
Mononuclear Pd(II) and Pt(II) complexes with an α-N-heterocyclic thiosemicarbazone: cytotoxicity, solution behaviour and interaction versus proven models from biological media†
Ana I.
Matesanz
 a,
Eva
Jimenez-Faraco
a,
María C.
Ruiz
b,
Lucia M.
Balsa
b,
Carmen
Navarro-Ranninger
a,
Ignacio E.
León
*b and
Adoracion G.
Quiroga
*a
a,
Eva
Jimenez-Faraco
a,
María C.
Ruiz
b,
Lucia M.
Balsa
b,
Carmen
Navarro-Ranninger
a,
Ignacio E.
León
*b and
Adoracion G.
Quiroga
*a
aDept. Química Inorgánica and IAdChem (Institute for Advanced Research in Chemical Science) Universidad Autónoma de Madrid, 28045 Madrid, Spain. E-mail: adoracion.gomez@uam.es
bCentro de Química Inorgánica (CEQUINOR, CONICET), Facultad de Ciencias Exactas, Universidad Nacional de La Plata, 47 y 115, 1900 La Plata, Argentina. E-mail: ileon@biol.unlp.edu.ar
First published on 18th October 2017
Abstract
Two Pd(II) and Pt(II) complexes with two pyrrol-2-carbaldehyde N-p-chlorophenylthiosemicarbazone ligands are designed and characterized showing mononuclear structures. An important pharmacological property for both compounds is the high selectivity for tumor cells and a lack of activity in healthy cells. The Pd(II) compound shows a higher antitumor activity and selectivity than the Pt(II) compound. Both complexes present a variety of biological interactions: with DNA models (pBR322 and CT DNA), proteins (lysozyme and RNase) and other biological targets like proteosome. Our results show that the Pd(II) complex is a more interesting candidate for potential anticancer therapies than the Pt(II) complex, and we provide new insight into the design and synthesis of palladium compounds as potential antitumor agents.
Introduction
Thiosemicarbazone (TSCN) ligands are molecules with an established history as pharmacological agents, not only for their biological properties but also because of their great capacity for binding with metals. Triapine® is a thiosemicarbazone containing an N-heterocyclic ring and is of special interest because of its multiple clinical trial studies.1,2 Triapine® is currently included in Phase 2 of an ongoing clinical trial for the treatment of non-localized cervical cancer at the National Cancer Institute.3 The most accepted hypothesis regarding the antitumoral action of TSCNs is based on their chelating properties, which indirectly lead one to study ribonucleotide reductase (RR) as the mechanism through which these molecules work.4 RR is an iron-dependent enzyme that promotes the reduction of ribose to deoxyribose. Some TSCNs affect RR inhibition and lead to the blockage of the synthesis phase of the cell cycle and eventually to cell death by apoptosis.5α-N-Heterocyclic thiosemicarbazones turned out to be the most potent inhibitors of RR so far,6 and the identification of their metal coordination was reported to afford more active species than free ligands.7 Recent studies have shown that the methylation of Triapine® results in a change of the mode of action, which might be associated with its possible interaction with copper caused by the balance of the intracellular copper concentration. However, these effects do not seem to be responsible for the increased cytotoxic activity of some derivatives in the nanomolar range.8
para-Substituted phenyl thiosemicarbazones are another example of strong antitumor compounds and their metallation using palladium and platinum are reported to afford complexes that highly enhance the antitumor action of the ligands. The data from these complexes showed a good correlation of their cytotoxic activity with their structures and mode of action.9 By changing the structure of the complex, their interaction with DNA varies from the cisplatin type of interaction to interstrand crosslinking.10,11 Heterocyclic TSCNs are good candidates which not only afford platinum and palladium complexes,12 but also complexes with some other metals such as copper13,14 and ruthenium.15 The most remarkable results have been achieved so far with copper,16,17 iron and gallium1 bound to Triapine®.
Following these results and trying to elucidate, if metal complexes from α-N-heterocyclic thiosemicarbazones can achieve more selectivity versus special tumor lines, we have developed a new series of metal complexes with pyrrol-2-carbaldehyde 4N-p-chlorophenylthiosemicarbazone (LH2) where the metal can be Pd(II) and Pt(II).
We seek not only selective and active compounds versus specific cancer cell lines such as the human osteosarcoma MG-63,18 but we also want our compounds to allow normal cell viability of non-tumoral cell lines “in vitro” (for example L929 fibroblasts). In this work, we present new compounds and the studies of their cytotoxicity, structure and stability in solution, demonstrating their potential as antitumor drugs. The binding and/or affinity to biological models such as two model proteins (lysozyme and RNase), proteasome and DNA, indicates a different mode of action to the classical metallodrugs, such as cisplatin.
Results and discussion
Chemistry
Scheme 1 shows the synthesis of the thiosemicarbazone and pyrrol-2-carbaldehyde 4N-p-chlorophenylthiosemicarbazone (LH2). The synthesis began with 4N-p-chlorophenyl thiosemicarbazide preparation, using stoichiometric amounts of p-chlorophenyl isothiocyanate and hydrazine monohydrate. The procedure to synthesize thiosemicarbazones was reported in the fifties and proceeds by the condensation of the aldehyde and the corresponding thiosemicarbazide.19 This procedure has been used for obtaining a large number of thiosemicarbazone derivatives.12 The Experimental section includes the detailed procedure used for the synthesis of LH2. The Pd(II) complex 1, was synthesized using Li2PdCl4, prepared in situ from palladium(II) chloride and lithium chloride (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) in MeOH,20 then reacted with the stoichiometric amount of LH2 at room temperature to afford complex 1.
:2) in MeOH,20 then reacted with the stoichiometric amount of LH2 at room temperature to afford complex 1.
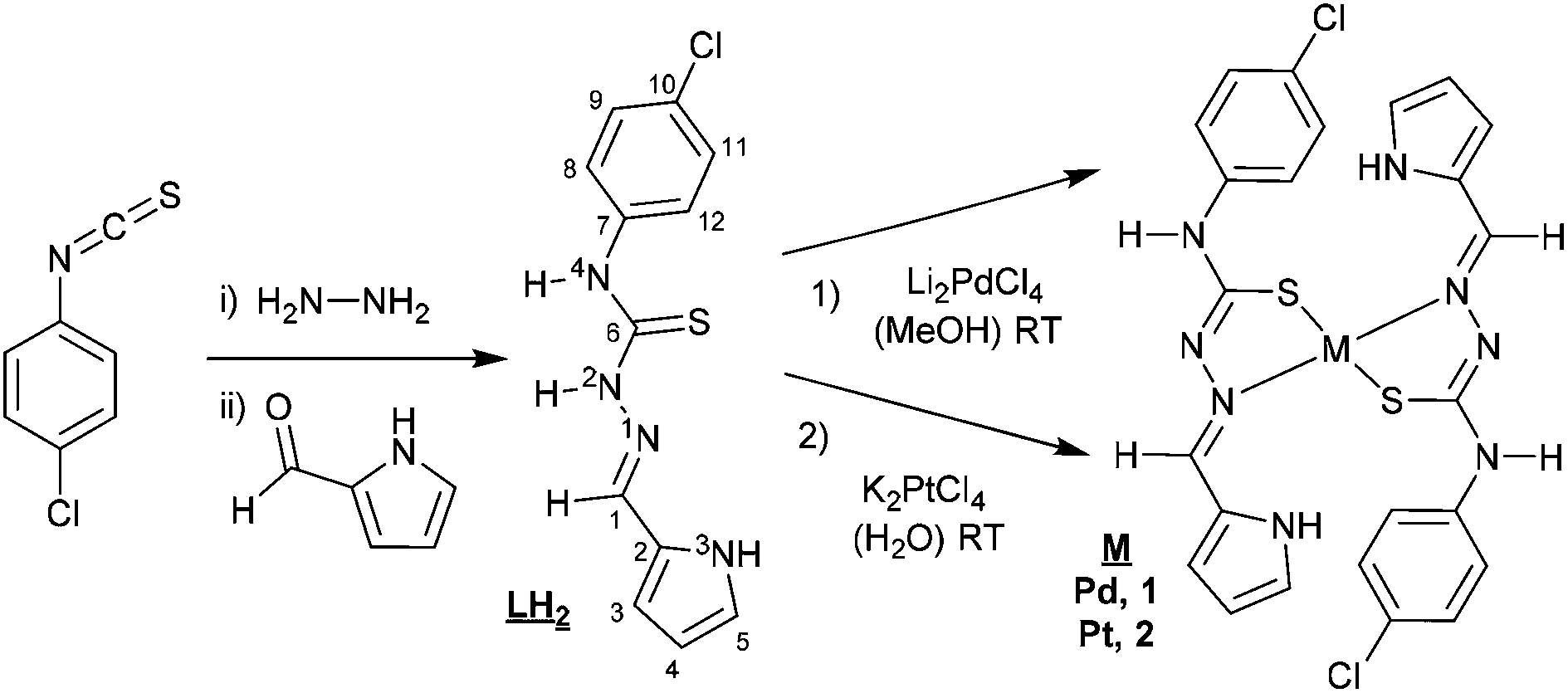 | ||
| Scheme 1 | ||
The low solubility of complex 1 allowed obtaining low concentrated DMSO solutions that easily led to single crystals suitable for X-ray diffraction structure resolution. We used crushed and vacuum dried single crystal samples for full characterization and biological assays. IR, NMR and analysis data are in agreement with the structure of the complex finally solved by X-ray diffraction (Fig. 1).
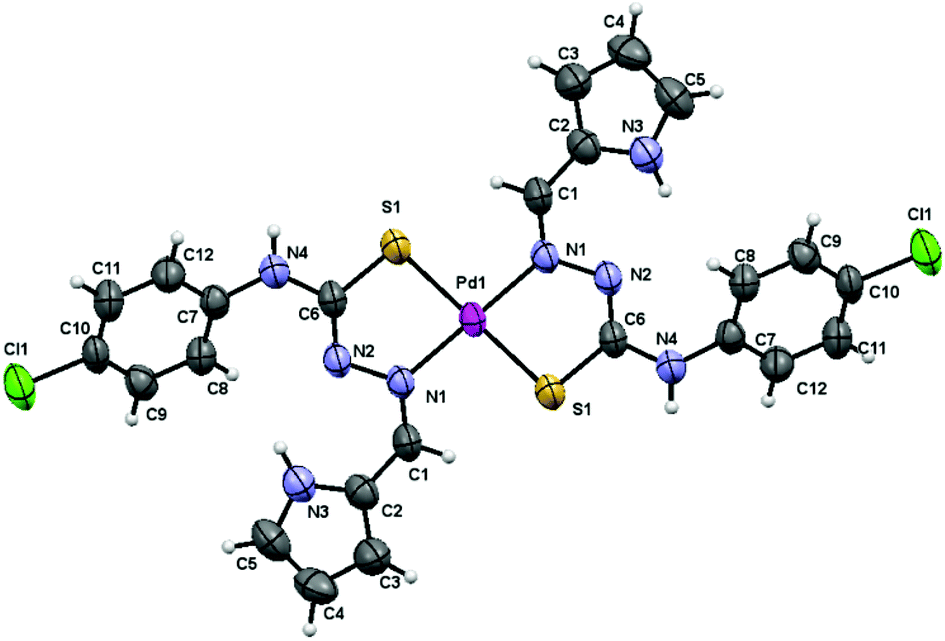 | ||
| Fig. 1 X-Ray structure of complex 1. | ||
The structure of complex 1 consists of discrete molecules that correspond to the [Pd(LH)2]·2DMSO unit. The geometry around the metal ion is square planar being the palladium atom bound to the sulfur and azomethine nitrogen atoms of the two mutually trans, deprotonated thiosemicarbazone ligands. The asymmetric unit only contains one-half of the complex 1 molecule, with the palladium atom located on a crystallographic inversion centre, together with one DMSO molecule. The distances and angles around the Pd atom are within the range expected for these kinds of mononuclear21 and other polynuclear complexes published previously by our group of research10 (Table 1).
| Bond distances (Å) | Bond distances (Å) | Bond angles (°) | |||
|---|---|---|---|---|---|
| Pd1–N1 | 2.023(3) | C6–N2 | 1.296(5) | N1–Pd1–N1 | 180.0 |
| Pd1–S1 | 2.2731(12) | C6–N4 | 1.357(5) | N1–Pd1–S1 | 83.33(8) |
| C1–N1 | 1.288(5) | C6–S1 | 1.736(4) | S1–Pd1–S1 | 180.0 |
| C2–N3 | 1.364(5) | C7–N4 | 1.410(5) | ||
| C5–N3 | 1.340(5) | N1–N2 | 1.415(4) | ||
The synthesis of the platinum complex 2 was similar to the one used for complex 1, as shown in Scheme 1, but required the use of water to dissolve the starting material K2PtCl4. The characterization of complex 2 by the usual techniques indicated a general formula: [Pt(LH)2], similar to complex 1. Unfortunately, none of the single crystals achieved were adequate for X-ray characterization.
The stability of both complexes was studied by 1H NMR (fresh sample and 24 h) in DMSO-d6 (Fig. SM1†) and by UV in Tris buffer:DMSO (95:5) (Fig. SM2 and SM3†). The behaviour of complexes 1 and 2 in solution is very similar. The UV spectra showed no significant changes (no shifts of the λmax and no new peaks) other than a small decrease in absorbance values after 24 h. Both solutions of complexes 1 and 2 are stable enough to be studied as potential metallodrugs within the pH range = 6–8.
Cytotoxicity
Following our expectations to achieve a novel drug with specific activity versus cancer cell lines, both compounds 1 and 2 showed antiproliferative activity values within 25–100 μM (Table 2) and 15–50 μM (Table 3) for the following cell lines: Jurkat (human leukemia), MG-63 (human osteosarcoma) and A549 (human lung adenocarcinoma), remarkably none of the complexes showed such effects in normal mouse L929 fibroblasts.| L929 (μM) | Jurkat (μM) | A549 (μM) | MG-63 (μM) | |
|---|---|---|---|---|
| 1, Pd(LH)2 | >100 | 47 ± 3 | 60 ± 3 | 34 ± 2 |
| 2, Pt(LH)2 | >100 | 63 ± 2 | >100 | 56 ± 5 |
| Cisplatin | 43 ± 4 | 8 ± 1 | 62 ± 5 | 33 ± 3 |
| L929 (μM) | Jurkat (μM) | A549 (μM) | MG-63 (μM) | |
|---|---|---|---|---|
| 1, Pd(LH)2 | 86 ± 4 | 17 ± 5 | 36 ± 3 | 20 ± 4 |
| 2, Pt(LH)2 | 81 ± 3 | 33 ± 4 | 79 ± 4 | 32 ± 3 |
| Cisplatin | 26 ± 3 | 4 ± 1 | 29 ± 5 | 17 ± 3 |
Cisplatin showed a much higher cytotoxicity in this normal cell line phenotype (L929) compared with the cytotoxicity of the new complexes, and this particular effect enhances the potential value of complexes 1 and 2 as new metallodrugs in anticancer therapy.
The best candidate is the palladium derivative, complex 1, which showed a lower IC50 in leukemic and osteosarcoma cell lines than the platinum derivative, complex 2. The IC50 value of complex 1 in the lung cancer cell line A549 (see Table 2, 60 μM) is a more remarkable value than that of the platinum complex (100 μM) and is similar to cisplatin (62 μM). Moreover, after 72 h of incubation, the IC50 value of complex 1 in A549 cells is 36 μM whilst for the platinum complex it is 79 μM (see Table 3).
Table 4 shows the higher selectivity of compound 1 for A549 and MG-63 cells in comparison with compound 2 and cisplatin. The SI values are 2.46, 1.02, 0.90 (A549 cells) and 4.3, 2.3 and 1.5 (MG-63 cells) for compounds 1, 2 and cisplatin, respectively. Nevertheless, cisplatin showed a better correlation of SI in Jurkat cells than compounds 1 and 2.
| Jurkat | A549 | MG-63 | |
|---|---|---|---|
| SI (Selectivity Index) is a comparison of the amount of a therapeutic agent that causes the therapeutic effect (on tumor cells) and the amount that causes toxicity (using normal cells). | |||
| 1, Pd(LH)2 | 5 | 2.46 | 4.3 |
| 2, Pt(LH)2 | 2.4 | 1.02 | 2.5 |
| Cisplatin | 6.5 | 0.90 | 1.5 |
To our knowledge, there are very few examples in the references where the palladium thiosemicarbazone derivative showed better cytotoxicity than the platinum analogue.22–24 In fact, this kind of response should not be that unusual in chelate complexes, based on the higher kinetic lability of the Pd(II) complexes,25 which should endow the complex with better interaction potential with DNA or other biological molecules usually overexpressed in the cancer cells.
Reactivity versus biological molecules
The affinity of both complexes for CT DNA was evaluated using UV spectroscopy titrations, as a preliminary step to obtain information concerning their possible targets and provide information about the mechanism. The typical β-form of DNA exhibits a characteristic π–π* band at 260 nm, which is sensitive to structural changes in the macromolecule and can become hyperchromic (increase in absorption of the DNA band at 260 nm) by perturbation resulting from non-covalent external interaction.31
First, the UV spectra of a CT DNA solution (3.2 × 10−5 M) were monitored using increasing concentrations of complexes 1–2 (following different r values in Table 5 and Fig. SM4†), allowing the sample to react for 10 minutes. The binding to CT DNA showed an increasing effect in the 260 nm band; typically described for a hyperchromic effect (Table 5 shows the values). All of these data might indicate interaction of the complexes with CT-DNA that could be interpreted as an external non-covalent interaction, and no bathochromic effect is detected.31
Secondly, the UV spectra of increasing concentrations of CT DNA solution (3.2 × 10−5 M) were monitored using a fixed concentration of complexes 1–2 (2.5 × 10−6 M, see Fig. SM5†), allowing the sample to react for 10 minutes. The DNA-binding constants (Kb) of complexes 1 and 2 (Tables 2–4) were determined by the plots [DNA]/(εa − εf) versus [DNA] (Fig. SM5†) using the Wolfe–Shimer equation.32 In brief, the Kb constants of complexes 1 and 2 are comparable to those observed for classical groove binders (Table 5), such as a Hoechst 32258 with 4.6 × 105 M−1 with an A3T3 duplex.33
These values, recorded in the first 10 minutes of reaction, seemed to indicate that the reactivity of these compounds could be interpreted as having a good affinity for CT DNA base pairs in a non-covalent binding mode of action. The reactivity observed is quite different to cisplatin, which is as a covalent binder reported to produce two effects: hyperchromic and bathochromic after 7 h of reaction.31
Interaction of the complexes with the plasmid DNA supercoiled pBR322
Based on the best cytotoxicity values, we selected complex 1 to evaluate its interaction with a more specific model of DNA. We performed a study of its interaction with a supercoiled DNA pBR322 (containing two isoforms) used as a model of the secondary structure of DNA. Cisplatin binds covalently to DNA and also to pBR322 producing a delay in its closed circular form (ccc) and slowing down the open circular form (oc) because of its unwinding.26,34Complex 1 was assayed at different ri concentrations after 24 h. The electrophoresis results showed that complex 1 did not seem to alter the electrophoretic mobility of the plasmid, but we could barely detect a blur band in between the oc and ccc forms (data not shown). Several repetitions did not improve the visibility of such a band that is why we allowed a longer time of reaction. After 48 h (Fig. 2), we could clearly detect a new band in lines 3 to 6 possibly caused by a nick in one of the pBR322 forms (marker and pBR322 control are in lines 1 and 2 respectively for better comparison). This assay manifests that complex 1 is able to produce a new band corresponding possibly to a new fragment. Complex 1 interaction with pBR322 showed a different mode of action compared to cisplatin.
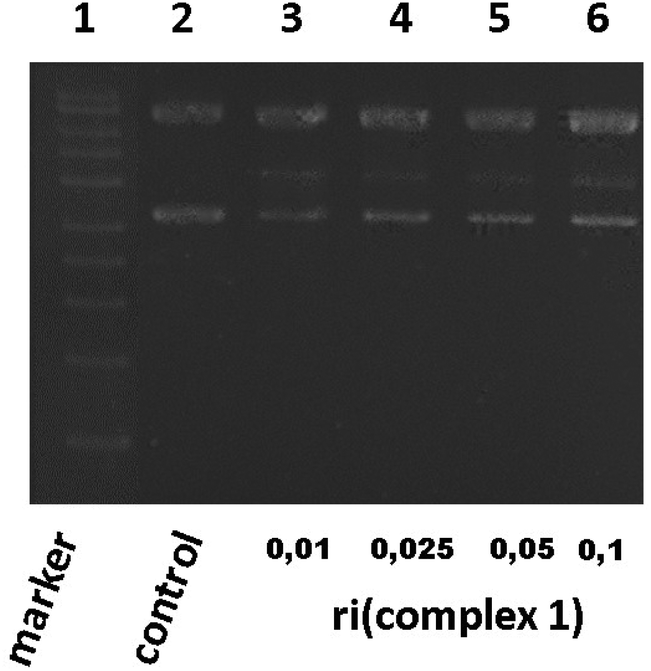 | ||
| Fig. 2 Electrophoresis in agarose gel of pBR322 plasmid treated with complex 1. ri: metal complex:DNA base pairs. Line 1: marker (DNA 1 kb ladder), line 2: C: control of incubated DNA plasmid pBR322, lines 3 to 6: complex 1: plasmid DNA incubated at ri = 0.01 to 0.1. | ||
Genotoxicity study
The genotoxic effects of complexes 1 and 2 were investigated through the induction of DNA damage. The single cell gel electrophoresis (SCGE) or comet assay is an important test used for the investigation of genotoxicity. It detects single and double strand DNA breaks. Sites where excision and repairs have occurred are detected under alkaline conditions.35 For both complexes, we evaluated the tail moment parameter, which is defined as the tail length × DNA amount in the tail. The distance of DNA migration is used to measure the extent of DNA damage. However, if DNA damage is relatively high, the tail increases in fluorescent staining intensity but not in length.36 Thus, for these reasons it is useful to use the tail moment as a genotoxic endpoint. As is shown in Fig. 3A, complex 1 produced a significant genotoxic effect in Jurkat cells from 2.5 to 5 μM with a dose–response effect (p < 0.001). From 10 μM the genotoxic effect is less pronounced. The decrease in DNA damage with the increase in complex concentration may be due to overt cytotoxicity exerted on this cell line. Moreover, the genotoxic effects of complex 1 are higher than that of bleomycin (positive control) showing following tail moment values: 16 ± 0.6, 28 ± 0.4 and 13 ± 0.6 for complex 1 and bleomycin, respectively.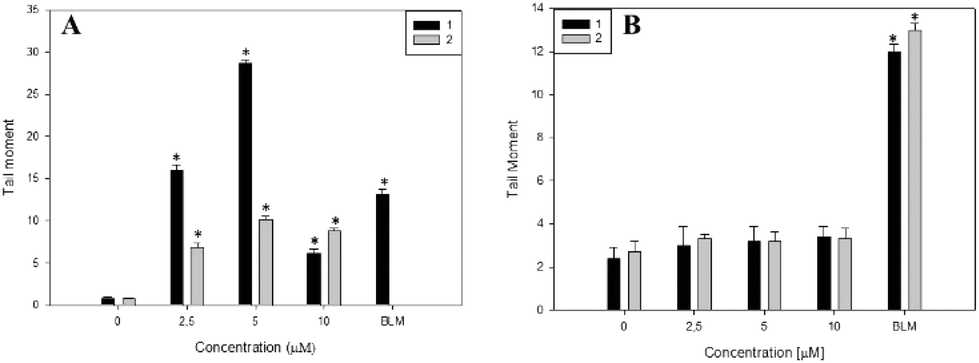 | ||
| Fig. 3 Genotoxicity of complexes 1 and 2 on Jurkat cells (A) and L929 cells (B) determined by SCGE (comet assay). DNA damage was evaluated by the tail moment. After incubation with both compounds for 48 h, cells were lysed and DNA fragments were processed by electrophoresis. After that, the nuclei were stained and analyzed. Results are expressed as mean ± SEM (n = 150), *p < 0.001. BLM stands for bleomycin (10 μg mL−1) used as a positive control. | ||
In addition, complex 2 exerts less genotoxic effects than complex 1. Besides this, both complexes did not exert genotoxic effects on normal L929 fibroblasts from 2.5 to 10 μM whilst cisplatin induced the break of cellular DNA in the same range of concentrations (see Fig. 3B and Fig. SM8†). These results may explain the low cytotoxicity of both compounds on L929 cells in comparison with the deleterious effects of cisplatin (see Tables 2–4). In this sense, cisplatin has a well-known mechanism of interaction with DNA.
Altogether, these results suggest that the genotoxic effects of complex 1 are higher than that of complex 2 in Jurkat cells, leading to a positive result in the comet assay and in agreement with the result observed in Fig. 2.
Proteins
The binding of complex 1 with lysozyme and RNase was evaluated with UV spectroscopy as described in the Experimental part. The k′ was calculated as a pseudo first order reaction (k′RNase: and k′Lys: 1.91 × 10−4 s−1 and 1.33 × 10−4 s−1) using the 3:1 stoichiometry (metal to protein) as the result was equal using the stoichiometry 10:1 (metal to protein) (Fig. SM6 and SM7†). The rate constant of complex 1 with lysozyme and RNase gives us an estimation of the compatibility and reactivity of complex 1 in the presence of representative proteins of cellular media. We have used cisplatin as a control, and its values with lysozyme (1.98 × 10−4 s−1) and RNase (1.88 × 10−4 s−1) are in agreement with those found with other models such as albumin, transferrin, and cytochrome c.37,38 The values of complex 1 are within the range found for cisplatin.
Interaction of the complexes with a proteasome target
In order to shed light on the mechanism of action for these complexes, and based on the lower reactivity with CT DNA, we evaluate a more specific target: proteasome. The ubiquitin-mediated proteasomal proteolysis is the main mechanism of degradation of proteins in human cells.39 The 20S proteasome, which is the proteolytic core of the multicatalytic 26S proteasome complex, has several proteolytic activities, including chymotrypsin-like, trypsin-like, etc.40Nevertheless, it has been shown that only inhibition of the chymotrypsin-like activity is tightly associated with the induction of tumor cell death programs.41 Proteasome inhibitors cause a buildup of unwanted proteins in the cell, inducing cell death rapidly and selectively, TSCN derivatives have also been reported to be inhibitors of this target.42
We performed a cell-free proteasome activity assay in the presence of each of these compounds at different concentrations (2.5 to 100 μM). As can be seen in Table 6, the gradual decrease in the fluorescence indicates the proteasome inhibition ability. Complexes 1 and 2 inhibit the chymotrypsin-like activity of purified 20S proteasome with different potencies since complex 2 was found to be the most potent inhibitor (see Table 6).
| Concentration (μM) | 1 (% Basal ± SD) | 2 (% Basal ± SD) |
|---|---|---|
| 2.5 | 97 ± 2 | 95 ± 1 |
| 5 | 94 ± 4 | 93 ± 1 |
| 10 | 73 ± 3 | 79 ± 3 |
| 25 | 73 ± 4 | 55 ± 4 |
| 50 | 69 ± 1 | 44 ± 2 |
| 100 | 51 ± 2 | 24 ± 2 |
In this order, Tundo and colleagues showed that cisplatin induces a dose dependent inhibition of the three activities of proteasome, at least over the concentration range investigated (2.5–15 μM). The described behaviors clearly demonstrate that cisplatin significantly affects the enzymatic properties of proteasome in vitro.43
On the other hand, to confirm the inhibitory effects of complexes 1 and 2 on the proteasome activity, we performed a cell proteasome activity experiment using Jurkat cells in the presence of 100 μM of complexes 1 and 2. The proteasomal activity was inhibited to similar levels by both compounds. As can be seen that complex 1 reduced the proteasome activity by 28% whilst complex 2 decreased the proteasome activity by 23% showing a similar proteasome inhibition effect.
ROS production
Oxidative stress is one of the main factors reported that trigger the deleterious actions of metal-based compounds.44,45For a better understanding of the possible mechanism involved in the cytotoxicity of both complexes in cancer cell lines, we evaluated the effect of complexes 1 and 2 on oxidative stress through the oxidation of the probe DHR-123. DHR-123 is a mitochondria-associated probe that selectively reacts with hydrogen peroxide.46 Incubation of Jurkat cells with complex 1 caused an increase in the production of ROS. At 10 μM, complex 1 increased ROS production after 48 h by generating 182% of the ROS level over the basal level (p < 0.01) whereas at 2.5 and 5 μM no production of ROS over the basal level could be observed (p < 0.01). Moreover, complex 2 does not exhibit ROS production in the range of concentrations tested (2.5–25 μM) (see Fig. SM9†).
Apoptosis
Apoptosis is a physiological process of cell death enhanced in the presence of injurious agents. As a consequence, a genetic program that leads to cell death is activated. Apoptosis is characterized by some morphological changes in the nucleus and the cytoplasm. Because of this, apoptosis can be assessed by using several characteristic features of programmed cell death. Independently of the cell type and the nature of the harmful agent, the externalization of phosphatidylserine is always present in the earlier apoptotic events. Annexin V-FITC is a fluorescent probe with high affinity for phosphatidylserine, allowing its determination by fluorescence assays. Fig. 4 depicts the flow cytometry results of the apoptotic process in the presence of complexes 1 and 2 (10, 25, 50 and 100 μM) after 48 h of incubation in Jurkat cells.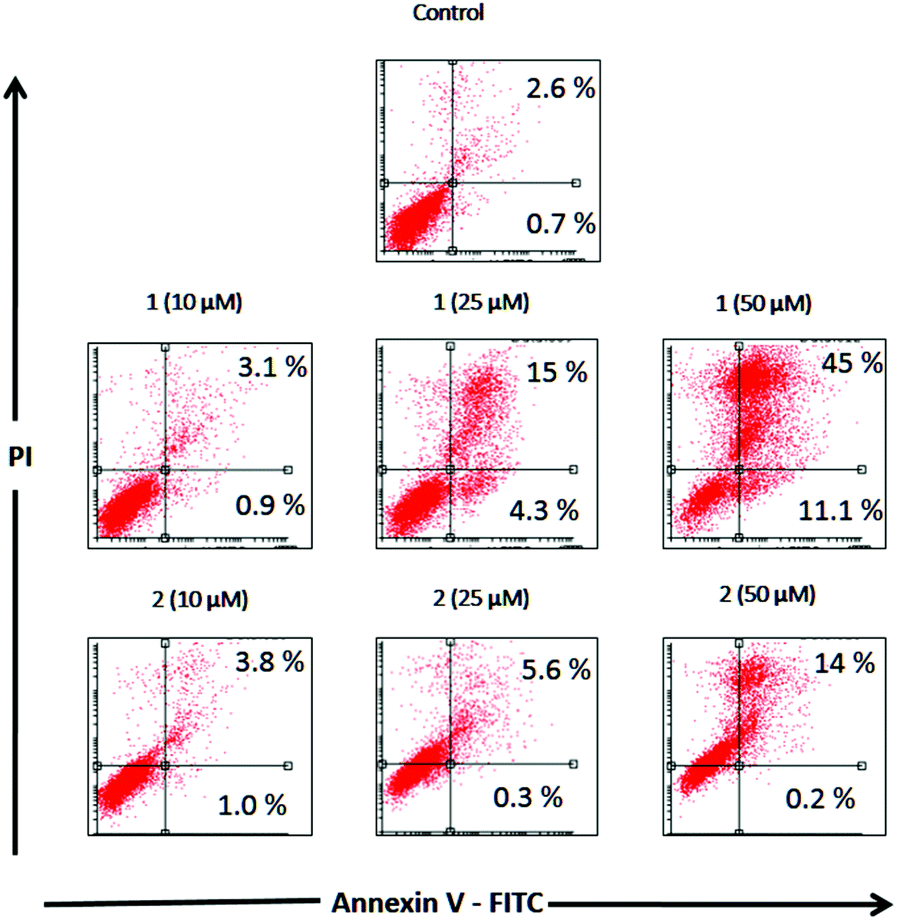 | ||
| Fig. 4 Induction of apoptosis in Jurkat cells. The panels show the cell distribution revealed by the intensity of PI-derived fluorescence and annexin V positive, in untreated cell cultures (Control) and cultures treated for 48 h in the presence of complexes 1 and 2 (10, 25, 50 and 100 μM). | ||
Table 7 displays the quantification of early and late stages of apoptosis obtained by flow cytometry in Jurkat cells. This table shows that the control cultures showed 0.7% early apoptotic cells and 2.6% late apoptotic cells. These results changed when the cells were incubated with 10, 25 and 50 μM of complexes 1 and 2, showing an increase in the early and late apoptotic cellular fractions.
| Concentration (μM) | Annexin V+/PI− | Annexin V+/PI+ | ||
|---|---|---|---|---|
| 1 (%) | 2 (%) | 1 (%) | 2 (%) | |
| 0 | 0.7 | 0.7 | 2.6 | 2.6 |
| 10 | 0.9 | 1 | 3.1 | 3.8 |
| 25 | 4.3 | 0.3 | 15.4 | 5.6 |
| 50 | 11.1 | 0.2 | 45.2 | 13.9 |
Complex 1 resulted in approximately 4.3% and 11.1% early apoptotic cells (annexin V positive) at 25 μM and 50 μM, respectively, whilst complex 2 did not show any changes in early apoptotic fractions over the basal level at 10, 25 and 50 μM.
Nevertheless, both compounds increased late apoptotic fractions, complex 1 produced 15.4% and 45.2% at 25 μM and 50 μM whilst complex 2 resulted in 5.6% and 13.9% at the same concentrations.
As can be seen, the percentages of apoptotic and apoptotic/necrotic cells increased with the concentration of both complexes. These results are in accordance with the viability assays, confirming that the deleterious action of complex 1 is higher than that of complex 2.
On the other hand, caspases (cysteine-requiring aspartate proteases) are a family of proteases that mediate cell death and are important to the process of apoptosis. Caspase 3 is one of the critical members of this family. It is an effector caspase that cleaves most of the caspase-related substrates involved in apoptosis regulation.47
In Fig. 5, it can be seen that after 48 h of incubation of the cells with complex 1 and complex 2, caspase 3 is activated at 25 and 50 μM for compound 1 and only at 50 μM for compound 2 (p < 0.01), demonstrating that the apoptotic action of both complexes is in agreement with the annexin V assay. The activation of caspase 3 is a good marker to confirm the results of annexin V for the detection of late apoptosis.
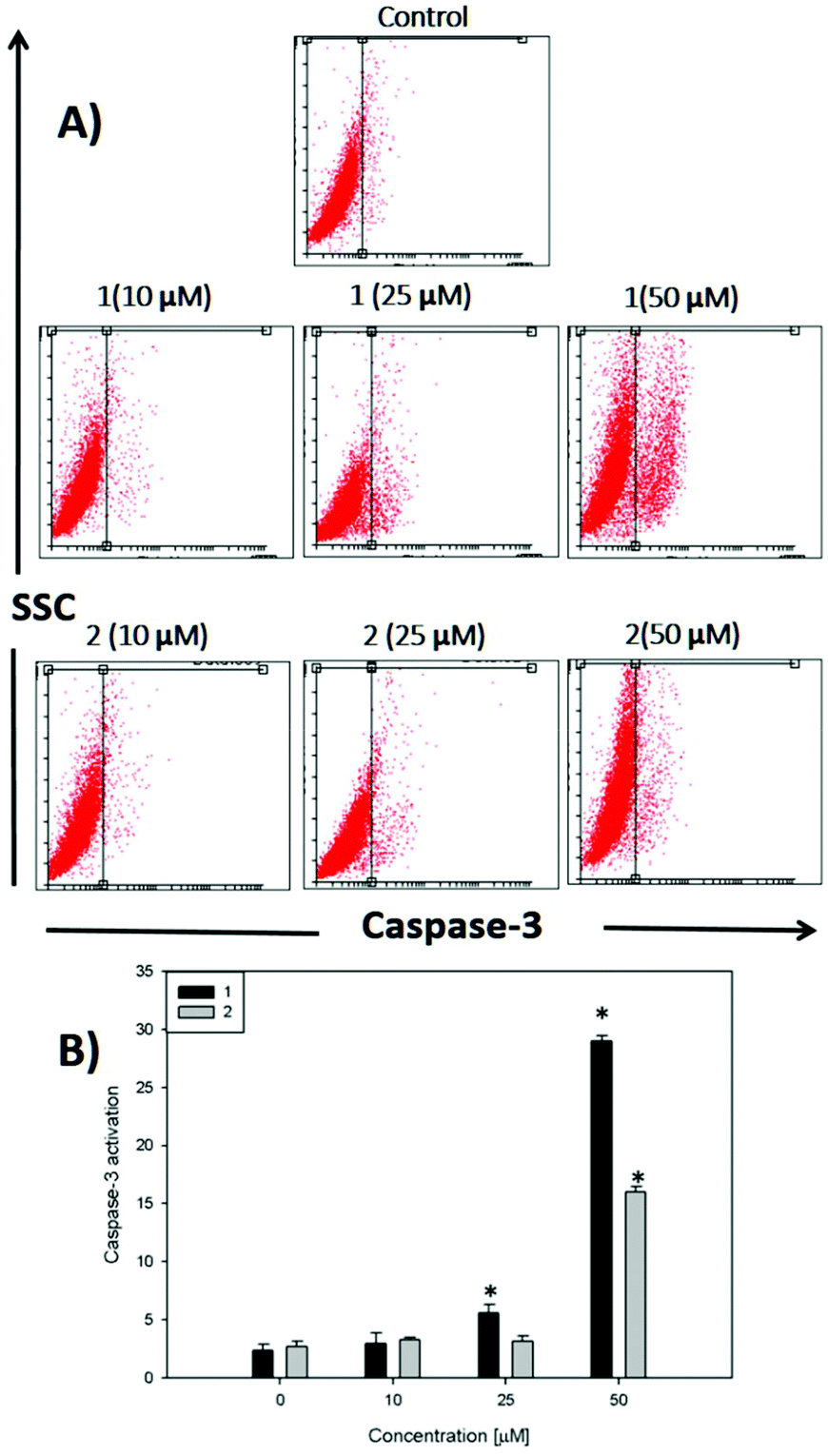 | ||
| Fig. 5 Induction of apoptosis in Jurkat cells. (A) The panels show the cell distribution with SSC and caspase-3 levels, in untreated cell cultures (Control) and cultures treated for 48 h in the presence of compounds 1 and 2 (10, 25 and 50 μM). (B) Caspase 3 activation versus the complex 1 and 2 concentration graph. | ||
Experimental
Materials and methods
In general, solvents and starting materials were purchased from commercial companies: VWR and Aldrich-Sigma. In particular, the proteasome was purchased from Merck and plasmid pBR322 from GenCust.Tissue culture materials were purchased from Corning, Dulbecco’ s Modified Eagle's Medium (DMEM), TrypLE™ from Gibco and fetal bovine serum (FBS) from Internegocios SA. Dihydrorhodamine 123 (DHR) was purchased from Molecular Probes (Eugene, OR). Syber Green and low melting point agarose were purchased from Invitrogen Corporation.
Mono-dimensional 13C-NMR and 1H-NMR experiments were performed in DMSO-d6 and D2O using a Bruker AMX-300 (300 MHz) spectrometer at room temperature (25 °C). Elemental analyses were performed on a PerkinElmer 2400 Series II microanalyzer. Fast atom bombardment (FAB) mass spectroscopy (MS) was performed on a VG AutoSpec spectrometer, IR spectroscopy was performed on a PerkinElmer Model 283 spectrophotometer with an ATR accessory (Miracle Single Reflection Horizontal) and UV-visible spectroscopy was performed on a Thermo Fisher Scientific Evolution 260 Bio spectrophotometer.
Chemistry
Mp 180 °C. Elemental analysis found, C, 41.70; H, 4.15; N, 20.80; S, 16.11; C12H11N4SCl requires C, 41.70; H, 4.00, N, 20.85; S, 15.85%. IR (KBr pellet): ν/cm−1 3293, 3178 (s, NH); 1635 (s, NH2); 820 (w, CS-thioamide IV). 1H NMR (d6-DMSO, ppm), δ = 9.19 [s, NH, 1H]; 7.70, 7.67 (d, J = 9.0 Hz, CH, 2H); 7.34; 7.31 (d, J = 9.0 Hz, CH, 2H).
Though the procedure was first reported in the fifties,19 it was later on slightly changed for α-N-heterocycle thiosemicarbazones and it proceeds as follows:49p-chlorophenylthiosemicarbazide LH2 (0.403 g, 2 mmol) was dissolved in 20 mL of ethanol and 5 mL of warm water at 40 °C. The clear solution was added dropwise to an ethanolic solution (10 mL) of pyrrol-2-carboxaldehyde (0.19 g, 2 mmol). The reaction mixture was heated to reflux for 5 h (∼78 °C), after this the reaction mixture was taken to a rotavapor and concentrated to half of the volume until a yellow solid precipitates which was isolated by filtration, washed with ethanol and dried.
Yield: 51% (0.284 g). Mp 195 °C (decomposes). Elemental analysis found, C, 51.20; H, 4.00; N, 19.70; S, 11.80; C12H11N4SCl requires C, 51.70; H, 3.95, N, 20.10; S, 11.50%. MS (FAB+ with mNBA: nitrobenzyl alcohol matrix) m/z 279 for [H2L + H]+. IR (KBr pellet): ν/cm−1 3291, 3225, 3151 (m, NH); 1609 (s, CN); 820 (w, CS-thioamide IV). 1H NMR (d6-DMSO, ppm), δ = 11.78 [s, 3NH, 1H]; 11.52 [s, 2NH, 1H]; 10.06 [s, 4NH, 1H]; 7.99 [s, 1CH, 1H]; 7.68, 7.65 (d, J = 9.3 Hz, 9CH, 11CH, 2H); 7.47; 7.44 (d, J = 9.3 Hz, 8CH, 12CH, 2H); 7.09 [s, 5CH, 1H]; 6.48 [s, 4CH, 1H]; 6.15 [s, 3CH, 1H]. 13C NMR (d6-DMSO, ppm), δ = 176.08 (C6); 139.29 (C7); 135.91 (C2); 130.38 (C1); 129.44 (C9, C11); 128.66 (C10); 127.97 (C8, C12); 123.57 (C5); 115.19 (C3); 110.85 (C4). UV/vis (DMSO): λ/nm 287 (IL π–π*), 347 (IL n-π*).
Yield: 86% (0.142 g). Mp >250 °C. Elemental analysis found, C, 40.15; H, 3.30; N, 15.35; S, 8.90; C24H20N8S2Cl2Pd·3H2O requires C, 40.25; H, 3.65, N, 15.65; S, 8.95%. MS (FAB+ with mNBA: nitrobenzyl alcohol matrix) m/z 663 for [H2L + H]+/[M]+. IR (KBr pellet): ν/cm−1 3228 (m, NH); 1598 (s, CN); 827 (w, CS-thioamide IV). 1H NMR (d6-DMSO, ppm), δ = 11.54 [s, 3NH, 1H]; 9.48 [s, 4NH, 1H]; 7.49 [s, 1CH, 1H]; 7.42–7.32 [m, 8CH, 9CH, 11CH, 12CH, 4H]; 7.17 [s, 5CH, 1H]; 6.91 [s, 4CH, 1H]; 6.26 [s, 3CH, 1H]; 13C NMR (d6-DMSO, ppm), δ = 169.44 (C6); 146.87 (C7); 140.28 (C2); 129.19 (C9, C11); 129.06 (C1); 127.37 (C10); 123.14 (C8, C12); 12213 (C5); 120.21(C3); 111.18 (C4). λ/nm 270 (IL π–π*), 389 (CT Metal-L). Single crystals, suitable for X-ray diffraction analysis, grown in a NMR tube from the d6-DMSO solution were used for NMR experiments.
Complex 2, [Pt(LH)2]. 5 mL of an aqueous solution of K2PtCl4 (0.104 g, 0.25 mmol) was added dropwise to a methanolic solution (20 mL) of LH2 (0.140 g, 0.5 mmol). The mixture was stirred at room temperature for 5 h. The final solid was filtered, washed with hot methanol and dried under vacuum.
Yield: 58% (0.187 g). Mp >250 °C. Elemental analysis found, C, 36.30; H, 3.05; N, 13.80; S, 8.00; C24H20N8S2Cl2Pt·2H2O requires C, 36.65; H, 3.05, N, 14.25; S, 8.15%. MS (FAB+ with mNBA: nitrobenzyl alcohol matrix) m/z 751 for [H2L + H]+. IR (KBr pellet): ν/cm−1 3226 (m, NH); 1608 (s, CN); 827 (w, CS-thioamide IV). 1H NMR (d6-DMSO, ppm), δ = 11.57 [s, 3NH, 1H]; 9.61 [s, 4NH, 1H]; 7.46 [s, 1CH, 1H]; 7.35–7.25 [m, 8CH, 9CH, 11CH, 12CH, 4H]; 7.07 [s, 5CH, 1H]; 6.50 [s, 4CH, 1H]; 6.16 [s, 3CH, 1H]; 13C NMR (d6-DMSO, ppm), δ = 175.15 (C6); 138.41 (C7); 135.01 (C2); 129.44 (C1); 128.52 (C9, C11); 127.76 (C10); 127.10 (C8, C12); 122.69 (C5); 114.29 (C3); 109.93 (C4). (DMSO): λ/nm 266(IL π–π*), 355(CT Metal-L).
Stability studies by UV and NMR
Complexes 1 and 2 have been monitored along 24 h in solution where the pH was adjusted to the physiological range, using appropriate buffer solutions (Tris-HCl 0.5 M Tris Base pH: 7.6).Crystallography
Data were collected on a Bruker Kappa Apex II diffractometer. A summary of the crystal data, experimental details and refinement results is listed in Table 8. The software package SHELXTL was used for space group determination, structure solution, and refinement.50 The structure was solved by direct methods, completed with difference Fourier syntheses, and refined with anisotropic displacement parameters.| [Pd(LH)2]·2DMSO | ||
|---|---|---|
| Chemical formula | C228H32Cl2N8O2PdS4 | |
| Formula weight | 409.08 g mol−1 | |
| Temperature | 296(2) K | |
| Wavelength | 0.71073 Å | |
| Crystal size | 0.050 × 0.060 × 0.190 mm | |
| Crystal habit | Orange needle | |
| Crystal system | Monoclinic | |
| Space group | P121/c1 | |
| Unit cell dimensions | a = 14.627(4) Å | α = 90° |
| b = 5.7150(18) Å | β = 4.995(10)° | |
| c = 20.939(7) Å | γ = 90° | |
| Volume | 1743.7(9) Å3 | |
| Z | 2 | |
| Density (calculated) | 1.558 g cm−3 | |
| Absorption coefficient | 0.964 mm−1 | |
| Theta range for data collection | 1.95 to 25.37° | |
| Index ranges | −17 ≤ h ≤ 17; −6 ≤ k ≤ 6; −25 ≤l ≤ 25 | |
| Reflections collected | 26868 |
|
| Independent reflections | 3189 [R(int) = 0.0538] | |
| Coverage of independent reflections | 99.9% | |
| Data/restraints/parameters | 3189/0/207 | |
| Goodness-of-fit on F2 | 1.083 | |
| Final R indices | I > 2σ(I): R1 = 0.0403, wR2 = 0.1064 | |
| All data: R1 = 0.0682, wR2 = 0.1297 | ||
| Largest diff. peak and hole | 0.700 and −0.665 e Å3 | |
Biology
Cytotoxicity by the MTT assay
The MTT assay was performed according to Mosmann et al.51 Briefly, the cells were seeded in a 96-multiwell dish, allowing attachment for 24 h and treated with different concentrations of complexes 1 and 2 at 37 °C for 48 and 72 h. After this, the medium was changed and the cells were incubated with 0.5 mg mL−1 MTT under normal culture conditions for 3 h. Cell viability was marked by the conversion of the tetrazolium salt MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium-bromide) to a coloured formazan by mitochondrial dehydrogenases. Colour development was measured spectrophotometrically in a microplate reader (7530, Cambridge Technology, Inc., USA) at 570 nm after cell lysis in DMSO (100 μL per well). Cell viability was plotted as the percentage of the control value.DNA and protein interaction
UV/Vis titration experiments
To investigate the potential binding ability of complexes 1–2 with DNA, spectrophotometric titrations (240–800 nm) were performed at room temperature by: (a) keeping the CT-DNA concentration constant (3.2 × 10−5 M) while varying the concentration of each complex (0–500 μM) and monitoring the changes in the typical absorbance of CT-DNA at 260 nm after equilibration (10 min at 37 °C), (b) keeping the complex concentration constant (2.5 × 10−6 M), varying the concentration of CT-DNA (0–500 μM) and monitoring the changes in the absorbance of one characteristic charge transfer band of the complex after equilibration (10 min at 37 °C). The first assay was used to estimate the nature of the supramolecular interactions while the latter assay was used to determine the DNA-binding constant of the complexes, Kb (in M−1) using the Wolfe–Shimer equation:32where [DNA] is the concentration of the nucleic acid in base pairs, εa is the apparent absorption coefficient obtained by calculating Aobs/[compound], and εf and εb are the absorption coefficients of the free and the fully bound compound, respectively.
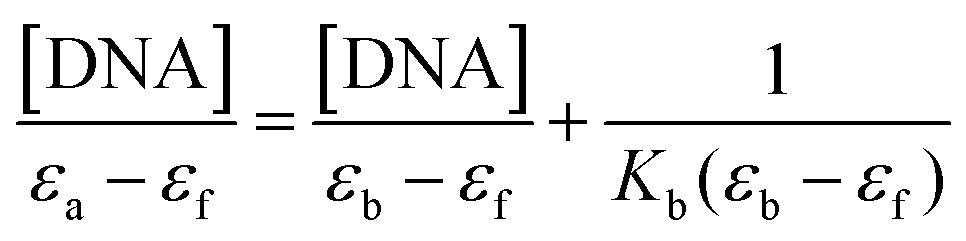
Agarose gel electrophoresis
Complex 1 was incubated at 37 °C with a concentration of 0.0625 μg μL−1 of pBR322 plasmid DNA, at different concentrations expressed as ri = complex:DNA(base pair) ratio. The ri used is from 0.01 to 0.2, in a total volume of 20 μL.
After an incubation period of 24 and 48 h, the mobility of the complex treated pBR322 samples was analyzed by gel electrophoresis at 70 V cm−1 in Tris-acetate/EDTA buffer. A control of pBR322 was also incubated, and a load of a 1 kb ladder (5 mL) was also loaded in lane 1 of the gel. The gel was stained with an ethidium bromide aqueous solution and DNA bands were visualized with a UV-transilluminator. pBR322 was purchased from GenCust and the 1 kb ladder from Sigma-Aldrich (D0428).
Single cell gel electrophoresis (SCGE) assay
For the detection of DNA strand breaks the single cell gel electrophoresis (‘comet’) assay was used in the alkaline version, based on the method of Singh et al.52 with minor modifications. Under alkaline conditions, DNA loops containing breaks lose supercoiling, unwind and are released from the nuclei and form a ‘comet-tail’ by gel electrophoresis. For this experiment, 2 × 104 cells were seeded in a twelve-well plate; 24 h later the cells were incubated with various concentrations of the complexes. After treatment, the cells were suspended in 0.5% low melting point agarose and were immediately poured onto glass microscope slides. The slides were immersed in an ice-cold prepared lysis solution in the dark for 1 h (4 °C) in order to lyse the cells, remove cellular proteins and to permit DNA unfolding. Immediately after this, the slides were put in a horizontal electrophoresis tank containing 1 mM Na2EDTA and 0.3 M NaOH (pH 12.7) and then electrophoresis was performed for 30 min at 25 V (4 °C). After this, there slides were neutralized and stained with Syber Green. Analysis of the slides was performed in an Olympus BX50 fluorescence microscope. Cellular images were acquired with a Leica IM50 Image Manager (Imagic Bildverarbeitung AG). A total of 50 randomly captured cells per experimental point of each experiment was used to determine the tail moment (the product of tail length by tail DNA percentage) using free comet scoring software (Comet Score version 1.5). Two parallel slides were taken for each experimental point. Independent experiments were repeated twice. A pulse of 20 minutes of 10 μg mL−1 bleomycin just before the cells were harvested was employed as the positive control.UV/Vis kinetics experiments
To investigate the interaction of complex 1 with plasma proteins, electronic spectra of the protein models, HEWL (hen egg white lysozyme) and RNase A at 10−5 M were recorded (monitoring the typical absorbance of proteins at 280 nm) before and after the addition of complex 1 at a stoichiometric ratio of 3:1 (metal to protein) for 24 h at R.T. The binding affinity constants were calculated as a pseudo first order based on the equal results obtained for stoichiometry: 10:1 and 3:1 for both cases.53
Proteasome activity assay
Proteasomal inhibition by complexes 1–2 was assayed using the 20S Proteasome Activity Assay Kit from Millipore. To determine whether both compounds inhibit proteasome functions directly, the provided 20S proteasome was diluted (1:60) in 100 μl assay buffer and incubated according to the manufacturer's instructions with the suc-LLVY-AMC substrate and the indicated concentrations of complexes 1–2 for 2 h at 37 °C. Fluorescence at 460 nm was read using a FluoSTAR OPTIMA microplate reader.
Moreover, Jurkat whole-cell extract (8 μg) was incubated with 10 μmol L−1 chymotrypsin-like-substrate (Suc-LLVY-AMC) in 100 μL assay buffer [50 mmol L−1 Tris-HCl (pH 7.5)] in the presence of both compounds (100 μM each) or solvent DMSO as the control. After a 2 h incubation at 37° C, the production of hydrolyzed AMC groups was measured using a Shimadzu RF-6000 spectrofluorophotometer with an excitation filter of 365 nm and an emission filter of 460 nm.
Determination of reactive oxygen species (ROS) production
Oxidative stress in Jurkat cells was evaluated by measurement of the intracellular production of reactive oxygen species (ROS) after incubation of the cells with different concentrations (2.5–25 μM) of complex 1 in water (from 20 mM of stock solution in DMSO) for 48 h at 37 °C. ROS generation was determined by the oxidation of DHR-123 to rhodamine by spectrofluorescence as we have previously described.54Measurements of externalization of phosphatidylserine by annexin V-FITC/PI staining
Cells in the early and late stages of apoptosis were detected with annexin V-FITC and PI staining. Cells were treated with different concentrations of compounds 1 and 2 and were incubated for 48 h prior to analysis. Cells were analyzed using a BD FACS Calibur™ flow cytometer and FlowJo 7.6 software. For each analysis, 10000 counts, gated on a forward scatter versus a side scatter dot plot, were recorded. Four subpopulations were defined in the dot plot: the undamaged vital (annexin V negative/PI negative), the vital mechanically damaged (annexin V negative/PI positive), the apoptotic (annexin V positive/PI negative), and the secondary necrotic (annexin V positive/PI positive) subpopulations.
Caspase 3 assay
The determination of caspase 3, one of the main effector caspases, was conducted with a commercial kit (Pharmingen™ caspase 3 assay kit, BD) following the recommendation of the manufacturer. The cells (10000 events) were analyzed using a BD FACS Calibur™ flow cytometer and FlowJo 7.6 software.
Conclusions
The synthesis of complexes using palladium and platinum as metals with a heterocyclic thiosemicarbazone derivative has afforded two new mononuclear structures, which in both cases, showed a good stability in solution and biological buffers. The compounds interact not only with DNA models (pBR322 and CT DNA) but also with proteins (lysozyme and RNase models) showing different values of interaction and following a different pattern than cisplatin. Both compounds caused cytotoxicity in a concentration-dependent manner on several tumor cell lines including leukemia and different solid tumors (lung and bone). The important pharmacological fact is that both compounds showed high selectivity for tumor cells and no activity versus healthy cells such as normal L929 fibroblasts.Moreover, the palladium complex 1 is not only endowed with a higher cytotoxicity than the platinum complex 2 but also with potent inhibition capacity of proteasome 20S. With these data on hand, we can establish that the mechanism of these complexes must be quite different to cisplatin.
As a whole, these results indicate that compound 1 is an interesting candidate for potential antitumor uses, and provide new insight into the development of palladium compounds as potential anticancer agents.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the following grants for the Spanish MINECO: SAF-2012-34424, CTQ2015-68779R and CTQ2015-70371-REDT. We thank Prof. J. Satrustegui from CBMSO for the use of the fluorescence equipment and the Secretary of University Policies (SPU, Argentina) for IEL's travel fellowship.References
- C. R. Kowol, R. Trondl, P. Heffeter, V. B. Arion, M. A. Jakupec, A. Roller, M. Galanski, W. Berger and B. K. Keppler, J. Med. Chem., 2009, 52, 5032–5043 CrossRef CAS PubMed.
- C. A. Kunos, T. Radivoyevitch, S. Waggoner, R. Debernardo, K. Zanotti, K. Resnick, N. Fusco, R. Adams, R. Redline, P. Faulhaber and A. Dowlati, Gynecol. Oncol., 2013, 130, 75–80 CrossRef CAS PubMed.
- Bethesta (USA), Clinical trials Coverment, NCT01835171, http://clinicaltrials.gov/show/NCT01835171.
- E. Colleen Moore and A. C. Sartorelli, Pharmacol. Ther., 1984, 24, 439–447 CrossRef.
- W. E. Antholine, J. M. Knight and D. H. Petering, J. Med. Chem., 1976, 19, 339–341 CrossRef CAS PubMed.
- M.-C. Liu, T.-S. Lin, A. C. Sartorelli, G. P. Ellis and D. K. Luscombe, in Prog Med Chem, Elsevier, 1995, vol. 32, pp. 1–35 Search PubMed.
- C. R. Kowol, R. Berger, R. Eichinger, A. Roller, M. A. Jakupec, P. P. Schmidt, V. B. Arion and B. K. Keppler, J. Med. Chem., 2007, 50, 1254–1265 CrossRef CAS PubMed.
- C. R. Kowol, W. Miklos, S. Pfaff, S. Hager, S. Kallus, K. Pelivan, M. Kubanik, Ã. v. A. Enyedy, W. Berger, P. Heffeter and B. K. Keppler, J. Med. Chem., 2016, 59, 6739–6752 CrossRef CAS PubMed.
- A. G. Quiroga and C. N. Ranninger, Coord. Chem. Rev., 2004, 248, 119–133 CrossRef CAS.
- A. G. Quiroga, J. M. Perez, I. Lopez-Solera, J. R. Masaguer, A. Luque, P. Roman, A. Edwards, C. Alonso and C. Navarro-Ranninger, J. Med. Chem., 1998, 41, 1399–1408 CrossRef CAS PubMed.
- A. G. Quiroga, J. M. Perez, E. I. Montero, J. R. Masaguer, C. Alonso and C. Navarro-Ranninger, J. Inorg. Biochem., 1998, 70, 117–123 CrossRef CAS PubMed.
- A. I. Matesanz, P. Albacete, J. Perles and P. Souza, Inorg. Chem. Front., 2015, 2, 75–84 RSC.
- K. A. Price, A. Caragounis, B. M. Paterson, G. Filiz, I. Volitakis, C. L. Masters, K. J. Barnham, P. S. Donnelly, P. J. Crouch and A. R. White, J. Med. Chem., 2009, 52, 6606–6620 CrossRef CAS PubMed.
- Y. Yu, D. S. Kalinowski, Z. Kovacevic, A. R. Siafakas, P. J. Jansson, C. Stefani, D. B. Lovejoy, P. C. Sharpe, P. V. Bernhardt and D. R. Richardson, J. Med. Chem., 2009, 52, 5271–5294 CrossRef CAS PubMed.
- A. I. Matesanz, C. Hernández, J. Perles and P. Souza, J. Organomet. Chem., 2016, 804, 13–17 CrossRef CAS.
- Y. Yu, J. Wong, D. B. Lovejoy, D. S. Kalinowski and D. R. Richardson, Clin. Cancer Res., 2006, 12, 6876–6883 CrossRef CAS PubMed.
- J. L. Hickey, J. L. James, C. A. Henderson, K. A. Price, A. I. Mot, G. Buncic, P. J. Crouch, J. M. White, A. R. White, T. A. Smith and P. S. Donnelly, Inorg. Chem., 2015, 54, 9556–9567 CrossRef CAS PubMed.
- A. Billiau, V. G. Edy, H. Heremans, J. Van Damme, J. Desmyter, J. A. Georgiades and P. De Somer, Antimicrob. Agents Chemother., 1977, 12, 11–15 CrossRef CAS PubMed.
- P. P. T. Sah and T. C. Daniels, Recl. Trav. Chim. Pays-Bas, 1950, 69, 1545–1556 CrossRef CAS.
- V. Martichonok, in Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, Ltd, 2001 Search PubMed.
- A. I. Matesanz, J. Perles and P. Souza, Dalton Trans., 2012, 41, 12538–12547 RSC.
- W. Hernandez, A. J. Vaisberg, M. Tobar, M. Alvarez, J. Manzur, Y. Echevarria and E. Spodine, New J. Chem., 2016, 40, 1853–1860 RSC.
- A. I. Matesanz, I. Leitao and P. Souza, J. Inorg. Biochem., 2013, 125, 26–31 CrossRef CAS PubMed.
- A. R. Kapdi and I. J. S. Fairlamb, Chem. Soc. Rev., 2014, 43, 4751–4777 RSC.
- D. A. Tocher, Appl. Organomet. Chem., 2000, 14, 172–173 CrossRef CAS.
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Chem. Rev., 2016, 116, 3436–3486 CrossRef CAS PubMed.
- A. Casini and J. Reedijk, Chem. Sci., 2012, 3, 3135–3144 RSC.
- S. Medici, M. Peana, V. M. Nurchi, J. I. Lachowicz, G. Crisponi and M. A. Zoroddu, Coord. Chem. Rev., 2015, 284, 329–350 CrossRef CAS.
- G. Psomas, J. Inorg. Biochem., 2008, 102, 1798–1811 CrossRef CAS PubMed.
- A. Meenongwa, R. F. Brissos, C. Soikum, P. Chaveerach, P. Gamez, Y. Trongpanich and U. Chaveerach, New J. Chem., 2016, 40, 5861–5876 RSC.
- K. Nakamoto, M. Tsuboi and G. D. Strahan, in Drug–DNA Interactions, John Wiley & Sons, Inc., 2008, pp. 303–356 Search PubMed.
- A. Wolfe, G. H. Shimer and T. Meehan, Biochemistry, 1987, 26, 6392–6396 CrossRef CAS PubMed.
- I. Haq, Arch. Biochem. Biophys., 2002, 403, 1–15 CrossRef CAS PubMed.
- G. L. Cohen, W. R. Bauer, J. K. Barton and S. J. Lippard, Science, 1979, 203, 1014–1016 CAS.
- A. R. Collins, V. L. Dobson, M. r. Dusinska, G. Kennedy and R. Stetina, Mutat. Res., 1997, 375, 183–193 CrossRef CAS PubMed.
- W. Liao, M. A. McNutt and W.-G. Zhu, Methods, 2009, 48, 46–53 CrossRef CAS PubMed.
- A. R. Timerbaev, C. G. Hartinger, S. S. Aleksenko and B. K. Keppler, Chem. Rev., 2006, 106, 2224–2248 CrossRef CAS PubMed.
- L. Messori, A. Casini, C. Gabbiani, E. Michelucci, L. Cubo, C. Rios-Luci, J. M. Padron, C. Navarro-Ranninger and A. G. Quiroga, ACS Med. Chem. Lett., 2010, 1, 381–385 CrossRef CAS PubMed.
- R. Z. Orlowski and E. C. Dees, Breast Cancer Res., 2003, 5, 1–7 CrossRef CAS PubMed.
- J. Sun, S. Nam, C.-S. Lee, B. Li, D. Coppola, A. D. Hamilton, Q. P. Dou and S. d. M. Sebti, Cancer Res., 2001, 61, 1280–1284 CAS.
- J. Adams, V. J. Palombella, E. A. Sausville, J. Johnson, A. Destree, D. D. Lazarus, J. Maas, C. S. Pien, S. Prakash and P. J. Elliott, Cancer Res., 1999, 59, 2615–2622 CAS.
- J. H. Gundelach, A. A. Madhavan, P. J. Wettstein and R. J. Bram, FASEB J., 2012, 27, 782–792 CrossRef PubMed.
- G. R. Tundo, D. Sbardella, C. Ciaccio, S. De Pascali, V. Campanella, P. Cozza, U. Tarantino, M. Coletta, F. P. Fanizzi and S. Marini, J. Inorg. Biochem., 2015, 153, 253–258 CrossRef CAS PubMed.
- I. E. Leon, A. L. Di Virgilio, D. A. Barrio, G. Arrambide, D. Gambino and S. B. Etcheverry, Metallomics, 2012, 4, 1287–1296 RSC.
- J. Rivadeneira, A. L. D. Virgilio, D. A. Barrio, C. I. Muglia, L. Bruzzone and S. B. Etcheverry, Med. Chem., 2010, 6, 9–23 CrossRef CAS.
- M. A. M. Capella, L. S. Capella, R. C. Valente, M. Gefa and A. G. Lopes, Cell Biol. Toxicol., 2007, 23, 413–420 CrossRef CAS PubMed.
- H. Sakahira, M. Enari and S. Nagata, Nature, 1998, 391, 96–99 CrossRef CAS PubMed.
- A. A. Hassan, E. M. El-Sheref and A. H. Abou-Zied, J. Heterocycl. Chem., 2011, 49, 38–58 CrossRef.
- F. E. Anderson, C. J. Duca and J. V. Scudi, J. Am. Chem. Soc., 1951, 73, 4967–4968 CrossRef CAS.
- SHELTXTL-NTversion 6.12, Structure Determination Package, Bruker-Nonius XS, Madison, Wishconsin USA, 1997–2001 Search PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- N. P. Singh, M. T. McCoy, R. R. Tice and E. L. Schneider, Exp. Cell Res., 1988, 175, 184–191 CrossRef CAS PubMed.
- T. Marzo, G. Bartoli, C. Gabbiani, G. Pescitelli, M. Severi, S. Pillozzi, E. Michelucci, B. Fiorini, A. Arcangeli, A. r. G. Quiroga and L. Messori, BioMetals, 2016, 29, 535–542 CrossRef CAS PubMed.
- I. E. Leon, A. L. Di Virgilio, V. Porro, C. I. Muglia, L. G. Naso, P. A. M. Williams, M. Bollati-Fogolin and S. B. Etcheverry, Dalton Trans., 2013, 42, 11868–11880 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: NMR and UV spectra, images of the genotoxicity effect, ROS production values in Jurkat cells. CCDC 1516216. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7qi00446j |
| This journal is © the Partner Organisations 2018 |