
Enhanced performance of the dye-sensitized solar cells by the introduction of graphene oxide into the TiO2 photoanode
Liguo
Wei
 abc,
Ping
Wang
c,
Yulin
Yang
*c,
Zhaoshun
Zhan
a,
Yongli
Dong
a,
Weina
Song
a and
Ruiqing
Fan
*c
abc,
Ping
Wang
c,
Yulin
Yang
*c,
Zhaoshun
Zhan
a,
Yongli
Dong
a,
Weina
Song
a and
Ruiqing
Fan
*c
aCollege of Environmental and Chemical Engineering, Heilongjiang University of Science and Technology, Harbin 150022, P.R. China
bPrecious Tyrone New Materials Co., Ltd, Qitaihe 154603, P.R. China
cMIIT Key Laboratory of Critical Materials Technology for New Energy Conversion and Storage, School of Chemistry and Chemical Engineering, Harbin Institute of Technology, Harbin 150001, P.R. China. E-mail: ylyang@hit.edu.cn; fanruiqing@hit.edu.cn; Fax: +86-451-86418270
First published on 25th October 2017
Abstract
In order to enhance the performance of the dye-sensitized solar cells (DSSCs), graphene oxide (GO) was introduced into the TiO2 photoanode to construct a GO-TiO2 composited photoanode. The photoelectric properties of the as-constructed photoanode were studied in comparison with those of the TiO2 photoanode and the effect of GO obtained by different methods on the performance of the GO-TiO2 composited photoanode was also compared. After constructed with GO, the photoanode displayed enhanced light and dye adsorption properties with a higher light harvesting efficiency, lower internal resistances, a faster electron transport, and a lower charge recombination rate, which resulted in a higher current density. At the optimal condition, the DSSCs based on the photoanode modified by GO obtained in our laboratory (GO1) exhibited a Jsc of 18.16 mA cm−2, a Voc of 0.68 V, and a FF of 0.59 with a energy conversion efficiency (η) of 7.30%, indicating an increased performance compared with that of the DSSCs based only on the pure TiO2 photoanode, which had a Jsc of 14.41 mA cm−2, a Voc of 0.66 V, and a FF of 0.60 with a η value of 5.67%. However, when GO that obtained from a production line in a factory (GO2) was used, the performance of the DSSCs was further improved, which showed a Jsc of 19.49 mA cm−2, a Voc of 0.68 V, and an FF of 0.59 with a η value of 7.83%. Different performances between GOs that obtained from the laboratory and the factory were mainly attributed to their distinct production processes, which distinguished their properties from each other. It is a potential method to enhance the performance of the DSSCs by constructing a GO-TiO2 photoanode with a suitable quality GO.
1. Introduction
In recent years, the dye-sensitized solar cells (DSSCs) have gained much attention as potential candidates to replace the traditional silicon-based solar cells due to their great advantages of lightweight, low-cost, and easy processing.1–6 A DSSCs-based device usually composes of a photoanode, a counter electrode, and an electrolyte. The photoanode is a dye-sensitized mesoporous TiO2 film coated on a fluorine-doped tin conducting oxide (FTO) glass. The mesoporous TiO2 film plays an important role in the operation of the DSSCs. It should not only adsorb sufficient dye molecules to ensure the generation of photoelectrons but also facilitate the dispersion of the electrolyte in the photoanode to ensure the regeneration of the oxidized dye by the redox couple in the electrolyte. Although the high surface area of a nanocrystalline TiO2 film meets the requirement of adsorbing a large amount of dyes in the monolayer, it has been found that the electron transport in the TiO2 nanoparticle network proceeds by a trap-limited diffusion process.7 The generated electrons encounter huge amounts of grain boundaries among nanoparticles as they travel through the nanostructured TiO2 networks inside the photoanode before reaching the collecting electrode. Such random walk mode can lead to the retardation in the electron transport, increase their probability to recombine with the oxidized dye molecules and the electron acceptor in the electrolyte, and, thus, decrease the efficiency of the DSSCs. In order to overcome this serious problem, designing a photoanode with an efficient transport pathway from the photoinjected charge carriers to the current collector is essential to enhance the device performance.8,9 Several attempts have been made to enhance the electron transport and prevent the charge recombination, including using composite metal oxides as a semiconductor with different band gaps,10 preparing the porous structure whose direction is perpendicular to the conducting substrate,11–13 and introducing charge carriers (such as carbonaceous materials) to direct the photogenerated electron.14–16Graphene, as a rising star carbonaceous material, has attracted a lot of attention because of its unique mechanical, thermal, electrical, and optical properties.17–19 As a 2D material, graphene is a zero band gap material with a single molecular layered structure.19 In graphene, each carbon atom uses 3 of its 4 outer orbital electrons to form 3 sigma bonds 120° apart with 3 adjacent carbon atoms in the same plane, leaving the 4th electron free to move. Therefore, the electrons in graphene behave just like massless relativistic particles without crystal lattice restrictions.20,21 As such, graphene possesses excellent electrical conductivities in two dimensions at room temperature (the charge carrier mobility is more than 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm2 V−1 s−1).22 Because of the intermolecular forces such as physisorption, electrostatic binding, or charge transfer interactions between graphene and TiO2 nanoparticles,23 TiO2 nanoparticles can anchor on graphene, thus, graphene could provide a fast channel for the electron transportation,24–28 which has accelerated the electron transport and lowered the recombination loss. Compared with graphene, graphene oxide (GO) with the presence of a lot of oxygen-containing groups can also easily combine with TiO2 to form a strong-coupling GO-TiO2 composite. Moreover, a porous structure may be formed by the escape of the GO nanosheet from the surface of the GO-TiO2 composited film when it is sintered, which will act as a scattering layer to further improve the light harvesting efficiency of the photoanode. Besides, the dye molecules could easily diffuse and transfer into the inside of the TiO2 photoanode via the porous structures, which will increase the dye loading mount of the photoanode. The adsorption of more dye molecules will be very helpful to increase the photoelectric conversion efficiency of the photoanodes. Therefore, on the basis of the above unique properties of GO, the GO-TiO2 composite could be applied as the photoanode material in the DSSCs and is an effective way to accelerate the electron transfer, retard the charge recombination, and improve the light harvesting efficiency of the cell. Although the application of GO in the photoanode was reported previously,29–33 most of them need to be reduced to reduced graphene oxide or graphene and they were only used in a layer to block the back-transport reactions or together with other components through a complex fabrication process.
000 cm2 V−1 s−1).22 Because of the intermolecular forces such as physisorption, electrostatic binding, or charge transfer interactions between graphene and TiO2 nanoparticles,23 TiO2 nanoparticles can anchor on graphene, thus, graphene could provide a fast channel for the electron transportation,24–28 which has accelerated the electron transport and lowered the recombination loss. Compared with graphene, graphene oxide (GO) with the presence of a lot of oxygen-containing groups can also easily combine with TiO2 to form a strong-coupling GO-TiO2 composite. Moreover, a porous structure may be formed by the escape of the GO nanosheet from the surface of the GO-TiO2 composited film when it is sintered, which will act as a scattering layer to further improve the light harvesting efficiency of the photoanode. Besides, the dye molecules could easily diffuse and transfer into the inside of the TiO2 photoanode via the porous structures, which will increase the dye loading mount of the photoanode. The adsorption of more dye molecules will be very helpful to increase the photoelectric conversion efficiency of the photoanodes. Therefore, on the basis of the above unique properties of GO, the GO-TiO2 composite could be applied as the photoanode material in the DSSCs and is an effective way to accelerate the electron transfer, retard the charge recombination, and improve the light harvesting efficiency of the cell. Although the application of GO in the photoanode was reported previously,29–33 most of them need to be reduced to reduced graphene oxide or graphene and they were only used in a layer to block the back-transport reactions or together with other components through a complex fabrication process.
Therefore, in this study, the GO-TiO2 composited photoanode was constructed through a simple way using GO-TiO2 as the whole photoanode film material and the photoelectric properties of the as-constructed photoanode were studied in comparison with those of the TiO2 photoanode. The effect of GO obtained in the laboratory and the production line of the factory was also compared. After the photoanode was constructed with GO, the performance of the DSSCs was improved and characterized by a dark current measurement, electrochemical impedance spectroscopy (EIS), and open-circuit voltage decay (OCVD). Different performances between GO1-TiO2 and GO2-TiO2 were mainly attributed to the distinct production processes of GOs, which distinguishes their properties from each other. It is a potential way to enhance the performance of the DSSCs by constructing a GO-TiO2 photoanode with a suitable quality graphene oxide.
2. Experimental
2.1 Materials and methods
Commercial TiO2 (P25) nanoparticles were purchased from Degussa Co., Ltd (Germany). Acetonitrile, I2, LiI, tert-butylpyridine, and lithium perchlorate were purchased from Tianli Chemical Co., Tianjin, China. F-doped SnO2-coated glass plate (FTO, 90% transmittance in the visible, and 15 Ω cm−2) was purchased from Acros Organics (Belgium) and cleaned by a standard procedure. cis–bis-(Isothiocyanato)bis(2,2-bipyridyl-4,4-dicarboxylato)ruthenium(II)bis-tetrabutylammonium (N719) was purchased from Solaronix Co., Switzerland. All other chemical reagents used in the experiments were analytical reagents and purchased from Sinopharm Chemical Reagent Beijing Co., Ltd without additional purification before use.2.2 Synthesis of GO
GO was prepared by the oxidation of graphite according to the Hummers method in the laboratory34 and denoted as GO1. Graphite powder (2 g) and sodium nitrate (1 g) were first mixed and stirred in concentrated sulfuric acid (50 mL) at 0 °C. Potassium permanganate (8 g) was added gradually to the above solution with vigorous mechanical stirring. The stirring was continued for 1.5 h at 0 °C and then 2 h at 35 °C. Followed by the addition of de-ionized water (100 mL), the temperature was raised to 98 °C and maintained for 15 min. Subsequently, de-ionized water (1 L) and hydrogen peroxide (30%, 10 mL) were added to terminate the reaction. Finally, the resulting suspension was filtered and washed with 10% HCl (500 mL) and de-ionized water. The obtained solid product was dried under vacuum at 40 °C for 24 h. For comparison, GO was also obtained from the production line in the Precious Tyrone New Materials Company of China and denoted as GO2.2.3 Preparation of the GO-TiO2 photoanode films and solar cell fabrication
Before the photoanode films were prepared, 100 mg solid state GO was dissolved in 100 mL ethanol and sonicated for 2 h until a suspended solution of 1 mg mL−1 was formed. The GO-TiO2 pastes were synthesized following the Grätzel method for preparing the TiO2 paste,35 except that 1.0 mL GO1 or GO2 solution was added. Each paste was printed onto the FTO conductive glass and then dried at 100 °C for 5 min. This printed process was repeated for seven times. The obtained photoanode film was then sintered at 500 °C for 15 min. It was dyed after immersed in a solution of 0.3 mM N719 in absolute ethanol for 24 h and then washed with ethanol. The composited photoanode printed with the paste containing GO1 was denoted as GO1-TiO2 and the composited photoanode printed with the paste containing GO2 was denoted as GO2-TiO2. For comparison, the photoanode printed with the pure TiO2 paste was also prepared and denoted as TiO2.A sandwich-type solar cell device was assembled by placing a platinum-coated conductive glass as a counter electrode on the dyed photoanode and then a drop of a liquid electrolyte containing 0.5 M LiI, 0.05 M I2, and 0.1 M 4-tert-butylpyridine was added to fill the void between two electrodes which were clipped together as the open cells for the measurement.
2.4 Characterization
The X-ray powder diffraction (XRD) measurement was carried out on a Bruker D8 Advance X-ray diffraction instrument with a Cu-Kα radiation. UV-Vis spectrum was recorded with an UV-2250 spectrophotometer (Shimadzu, Japan). Scanning electron microscopy (SEM) images were taken using a Phenom ProX SEM. The dye loading amount was measured by desorbing the dye from the dye-sensitized films in a 0.1 M NaOH solution in water and ethanol (1:1, v/v) and then the concentration of the desorbed dye in the NaOH solution was measured using an UV-visible spectrophotometer. N2 adsorption–desorption of the samples was conducted on a Micromeritics ASAP2020 surface area and pore size analyzer at 196 °C (liquid nitrogen temperature) using an accompanying software from Micromertitics. The surface areas were calculated by using the conventional BET method and the pore size diameters were calculated by using BJH method. The Raman spectra were determined on a Renishaw inVia micro Raman spectroscopy system, using the TE air-cooled 576 × 400 CCD array in a confocal Raman system (wavelength: 532 nm). The incident laser power was kept at 0.1 mW and a total accumulation time of 5 s was employed. Dark current, EIS, and OCVD were recorded on a CHI660D Electrochemical Analyzer. The current density–voltage (J–V) curves of the DSSCs were recorded on a Keithley model 2400 digital source meter under an AM 1.5G irradiation. The incident light intensity was 100 mW cm−2 calibrated by a standard silicon solar cell. The working areas of the cells were masked to 0.16 cm2. Based on the J–V curve, (1) the fill factor (FF) is defined as: FF = (Jmax× Vmax)/(Jsc× Voc), where Jmax and Vmax are the photocurrent density and the photovoltage at the maximum power output, Jsc and Voc are the short-circuit photocurrent density and the open-circuit photovoltage, respectively, and (2) the overall energy conversion efficiency η is defined as: η = (FF × Jsc× Voc)/Pin, where Pin is the power of the incident light. The measurement of the incident photon-to-current conversion efficiency (IPCE) was performed on an EQE/IPCE spectral response system (Newport).
3. Results and discussion
The XRD patterns of GO obtained in the laboratory (GO1) and the factory (GO2) are shown in Fig. 1a. It was found that GO1 showed a very strong peak at 2θ = 10.6° and a small and broad peak at 2θ = 21.6°, which should be stronger and sharper in raw graphite. The change intensity of the peak at 2θ = 21.6° could be attributed to the fact that the GO1 sheets were thicker than raw graphite due to the oxygen-containing functional groups attached on both sides of GO1.36 As for GO2, the diffraction peak at 2θ = 10.6° was weak and the peak at 2θ = 21.6° was not obvious. However, the existence of the peak at 2θ = 21.6° of GO2 was confirmed as seen in the inset of Fig. 1a, indicating the similar crystal structure but with a lower crystalline degree of GO2 to GO1.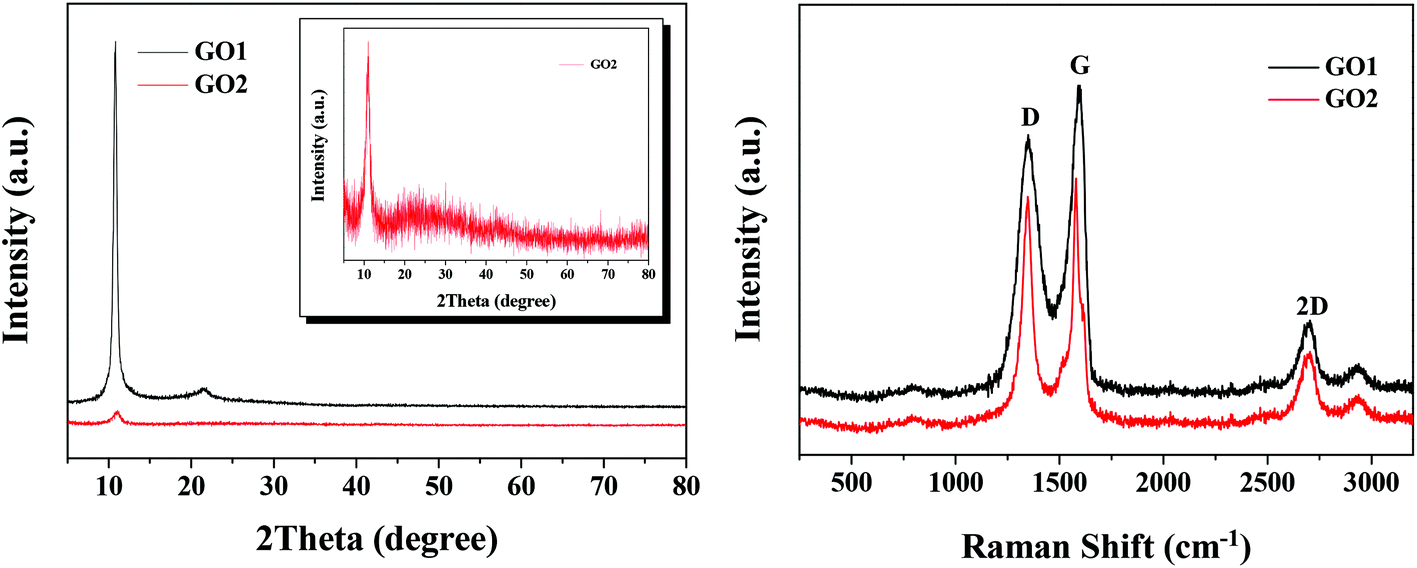 | ||
| Fig. 1 XRD patterns (a) and Raman spectra (b) of GO1 and GO2. | ||
The Raman spectra of GO1 and GO2 in Fig. 1b indicate that they possessed the same carbon framework. There were two characteristic peaks in the Raman spectra of GO1 and GO2, which were the D band at 1348 cm−1 and the G band at 1598 cm−1. The D band was ascribed to the defects and disorder in the hexagonal lattice, whereas the G band resulted from the vibration of the sp2-bonded carbon atoms in the two-dimensional hexagonal lattice.37 Moreover, a 2D peak was observed in the Raman spectra of GO1 and GO2, which indicates that GOs with a decreasing number of layers were synthesized.38
The morphology of GO1 and GO2 were also observed by SEM with different magnifications. As shown in Fig. 2, GO1 showed a silk-like morphology with some wrinkles on the surface. However, the nanosheets of GO1 were continuous without obvious fringe, while the nanosheets of GO2 were not continuous and many fringes existed on its top view. In addition, the GO2 nanosheets were much thinner and curl. All of these results indicate that GOs obtained by different synthesis processes were much different from each other, which may cause different properties when they were applied in a certain area.
 | ||
| Fig. 2 SEM imagines of GO1 and GO2 with different magnifications. | ||
After the photoanodes were constructed with GO1 and GO2, they showed many different effects on the properties compared to those of the TiO2 photoanode. The typical SEM images of different photoanodes are shown in Fig. 3. From the top-view images of the TiO2 film in Fig. 3a, it was seen that many small TiO2 nanoparticles were dispersed randomly on the surface of the FTO glass. Comparatively, the top-view images of GO1-TiO2 and GO2-TiO2 (Fig. 3b and c) showed the porous structure which caused by decomposing of GO from the surface of the films into the gaseous CO2 after high-temperature calcination in the air. It was also found that the GO2-TiO2 film composed of more mesopores than those of the GO1-TiO2 film. This may be attributed to the fact that GO2 possessed a thinner and smaller nanosheet than that of GO1, which makes it easier to decompose. Based on the cross-sectional images of GO1-TiO2 and GO2-TiO2 (Fig. 3e and f), the thickness of the GO1-TiO2 and GO2-TiO2 films was about 14.2 μm, which is a comparable value to that of the pure TiO2 film (Fig. 3d).
 | ||
| Fig. 3 SEM images of the surface: (a) TiO2, (b) GO1-TiO2, and (c) GO2-TiO2 and SEM images of the cross-section: (d) TiO2, (e) GO1-TiO2, and (f) GO1-TiO2 photoanodes. | ||
Furthermore, the pore structures of the GO1-TiO2 and GO2-TiO2 composited photoanodes were also characterized by nitrogen adsorption–desorption isotherms, and the results are shown in Fig. 4. According to Brunauer–Deming–Deming–Teller (BDDT) classification,39 the N2 adsorption–desorption isotherms of TiO2, GO1-TiO2, and GO2-TiO2 shown in Fig. 4 were similar to type I in the low-pressure range (p/p0 < 0.4), where the isotherm exhibited a high adsorption, indicating the presence of micropores (type I). However, the isotherms of TiO2, GO1-TiO2, and GO2-TiO2 were similar to those of type III in the higher pressure range (p/p0 > 0.4) and the curve exhibited a hysteresis loop in the high relative pressure range of 0.7–1, indicating the presence of mesopores. It can be found from the inset parameters in Fig. 4 that (1) the BET surface area of GO1-TiO2 (44.92 m2 g−1) and GO2-TiO2 (46.01 m2 g−1) was higher than that of TiO2 (43.75 m2 g−1), (2) the pore volume of GO1-TiO2 and GO2-TiO2 was about 0.35 cm3 g−1 and 0.37 cm3 g−1, respectively, which was larger than that of TiO2 (0.32 cm3 g−1); and (3) the pore size of GO1-TiO2 (31.15 nm) and GO2-TiO2 (32.22 nm) was also higher than that of TiO2 (27.59 nm). These results further confirm that the porous structure was formed by the escape of GO from the surface of the films and it was more obvious in the case of the GO2-TiO2 photoanode than that in the case of the GO1-TiO2 photoanode.
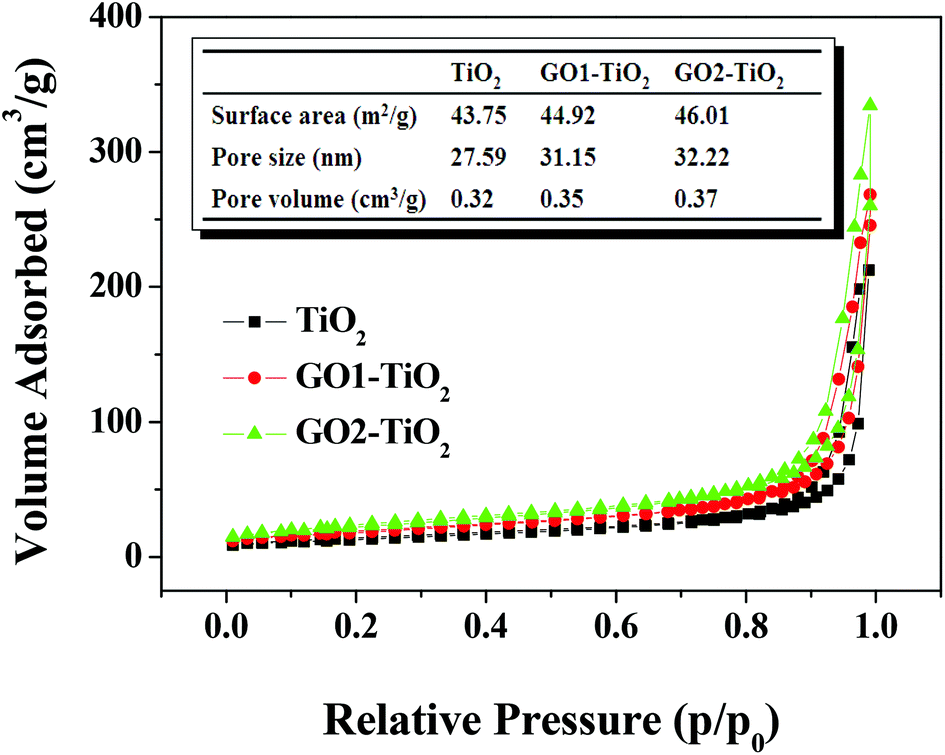 | ||
| Fig. 4 Nitrogen adsorption–desorption isotherm and the corresponding parameters of the TiO2, GO1-TiO2, and GO2-TiO2 photoanodes. | ||
The formation of the porous structure on the surface of the GO-TiO2 photoanodes can make it easy for the dye molecules to diffuse and transfer into the inside of the photoanodes, which will increase its dye loading amount. To confirm this hypothesis, the loading amounts of the dye on the GO-TiO2 photoanodes were measured by desorbing the dye from the dye-sensitized films in the 0.1 M NaOH solution in water and ethanol (1:1, v/v) and the results are listed in Table 1. As shown in Table 1, the dye loading amount for the TiO2, GO1-TiO2 and GO2-TiO2 photoanodes was 1.22 × 10−7, 1.92 × 10−7, and 1.98 × 10−7 mol cm−2, respectively. The adsorption ability of the TiO2 photoanode increased with the introduction of GO and this effect was more obvious for the GO2-TiO2 photoanode.
| Photoanode | Dye loading amount (10−7 mol cm−2) |
|---|---|
| TiO2 | 1.22 |
| GO1-TiO2 | 1.92 |
| GO2-TiO2 | 1.98 |
More dye loading amount is a benefit for improving the sunlight absorbance of the photoanode. Fig. 5 shows the absorption ability of different photoanodes after dye sensitization. It was found that the GO-TiO2 photoanodes showed an obvious enhancement for the visible-light absorption compared with that of the TiO2 photoanode. This confirmed that the constructed GO-TiO2 photoanodes had a higher ability for the effective adsorption of the dye and the absorption of light, which was very helpful for enhancing the energy conversion efficiency of the DSSCs system.
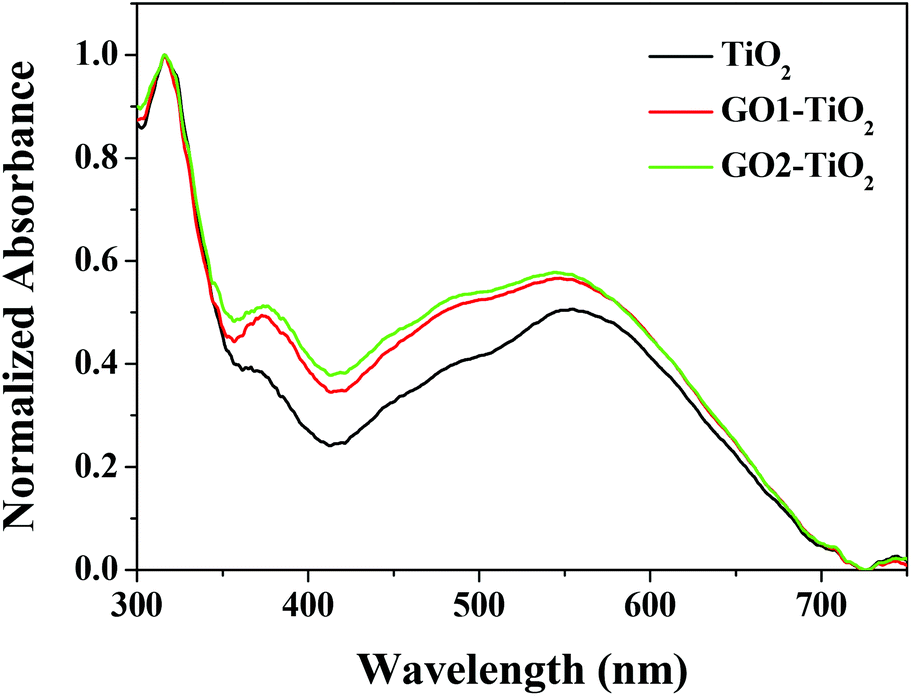 | ||
| Fig. 5 UV-visible absorption spectra of different photoanodes. | ||
The photovoltaic performance of the DSSCs based on different photoanodes were investigated under a simulated solar irradiation of 100 mW cm−2 (AM 1.5G). The J–V curves of the DSSCs are shown in Fig. 6a (under irradiation) and the corresponding parameters are summarized in Table 2. The device based on the TiO2 photoanode was found to exhibit a η value of 5.67% (with Jsc = 14.41 mA cm−2, Voc = 0.66 V, and FF = 0.60), while the GO1-TiO2 and GO2-TiO2 devices exhibited a η value of 7.30% (with Jsc = 18.16 mA cm−2, Voc = 0.68 V, and FF = 0.59) and 7.83% (with Jsc = 19.49 mA cm−2, Voc = 0.68 V, and FF = 0.59), respectively. It was found that the photovoltaic performance of the DSSCs was promoted significantly with the constructed GO-TiO2 photoanodes. Furthermore, the DSSCs based on the GO2-TiO2 photoanode obtained a much higher Jsc and η than those of the DSSCs based on the GO1-TiO2 photoanode, which was mainly due to the different properties of GOs causing by different preparation processes.
 | ||
| Fig. 6 (a) J–V curves and (b) IPCE spectra of the DSSCs based on different photoanodes. | ||
| Photoanode | J sc (mA cm−2) | V oc (V) | FF | η (%) | J sc (Int.) (mA cm−2) |
|---|---|---|---|---|---|
| TiO2 | 14.41 | 0.66 | 0.60 | 5.67 | 13.18 |
| GO1-TiO2 | 18.16 | 0.68 | 0.59 | 7.30 | 16.85 |
| GO2-TiO2 | 19.49 | 0.68 | 0.59 | 7.83 | 17.76 |
The higher Jsc of the DSSCs based on the GO-TiO2 photoanodes is mainly ascribed to a better light harvesting due to an increased dye loading amount. It can be illustrated clearly by the IPCE measurement since the precise Jsc value can be described by integrating the IPCE spectra over the spectral distribution of the standard AM 1.5G solar photon flux ΦAM 1.5G(λ):
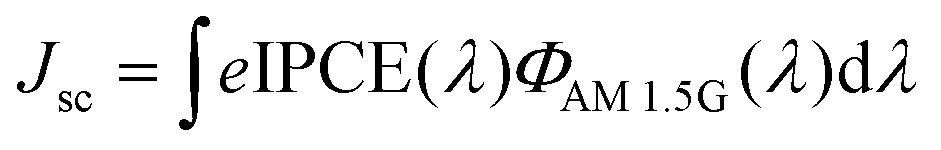
Besides enhanced light harvesting, the improved electron transport through a fast and effective electron transport pathway provided by GO was also contributed to the enhancement of the Jsc value. In an effort to get information about the electron transport in the devices based on different photoanodes, electrochemical impedance spectroscopy (EIS) of different devices were measured under the standard AM 1.5G solar irradiation by applying a forward bias of −0.75 V. As shown in Fig. 7a, three semicircles located in high-, middle- and low-frequency regions (from left to right) were attributed to the electrochemical reaction at the Pt/electrolyte interface, the electron transfer at the TiO2/dye/electrolyte interface and the charge transfer in the electrolyte (Nernst diffusion).41 The radius of the large semicircle located in the middle-frequency region in the Nyquist plots decreased after the GO-TiO2 photoanodes were used and the values were in the order of GO2-TiO2 < GO1-TiO2 < TiO2, which indicates a decrease of the electron transfer impedance (Rct) and an increase of the electron transfer rate at this interface after the GO-TiO2 photoanodes were used, resulting in the increase of the Jsc value. The smaller Rct of GO2-TiO2 than that of GO1-TiO2 was attributed to the smaller and thinner nanosheet of GO2, which could composite better with TiO2 and provide more electron transport pathway. Based on the Bode plot (Fig. 7b), the frequency was shifted to the higher-frequency region with the application of the GO-TiO2 photoanodes. Since fmax is inversely associated with the electron transport time in illumination condition τs = 1/(2πfmax),42 the increase in fmax indicates an increased rate of the charge-transport process in the DSSCs. Hence, the electron with a shorter τs value indicates that the electrons are collected at the FTO electrode at a faster rate.
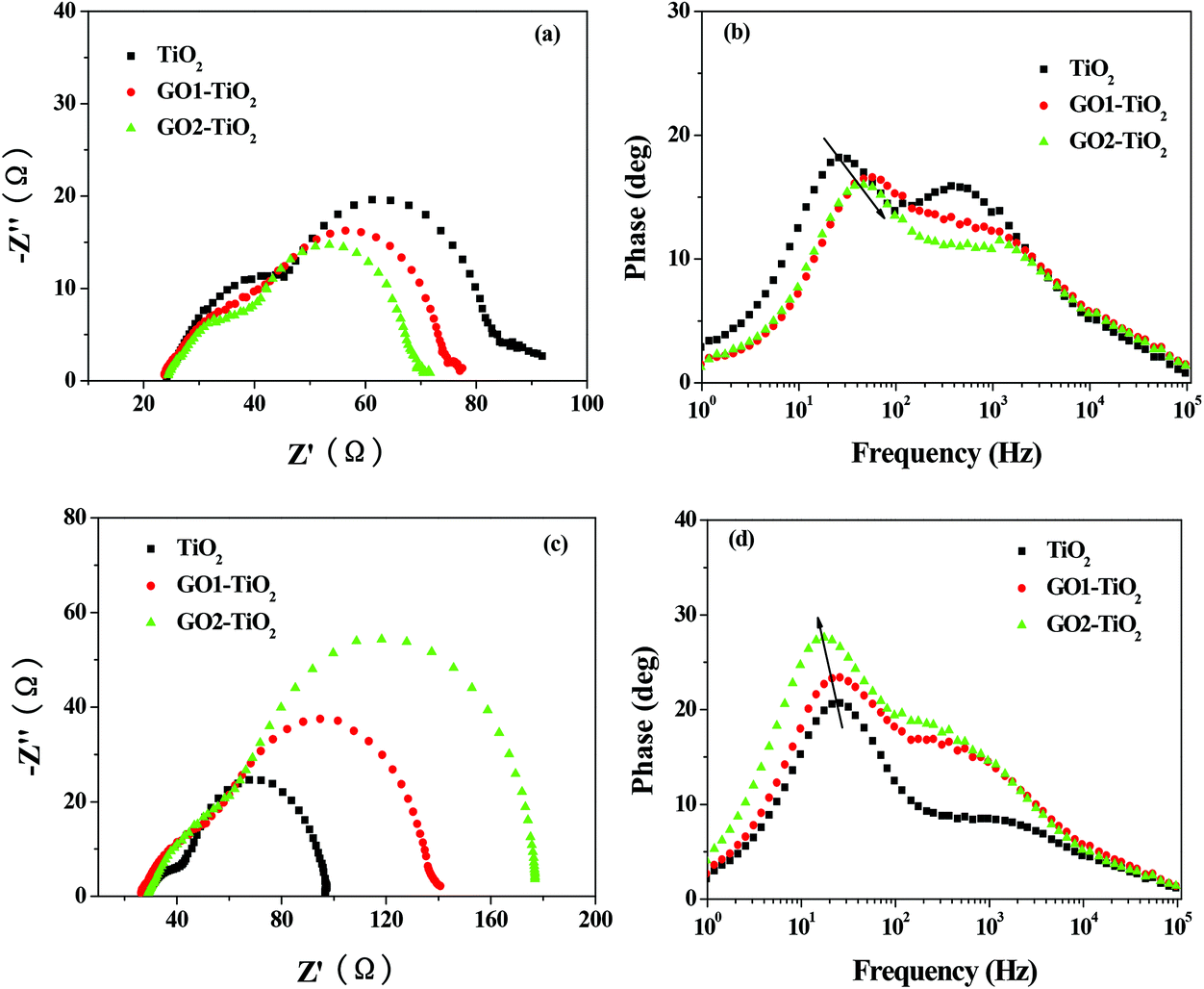 | ||
| Fig. 7 (a) Nyquist plots and (b) Bode plots of the DSSCs based on different photoanodes measured under the standard AM 1.5G solar irradiation and (c) Nyquist plots and (d) Bode plots of the DSSCs based on different photoanodes measured in dark. | ||
Meanwhile, in an effort to investigate the kinetics of the recombination processes, EIS of the devices with different photoanodes were also measured in the dark conditions by applying a forward bias of −0.75 V. In the dark conditions, as shown in Fig. 7c, similar to under the illumination, three semicircles located in high-, middle- and low-frequency regions (from left to right) of the Nyquist plots were attributed to the redox reaction at the Pt counter electrode, the electron transfer at the TiO2/dye/electrolyte interface, and the charge transfer in the electrolyte (Nernst diffusion). Therefore, the larger semicircle observed in the middle-frequency region represented the resistances of the charge transfer from TiO2 to the electrolyte (back recombination resistance Rrec). The radius of this semicircle increased after the GO-TiO2 photoanodes were used, indicating an increase of Rrec. A large Rrec means the small charge recombination rate and vice versa. The radius of the semicircle observed in the middle-frequency range lied in the order of GO2-TiO2 > GO1-TiO2 > TiO2, indicating also the sequence of Rrec at the TiO2/dye/electrolyte interface. The increased value of Rrec for the DSSCs implies the retardation of the charge recombination between the injected electron and I3− ions in the electrolyte, resulting in a smaller dark current. Based on the Bode plot (Fig. 7d), the frequency was shifted to the lower-frequency region with the application of the GO-TiO2 photoanodes. Since fmax was inversely associated with the electron lifetime in the dark condition τe = 1/(2πfmax),42,43 the decrease in fmax indicated a reduced rate of the charge-recombination process in the DSSCs. Hence, the electrons with longer τe values were prevented from recombining, characterized by a larger Rrec and a reduced dark current. The reduced dark current was confirmed by dark current–voltage (J–V) measurements of different devices. The dark current–voltage characteristics of the DSSCs based on different photoanodes with the applied bias from 0 to +0.80 V was also shown in Fig. 6a (in dark). The onset of the dark current for the device based on the TiO2 photoanode occurred at a bias about +0.25 V with a subsequent dramatic increase of the dark current with the increase of the potential. In contrast, for the devices with the GO-TiO2 photoanodes, the onset potential shifted gradually to about +0.30 V. Furthermore, the dark current of these DSSCs with the GO-TiO2 photoanodes increased much slower than that of the TiO2 device when the potential was greater than +0.25 V. In other words, under the same potential bias, when the potential was ≥0.25 V, the dark current for the cells with the GO-TiO2 photoanodes was noticeably smaller than that for the device based on the TiO2 photoanode. The increase of the onset potential and the reduction of the dark current further demonstrated that the electron back reaction with I3− in the electrolyte was successfully suppressed by the constructed GO-TiO2 photoanodes.
The open-circuit voltage decay (OCVD) curves were also recorded to further probe the recombination kinetics of the devices. The decay of the photovoltage reflects the decrease of the electron concentration at the FTO surface, which is mainly caused by the charge recombination. In other words, the recombination velocity of the photoelectron is proportional to the response of the OCVD.44Fig. 8a shows the OCVD decay curves of different devices. It was observed that the OCVD responses of the devices with the GO-TiO2 photoanodes were much slower than that of the device with the TiO2 photoanode, especially in the shorter time domain (within 20 s). The correlation between Voc decay and electron lifetime (τe) can be expressed by the equation:
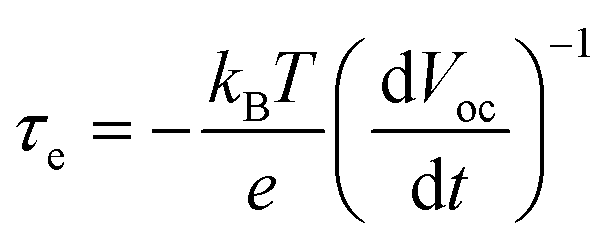
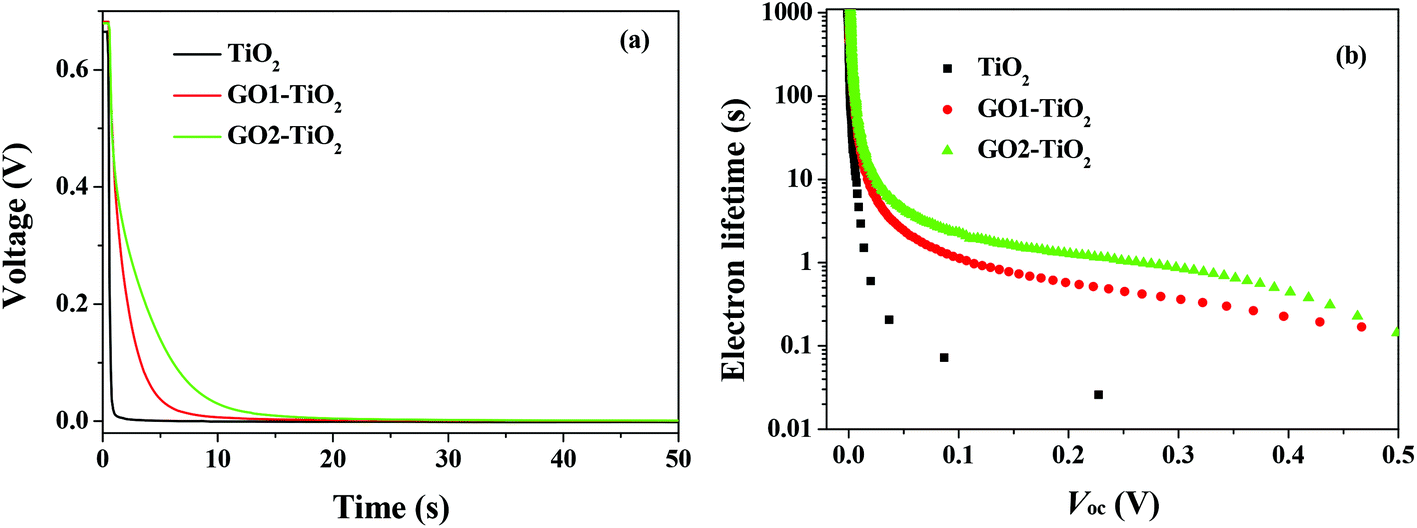 | ||
| Fig. 8 (a) The OCVD curves of the DSSCs with different photoanodes and (b) the electron lifetime as a function of Voc of the DSSCs with different photoelectrodes. | ||
4. Conclusions
In conclusion, the effect of graphene oxide on the performance of the dye-sensitized solar cells was investigated by constructing the GO-TiO2 photoanodes. After the photoanode was constructed with GO, it displayed the enhanced light and dye adsorption properties, lower internal resistances, a faster electron transport and a lower charge recombination rate, which resulted in a high current density. At the optimal condition, the DSSCs based on the GO-TiO2 photoanodes exhibited a Jsc of 19.49 mA cm−2, a Voc of 0.68 V, and a FF of 0.59 with a energy conversion efficiency (η) of 7.83%, indicating an increase in Jsc and η compared to those of the DSSCs based only on the pure TiO2 photoanode, which had a Jsc of 14.41 mA cm−2, a Voc of 0.66 V, and a FF of 0.60 with a η of 5.67%. Different performances between GOs obtained from the laboratory and the factory were mainly attributed to their distinct production processes, which distinguishes their properties from each other. It is a potential way to enhance the performance of the DSSCs by constructing the GO-TiO2 photoanode with a suitable quality graphene oxide.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Natural Science Foundation of China (Grant 21571042, 21371040, 61705063, and 21203058), the Science Foundation for Youths of Heilongjiang Province of China (Grant No. QC2016013), the Foundation of Educational Commission of Heilongjiang Province of China (No. 12531579), the Innovative Talents Program of Heilongjiang University of Science and Technology (Q20130202), and the Postdoctoral Foundation of Heilongjiang Province of China (LBH-Z16203).Notes and references
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
- M. Grätzel, Acc. Chem. Res., 2009, 42, 1788–1798 CrossRef PubMed.
- Y. J. Wang, W. L. Chen, L. Chen, X. T. Zheng, S. S. Xu and E. B. Wang, Inorg. Chem. Front., 2017, 4, 559–565 RSC.
- X. W. Guo, X. H. Li, Z. J. Liu, W. L. Chen, X. T. Zheng, E. B. Wang and Z. M. Su, Inorg. Chem. Front., 2017, 4, 1187–1191 RSC.
- P. Y. Su, J. M. Liu, X. L. Lin, Y. F. Chen, Y. Shen, L. M. Xiao, D. B. Kuang and C. Y. Su, Inorg. Chem. Front., 2015, 2, 1040–1044 RSC.
- G. E. Zervaki, P. A. Angaridis, E. N. Koukaras, G. D. Sharma and A. G. Coutsolelos, Inorg. Chem. Front., 2014, 1, 256–270 RSC.
- X. Fang, M. Li, K. Guo, X. Liu, Y. Zhu, B. Sebo and X. Zhao, Sol. Energy, 2014, 101, 176–181 CrossRef CAS.
- M. S. Ahmad, A. K. Pandey and N. A. Rahim, Renewable Sustainable Energy Rev., 2017, 77, 89–108 CrossRef.
- U. Mehmood, I. A. Hussein, K. Harrabi, M. B. Mekki, S. Ahmed and N. Tabet, Sol. Energy Mater. Sol. Cells, 2015, 140, 174–179 CrossRef CAS.
- H. M. A. Javed, W. Que, X. Yin, Y. Xing, J. Shao and L. B. Kong, J. Alloys Compd., 2016, 685, 610–618 CrossRef CAS.
- P. Wang, F. He, J. Wang, H. Yu and L. Zhao, Appl. Surf. Sci., 2015, 358, 175–180 CrossRef CAS.
- J. Y. Bae, J. Nanosci. Nanotechnol., 2016, 16, 10495–10498 CrossRef CAS.
- J. R. Jennings, A. Ghicov, L. M. Peter, P. Schmuki and A. B. Walker, J. Am. Chem. Soc., 2008, 130, 13364–13372 CrossRef CAS PubMed.
- J. Huang, K. Fu, N. Yao, X. Deng, M. Ding, M. Shao and X. Zhao, J. Nanosci. Nanotechnol., 2016, 16, 1477–1482 CrossRef CAS PubMed.
- S. Mallakpour and E. Khadem, Chem. Eng. J., 2016, 302, 344–367 CrossRef CAS.
- N. Yang, J. Zhai, D. Wang, Y. Chen and L. Jiang, ACS Nano, 2010, 4, 887–894 CrossRef CAS PubMed.
- C. H. Ng, H. N. Lim, S. Hayase, I. Harrison, A. Pandikumar and N. M. Huang, J. Power Sources, 2015, 296, 169–185 CrossRef CAS.
- Y. H. Ng, I. V. Lightcap, K. Goodwin, M. Matsumura and P. V. Kamat, J. Phys. Chem. Lett., 2010, 1, 2222–2227 CrossRef CAS.
- R. Nair, P. Blake, A. Grigorenko, K. Novoselov, T. Booth, T. Stauber, N. Peres and A. Gein, Science, 2008, 320, 1308–1308 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, G. H. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282–286 CrossRef CAS PubMed.
- D. Li and R. B. Kaner, Science, 2008, 320, 1170–1171 CrossRef CAS PubMed.
- Y. Si and E. T. Samulski, Nano Lett., 2008, 8, 1679–1682 CrossRef CAS PubMed.
- G. Williams, B. Seger and P. V. Kamat, ACS Nano, 2008, 2, 1487–1491 CrossRef CAS PubMed.
- S. A. Kazmi, S. Hameed, A. S. Ahmed, M. Arshad and A. Azam, J. Alloys Compd., 2017, 691, 659–665 CrossRef CAS.
- B. Tang, G. Hu, H. Gao and Z. Shi, J. Power Sources, 2013, 234, 60–68 CrossRef CAS.
- Z. Xiang, X. Zhou, G. Wan, G. Zhang and D. Cao, ACS Sustainable Chem. Eng., 2014, 2, 1234–1240 CrossRef CAS.
- C. H. Shan, H. Zhan, W. L. Chen, Z. M. Su and E. B. Wang, J. Mater. Chem. A, 2016, 4, 3297–3303 CAS.
- E. Singh and H. S. Nalwa, Sci. Adv. Mater., 2015, 7, 1863–1912 CrossRef CAS.
- C. B. Song, Y. H. Qiang, Y. L. Zhao, X. Q. Gu, L. Zhu, J. Song and X. Liu, Int. J. Electrochem. Sci., 2014, 9, 8090–8096 Search PubMed.
- R. Raja, M. Govindaraj, M. D. Antony, K. Krishnan, E. Velusamy, A. Sambandam, M. Subbaiah and V. W. Rayar, J. Solid State Electrochem., 2017, 21, 891–903 CrossRef CAS.
- S. R. Kim, M. K. Parvez and M. Chhowalla, Chem. Phys. Lett., 2009, 483, 124–127 CrossRef CAS.
- M. Motlak, N. A. M. Barakat, M. S. Akhtar, A. G. El-Deen, M. Obaid, C. S. Kim, K. A. Khalil and A. A. Almajid, Chem. Eng. J., 2015, 268, 153–161 CrossRef CAS.
- R. Agarwal, S. Sahoo, O. Resto and R. S. Katiyar, ECS J. Solid State Sci. Technol., 2015, 4, M64–M68 CrossRef CAS.
- W. Song, C. He, W. Zhang, Y. Gao, Y. Yang, Y. Wu, Z. Chen, X. Li and Y. Dong, Carbon, 2014, 77, 1020–1030 CrossRef CAS.
- P. Y. Reddy, L. Giribabu, C. Lyness, H. J. Snaith, C. Vijaykumar, M. Chandrasekharam, M. Lakshmikantam, J. H. Yum, K. Kalyanasundaram, M. Grätzel and M. K. Nazeeruddin, Angew. Chem., Int. Ed., 2007, 46, 373–376 CrossRef CAS PubMed.
- D. A. Dikin, S. Stankovich, E. J. Zimney, R. D. Piner, G. H. B. Dommett, G. Evmenenko, S. T. Nguyen and R. S. Ruoff, Nature, 2007, 448, 457–460 CrossRef CAS PubMed.
- S. P. Lim, N. M. Huang and H. N. Lim, Ceram. Int., 2013, 39, 6647–6655 CrossRef CAS.
- A. Siokou, F. Ravani, S. Karakalos, O. Frank, M. Kalbac and C. Galiotis, Appl. Surf. Sci., 2011, 257, 9785–9790 CrossRef CAS.
- J. Fan, S. Liu and J. Yu, J. Mater. Chem., 2012, 22, 17027–17036 RSC.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- D. Kuang, S. Uchida, R. Humphry-Baker, S. M. Zakeeruddin and M. Grätzel, Angew. Chem., Int. Ed., 2008, 47, 1923–1927 CrossRef CAS PubMed.
- S. P. Lim, A. Pandikumar, N. M. Huang and H. N. Lim, RSC Adv., 2015, 5, 44398–44407 RSC.
- S. P. Lim, A. Pandikumar, N. M. Huang, H. N. Lim, G. Gu and T. L. Ma, RSC Adv., 2014, 4, 48236–48244 RSC.
- A. Zaban, M. Greenshtein and J. Bisquert, ChemPhysChem, 2003, 4, 859–864 CrossRef CAS PubMed.
- A. Zaban, A. Meier and B. A. Gregg, J. Phys. Chem. B, 1997, 101, 7985–7990 CrossRef CAS.
| This journal is © the Partner Organisations 2018 |