
Morphology-controlled synthesis of TiO2/MoS2 nanocomposites with enhanced visible-light photocatalytic activity†
Yuanqing
Sun
ac,
Huihui
Lin
b,
Chuanxi
Wang
 *a,
Qian
Wu
b,
Xiaojie
Wang
b and
Minghui
Yang
*a
*a,
Qian
Wu
b,
Xiaojie
Wang
b and
Minghui
Yang
*a
aInstitute of New Energy Technology, Ningbo Institute of Industrial Technology, Chinese Academy of Sciences, Ningbo, 315201, P. R. China. E-mail: wangcx@nimte.ac.cn; myang@nimte.ac.cn
bSchool of Chemical & Material Engineering, Jiangnan University, Wuxi 214122, P. R. China
cState Key Laboratory of Heavy Oil Processing, College of Science, China University of Petroleum, Beijing, 102249, P. R. China
First published on 6th November 2017
Abstract
The development of photocatalysts that utilize sunlight is very important for solving the energy crisis and reducing environmental pollution. In this report, two kinds of TiO2/MoS2 cocatalyst with novel morphologies (hollow and yolk–shell structure) are prepared via a polymer assisted targeted-etching method. MoS2, as a typical two-dimensional (2D) layered transition metal sulfide, can accept electrons and provide active sites for photocatalytic reactions. Besides, with the assistance of MoS2, the absorption band of the resultant heterostructures becomes broader and covers the entire visible region. Thus the photocatalytic activity of TiO2 in the visible light region can be improved. Moreover, the hollow and yolk–shell heterostructures of TiO2/MoS2 possess a high specific surface area, and the excellent interface of the heterostructures enables the easy separation of holes and electrons, which can enhance the photodegradation of dye. The results demonstrate that the as-prepared TiO2/MoS2 nanocomposites with hollow and yolk–shell structures show outstanding catalytic activity which is tested through the degradation of methylene blue and rhodamine B under visible light. Our design provides a new strategy to acquire novel photocatalysts, with various nanostructures, that have remarkable potential applications in environmental protection, e.g. water treatment and dye pollutant degradation.
Introduction
The development of semiconductor photocatalysts is attracting more attention recently due to their capacity to solve the energy crisis and reduce environmental pollution.1 To date, a series of semiconductor photocatalysts have been exploited, such as ultraviolet active TiO2 and SrTiO3, and visible active C3N4, Cu2O and CdS.2 Among these photocatalytic materials, nanostructured TiO2 has been intensively investigated as an n-type semiconductor photocatalytic material, in self-cleaning and the removal of hazardous compounds, due to its superior photocatalytic activity, chemical stability, low cost and nontoxicity.3 However, a major drawback of TiO2 is its large band gap (3.2 eV), and thus only UV light, which constitutes merely 2–3% of the solar spectrum, can be utilized, which significantly limits the use of sunlight in photocatalysis.4 Besides, the low charge separation rate of pure TiO2 also reduces its photocatalytic activity. To overcome these shortcomings, many controllable synthesis efforts have already been used to synthesize particles with well-defined sizes and shapes, such as monodispersed nanoparticles, hierarchical microspheres, hollow spheres, nanorods, nanotubes and porous networks, which result in improving the photocatalytic performance.2a,5 Among these nanostructures, hollow or yolk–shell spheres,6 as special core–shell nanostructures, have caught the eye of scientists due to their properties such as low density, large surface area and surface permeability.Moreover, researchers have reported a lot of work developing materials as cocatalysts with TiO2-based photocatalysts, such as Ag2O/TiO2, C/TiO2, and nanocluster doped TiO2,7 that could broaden their light harvesting window to the visible range and improve charge separation. Specifically, as a typical two-dimensional (2D) layered transition metal sulfide, molybdenum disulfide (MoS2), with a structure composed of three stacked atom layers (S–Mo–S) held together by van der Waals forces, has become a focus in photocatalysis research activities as a cocatalyst due to its distinctive electronic, optical and catalytic properties.8 Besides, MoS2 can also accept electrons and provide active sites for photocatalytic reactions.9 Recently, various morphological MoS2 nanomaterials (nanoparticles, hollow microspheres, nanotubes, nanorods, nanowires, nanoflowers) have been prepared by gas-phase synthesis, electrochemical deposition, thermal decomposition, sonochemical synthesis and hydrothermal methods.2a,e,8a,10 However, irregular aggregates of nanoparticles or stacked multilayers limit the photocatalytic performance of the as-prepared MoS2.11 Therefore, the challenge still remains to prepare heterostructure photocatalysts based on MoS2 nanomaterials with enhanced photocatalytic activity. Recently, CdS/MoS2 and graphene/MoS2 heterostructures have been prepared.2e,12 In particular, TiO2/MoS2 heterostructures with diverse morphologies such as nanoparticles, nanobelts and nanowires have shown high activities as co-catalysts in the visible-light region.11b,13 Meanwhile, constructing novel nanostructures of TiO2/MoS2 (hollow and yolk–shell structures) turns out to be an effective way of developing photocatalysts with both large surface areas and high numbers of active sites.
Herein, we design a targeted etching route to prepare TiO2/MoS2 nanocomposites with novel nanostructures (hollow and yolk–shell structures). The monodispersed TiO2 microspheres are formed by a typical sol–gel method using tetrabutyl titanate (TBOB) as a precursor.14 Through a simple solvent-thermal process, the TiO2/MoS2 core–shell nanostructure can be synthesized.11b,15 Then, the novel hollow and yolk–shell structure of TiO2/MoS2 can be formed using a polymer assisted targeted-etching method. In this process, the sacrificial core can be easily removed with alkaline and acid solution, or even with water, and the shell forms while the dissolution of the analogue species proceeds. In this case, the problems associated with the coating and removing of templates in the conventional template method do not exist. Thus, the targeted-etching process is highly reproducible and scalable. The resultant nanocomposites show uniform morphologies with few-layer MoS2, large surface areas and high adsorption abilities. Moreover, as a proof of concept, we demonstrate that the novel TiO2/MoS2 heterostructures are excellent photocatalysts for the degradation of organic dyes, e.g. methylene blue (MB) and rhodamine B (RhB). Therefore, this novel photocatalyst may be used in the treatment of polluted water, the degradation of dye pollutants and environmental cleaning, with scope for continuous exploration in the future. Moreover, it can be expected that this design of controllable morphologies will be effective for preparing other nanoparticles.
Experimental
Materials
Tetrabutyl titanate (TBOB), ammonium tetrathiomolybdate ((NH4)2MoS4), methylene blue (MB) and rhodamine B (RhB) were purchased from Sinopharm. Polyvinyl pyrrolidone (PVP, molecular weight of 10 K) and polyethylene glycol (PEG, molecular weight of 2.5 K) were purchased from Sigma-Aldrich. Aqueous ammonia (NH3·H2O, 28%), sodium hydroxide (NaOH) and sodium fluoride (NaF) were analytical grade. All reagents were used as received without further purification. De-ionized water was used in all experiments.The synthesis of TiO2 microspheres
The monodispersed TiO2 microspheres (size: ∼180 nm) were synthesized by following the previous procedures.11b,14,15 In a typical process, NH3·H2O (28%, 0.38 mL) solution and H2O (0.91 mL) were added into a mixed solution (250 mL) containing ethanol and acetonitrile (in a volume ratio of 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4) under magnetic stirring. Then TBOB (3 mL) was promptly injected into the solution under vigorous stirring for 6 h. The resultant TiO2 microspheres were centrifuged and washed with ethanol and DI water three times in order to remove the unreacted precursors.
:4) under magnetic stirring. Then TBOB (3 mL) was promptly injected into the solution under vigorous stirring for 6 h. The resultant TiO2 microspheres were centrifuged and washed with ethanol and DI water three times in order to remove the unreacted precursors.
The preparation of TiO2/MoS2 core–shell nanostructures
The TiO2/MoS2 core–shell nanostructures were prepared by a simple solvent-thermal process.11b In a typical process, 2 mg mL−1 of (NH4)2MoS4 and 2 mg mL−1 of TiO2 microspheres were dispersed in DMF (10 mL). The mixture was sonicated at room temperature for approximately 30 min until a clear and homogeneous solution was achieved. Then, the solution was transferred to a 20 mL Teflon-lined autoclave and heated in an oven at 180 °C for 24 h. The resultant product was collected by centrifugation at 8000 rpm for 5 min, washed with DI water and recollected by centrifugation.The synthesis of TiO2/MoS2 hollow microspheres
Typically, the TiO2/MoS2 core–shell microspheres (20 mg) were re-dispersed in water (10 mL). Then NaF (10 mg) was added into the suspension as the etching agent. After stirring for 1 h, PVP (10 mg) with an average molecular weight (AMW) of 10 K was added. The suspension was stirred for another 1 h before transferring to a Teflon-lined autoclave (20 mL). A hydrothermal reaction was carried out at 120 °C for 2 h to conduct the crystallization and targeted etching processes. The resultant TiO2/MoS2 hollow microspheres were collected. After washing with dilute NaOH solution (10 mmol L−1) and water three times, the final samples were obtained by drying at 60 °C for 10 h.The synthesis of TiO2/MoS2 yolk–shell microspheres
TiO2/MoS2 core–shell microspheres (20 mg) were re-dispersed in water (10 mL). PEG (AMW, 2.5 K; 10 mg) was added into the solution. The solution was stirred and ultrasonicated to fill PEG into the pores of TiO2/MoS2. Then the product was mixed with NaF (10 mg). After stirring for 1 h, PVP (5 mg) was added. The resulting suspension was stirred for a further 1 h before being transferred to a Teflon-lined autoclave (20 mL). A hydrothermal reaction was carried out at 120 °C for 1 h. The white products were collected and washed with dilute NaOH solution (10 mmol L−1) and water three times, then dried at 60 °C for 10 h.Photocatalytic degradation of methylene blue (MB) under visible light
The photocatalytic activity of the samples was determined by the degradation of MB under visible light irradiation at room temperature. A 500 W Xe-illuminator was used as a light source and set about 10 cm from the reactor. In each run, 3 mg of the photocatalyst was added to the MB solution (100 mL, 5 mg L−1) and the mixture was sonicated for 1 min. Before irradiation, the suspension was stirred for 60 min in the dark to reach the adsorption–desorption equilibrium between the MB and the photocatalyst. During given time intervals, the photo-reacted solution (3 mL) was extracted and then separated by membrane filtration (pore size 0.22 μm) to remove essentially all the photocatalyst particles. Finally, a UV-vis spectrophotometer was used to determine the degradation efficiency: (1 − C/C0) × 100%.Characterization methods
UV–Vis absorption spectra of dyes were obtained using a TU-1991 UV–Vis spectrophotometer. UV–vis absorption spectra of the products (solid state) were obtained using a Shimadzu UV-3600 plus PC spectrofluorimeter. X-ray photoelectron spectroscopy (XPS), using Mg Kα excitation (1253.6 eV), was performed using a VG ES-CALAB MKII spectrometer. Binding energy calibration was based on the C 1s peak at 284.6 eV. X-ray diffraction (XRD) data were collected on a Siemens D-5005 X-ray diffractometer with Cu Kα radiation (λ = 1.5418 Å). The morphologies and structures of the samples were characterized by scanning electron microscopy (SEM, Hitachi S4800) and transmission electron microscopy (TEM, JEM-2100).Results and discussion
The process of preparing TiO2/MoS2 nanocomposites with various morphologies is shown in Scheme 1. The monodispersed TiO2 microspheres were prepared by a typical sol–gel method using tetrabutyl titanate (TBOB) as the precursor.15 The prepared TiO2 microspheres showed a good dispersion and spherical morphology with an average diameter of 180 nm (Fig. S1†). Using the resultant TiO2 microspheres as seeds, TiO2/MoS2 core–shell nanostructures were formed through a facile solvent-thermal route, which was similar to that of the previous research.11b In this process, the (NH4)2MoS4 worked as a Mo and S source simultaneously, and has showed numerous applications in the preparation of MoS2 nanomaterials.16 Through adjusting the experimental conditions, uniform TiO2/MoS2 core–shell microspheres were formed. Then, with the assistance of PVP and PEG, the TiO2 core was removed selectively by NaF, and some novel nanostructures of TiO2/MoS2 nanocomposites were formed, such as the hollow and yolk–shell structures. Hence, a targeted etching route is developed to control the morphology of TiO2/MoS2 nanocomposites, which is an effective way to improve the performance of semiconductor photocatalysts.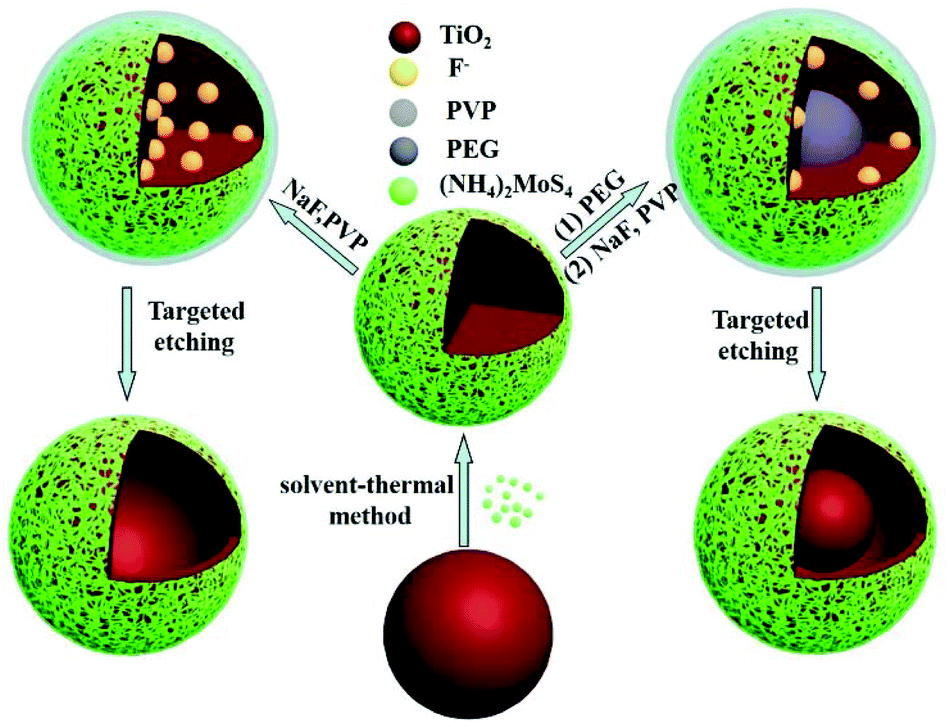 | ||
| Scheme 1 Scheme illustrating the formation of multiple morphologies of MoS2/TiO2 nanocomposites including core–shell microspheres, yolk–shell microspheres and hollow microspheres. | ||
Fig. 1a shows the transmission electron microscopy (TEM) image of the as-prepared TiO2 microspheres, which are 180 nm in diameter and have a smooth surface. After the solvent-thermal process with (NH4)2MoS4, the MoS2 nanosheets can grow on the surface of TiO2 and the TiO2/MoS2 core–shell structure formed (Fig. 1b). The resultant core–shell structure showed a uniformly spherical morphology and the size was larger than that of the pure TiO2 microspheres as shown in Fig. 1b. Moreover, such core–shell structures look like flowers with some thin MoS2 nanosheets coating the surface of the TiO2 microspheres (inset of Fig. 1b). The high-resolution TEM (HRTEM) image determined the growth direction of the TiO2 microspheres to be [010], and the lattice fringes perpendicular to the growth direction have a spacing of 0.35 nm,2a which is equal to the lattice parameter of the (101) facet (Fig. 1c). Besides, the HRTEM image also revealed a highly crystalline structure with continuous lattice fringes between the thin nanosheets separated by about 0.62 nm, corresponding to the (002) faces of the MoS2 crystals (Fig. 1c).16b The element mapping suggested that the resultant heterostructure contained four elements: Mo, S, Ti and O, simultaneously (Fig. 1d–f). In particular, considering the features of the spatial distribution of the different elements, the data also confirmed that the TiO2/MoS2 microspheres were hierarchical in structure, containing TiO2 microspheres as the core and MoS2 nanosheets as the shell (Fig. 1f). It should be noted that the TiO2/MoS2 microsphere in the dashed circle was incompletely covered by MoS2 nanosheets, as shown in Fig. 1d and e, so it possesses a strong Ti and O signal, while the others were weak due to signal interference with the MoS2 shell (Fig. 1f). After comprehensive consideration of these verified results, we can claim that the TiO2/MoS2 core–shell structure was prepared. Moreover, similar to previous mechanisms,11b this uniform structure was controlled by the ratio of the seeds to (NH4)2MoS4 as shown in Fig. S2.† When the amount of seed (1 mg mL−1) was low, TiO2/MoS2 core–shell structures with some irregular MoS2 nanosheets were synthesized (Fig. S2a†). In contrast, if the amount of seed was high, the MoS2 nanosheets would not coat the TiO2 completely to form the uniform core–shell morphology (Fig. S2c and d†). The perfect core–shell structures with uniform size and regular morphology were acquired when the amount of TiO2 was 2 mg mL−1 (Fig. 1b and Fig. S2b†).
 | ||
| Fig. 1 TEM images of (a) monodispersed TiO2 microspheres and (b) MoS2/TiO2 core–shell microspheres. (c) and inset image of (b) high-resolution TEM images of core–shell microspheres. (d–f) Elemental mapping images of (e) Mo/S and (f) Ti/O. | ||
To obtain hollow (h-TiO2/MoS2) and yolk–shell structures (y-TiO2/MoS2) of TiO2/MoS2, a polymer assisted method was used in the targeted etching process. Polyvinyl pyrrolidone (PVP), which has played an important role in forming novel heterostructures, was introduced in the system.17 In the targeted etching process, PVP protects the shell structure of MoS2,14 and restrains the diffusion of the dissolved Ti species into the solvent. Then, another polymer (polyethylene glycol (PEG)) was added, that filled the inside of the TiO2/MoS2 structure and possesses the capability to effectively partially decrease the amount of inner TiO2. Thus this process was favorable for preparing y-TiO2/MoS2. With the help of a hydrothermal reaction, uniform y-TiO2/MoS2 was synthesized. As shown in Fig. 2a, after etching, the TiO2 disappeared and hollow spheres were formed, but the center was a solid sphere with a size of 135 nm. This nanostructure of reported materials is defined as a yolk–shell structure. Moreover, MoS2 nanosheets were on the surface and their crystal morphology was almost undamaged during the targeted etching process (Fig. 2b). The elemental mappings showed a homogeneous hierarchical distribution of Mo and Ti (Fig. 2c–e). The results confirmed that the inside was TiO2, forming the yolk, and MoS2 was on the surface as the shell. Thus uniform y-TiO2/MoS2 was formed, but this structure was controlled by the etching time. As shown in Fig. S3,† big yolks start to appear inside the sphere after an etching reaction for 0.5 h (Fig. S3a and c†), and do not have a clear structure like the samples in Fig. 2a. When the etching time was prolonged to 2 h, an irregular yolk–shell structure with a broken MoS2 shell was acquired (Fig. S3b and d†).
 | ||
| Fig. 2 (a) TEM and (b) high-resolution TEM images of MoS2/TiO2 yolk–shell microspheres synthesized by targeted etching for 1 h; (c–e) elemental mapping images of (d) Mo and (e) Ti. | ||
Moreover, PEG contributed to the formation of the yolk–shell structure. Since PEG could fill the inner space of TiO2/MoS2, and hinder the penetration of fluoride and water upon soaking in fluoride solution, the inner part could be protected from etching.14,18 If the experiment was done in the absence of PEG, the h-TiO2/MoS2 structure was prepared. As shown in Fig. 3a, after targeted etching for 2 h, the uniform h-TiO2/MoS2 structure appeared, with a size of 203 nm and a shell thickness of 11 nm. The outside layer of h-TiO2/MoS2 was composed of MoS2 nanosheets which can be observed in the HRTEM image (Fig. 3b). However, the shell of h-TiO2/MoS2 was composed of MoS2 and TiO2 nanocomposites, which agreed with the results of the elemental mappings in Fig. 3c–e. In addition, it is easily understood that the extent of the hollow evolution process was time dependent, products with different sizes of hollow structures were synthesized by changing the etching time. At the beginning of the etching (1 h), the hollow structure appeared, but when increasing the etching time up to 3 h, the shell of the hollow structure was damaged (Fig. S4†).
 | ||
| Fig. 3 (a) TEM and (b) high-resolution TEM images of MoS2/TiO2 hollow microspheres synthesized by targeted etching for 2 h; (c–e) elemental mapping images of (d) Mo and (e) Ti. | ||
The XRD patterns of the as-prepared TiO2-anatase, TiO2/MoS2 core–shell microspheres, h-TiO2/MoS2, y-TiO2/MoS2, pure MoS2 and pure TiO2 are shown in Fig. 4. TiO2 microspheres prepared by sol–gel methods were amorphous as shown in Fig. 4f. After hydrothermal treatment, TiO2-anatase was formed (Fig. 4a), which matched with the standard peaks of a pure anatase phase (JCPDS card no. 21-1272).19 As for the pure MoS2 sample (Fig. 4e), the detected peaks can be assigned to the hexagonal phase of MoS2 (JCPDS card no. 37-1492).20 The weak (002) peak at 14.5° demonstrated that MoS2 was composed of few layer sheets (the MoS2 sample was acquired by freeze drying to avoid irregular aggregation).21 After forming the TiO2/MoS2 composites, the diffraction peaks of TiO2 were unchanged compared with those of TiO2-anatase, while the diffraction peaks of MoS2 were almost not observed (Fig. 4b–d).11 The weak peaks may be attributed to the presence of few layer MoS2 sheets on the surface of the composites that were too thin to be detected.2a However, the chemical composition of the resultant samples was confirmed by XPS spectra. The binding energies of the Mo 3d5/2 and Mo 3d3/2 peaks at 228.9 and 232.2 eV, and the S 2p3/2 and S 2p1/2 peaks at 162.5 and 163.6 eV, indicated that Mo4+ and S2− were the dominant oxidation states (Fig. S5†).22 The asymmetric peaks and tailing spectra (at about 236 eV) for Mo in the heterostructures were likely due to the existence of a small amount of Mo6+ (Fig. S5†).2e Moreover, the amount of Ti in TiO2/MoS2, y-TiO2/MoS2 and h-TiO2/MoS2 decreased during the etching process (Table S1†). These results indicated that the various morphologies of the TiO2/MoS2 nanocomposites were synthesized through the polymer assisted targeted-etching method. The composition of the nanocomposites includes TiO2 and MoS2 simultaneously. These heterostructures, which are composed of TiO2 and MoS2 simultaneously, may provide a large number of interfaces, which favors the separation of electrons and holes when the heterostructures are under visible light, and enhances the photocatalytic performance.
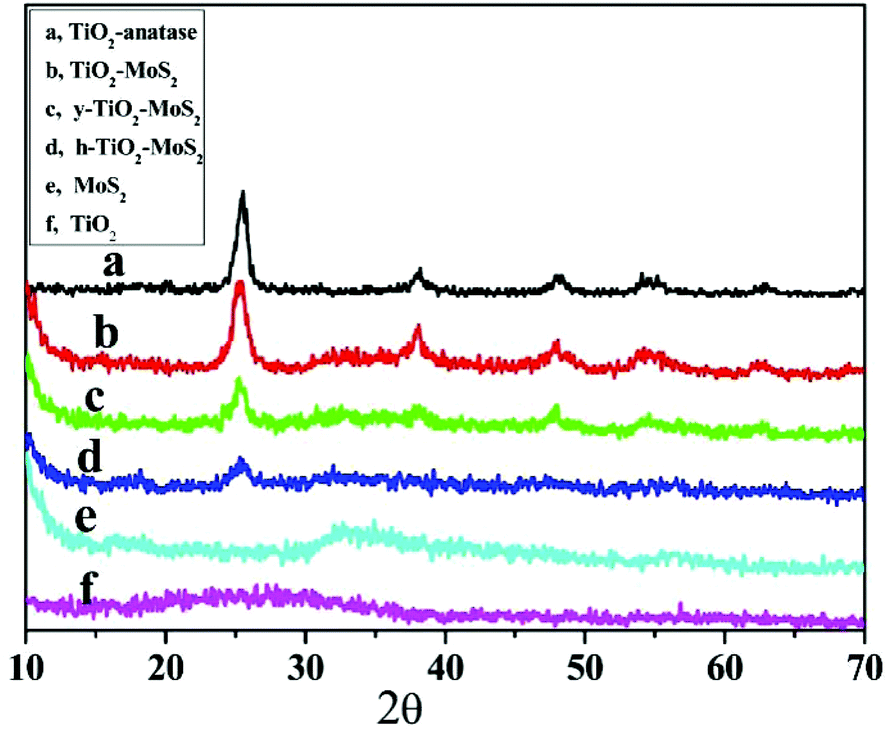 | ||
| Fig. 4 XRD patterns of (a) TiO2-anatase; (b) MoS2/TiO2 core–shell microspheres; (c) MoS2/TiO2 yolk–shell microspheres; (d) MoS2/TiO2 hollow microspheres; (e) pure MoS2 (f) and pure TiO2. | ||
Moreover, the coexistence of TiO2 and MoS2 in the resultant heterostructures enhances the absorption ability of TiO2 in the visible light region, which might increase the photocatalytic activity. As shown in Fig. S6,† the characteristic absorption band of TiO2 was in the ultraviolet range (<400 nm), while MoS2 nanosheets absorb in the range 300–800 nm. After forming heterostructures, the integral areas of TiO2/MoS2, y-TiO2/MoS2, h-TiO2/MoS2 in the ultraviolet range decreased with removal of the inner TiO2, which is extremely useful for improving the activity of the catalysts under visible light. As in previous reports,23 with the help of MoS2, the absorption band of the resultant heterostructures became broader and covered the entire visible region. Additionally, the photoluminescence (PL) spectra of TiO2, h-TiO2/MoS2 and y-TiO2/MoS2 are given in Fig. S7.† After forming heterostructures, the PL intensity decreases obviously. Generally, the PL signal is generated from the radioactive recombination of electrons and holes; thus, a low PL emission peak shows a slow recombination rate of charge carriers which means the photocatalytic activity can be improved.24
The photocatalytic activity of the as-prepared samples was evaluated via the photocatalytic degradation of MB in aqueous solution under visible-light (λ > 400 nm) irradiation as shown in Fig. 5, where C and C0 denote the remnant and initial concentrations of MB, respectively. Control experiments indicated that no degradation of MB was measured in the absence of the photocatalyst (Fig. 5a). Before the light irradiation (0 min), the suspensions were stirred (1 h) in the dark to establish the adsorption/desorption equilibrium between the dye and the catalyst. The adsorption values of pure TiO2, TiO2-anatase, pure MoS2, TiO2/MoS2, y-TiO2/MoS2 and h-TiO2/MoS2 were 189.7, 130.4, 396.6, 299.9, 498.5 and 498.5 mg g−1, respectively (Table S2†). As in previous work,2e,11b MoS2 as a two-dimensional layered nanomaterial has an excellent anchoring surface for adsorbing dye molecules, and it possessed a high adsorption value of about 396.6 mg g−1. This suggested that, compared with TiO2 or TiO2-anatase, the adsorption values of TiO2/MoS2 nanocomposites were improved, which was confirmed by the BET measurement (Table S1†). The BET area of the nanocomposites decreased rapidly. MoS2, with an anchoring surface for adsorbing dye molecules, filled the pores of TiO2,11b forming heterojunctions on the surface and near-surface of TiO2 under hydrothermal conditions. The resultant heterostructures had highly porous structures (Fig. S8†). However, it should be noted that it is impossible to form heterosjunctions in the TiO2 core, where the MoS2 precursors could not diffuse in. With etching, pure TiO2 in TiO2/MoS2 was removed and the proportion of TiO2/MoS2 at the interface was improved. Mesoporous structured TiO2 was the main contribution to the BET area of the composites. But the adsorption values of TiO2/MoS2, y-TiO2/MoS2 and h-TiO2/MoS2 increased with decreasing BET area.
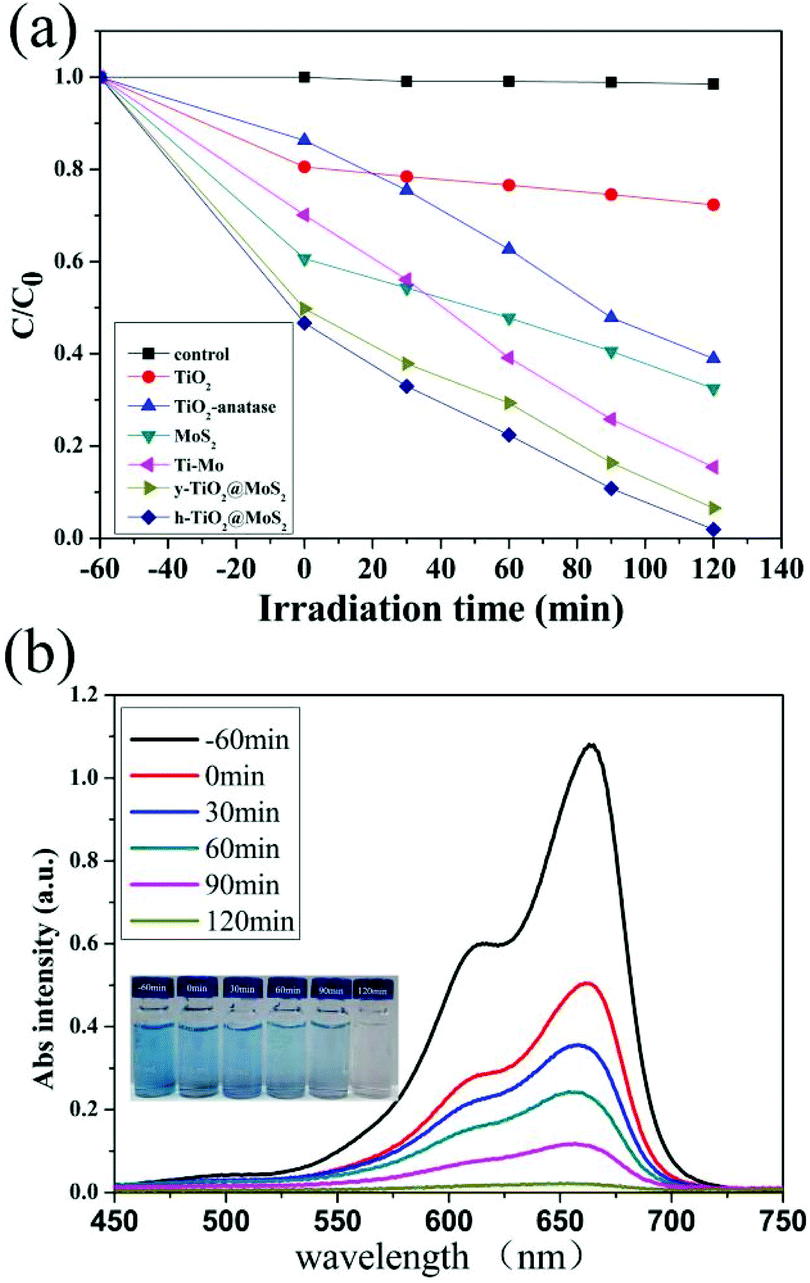 | ||
| Fig. 5 (a) The photodegradation of MB by different photocatalysts; (b) the photodegradation of MB in the presence of MoS2/TiO2 hollow microspheres; the inset is a series of photographs of the photodegradation of MB at various times using MoS2/TiO2 hollow microspheres as photocatalysts. | ||
As shown in Fig. 5a, the photocatalytic performances of all the composites was enhanced due to the formation of heterojunctions as catalytic active sites. The degradation rates of TiO2/MoS2, y-TiO2/MoS2 and h-TiO2/MoS2 were measured to be 84.56%, 93.39% and 98.28% under visible-light irradiation for 120 min. Similar to previous reports,2a,e the photocatalytic activity was controlled by the proportion of heterojunctions. h-TiO2/MoS2 showed the highest proportion of heterojunctions obviously, and as a result possessed excellent photocatalytic performance. Fig. 5b shows the corresponding UV-vis spectra of MB in the presence of TiO2/MoS2 hollow microspheres as catalysts. The continuous reduction of the absorption intensity at about 650 nm indicates the gradually decreasing concentration of MB with increasing irradiation time; it took about 120 min to completely degrade MB. The change in color of MB can be clearly observed from the corresponding photographs (inset of Fig. 5b). The corresponding UV-vis spectra of MB degraded by other catalysts are shown in Fig. S9.†
In addition, Fig. 6a reveals that the h-TiO2/MoS2 was very stable during a five-cycle experiment. In the fifth cycle, the amount of MB degraded in 2 h remains more than 88.4% (Fig. S10a†). Only a slight decrease in the photocatalytic activity occurred, which could partly be caused by a loss of the photocatalyst during each cycle of collection and rinsing. Fig. S10b† shows a TEM image of the TiO2/MoS2 hollow microspheres after photocatalytic degradation, and the hollow structure is almost undamaged which contributes to the persistently high photocatalytic activity and efficiency. The degradation of RhB with the same catalyst was also explored (100 mL, 5 mg L−1 RhB solution with 5 mg h-TiO2/MoS2 heterostructures) as shown in Fig. 6b. The surprising photocatalytic activity indicates that the as-prepared samples have good potential applications in the degradation of multiple dyes in the future. Moreover, the as-prepared sample showed a high photocatalytic hydrogen evolution activity (Fig. S11†). This result demonstrates that the resultant heterostructures are promising for applications in polluted water treatment, catalysis and energy conversion.
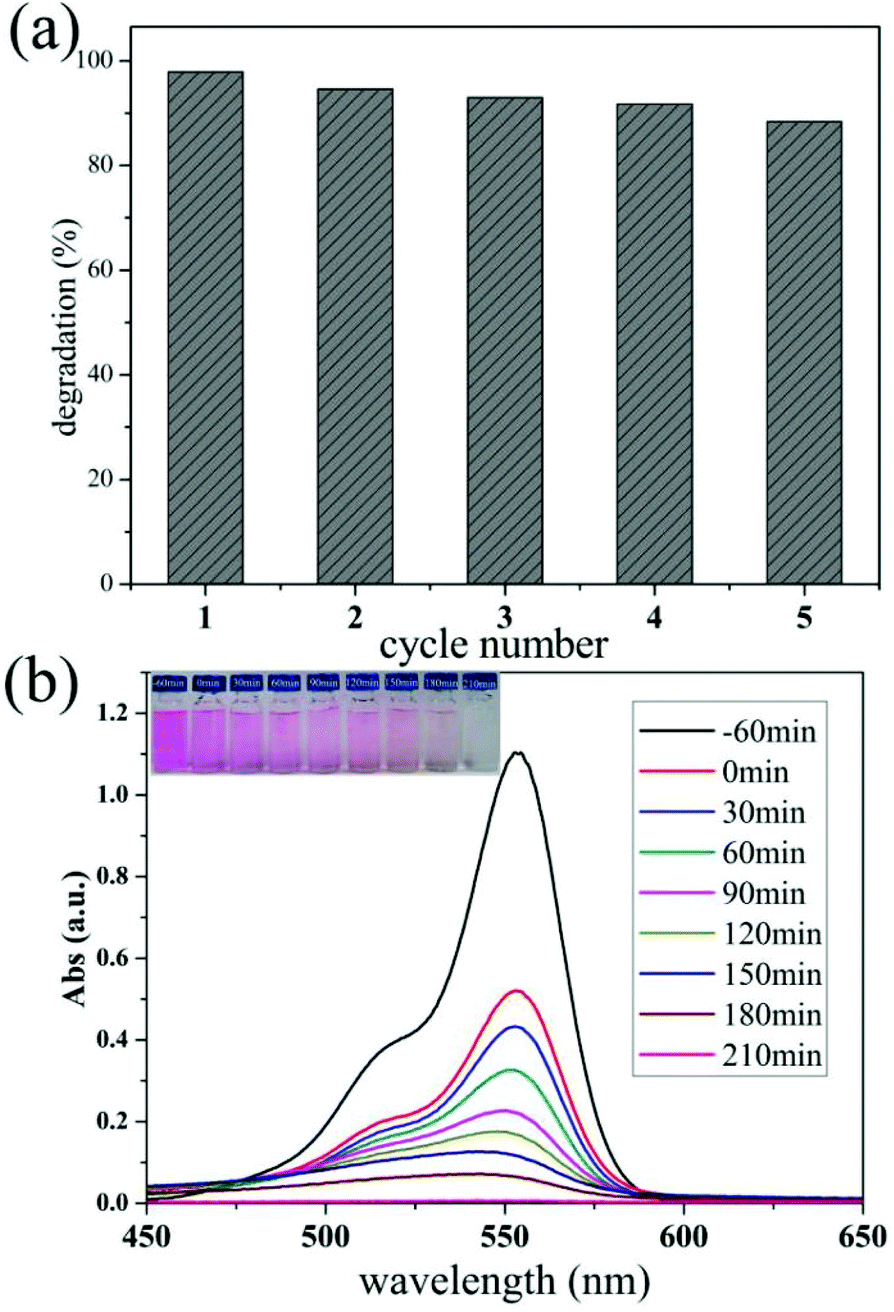 | ||
| Fig. 6 (a) The cycles of degradation of MB using MoS2/TiO2 hollow microspheres as the photocatalysts under visible light irradiation for 2 h; (b) the photodegradation of RhB in the presence of MoS2/TiO2 hollow microspheres; the inset is a series of photographs of the photodegradation of RhB after various times, using MoS2/TiO2 hollow microspheres as photocatalysts. | ||
The photocatalytic mechanism for the enhanced photocatalytic activity of the heterostructures is shown in Fig. 7. As in previous results,2a,21b the redox potentials of MoS2 are changed and its band gap is close to 1.9 eV, when the MoS2 nanosheets are too thin. Thus, both the conduction band (CB) and valence band (VB) positions of MoS2 are higher than those of TiO2 and the energy bands between MoS2 and TiO2 match (Fig. 7).25 Under light irradiation, the photoexcited electrons in the CB of the MoS2 nanosheets are transferred to the CB of TiO2, and eventually react with oxygen to produce superoxide anion radicals.11b Simultaneously, the holes left in the VB of the MoS2 nanosheets, and those transferred from the VB of TiO2, can react with H2O or OH− to form hydroxyl radicals. Hydroxyl radicals are powerful oxidants that start a series of oxidation reactions, and can ultimately totally degrade the dyes. Heterojunctions between TiO2 and MoS2 accelerated the separation of the photoexcited electrons and holes,2a and increased the physical distance. Therefore, this kind of heterostructure can degrade dyes effectively under visible light. Moreover h-TiO2/MoS2, with the highest proportion of heterojunctions, shows the best photocatalytic activity.
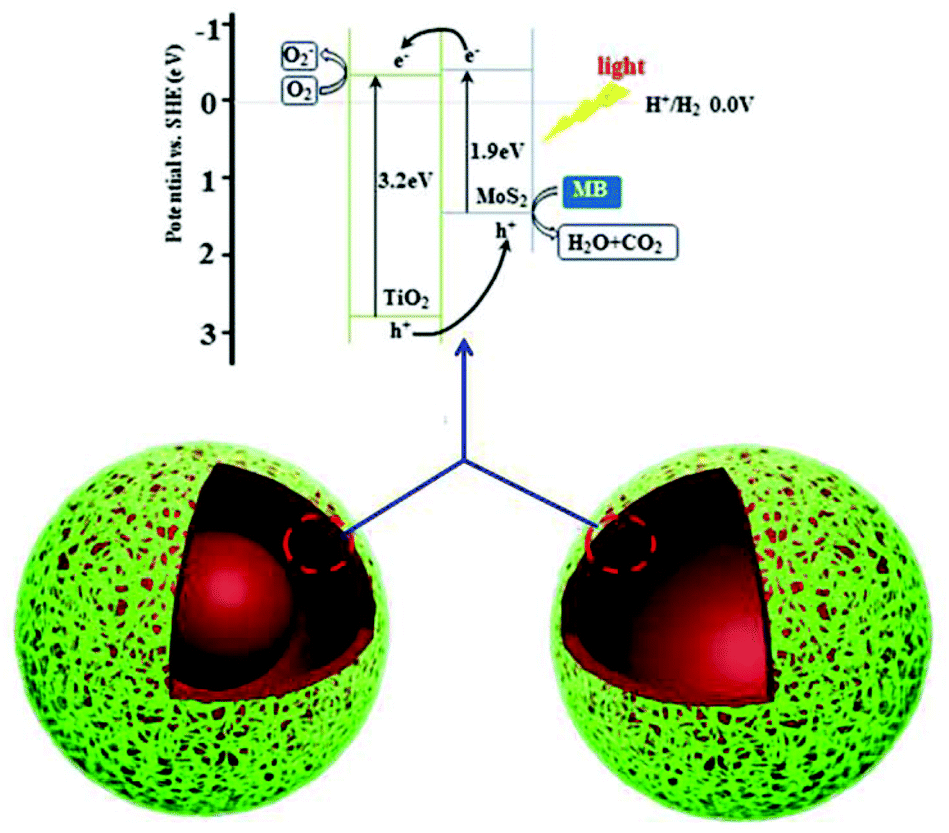 | ||
| Fig. 7 Schematic illustration of the charge-transfer in TiO2/MoS2 induced by light irradiation for photocatalytic activity. | ||
Conclusions
A targeted etching route to prepare TiO2/MoS2 nanocomposites with novel nanostructures (hollow structure and yolk–shell structure) has been developed. Through a simple solvent-thermal process, the TiO2/MoS2 core–shell nanostructure can be synthesized. Then, the hollow structure and yolk–shell structure of TiO2/MoS2 can be obtained by targeted etching using a polymer assisted method. Interestingly, the proportion of interface in TiO2/MoS2, y-TiO2/MoS2 and h-TiO2/MoS2 increases in order, and we achieved the highest photocatalytic activity with h-TiO2/MoS2. Therefore, this novel photocatalyst can be used in the treatment of polluted water, the degradation of dye pollutants, and environmental cleaning with scope for continuous exploration in the future. Moreover, it can be expected that this design of controllable morphologies will be a useful method to prepare other nanoparticles with complex structures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 51503085), open project of state key laboratory of supramolecular structure and materials (sklssm201724), Science Foundation of China University of Petroleum, Beijing (no. 2462017YJRC027).Notes and references
- (a) Q. Xiang, J. Yu and M. Jaroniec, J. Am. Chem. Soc., 2012, 134, 6575–6578 CrossRef CAS PubMed; (b) Y. Tan, K. Yu, T. Yang, Q. Zhang, W. Cong, H. Yin, Z. Zhang, Y. Chen and Z. Zhu, J. Mater. Chem. C, 2014, 2, 5422–5430 RSC; (c) Y. Chao, J. Zheng, J. Chen, Z. Wang, S. Jia, H. Zhang and Z. Zhu, Catal. Sci. Technol., 2017, 7, 2798–2804 RSC; (d) S. H. Li, S. Q. Liu, J. C. Colmenares and Y. J. Xu, Green Chem., 2016, 18, 594–607 RSC.
- (a) W. Zhou, Z. Yin, Y. Du, X. Huang, Z. Zeng, Z. Fan, H. Liu, J. Wang and H. Zhang, Small, 2013, 9, 140–147 CrossRef CAS PubMed; (b) L. Gao, W. Ren, B. Liu, Z.-S. Wu, C. Jiang and H.-M. Cheng, J. Am. Chem. Soc., 2009, 131, 13934–13936 CrossRef CAS PubMed; (c) Q. Xiang, J. Yu and M. Jaroniec, J. Phys. Chem. C, 2011, 115, 7355–7363 CrossRef CAS; (d) J. Y. Ho and M. H. Huang, J. Phys. Chem. C, 2009, 113, 14159–14164 CrossRef CAS; (e) C. X. Wang, H. Lin, Z. Xu, H. Cheng and C. Zhang, RSC Adv., 2015, 5, 15621–15626 RSC; (f) J. Matos, S. Miralles-Cuevas, A. Ruiz-Delgado, I. Oller and S. Malato, Carbon, 2017, 122, 361–373 CrossRef CAS.
- Z. Yin, Z. Wang, Y. Du, X. Qi, Y. Huang, C. Xue and H. Zhang, Adv. Mater., 2012, 24, 5374–5378 CrossRef CAS PubMed.
- X. Chen, L. Liu, Y. Y. Peter and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS PubMed.
- W. Zhou, H. Liu, J. Wang, D. Liu, G. Du and J. Cui, ACS Appl. Mater. Interfaces, 2010, 2, 2385–2392 CAS.
- (a) I. Lee, J. B. Joo, Y. Yin and F. Zaera, Angew. Chem., Int. Ed., 2011, 123, 10390–10393 CrossRef; (b) Q. Xiang, J. Yu, B. Cheng and H. Ong, Chem. – Asian J., 2010, 5, 1466–1474 CAS.
- (a) D. Sarkar, C. K. Ghosh, S. Mukherjee and K. K. Chattopadhyay, ACS Appl. Mater. Interfaces, 2012, 5, 331–337 CrossRef PubMed; (b) S. M. Oh, J. Y. Hwang, C. Yoon, J. Lu, K. Amine, I. Belharouak and Y. K. Sun, ACS Appl. Mater. Interfaces, 2014, 6, 11295–11301 CrossRef CAS PubMed; (c) Y. Shiraishi, S. Shiota, H. Hirakawa, S. Tanaka, S. Ichikawa and T. Hirai, ACS Catal., 2017, 7, 293–300 CrossRef CAS; (d) Y. Z. Gong, C. Yang, H. W. Ji, C. C. Chen, W. H. Ma and J. C. Zhao, Chem. – Asian J., 2016, 11, 3568–3574 CrossRef CAS PubMed.
- (a) D. Gopalakrishnan, D. Damien and M. M. Shaijumon, ACS Nano, 2014, 8, 5297–5303 CrossRef CAS PubMed; (b) J. Xie, H. Zhang, S. Li, R. Wang, X. Sun, M. Zhou, J. Zhou, X. W. D. Lou and Y. Xie, Adv. Mater., 2013, 25, 5807–5813 CrossRef CAS PubMed; (c) Y. Cheng, S. Lu, F. Liao, L. Liu, Y. Li and M. Shao, Adv. Funct. Mater., 2017, 27, 1700359 CrossRef; (d) S. Zhao, R. Jin, Y. Song, H. Zhang, S. D. House, J. C. Yang and R. Jin, Small, 2017, 13, 1701519 CrossRef PubMed.
- Q. Liu, X. Li, Q. He, A. Khalil, D. Liu, T. Xiang, X. Wu and L. Song, Small, 2015, 11, 5556–5564 CrossRef CAS PubMed.
- S. Kanda, T. Akita, M. Fujishima and H. Tada, J. Colloid Interface Sci., 2011, 354, 607–610 CrossRef CAS PubMed.
- (a) G. Tang, Y. Wang, W. Chen, H. Tang and C. Li, Mater. Lett., 2013, 100, 15–18 CrossRef CAS; (b) C. X. Wang, H. Lin, Z. Liu, J. Wu, Z. Xu and C. Zhang, Part. Part. Syst. Charact., 2016, 33, 221–227 CrossRef CAS.
- (a) J. Chen, X. J. Wu, L. Yin, B. Li, X. Hong, Z. Fan, B. Chen, C. Xue and H. Zhang, Angew. Chem., Int. Ed., 2015, 54, 1210–1214 CrossRef CAS PubMed; (b) Y. Min, G. He, Q. Xu and Y. Chen, J. Mater. Chem. A, 2014, 2, 2578–2584 RSC; (c) K. Chang, Z. Mei, T. Wang, Q. Kang, S. Ouyang and J. Ye, ACS Nano, 2014, 8, 7078–7087 CrossRef CAS PubMed; (d) Z. Ye, J. Yang, B. Li, L. Shi, H. Ji, L. Song and H. Xu, Small, 2017, 13, 1700111 CrossRef PubMed.
- (a) H. Xia, W. Xiong, C. K. Lim, Q. Yao, Y. Wang and J. Xie, Nano Res., 2014, 7, 1797–1808 CrossRef CAS; (b) H. Li, Y. Wang, G. Chen, Y. Sang, H. Jiang, J. He, X. Li and H. Liu, Nanoscale, 2016, 8, 6101–6109 RSC; (c) X. Liu, Z. Xing, H. Zhang, W. Wang, Y. Zhang, Z. Li, X. Wu, X. Yu and W. Zhou, ChemSusChem, 2016, 9, 1118–1124 CrossRef CAS PubMed.
- J. H. Pan, X. Z. Wang, Q. Huang, C. Shen, Z. Y. Koh, Q. Wang, A. Engel and D. W. Bahnemann, Adv. Funct. Mater., 2014, 24, 95–104 CrossRef CAS.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- (a) H. Lin, X. C. Wang, J. Wu, Z. Xu, Y. Huang and C. Zhang, New J. Chem., 2015, 39, 8492–8497 RSC; (b) Y. Li, H. Wang, L. Xie, Y. Liang, G. Hong and H. Dai, J. Am. Chem. Soc., 2011, 133, 7296–7299 CrossRef CAS PubMed.
- (a) Q. Zhang, T. Zhang, J. Ge and Y. Yin, Nano Lett., 2008, 8, 2867–2871 CrossRef CAS PubMed; (b) Q. Zhang, I. Lee, J. Ge, F. Zaera and Y. Yin, Adv. Funct. Mater., 2010, 20, 2201–2214 CrossRef CAS.
- J. Liu, S. Z. Qiao, J. S. Chen, X. W. D. Lou, X. Xing and G. Q. M. Lu, Chem. Commun., 2011, 47, 12578–12591 RSC.
- (a) A. L. Linsebigler, G. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735–758 CrossRef CAS; (b) T. L. Thompson and J. T. Yates, Chem. Rev., 2006, 106, 4428–4453 CrossRef CAS PubMed.
- (a) P. Joensen, E. Crozier, N. Alberding and R. Frindt, J. Phys. C: Solid State Phys., 1987, 20, 4043 CrossRef CAS; (b) Y. Peng, Z. Meng, C. Zhong, J. Lu, W. Yu, Y. Jia and Y. Qian, Chem. Lett., 2001, 772–773 CrossRef CAS; (c) H. Ramakrishna Matte, A. Gomathi, A. K. Manna, D. J. Late, R. Datta, S. K. Pati and C. Rao, Angew. Chem., Int. Ed., 2010, 122, 4153–4156 CrossRef.
- (a) P. Joensen, R. Frindt and S. R. Morrison, Mater. Res. Bull., 1986, 21, 457–461 CrossRef CAS; (b) G. Eda, H. Yamaguchi, D. Voiry, T. Fujita, M. Chen and M. Chhowalla, Nano Lett., 2011, 11, 5111–5116 CrossRef CAS PubMed.
- Y. Wang, J. Z. Ou, S. Balendhran, A. F. Chrimes, M. Mortazavi, D. D. Yao, M. R. Field, K. Latham, V. Bansal and J. R. Friend, ACS Nano, 2013, 7, 10083–10093 CrossRef CAS PubMed.
- (a) X. Zhang, U. Veikko, J. Mao, P. Cai and T. Peng, Chem. – Eur. J., 2012, 18, 12103–12111 CrossRef CAS PubMed; (b) J. Tao, J. Chai, L. Guan, J. Pan and S. Wang, Appl. Phys. Lett., 2015, 106, 081602 CrossRef.
- N. Q. Wu, J. Wang, D. N. Tafen, H. Wang, J. G. Zheng, J. P. Lewis, X. G. Liu, S. S. Leonard and A. Manivannan, J. Am. Chem. Soc., 2010, 132, 6679–6685 CrossRef CAS PubMed.
- W. Ho, J. C. Yu, J. Lin, J. G. Yu and P. S. Li, Langmuir, 2004, 20, 5865 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7qi00491e |
| This journal is © the Partner Organisations 2018 |