solvents with water because they overcome the inherent SET-LRP requirements to use polar disproportionating solvents. To support this statement, we reported relevant examples of SET-LRP synthesis of hydrophobic polymers in biphasic aqueous mixtures of non-polar and polar-non-disproportionation solvents.35–38 This approach can also be applied to solvents with good disproportionation capabilities such as alcohols as well as dipolar aprotic solvents (i.e. DMSO, sulfolane, NMP, DMAC and DMF).39–41 Most of these systems were developed applying a multicomponent catalytic SET-LRP methodology where Cu(0) is generated “in situ” via the reduction of Cu(II)Br2 with NaBH4 although the simplest Cu(0) wire catalytic system was also successfully employed.5,42,43
Recently our laboratory reported the replacement of Me6-TREN, the common ligand in the first generation of “programmed” multiphasic SET-LRP systems, by TREN, which is 80 times less expensive.40 This attractive alternative provided excellent results when applied to the aqueous polymerization of vinyl chloride44–46 and also to the Cu(0)-catalyzed SET-LRP in organic media.1,47 Biphasic-binary water–organic solvent mixtures containing traditional disproportionating and non-disproportionating organic solvents were reinvestigated using TREN and non-activated Cu(0) wire to further reduce the cost of these “programmed” biphasic SET-LRP systems for large-scale applications. The work reported in this manuscript expands the scope of this biphasic SET-LRP methodology to mixtures of 2,2,2-trifluoroethanol (TFE) and 2,2,3,3-tetrafluoropropanol (TFP) with water. These fluorinated alcohols carry both fluorinated hydrophobic and hydroxyl hydrophilic sites, which help to mediate the disproportionation of Cu(I)X, solubilize monomers and polymers with a wide polarity range, and consequently have the potential to serve as “universal solvents” for the SET-LRP of a larger diversity of monomers than any other solvent used before.2,4,5,8 These fluorinated alcohols facilitated efficient SET-LRP to prepare near-perfect chain-end functional hydrophilic, hydrophobic, amphiphilic and also semifluorinated acrylates20,21,27 and methacrylates.22,23 Moreover, they possess a self-activation property by which commercial Cu(0) wire used as a catalyst for SET-LRP is activated in situ.24 This avoids pre-activation treatments,48,49 with reducing agents or acids, to remove the oxide layer on the surface of commercial wires and paves the way to practice SET-LRP without undesirable induction periods.5
Results and discussion
Previous experiments that inspired “programmed” biphasic SET-LRP systems
The scope of polymers and macromolecular architectures accessible by SET-LRP was originally thought to be determined by the solubility of the targeted monomers and polymers in solvents with good disproportionating properties. However, it has to be noted that a certain homogeneous SET-LRP process even succeeded when transitioned to biphasic systems due to the precipitation of the polymer out of the reaction mixture beyond a certain degree of polymerization. This was first observed and reported by our laboratory during the synthesis of ultrahigh molar mass poly(methyl acrylate) (PMA) by the Cu(0) powder-catalyzed SET-LRP of methyl acrylate (MA) in DMSO.1 Our laboratory also reported the formation of a biphasic system for the SET-LRP of butyl acrylate (BA) in DMSO.14 Later, the Cu(0) wire-catalyzed SET-LRP of challenging long hydrocarbon chain hydrophobic monomers such as lauryl acrylate and stearyl acrylate in DMSO and isopropanol also furnished “self-generated” biphasic systems.50 Moreover, the “self-generated” system during the SET-LRP of BA in DMSO was investigated in more detail by the laboratory of Haddleton that reported appealing advantages in terms of enhanced end-group fidelity via suppression of bimolecular termination and in situ purification of the resulting polymer from copper species.8,51–53 The SET-LRP of BA also resulted in biphasic systems when dimethyl lactamide was used as a solvent.54 Meanwhile, a biphasic reaction mixture containing Cu(II)Br2 solution in alcohol separated from the immiscible polymer solution in alcohol was observed during the SET-LRP of 2-ethylhexyl acrylate in TFE.20 These developments together with the development of SET-LRP of N-isopropylacrylamide catalyzed by Cu(0) generated in situ by the reduction of Cu(II)Br2 with NaBH4 in water led to the elaboration of “programmed” biphasic SET-LRP systems that are produced by design.42,43
SET-LRP of MA catalyzed by non-activated Cu(0) wire/Me6-TREN in TFE/water and TFP/water mixtures
Previously, we reported the SET-LRP of MA and BA, using Cu(0) generated in situ by the reduction of Cu(II)Br2 with NaBH4, in “programmed” bi(multi)phasic alcohol/water mixtures.35,41 The selection of an alcohol solvent was determinant for the evolution of the SET-LRP reaction mixture. Whereas systems based on higher alcohols remained biphasic up to complete conversion, compositions based on methanol and ethanol transitioned, above a certain conversion and molecular weight, to a triphasic system. Other “programmed” biphasic SET-LRP systems based on both disproportionating and non-disproportionating solvents were also successfully developed using a more convenient methodology based on Cu(0) wire.36,39,40 The driving force of these “programmed” bi(multiphasic) SET-LRP systems is the immiscibility of water containing Cu(II)Br2 and the ligand with even water-soluble solvents containing the monomer and polymer. This effect has dramatic implications even in the presence of traces of water.
However, neither of these catalytic systems has ever been used to study in detail mixtures of water with one of the most versatile classes of SET-LRP solvents such as TFE and TFP. Hence, the first series of experiments reported here focus on the Me6-TREN-mediated SET-LRP of MA in mixtures of these fluorinated alcohols with water in the presence of non-activated Cu(0) wire (Scheme 1a). A series of kinetic experiments were performed in various alcohol/water ratios using the monofunctional initiator methyl 2-bromopropionate (MBP) under the following conditions [MA]0/[MBP]0/[Me6-TREN]0 = 222/1/0.1. Fig. 1 and 2 show all the kinetic experiments while more relevant data from these experiments are summarized in Table 1. These experiments were performed under conditions comparable to those reported under homogeneous conditions in TFE and TFP.20–24 When biphasic mixtures of TFE and TFP with water were employed, the ratio between TFE and TFP to water was varied from 9/1 to 8/2 and to 7/3 (Fig. 1, 2 and Table 1) in order to maintain comparable conditions with similar experiments performed with acetone/water36 acetonitrile/water38 and alcohols/water,41 although a similar trend of the rate of reaction was observed up to solvent/water = 6/4, v/v.36,38,41 At lower ratios between solvent and water corresponding to less solvent in the reaction mixture the concentration of the solvent becomes too low to maintain a biphasic system41 and the mechanism of SET-LRP changes from biphasic to triphasic. The second part of this mechanism will be investigated and be reported in different publications.
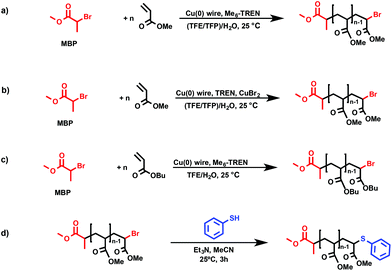 |
| | Scheme 1 (a) Me6-TREN and (b) TREN-mediated SET-LRP of MA in binary mixtures of TFE or TFP with water in the presence of non-activated Cu(0) wire. (c) Me6-TREN-mediated SET-LRP of BA in binary mixtures of TFE with water in the presence of non-activated Cu(0) wire. (d) “thio-bromo click” modification of the corresponding PMA with thiophenol. | |
 |
| | Fig. 1 Kinetic plots, molecular weight and dispersity evolution for the SET-LRP of MA in TFE and TFE/water mixtures initiated with MBP and catalyzed by nonactivated Cu(0) wire at 25 °C. (a) TFE; (b) TFE/water (9/1, v/v); (c) TFE/water (8/2, v/v) and (d) TFE/water (7/3, v/v). The v/v ratio must be multiplied by 10 to obtain % solvent/% water. The value of v + v must be divided by 10 to obtain the total volume of solvents, 1 mL. Reaction conditions: MA = 1 mL, (a) TFE = 0.5 mL, (b, c, d) TFE + water = 0.5 mL, [MA]0/[MBP]0/[Me6-TREN]0 = 222/1/0.1. Experimental data in different colors were obtained from different kinetic experiments sometimes performed by different researchers. | |
 |
| | Fig. 2 Kinetic plots, molecular weight and dispersity evolution for the SET-LRP of MA in TFP and TFP/water mixtures initiated with MBP and catalyzed by nonactivated Cu(0) wire at 25 °C. (a) TFP; (b) TFP/water (9/1, v/v); (c) TFP/water (8/2, v/v) and (d) TFP/water (7/3, v/v). The v/v ratio must be multiplied by 10 to obtain % solvent/% water. The value of v + v must be divided by 10 to obtain the total volume of solvents, 1 mL. Reaction conditions: MA = 1 mL, (a) TFP = 0.5 mL, (b, c, d) TFE + water = 0.5 mL, [MA]0/[MBP]0/[Me6-TREN]0 = 222/1/0.1. Experimental data in different colors were obtained from different kinetic experiments sometimes performed by different researchers. | |
Table 1 SET-LRP of MA in biphasic mixtures of water with 2,2,2-trifluoroethanol (TFE) and 2,2,3,3-tetrafluoro-1-propanol (TFP) mediated with Me6-TREN and catalyzed by nonactivated 4.5 cm of 20 gauge Cu(0) wire. Reaction conditions: Monomer = 1 mL; solvent + water = 0.5 ml. The v/v ratio must be multiplied by 10 to obtain % solvent/% water. The value of v + v must be divided by 20 to obtain the total volume of solvents, 0.5 mL
| Entry |
Reaction medium |
Reaction conditions |
k
appp (min−1) |
k
appp (s+w)/kappp (s) |
Reaction timea (min) |
Conv. (%) |
(Mw/Mn) |
I
eff![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b (%) b (%) |
|
The reaction time to the monomer conversion (%) is reported in the next column.
Initiator efficiency.
|
| 1 |
TFE |
[MA]/[MBP]/[Me6-TREN] 222/1/0.1 |
0.013 |
— |
155 |
86 |
1.14 |
94 |
| 2 |
TFE/H2O (9/1, v/v) |
[MA]/[MBP]/[Me6-TREN] 222/1/0.1 |
0.019 |
1.46 |
92 |
84 |
1.17 |
92 |
| 3 |
TFE/H2O (8/2, v/v) |
[MA]/[MBP]/[Me6-TREN] 222/1/0.1 |
0.025 |
1.92 |
75 |
85 |
1.16 |
100 |
| 4 |
TFE/H2O (7/3, v/v) |
[MA]/[MBP]/[Me6-TREN] 222/1/0.1 |
0.028 |
2.15 |
60 |
79 |
1.22 |
98 |
| |
| 5 |
TFP |
[MA]/[MBP]/[Me6-TREN] 222/1/0.1 |
0.017 |
— |
120 |
85 |
1.13 |
95 |
| 6 |
TFP/H2O (9/1, v/v) |
[MA]/[MBP]/[Me6-TREN] 222/1/0.1 |
0.022 |
1.35 |
90 |
85 |
1.14 |
92 |
| 7 |
TFP/H2O (8/2, v/v) |
[MA]/[MBP]/[Me6-TREN] 222/1/0.1 |
0.031 |
1.82 |
60 |
82 |
1.22 |
100 |
| 8 |
TFP/H2O (7/3, v/v) |
[MA]/[MBP]/[Me6-TREN] 222/1/0.1 |
0.040 |
2.35 |
45 |
82 |
1.22 |
100 |
| |
| 9 |
TFE |
[BA]/[MBP]/[Me6-TREN] 222/1/0.1 |
0.006 |
— |
330 |
84 |
1.15 |
100 |
| 10 |
TFE/H2O (9/1, v/v) |
[BA]/[MBP]/[Me6-TREN] 222/1/0.1 |
0.015 |
2.50 |
120 |
82 |
1.18 |
100 |
| 11 |
TFE/H2O (8/2, v/v) |
[BA]/[MBP]/[Me6-TREN] 222/1/0.1 |
0.020 |
3.33 |
90 |
82 |
1.20 |
100 |
| 12 |
TFE/H2O (7/3, v/v) |
[BA]/[MBP]/[Me6-TREN] 222/1/0.1 |
0.028 |
4.66 |
65 |
83 |
1.17 |
100 |
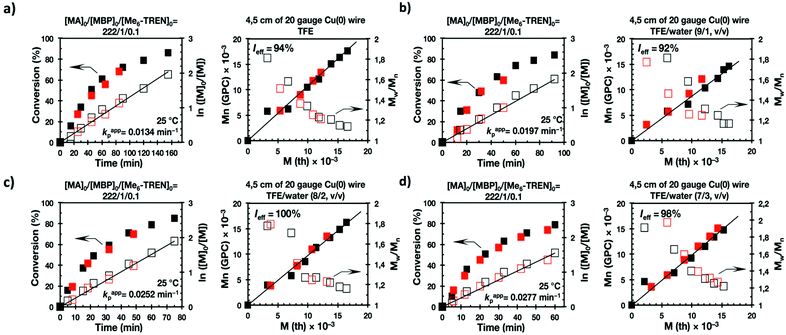
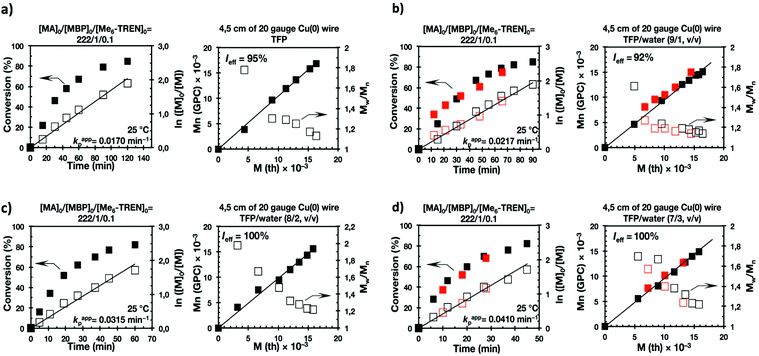
The first two experiments were performed in pure TFE and TFP for comparison purposes (Fig. 1a and 2a, respectively). As described previously,20–24,27 both fluorinated alcohols behaved excellently as SET-LRP solvents in the non-activated Cu(0) wire/Me6-TREN-mediated polymerization of MA. The living behavior of these polymerizations was validated through the linear evolution of ln([M]0/[M]) vs. polymerization time, narrow dispersity and almost quantitative chain end functionality. Note that no significant differences in terms of kappp and molecular weight control were observed between the polymerizations carried out in these two alcohols. In both cases kappp was almost 0.015 min−1, Ieff was close to 100%, and Mw/Mn at the end of the polymerization reached values <1.15 in all systems (Table 1, entries 1 and 5).
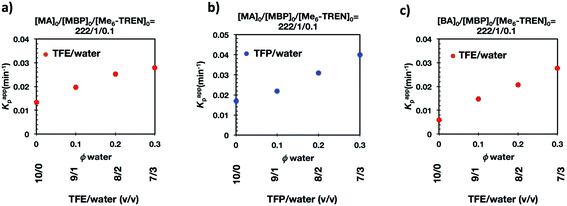
It is important to point out that SET-LRP of MA in both fluorinated alcohols proceeds in a one phase through the entire reaction, resulting in a clear, homogeneous and transparent reaction mixture at high conversion (Fig. 3a and e). Next experiments were carried out in mixtures of TFE and TFP containing 10%, 20% and 30% water (Fig. 1b–d and 2b–d, respectively). All kinetic experiments showed high reproducibility and accomplish the expected features of a living polymerization system. This statement is supported by first order kinetic experiments showing also experimental Mn increasing linearly with conversion, regardless of the alcohol/water ratio. Table 1 entries 2–4 and 6–8 collect the more relevant data obtained from these experiments. Remarkably, kappp increases continuously with the water content. The highest kappp was observed at an alcohol/water volume ratio 7/3 (kappp = 0.028 min−1 in TFE/water and 0.040 min−1 in TFP/water). The addition of only 10% water to TFE increased the rate constant from kappp = 0.013 min−1 to kappp = 0.019 min−1 (compare Fig. 1a with b and Table 1 entries 1 and 2). The increment of kappp continues up to the mixture containing 30% water. Thus, in the presence of 20% water kappp increases to 0.025 min−1 whereas in TFE containing 30% water the rate constant increases by a factor of 2.15 in comparison to the experiment carried out in pure TFE (kappp = 0.013 min−1 in pure TFE to kappp = 0.019 min−1 in TFE/water 7/3, v/v). The same trend was observed for the homologous TFP/water mixtures (compare Table 1, entries 5–8). As can be seen in Fig. 4a and b, kappp increases linearly as the volume fraction of water (ΦH2O) increases in both fluorinated alcohol/water mixtures. Butyl acrylate (BA) was used to expand the monomer scope of these biphasic SET-LRP systems to more hydrophobic monomers (Fig. 5). The polymerization of BA was investigated in various TFE/water mixtures (Table 1 entries 9–12). As can be seen in Fig. 3, the polymerization of BA follows the expected characteristics of a living polymerization reaction. In this case, kappp also increases linearly as the volume fraction of water (ΦH2O) increases (Fig. 4c).
 |
| | Fig. 3 Visualization of the reaction mixture after SET-LRP of MA initiated with MBP and catalyzed by non-activated Cu(0) wire at 25 °C in (a) TFE; (b) TFE/water (9/1, v/v); (c) TFE/water (8/2, v/v); (d) TFE/water (7/3, v/v); (e) TFP; (f) TFP/water (9/1, v/v); (g) TFP/water (8/2, v/v) and (h) TFP/water (7/3, v/v). Reaction conditions: MA = 1 mL, fluorinated solvent (TFE/TFP) + water = 0.5 mL, [MA]0/[MBP]0/[Me6-TREN]0 = 222/1/0.1. | |
 |
| | Fig. 4 The dependence of kappp with the volume fraction of water (ΦH2O) on the SET-LRP of MA and BA initiated with MBP mediated by Me6-TREN and catalyzed by 4.5 cm of non-activated Cu(0) wire in (a and c) TFE/water and (b) TFP/water mixtures. | |
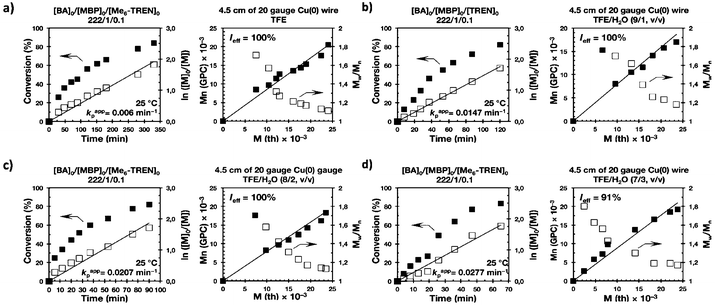 |
| | Fig. 5 Kinetic plots, molecular weight and dispersity evolution for the SET-LRP of BA in TFE and TFE/water mixtures initiated with MBP and catalyzed by non-activated Cu(0) wire at 25 °C. (a) TFE; (b) TFE/water (9/1, v/v); (c) TFE/water (8/2, v/v) and (d) TFE/water (7/3, v/v). The v/v ratio must be multiplied by 10 to obtain % solvent/% water. The value of v + v must be divided by 10 to obtain the total volume of solvents, 1 mL. Reaction conditions: MA = 1 mL, (a) TFE = 0.5 mL, (b, c, d) TFE + water = 0.5 mL, [BA]0/[MBP]0/[Me6-TREN]0 = 222/1/0.1. | |
The biphasic nature of the TFE/water and TFP/water systems is supported by the series of digital images shown in Fig. 3b–d and f–h, respectively. At the transition from pure alcohol to the system containing 10% water the reaction mixture became slightly turbid in both TFE and TFP (Fig. 3b and f) rather than transparent (Fig. 3a and e). This turbidity was more evident at a ratio 8/2 v/v, especially in the TFP/water system. The system at 8/2, v/v shows some bluish water droplets, containing mainly Cu(II)Br2/Me6-TREN complexes and probably some traces of TFP and monomer, which are not miscible with the organic phase containing TFP, PMA and the residual MA. As can be seen in Fig. 3d and h, a clear biphasic system evolved in both systems at ΦH2O = 0.3. It is remarkable that the transition from one phase to the biphasic system did provide only a small increase in Mw/Mn from ∼1.15 to ∼1.20 and did not affect the Ieff (Table 1). Representative gel permeation chromatography (GPC) traces of PMA obtained during the SET-LRP process TFE/water at a ratio 8/2, v/v as a function of conversion are shown in Fig. 6a.
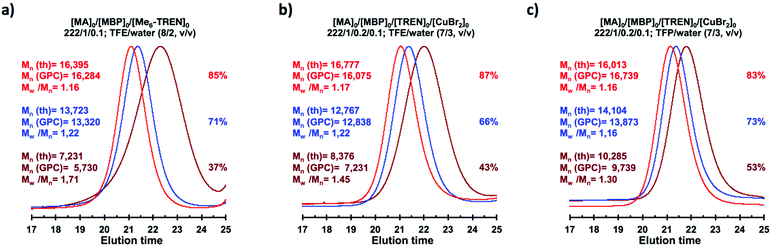 |
| | Fig. 6 GPC traces of PMA obtained by SET-LRP of MA initiated with MBP and catalyzed by nonactivated Cu (0) wire at 25 °C in (a) TFE/water (8/2, v/v); (b) TFE/water (7/3, v/v) and (c) TFP/water (7/3, v/v). Reaction conditions: (a) MA = 1 mL, TFE + water = 0.5 mL, [MA]0/[MBP]0/[Me6-TREN]0 = 222/1/0.1 and (b and c) [MA]0/[MBP]0/[TREN]0/[CuBr2]0 = 222/1/0.2/0.1. | |
SET-LRP of MA catalyzed by non-activated Cu(0) wire/TREN/Cu(II)Br2 in TFE/water and TFP/water mixtures
Recently our laboratory reported the replacement of Me6-TREN, the common ligand in the first generation of “programmed” multiphasic SET-LRP systems, by TREN,40 which is 80 times less expensive and at least in biphasic systems it has been shown to be as efficient as Me6-TREN. Importantly, the external addition of the Cu(II)Br2 deactivator to the water phase was necessary to produce well-defined polyacrylates when switching from Me6-TREN to TREN. In the next series of experiments, the above first time presented “programmed” biphasic systems based on the mixtures of TFE and TFP with water were reinvestigated using TREN as the ligand (Scheme 1b).
Fig. 7 and 8 depict the kinetic experiments in both alcohol/water systems and Table 2 summarizes the obtained kinetic data. Fig. 7a demonstrates that the straightforward replacement of Me6-TREN by TREN was not successful in a TFE/water 9/1, v/v system because it resulted in the loss of all the living features described above (Fig. 7avs.Fig. 1b). However, all the tested TFE/water and TFP/water compositions recover first order kinetics and a linear evolution of experimental Mn with conversion in the presence of externally added Cu(II)Br2 under the following conditions: [MA]/[MBP]/[TREN]/[CuBr2] = 222/1/0.2/0.1. Based on previous studies reported by our laboratory,40,47 [TREN]/[CuBr2] molar ratio 2/1 was used to complex the added Cu(II)Br2 and the potentially formed Cu(I)Br that must be partitioned by the ligand between the organic and aqueous phases and also be disproportionated in the water phase. It is important to point out that the use of TREN and externally added Cu(II)Br2 instead of Me6-TREN resulted in slower polymerizations. For example, the polymerization of MA in a TFP/water 8/2, v/v showed kappp = 0.031 min−1 for the Me6-TREN systems but decreased to 0.019 min−1 when Me6-TREN was replaced by TREN (compare Table 1, entry 7 and Table 2, entry 6). Nevertheless, comparable levels of control were observed in both systems as indicated by systematic Ieff values close to 100% and Mw/Mn values at around ∼1.15, indistinctly (Table 2, entries 2–7). Fig. 6b and c show representative GPC traces of PMA obtained in TFE/water and TFP/water 7/3, v/v systems. As expected, the increment of the volume ratio of water from 9/1 to 8/2 and to 7/3 was accompanied in both systems by a dramatic increase of kappp. Remarkably, an increase in the TFP/water volume ratio from 9/1 to 7/3 increases the kappp by a factor of 4.00 (0.0032 min−1vs. 0.008 min−1). Notice also that a linear increase of the kappp with the increase of the ΦH2O was observed in both TFE/water and TFP/water SET-LRP systems (Fig. 9a and b, respectively). An additional experiment shown in Fig. 8d demonstrates that the living behavior in these series of experiments is also retained when using [TREN]/[CuBr2] molar ratio 1/1 (Table 2 entry 8). However, a detailed series of experiments with different ratios of [TREN]/[CuBr2] molar ratios will be discussed in a publication dedicated entirely to this topic.
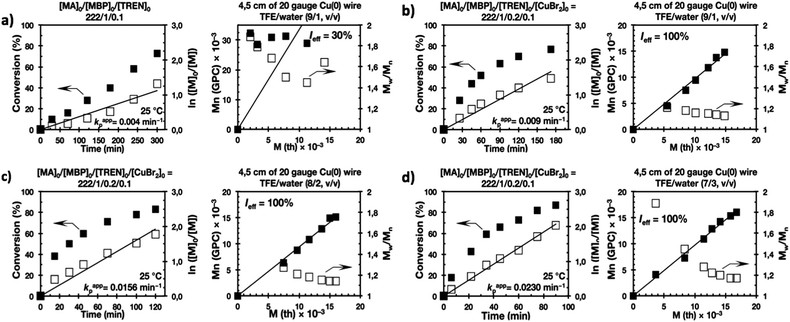 |
| | Fig. 7 Kinetic plots, molecular weight and dispersity evolution for the SET-LRP of MA in TFE/water mixtures initiated with MBP and catalyzed by non-activated Cu(0) wire at 25 °C. (a, b) TFE/water (9/1, v/v); (c) TFE/water (8/2, v/v) and (d) TFE/water (7/3, v/v). The v/v ratio must be multiplied by 10 to obtain % solvent/% water. The value of v + v must be divided by 10 to obtain the total volume of solvents, 1 mL. Reaction conditions: MA = 1 mL, TFP + water = 0.5 mL, (a) [MA]0/[MBP]0/[TREN]0 = 222/1/0.1, (b–d) [MA]0/[MBP]0/[TREN]0/[CuBr2]0 = 222/1/0.2/0.1. | |
 |
| | Fig. 8 Kinetic plots, molecular weight and dispersity evolution for the SET-LRP of MA in TFP/water mixtures initiated with MBP and catalyzed by non-activated Cu(0) wire at 25 °C. (a) TFP/water (9/1, v/v); (b) TFP/water (8/2, v/v) and (c and d) TFP/water (7/3, v/v). The v/v ratio must be multiplied by 10 to obtain % solvent/% water. The value of v + v must be divided by 10 to obtain the total volume of solvents, 1 mL. Reaction conditions: MA = 1 mL, TFP + water = 0.5 mL, [MA]0/[MBP]0/[TREN]0/[CuBr2]0 = 222/1/0.2/0.1 (a–c), [MA]0/[MBP]0/[TREN]0/[CuBr2]0 = 222/1/0.1/0.1 (d). Experimental data in different colors were obtained from different kinetics experiments sometimes performed by different researchers. | |
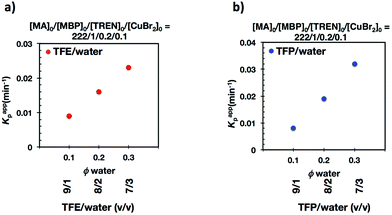 |
| | Fig. 9 The dependence of kappp with the volume fraction of water (ΦH2O) on the SET-LRP of MA initiated with MBP mediated TREN/Cu(II)Br2 and catalyzed by 4.5 cm of non-activated Cu(0) wire in (a) TFE/water and (b) TFP/water mixtures. | |
Table 2 SET-LRP of MA in biphasic mixtures of water with 2,2,2-trifluoroethanol (TFE) and 2,2,3,3-tetrafluoro-1-propanol (TFP) mediated with TREN and CuBr2, catalyzed by nonactivated 4.5 cm of 20 gauge Cu(0) wire. Reaction conditions: Monomer = 1 mL; solvent + water = 0.5 ml. The v/v ratio must be multiplied by 10 to obtain % solvent/% water. The value of v + v must be divided by 20 to obtain the total volume of solvents, 0.5 mL
| Entry |
Reaction medium |
Reactions conditions |
k
appp (min−1) |
k
appp (s+w)/kappp (s+w)a |
Reaction timeb (min) |
Conv. (%) |
(Mw/Mn) |
I
effc (%) |
|
Correspond to the kappp of (9/1 v/v) of SET-LRP experiments mediated by TREN.
The reaction time to the monomer conversion (%) is reported in the next column.
Initiator efficiency.
2.0 cm of 20 gauge non-activated Cu(0) wire was used.
12.5 cm of 20 gauge non-activated Cu(0) wire was used.
|
| 1 |
TFE/H2O (9/1, v/v) |
[MA]/[MBP]/[TREN] 222/1/0.1 |
0.004 |
— |
300 |
73 |
1.64 |
30 |
| |
| 2 |
TFE/H2O (9/1, v/v) |
[MA]/[MBP]/[TREN]/[CuBr2] 222/1/0.2/0.1 |
0.009 |
— |
172 |
77 |
1.13 |
100 |
| 3 |
TFE/H2O (8/2, v/v) |
[MA]/[MBP]/[TREN]/[CuBr2] 222/1/0.2/0.1 |
0.016 |
1.77 |
120 |
83 |
1.14 |
100 |
| 4 |
TFE/H2O (7/3, v/v) |
[MA]/[MBP]/[TREN]/[CuBr2] 222/1/0.2/0.1 |
0.023 |
2.55 |
90 |
87 |
1.17 |
100 |
| |
| 5 |
TFP/H2O (9/1, v/v) |
[MA]/[MBP]/[TREN]/[CuBr2] 222/1/0.2/0.1 |
0.008 |
— |
135 |
96 |
1.13 |
96 |
| 6 |
TFP/H2O (8/2, v/v) |
[MA]/[MBP]/[TREN]/[CuBr2] 222/1/0.2/0.1 |
0.019 |
2.37 |
100 |
80 |
1.15 |
100 |
| 7 |
TFP/H2O (7/3, v/v) |
[MA]/[MBP]/[TREN]/[CuBr2] 222/1/0.2/0.1 |
0.032 |
4.00 |
50 |
83 |
1.16 |
100 |
| 8 |
TFP/H2O (7/3, v/v) |
[MA]/[MBP]/[TREN]/[CuBr2] 222/1/0.1/0.1 |
0.033 |
4.12 |
60 |
82 |
1.15 |
100 |
| |
| 9d |
TFE/H2O (8/2, v/v) |
[MA]/[MBP]/[TREN]/[CuBr2] 222/1/0.2/0.1 |
0.015 |
— |
120 |
82 |
1.16 |
96 |
| 10e |
TFE/H2O (8/2, v/v) |
[MA]/[MBP]/[TREN]/[CuBr2] 222/1/0.2/0.1 |
0.013 |
— |
120 |
75 |
1.18 |
87 |
| |
| 11d |
TFE/H2O (8/2, v/v) |
[MA]/[MBP]/[TREN]/[CuBr2] 222/1/0.1/0.05 |
0.019 |
— |
90 |
81 |
1.15 |
89 |
| 12d |
TFE/H2O (8/2, v/v) |
[MA]/[MBP]/[TREN]/[CuBr2] 222/1/0.4/0.2 |
0.007 |
— |
150 |
63 |
1.20 |
97 |
All the tested compositions were biphasic as can be seen from the digital images taken after the SET-LRP process (Fig. 10). However, the analysis of these images suggests better dispersion of the water phase containing Cu(II)Br2 and TREN in the organic phase at low ΦH2O. Nevertheless, there is no doubt that the SET-LRP process takes place in a biphasic regime when the alcohol/water volume ratio is 7/3, v/v.
 |
| | Fig. 10 Visualization of the reaction mixture after SET-LRP of MA initiated with MBP and catalyzed by non-activated Cu(0) wire at 25 °C in (a) TFE/water (9/1, v/v); (b) TFE/water (8/2, v/v); (c) TFE/water (7/3, v/v); (e) TFP/water (9/1, v/v); (e) TFP/water (8/2, v/v) and (f) TFP/water (7/3, v/v). Reaction conditions: MA = 1 mL, Fluorinated solvent (TFE/TFP) + water = 0.5 mL, [MA]0/[MBP]0/[TREN]0/[CuBr2]0 = 222/1/0.2/0.1. | |
Chain-end analysis of PMA obtained by the SET-LRP process catalyzed by non-activated Cu(0) wire mediated by Me6-TREN and TREN in a TFE containing 20% water
A “programmed” biphasic system based on TFE was selected to perform a representative experiment directed to analyze the chain ends of PMA isolated from the above presented biphasic SET-LRP systems catalyzed by non-activated Cu(0) wire using Me6-TREN and TREN. TFE is technologically more attractive than TFP because it is commercially available at lower prices and has a lower boiling point (74.05 °C vs. 107–108 °C). Low molar mass PMA samples were isolated at around 90% monomer conversion after the SET-LRP process in a TFE/water mixture 8/2, v/v at a [MA]0/[MBP]0 = 50. The analysis of the polymer chain-ends before and after “thio-bromo click”55,56 functionalization by a combination of 500 MHz 1H-NMR and MALDI-TOF is a powerful combination of analytical methods to assess the chain-end functionality of polymers prepared by any LRP technique. Fig. 11 shows the 500 MHz 1H-NMR spectra of PMA isolated at 86% conversion after the Me6-TREN-mediated SET-LRP before and after the modification of the bromine chain ends with thiophenol via a “thio-bromo click” thioetherification reaction (see Scheme 1d).55,56
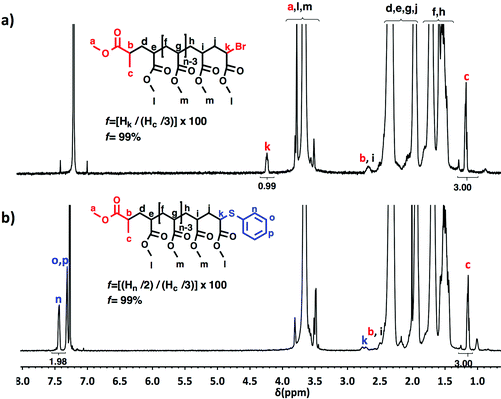 |
| | Fig. 11
1H-NMR (500 MHz) recorded in CDCl3, of PMA-Br isolated at 86% conversion from SET-LRP of MA in TFE/water (8/2, v/v) mixtures initiated with MBP and catalyzed by nonactivated Cu(0) wire at 25 °C. (a) Before the “thio-bromo click” reaction. (b) After the “thio-bromo click” reaction. Reaction conditions: MA = 1 mL, TFE + water = 0.5 mL, [MA]0/[MBP]0/[Me6-TREN]0 = 50/1/0.1, 4.5 cm of 20 gauge Cu(0) wire. | |
Taking into account the experimental error, the high degree of bromine functionality (f) determined for the isolated PMA (f = 99%) endorses the excellent SET-LRP conditions to this combination of catalyst, solvents and ligand. Fig. 12 supports that the transition from the non-activated Cu(0) wire/Me6-TREN to non-activated Cu(0) wire/TREN/Cu(II)Br2 catalytic system does not compromise the fraction of chains that are functionalized with a bromine atom at high conversion. In this case chain-end functionality was determined to be 96%. Hence, both catalytic systems ensure near perfect bromine chain end functionality of the PMA regardless of the biphasic nature of the reaction mixture generated in the presence of TFE containing 20% water. Modification of the bromine chain ends of both PMA samples with thiophenol was also monitored by MALDI-TOF spectrometry (Fig. 13 and 14). The presence of only one distribution after the thioetherification step, which appears shifted 29 mass units above the original series of PMA-Br, also supports high chain end fidelity for both catalytic systems in this mixture of solvents. These results are comparable to those obtained using previously described “programmed” biphasic SET-LRP systems based on both disproportionating and non-disproportionating solvents as well as homogeneous systems based on TFE and TFP in the absence of water.
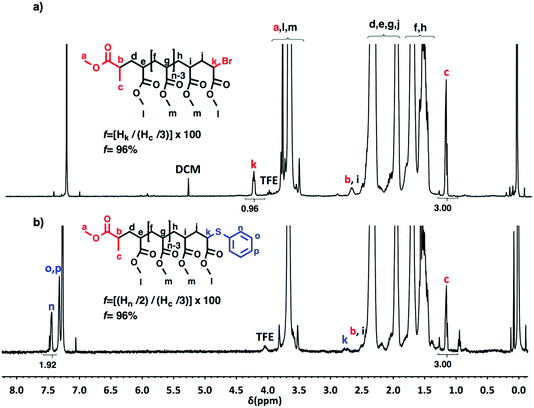 |
| | Fig. 12
1H-NMR (500 MHz) recorded in CDCl3, of PMA-Br isolated at 88% conversion from SET-LRP of MA in TFE/water (8/2, v/v) mixtures initiated with MBP and catalyzed by nonactivated Cu(0) wire at 25 °C. (a) Before the “thio-bromo click” reaction. (b) After the “thio-bromo click” reaction. Reaction conditions: MA = 1 mL, TFE + water = 0.5 mL, [MA]0/[MBP]0/[TREN]0/[CuBr2]0 = 50/1/0.2/0.1, 4.5 cm of 20 gauge Cu(0) wire. | |
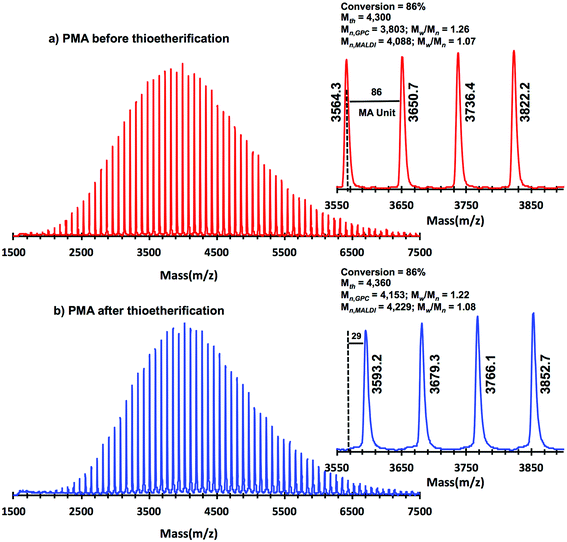 |
| | Fig. 13 MALDI-TOF of PMA-Br isolated at 86% from SET-LRP of MA in TFE/water (8/2, v/v) mixtures initiated with MBP and catalyzed by nonactivated Cu(0) wire at 25 °C. (a) Before “thio-bromo click” reaction. (b) After “thio-bromo click” reaction. Reaction conditions: MA = 1 mL, TFE + water = 0.5 mL, [MA]0/[MBP]0/[Me6-TREN]0 = 50/1/0.1, 4.5 cm of 20 gauge Cu(0) wire. The dotted line in expansion after thioeterification shows the original peak from before thioeterification, while 29 represents the increase in molar mass after thioeterification i.e., SC6H5 (109, 2)–Br (79, 9) = 29.3 for each chain end. | |
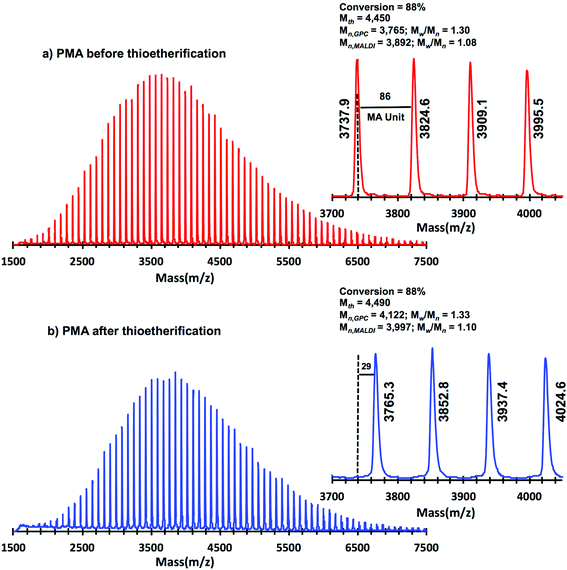 |
| | Fig. 14 MALDI-TOF of PMA-Br isolated at 88% from SET-LRP of MA in TFE/water (8/2, v/v) mixtures initiated with MBP, catalyzed by nonactivated Cu(0) wire at 25 °C. (a) Before the “thio-bromo click” reaction. (b) After the “thio-bromo click” reaction. Reaction conditions: [MA]0/[MBP]0/[TREN]0/[CuBr2]0 = 50/1/0.2/0.1, MA = 1 mL, TFE + water = 0.5 mL. 4.5 cm of 20 gauge Cu(0) wire. The dotted line in expansion after thioeterification shows the original peak from before thioeterification, while 29 represents the increase in molar mass after thioeterification i.e., SC6H5 (109, 2)–Br (79, 9) = 29.3 for each chain end. | |
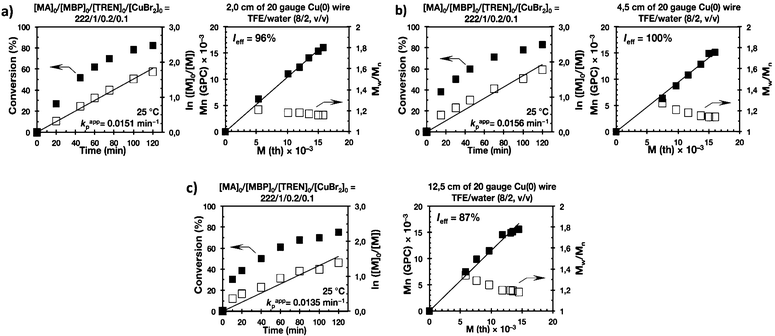
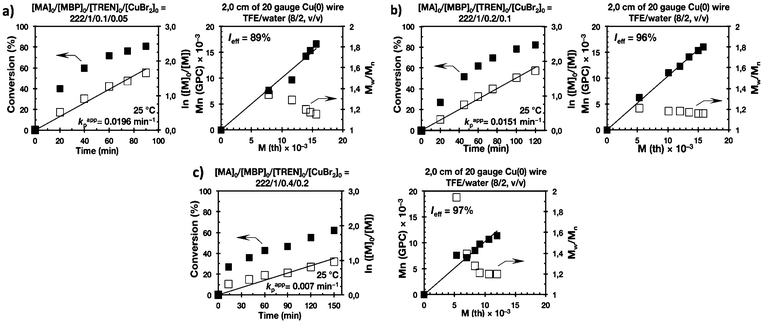
Biphasic SET-LRP processes tolerate both the use of excess of Cu(0) wire and of Cu(II)Br2
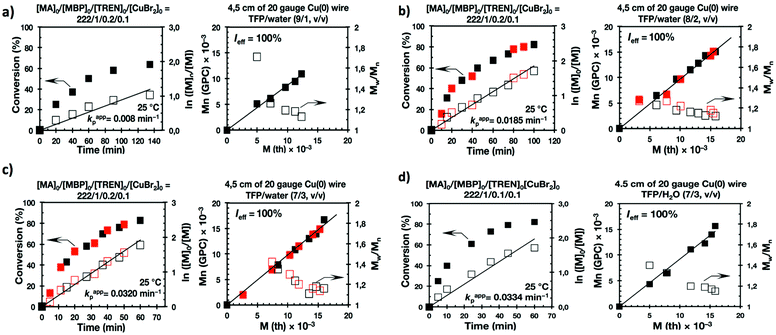
The last series of experiments were performed to bring out one of the most important features of biphasic SET-LRP processes. Biphasic SET-LRP provides a “self-controlled” reversible deactivation that does not provide the side reactions encountered in homogeneous systems when an excess of Cu(II)Br2 is used during the polymerization.15,16 This important characteristic relies on the partitioning of reagents between phases because reversible deactivation occurs at the interface of the two phases. The same is the case also for the amount of Cu(0) wire. To support these statements, the SET-LRP of MA was investigated using the TREN/Cu(II)Br2 catalytic system in a TFE containing 20% water in the presence of different amounts of Cu(0) wire and Cu(II)Br2 (Fig. 15 and 16, respectively). Table 2 summarizes all experimental data collected from both series of experiments.
 |
| | Fig. 15 Kinetic plots, molecular weight and dispersity evolution for the SET-LRP of MA in TFE/water mixtures initiated with MBP and catalyzed by nonactivated Cu(0) wire at 25 °C. (a–c) TFE/water (8/2, v/v). The v/v ratio must be multiplied by 10 to obtain % solvent/% water. The value of v + v must be divided by 10 to obtain the total volume of solvents, 1 mL. Reaction conditions: MA = 1 mL, TFP + water = 0.5 mL, [MA]0/[MBP]0/[TREN]0/[CuBr2]0 = 222/1/0.2/0.1. | |
 |
| | Fig. 16 Kinetic plots, molecular weight and dispersity evolution for the SET-LRP of MA in TFE/water mixtures initiated with MBP and catalyzed by nonactivated Cu(0) wire at 25 °C. (a–c) TFE/water (8/2, v/v). The v/v ratio must be multiplied by 10 to obtain % solvent/% water. The value of v + v must be divided by 10 to obtain the total volume of solvents, 1 mL. Reaction conditions: MA = 1 mL, TFP + water = 0.5 mL, (a) [MA]0/[MBP]0/[TREN]0/[CuBr2]0 = 222/1/0.1/0.05, (b) [MA]0/[MBP]0/[TREN]0/[CuBr2]0 = 222/1/0.2/0.1, (c) [MA]0/[MBP]0/[TREN]0/[CuBr2]0 = 222/1/0.4/0.2. | |
Experiments with Cu(0) wire lengths of 2.0, 4.5 and 12.5 cm were carried under the following conditions [MA]0/[MBP]0/[TREN]0/[Cu(II)Br2]0 = 222/1/0.2/0.1. All kinetic experiments reach about 80% conversion in 120 min reaction time and exhibit linear first order kinetics. Regardless of the Cu(0) wire length, the resulting PMA at the end of the polymerization exhibits Mw/Mn values ranging from 1.14 to 1.18 and kappp is almost 0.015 min−1 (Table 2, entries 3, 9 and 10). These results support the tolerance of this biphasic system to a large excess of Cu(0) in the form of wire. The tolerance of these “interfacial” or “on water” SET-LRP processes35–41 to high externally added Cu(II)Br2 deactivator loadings was investigated by increasing the amount of Cu(II)Br2 relative to the initiator from 5% to 10% and to 20% while maintaining a constant ratio [TREN]0/[CuBr2]0 = 2/1. As can be seen in Fig. 16, in all cases first order kinetics with excellent control over the polymerization of MA was observed. The kappp only decreased at the highest concentration of Cu(II)Br2 while maintaining the same level of control as the experiments performed in the presence of lower Cu(II)Br2 loadings (compare Table 2, entries 3, 11 and 12).
It is important to mention at this point that regardless of the level of improvement of the SET-LRP methodology, it does not yet compete with the perfection of the living cationic polymerization of functional vinyl ethers employed in our laboratory in the late 1980s and early 1990s,57–61 with the living metathesis ring opening polymerization employed in the mid 1990s,62,63 group transfer polymerization in the late 1980s,64 reactions on narrow dispersity polymers in the late 1980s,65 living polymerization of acetylenes66 and living cationic ring opening polymerization of cyclic imino ethers67 both in the late 1990s that were used in the construction of complex macromolecular systems. Neither ATRP68 nor other metal catalyzed living radical polymerizations69,70 are competitive with the previously mentioned methods in spite of the very large research efforts dedicated to the field of living radical polymerization for the last more than 25 years. Modest progress made so far by SET-LRP was reported only recently.2,4–7,9,55,56,71 Iterative methods for the synthesis of sequence defined monodisperse macromolecules and their use in the construction of complex macromolecular systems became successful only in 2015.72,73 Therefore, conventional radical polymerization74–78 has remained so far the method of choice employed in the synthesis of complex macromolecular systems. We believe that rather than debating mechanisms, improving methodologies by elucidating in a constructive way their mechanism up to the point that will transform living radical polymerization into a technique competitive with much older living polymerization methods would be, in our opinion, more efficient and desirable.
Conclusions
This report demonstrates the synthesis of PMA via SET-LRP initiated with MBP in biphasic mixtures of water with fluorinated alcohols. Mixtures of TFE and TFP containing 10, 20, and 30% water provided a living polymerization in the presence of two catalytic systems: non-activated Cu(0) wire/Me6-TREN and non-activated Cu(0) wire/TREN/Cu(II)Br2. SET-LRP of MA in this combination of solvents evolved into biphasic reaction mixtures due to the immiscibility between the aqueous phase containing Cu(II)Br2 and the ligand and the alcohol phase containing the monomer and polymer. This was validated through kinetic experiments showing linear evolution of ln([M]0/[M]) vs. polymerization time, linear dependence of experimental Mn on conversion, narrow molecular weight distribution and almost quantitative chain end functionality at high conversion. For example, low Mn PMA samples showed near perfect retention of end group fidelity regardless of the catalytic system used. SET-LRP of MA in this combination of fluorinated alcohols and water evolved into biphasic reaction mixtures. According to the required disproportionation of Cu(I)X into Cu(0) and Cu(II)X2, a linear relationship between the kappp of the SET-LRP process and the ΦH2O was observed. Thus, simply changing the polarity balance of the reaction mixture a rate enhancement of up to four times was achieved. Also important is to highlight that the SET-LRP of MA in a TFE/water 8/2, v/v performed well also in the presence of excess of Cu(0) wire or Cu(II)Br2, unlike the corresponding single-phase systems. This important characteristic relies on the partitioning of reagents between phases occurring during biphasic SET-LRP that provides a “self-controlled” reversible deactivation at the interface of the two phases that avoids side reactions encountered in the homogeneous state when an excess of Cu(II)X2 is used. The same is the case also about the activation step.
Experimental
Materials
MA (99%) (from Acros) was passed over a short column of basic Al2O3 before use in order to remove the radical inhibitor. Tris(2-aminoethyl)amine (TREN) (99% Acros), Cu(0) wire (20 gauge wire, 0.812 mm diameter from Fischer), CuBr2 (99%, Alfa Aesar), methyl 2-bromopropionate (MBP) (99% Acros), 2,2,3,3-tetrafluoropropanol (TFP) (SynQuest Laboratories), 2,2,2-trifluoroethanol (TFE) (SynQuest Laboratories), and thiophenol (99%, Acros) were used as received. Triethylamine (NEt3, 99.9%, Chemimpex) was destilled under N2 over CaH2. Tris[2-(dimethylamino)ethyl]amine (Me6-TREN) was synthesized according to a literature procedure.79
Techniques
500 MHz 1H-NMR spectra were recorded on a Bruker UNI500 NMR instrument at 25 °C in CDCl3 containing tretamethylsilane (TMS) as an internal standard. For the chain end analysis of PMA, the delay time (D1) applied was 10 s and the number of scans (nt) was 80. Gel permeation chromatography (GPC) analysis of the polymer samples was performed using a PerkinElmer Series LC 100 column oven containing three AM gel columns (a guard column, a 500 Å, 10 μm column and a 104 Å, 10 μm column), a Shimadzu LC-20AD high performance liquid chromatograph pump, a PE Nelson Analytical 900 Series integration data station, a Shimadzu RID-10A refractive index (RI) detector and a Shimadzu SIL-10ADvp Autoinjector. THF (Fischer, HPLC grade) was used as an eluent at a flow rate of 1 mL min−1. The number-average (Mn) and weight-average (Mw) molecular weights of PMA samples were determined using poly(methyl methacrylate) (PMMA) standards purchased from American Polymer Standards. MALDI-TOF spectra were recorded in reflection mode on a Voyager DE (Applied Biosystems) equipped with a 337 nm nitrogen laser (3 ns pulse width). The accelerating potential was 25 kV, the grid was 88%, the laser power was 1950 arbitrary units, and a positive mode was employed. The sample analysis was carried out with 2-(4-hydroxyphenylazo)benzoic acid as the matrix. THF solutions of the matrix (30 mg mL−1), NaCl (10 mg mL−1) and polymer (10 mg mL−1) were prepared separately. The final solution for MALDI-TOF analysis was obtained by mixing the matrix, polymer and the cationization agent solution at a 9/1/1 volumeric ratio. Then 1 μL of the solution mixture were deposited onto six wells of the sample plate and dried in air at room temperature before being subjected to the MALDI-TOF analysis.
Typical procedure for TREN and Me6-TREN-mediated SET-LRP in biphasic fluorinated solvent–water mixtures
An organic solvent (TFE or TFP), monomer (MA), water (stock solution containing the ligand (TREN) and CuBr2) and an initiator (MBP) were added to a 25 mL Schlenk tube in the order mentioned. In the case of using Me6-TREN as a ligand, water and the ligand were added separately, following the order: organic solvent, monomer, water, ligand and initiator. The reaction mixture was then deoxygenated by six freeze–pump–thaw cycles. After these cycles, the Schlenk tube was opened under a positive flow of nitrogen to add the Cu(0) wire wrapped around a Teflon-coated stir bar. Two more freeze–pump–thaw cycles were carried out during which the Cu(0) wrapped in the stir bar was held above the reaction mixture using an external magnet. After that, the Schlenk tube was filled with nitrogen and the reaction mixture was placed in a water bath thermostated at 25 °C. Then, the stir bar wrapped with the Cu(0) wire was dropped gently into the reaction mixture. The introduction of the Cu(0) wire defines t = 0. Samples were taken at different reaction times by purging the side arm of the Schlenk tube with nitrogen for 2 min using a deoxygenated glass syringe and stainless steel needles. The collected samples were dissolved in CDCl3 and quenched by air bubbling. After that, the monomer conversion was measured by 1H-NMR spectroscopy. In order to determine the molecular weight and polydispersity of the samples, the solvent and the residual monomer were removed under vacuum. Finally the samples were dissolved in THF and passed through a short small basic Al2O3 chromatographic column to remove any residual copper and subsequently were analyzed by GPC. The resulting PMA was precipitated in cold methanol and dried under vacuum until constant weight to perform chain end analysis by 1H-NMR spectroscopy, before and after the thioeterification reaction.
General procedure for the chain modification via the “thio-bromo” click reaction
In a 10 mL test tube sealed with a rubber septum, thiophenol (0.05 equiv.) and distilled triethylamine (NEt3, 0.05 equiv.) were added into a solution of the corresponding polymer (0.01 equiv.) in acetonitrile (1 mL) under a nitrogen flow. The reaction mixture was stirred at room temperature for 3 h. Then, the resulting modified PMA was precipitated in cold methanol and washed with methanol several times. The resulting modified polymers were dried under vacuum until constant weight.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
Financial support by the National Science Foundation (DMR-1066116 and DMR-1120901) and P. Roy Vagelos Chair at the University of Pennsylvania is greatly acknowledged. G. L. and M. G. acknowledge support from the Spanish Ministerio de Economía, Industria y Competitividad (MINECO) through projects MAT2014-53652-R and MAT2017-82669-R. G. L. also thanks the Serra Húnter Programme. A. M. was supported by an FPI grant (BES-2015-072662) and a mobility grant (BES-2015-072) from the MINECO. T. L. acknowledges financial support from NanKai University, China. The European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement (No. 642687 to I. B.) is also acknowledged. L. D. thanks the National Natural Science Foundation of China (No 21774107, 21774029).
References
- V. Percec, T. Guliashvili, J. S. Ladislaw, A. Wistrand, A. Stjerndahl, M. J. Sienkowska, M. J. Monteiro and S. Sahoo, J. Am. Chem. Soc., 2006, 128, 14156–14165 CrossRef CAS PubMed.
- B. M. Rosen and V. Percec, Chem. Rev., 2009, 109, 5069–5119 CrossRef CAS PubMed.
- N. Zhang, S. R. Samanta, B. M. Rosen and V. Percec, Chem. Rev., 2014, 114, 5848–5958 CrossRef CAS PubMed.
- G. Lligadas, S. Grama and V. Percec, Biomacromolecules, 2017, 18, 1039–1063 CrossRef CAS PubMed.
- G. Lligadas, S. Grama and V. Percec, Biomacromolecules, 2017, 18, 2981–3008 CrossRef CAS PubMed.
- A. Anastasaki, V. Nikolaou, G. Nurumbetov, P. Wilson, K. Kempe, J. F. Quinn, T. P. Davis, M. R. Whittaker and D. M. Haddleton, Chem. Rev., 2016, 116, 835–877 CrossRef CAS PubMed.
- C. Boyer, N. A. Corrigan, K. Jung, D. Nguyen, T. K. Nguyen, N. N. Adnan, S. Oliver, S. Shanmugam and J. Yeow, Chem. Rev., 2016, 116, 1803–1949 CrossRef CAS PubMed.
- A. Anastasaki, V. Nikolaou and D. M. Haddleton, Polym. Chem., 2016, 7, 1002–1026 RSC.
- N. H. Nguyen, C. Rodriguez-Emmenegger, E. Brynda, Z. Sedlakova and V. Percec, Polym. Chem., 2013, 4, 2424–2427 RSC.
- X. Leng, N. H. Nguyen, B. van Beusekom, D. A. Wilson and V. Percec, Polym. Chem., 2013, 4, 2995–3004 RSC.
- Q. Zhang, P. Wilson, Z. Li, R. McHale, J. Godfrey, A. Anastasaki, C. Waldron and D. M. Haddleton, J. Am. Chem. Soc., 2013, 135, 7355–7363 CrossRef CAS PubMed.
- N. H. Nguyen, B. M. Rosen, X. Jiang, S. Fleischmann and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5577–5590 CrossRef CAS.
- N. H. Nguyen, B. M. Rosen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1752–1763 CrossRef CAS.
- G. Lligadas and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4684–4695 CrossRef CAS.
- N. H. Nguyen, M. E. Levere and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 860–873 CrossRef CAS.
- N. H. Nguyen, M. E. Levere, J. Kulis, M. J. Monteiro and V. Percec, Macromolecules, 2012, 45, 4606–4622 CrossRef CAS.
- S. Fleischmann and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2243–2250 CrossRef CAS.
- G. Lligadas and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2745–2754 CrossRef CAS.
- S. Fleischmann and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2236–2242 CrossRef CAS.
- S. R. Samanta, M. E. Levere and V. Percec, Polym. Chem., 2013, 4, 3212–3224 RSC.
- S. R. Samanta, A. Anastasaki, C. Waldron, D. M. Haddleton and V. Percec, Polym. Chem., 2013, 4, 5555–5562 RSC.
- S. R. Samanta, A. Anastasaki, C. Waldron, D. M. Haddleton and V. Percec, Polym. Chem., 2013, 4, 5563–5569 RSC.
- S. R. Samanta, R. Cai and V. Percec, Polym. Chem., 2014, 5, 5479–5491 RSC.
- S. R. Samanta, H. J. Sun, A. Anastasaki, D. M. Haddleton and V. Percec, Polym. Chem., 2014, 5, 89–95 RSC.
- A. Moreno, D. Garcia, M. Galià, J. C. Ronda, V. Cádiz, G. Lligadas and V. Percec, Biomacromolecules, 2017, 18, 3447–3456 CrossRef CAS PubMed.
- X. Jiang, S. Fleishmann, N. H. Nguyen, B. M. Rosen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5591–5605 CrossRef CAS.
- S. R. Samanta and V. Percec, Polym. Chem., 2014, 5, 169–174 RSC.
- B. M. Rosen, X. Jiang, X. C. J. Wilson, N. H. Nguyen, M. J. Monteiro and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5606–5628 CrossRef CAS.
- B. M. Rosen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4950–4964 CrossRef CAS.
- X. Jiang, B. M. Rosen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 403–409 CrossRef CAS.
- M. E. Levere, N. H. Nguyen, X. Leng and V. Percec, Polym. Chem., 2013, 4, 1635–1647 RSC.
- N. H. Nguyen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4227–4240 CAS.
- G. Lligadas, B. M. Rosen, M. J. Monteiro and V. Percec, Macromolecules, 2008, 41, 8360–8364 CrossRef CAS.
- G. Lligadas and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6880–6895 CrossRef CAS.
- M. Enayati, R. B. Smail, S. Grama, R. L. Jezorek, M. J. Monteiro and V. Percec, Polym. Chem., 2016, 7, 7230–7241 RSC.
- R. B. Smail, R. L. Jezorek, J. Lejnieks, M. Enayati, S. Grama, M. J. Monteiro and V. Percec, Polym. Chem., 2017, 8, 3102–3123 RSC.
- R. L. Jezorek, M. Enayati, R. B. Smail, J. Lejnieks, S. Grama, M. J. Monteiro and V. Percec, Polym. Chem., 2017, 8, 3405–3424 RSC.
- M. Enayati, R. L. Jezorek, M. J. Monteiro and V. Percec, Polym. Chem., 2016, 7, 5930–5942 RSC.
- S. Grama, J. Lejnieks, M. Enayati, R. B. Smail, L. Ding, G. Lligadas, M. J. Monteiro and V. Percec, Polym. Chem., 2017, 8, 5865–5874 RSC.
- A. Moreno, S. Grama, T. Liu, M. Galià, G. Lligadas and V. Percec, Polym. Chem., 2017, 8, 7559–7574 RSC.
- M. Enayati, R. L. Jezorek, M. J. Monteiro and V. Percec, Polym. Chem., 2016, 7, 3608–3621 RSC.
- M. Gavrilov, T. J. Zerk, P. V. Bernhardt, V. Percec and M. J. Monteiro, Polym. Chem., 2016, 7, 933–939 RSC.
- M. Gavrilov, Z. Jia, V. Percec and M. J. Monteiro, Polym. Chem., 2016, 7, 4802–4809 RSC.
- V. Percec, A. V. Popov, E. Ramirez-Castillo, M. Monteiro, B. Barboiu, O. Weichold, A. D. Asandei and C. M. Mitchell, J. Am. Chem. Soc., 2002, 124, 4940–4941 CrossRef CAS PubMed.
- V. Percec, A. V. Popov, E. Ramirez-Castillo and O. Weichold, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3283–3299 CrossRef CAS.
- A. D. Asandei and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 3392–3418 CrossRef CAS.
- N. H. Nguyen, M. E. Levere and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 35–46 CrossRef CAS.
- N. H. Nguyen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5109–5119 CrossRef CAS.
- M. Enayati, R. L. Jezorek and V. Percec, Polym. Chem., 2016, 7, 4549–4558 RSC.
- A. Anastasaki, C. Waldron, V. Nikolaou, P. Wilson, R. McHale, T. Smith and D. M. Haddleton, Polym. Chem., 2013, 4, 4113–4119 RSC.
- C. Waldron, A. Anastasaki, R. McHale, P. Wilson, Z. Li, T. Smith and D. M. Haddleton, Polym. Chem., 2014, 5, 892–898 RSC.
- C. Boyer, A. Atme, C. Waldron, A. Anastasaki, P. Wilson, P. B. Zetterlund, D. M. Haddleton and M. R. Whittaker, Polym. Chem., 2013, 4, 106–112 RSC.
- A. Simula, G. Nurumbetov, A. Anastasaki, P. Wilson and D. M. Haddleton, Eur. Polym. J., 2015, 62, 294–303 CrossRef CAS.
- O. Bertrand, P. Wilson, J. A. Burns, G. A. Bell and D. M. Haddleton, Polym. Chem., 2015, 6, 8319–8324 RSC.
- B. M. Rosen, G. Lligadas, C. Hahn and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3931–3939 CrossRef CAS.
- B. M. Rosen, G. Lligadas, C. Hahn and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3940–3948 CrossRef CAS.
- V. Percec and M. Lee, Macromolecules, 1991, 24, 1017–1024 CrossRef CAS.
- V. Percec and D. Tomazos, Adv. Mater., 1992, 4, 548–561 CrossRef CAS.
- V. Percec, M. Lee and H. Jonsson, J. Polym. Sci., Part A: Polym. Chem., 1991, 29, 327–337 CrossRef CAS.
- V. Percec and M. Lee, J. Macromol. Sci., Pure Appl. Chem., 1992, 29, 723–740 CrossRef.
- V. Percec, J. Heck, M. Lee, G. Ungar and A. Alvarez-Castillo, J. Mater. Chem., 1992, 2, 1033–1039 RSC.
- V. Percec, D. Schlueter and G. Ungar, Macromolecules, 1997, 30, 645–648 CrossRef CAS.
- V. Percec, D. Schlueter, G. Ungar, S. Z. D. Cheng and A. Zhang, Macromolecules, 1998, 31, 1745–1762 CrossRef CAS.
- V. Percec, D. Tomazos and C. Pugh, Macromolecules, 1989, 22, 3259–3267 CrossRef CAS.
- V. Percec and B. Hahn, Macromolecules, 1989, 22, 1588–1599 CrossRef CAS.
- V. Percec, J. G. Rudick, M. Peterca, M. Wagner, M. Obata, C. M. Mitchell, W. D. Cho, V. S. K. Balagurusamy and P. A. Heiney, J. Am. Chem. Soc., 2005, 127, 15257–15264 CrossRef CAS PubMed.
- M. N. Holerca, D. Sahoo, M. Peterca, B. E. Partridge, P. A. Heiney and V. Percec, Macromolecules, 2017, 50, 375–385 CrossRef CAS.
- Z. Zeng, M. F. Wen, G. Ye, X. M. Huo, F. C. Wu, Z. Wang, J. J. Yang, K. Matyjaszewski, Y. X. Lu and J. Chen, Chem. Mater., 2017, 29, 10212–10219 CrossRef CAS.
- M. Ouchi, T. Terashima and M. Sawamoto, Chem. Rev., 2009, 109, 4963–5050 CrossRef CAS PubMed.
- G. Hattori, Y. Hirai, M. Sawamoto and T. Terashima, Polym. Chem., 2017, 8, 7248–7259 RSC.
- K. A. Andreopoulou, M. Peterca, D. A. Wilson, B. E. Partridge, P. A. Heiney and V. Percec, Macromolecules, 2017, 50, 5271–5284 CrossRef CAS.
- S. Zhang, Q. Xiao, S. E. Sherman, A. Muncan, A. D. M. Ramos Vicente, Z. Wang, D. A. Hammer, D. Williams, Y. Chen, D. J. Pochan, S. Vértesy, S. André, M. L. Klein, H.-J. Gabius and V. Percec, J. Am. Chem. Soc., 2015, 137, 13334–13344 CrossRef CAS PubMed.
- Q. Xiao, S. Zhang, Z. Wang, S. E. Sherman, R.-O. Moussodia, M. Peterca, A. Muncan, D. R. Williams, D. A. Hammer, S. Vértesy, S. André, H.-J. Gabius, M. L. Klein and V. Percec, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 1162–1167 CrossRef CAS PubMed.
- V. Percec, D. Schlueter, Y. K. Kwon, J. Blackwell, M. Moeller and P. J. Slangen, Macromolecules, 1995, 28, 8807–8818 CrossRef CAS.
- Y. K. Kwon, S. N. Chvalun, J. Blackwell, V. Percec and J. A. Heck, Macromolecules, 1995, 28, 1552–1558 CrossRef CAS.
- Y. K. Kwon, S. Chvalun, A. I. Schneider, J. Blackwell, V. Percec and J. A. Heck, Macromolecules, 1994, 27, 6129–6132 CrossRef CAS.
- V. Percec, D. Tomazos, J. Heck, H. Blackwell and G. Ungar, J. Chem. Soc., Perkin Trans. 2, 1994, 31–44 RSC.
- V. Percec, J. A. Heck, D. Tomazos and G. Ungar, J. Chem. Soc., Perkin Trans. 2, 1993, 2381–2388 RSC.
- M. Ciampolini and N. Nardi, Inorg. Chem., 1996, 5, 41–44 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Tong
Liu
ab,
Tong
Liu
















