
Synthesis and photophysical properties of a porphyrin–BODIPY dyad and a porphyrin–o-carborane–BODIPY triad†
Ekin
Berksun
 a,
Ilgın
Nar
a,
Armağan
Atsay
a,
İbrahim
Özçeşmeci
a,
Ali
Gelir
b and
Esin
Hamuryudan
*a
a,
Ilgın
Nar
a,
Armağan
Atsay
a,
İbrahim
Özçeşmeci
a,
Ali
Gelir
b and
Esin
Hamuryudan
*a
aIstanbul Technical University, Chemistry Department, 34469, Maslak, Istanbul, Turkey. E-mail: esin@itu.edu.tr
bIstanbul Technical University, Physics Department, 34469, Maslak, Istanbul, Turkey
First published on 1st December 2017
Abstract
A novel porphyrin–BODIPY dyad, where BODIPY acts as the central antenna, linked via the Sonogashira coupling reaction, and a novel porphyrin–o-carborane–BODIPY triad, where both porphyrin and BODIPY are covalently attached to the o-carborane, have been synthesised and characterised. X-ray crystallography confirmed a V-shaped triad molecule. Detailed studies on the photophysical properties revealed the excitation of the BODIPY dyad system, which selectively triggered an efficient resonance energy transfer to the porphyrin unit. The presence of the o-carborane group in the triad system significantly diminished the energy transfer efficiency from BODIPY to the porphyrin moiety, because of its quenching properties. Besides, the triad system displayed aggregation-enhanced emission in THF/water systems, due to the presence of the o-carborane unit. The in-depth investigation of the electrochemical properties demonstrated that o-carborane insertion extended the initial reduction potentials and shifted half waves from the anodic to the cathodic site.
Introduction
Resonance energy transfer (RET) plays a key role in the understanding of natural processes and also in the building of new artificial devices. Consequently, many recent studies have been focussing on energy transfer processes and several synthetic model compounds have been investigated.1–5Porphyrin and boron dipyrromethene (BODIPY) derivatives are two versatile classes of functional materials for energy transfer processes.6,7 Porphyrins can be readily modified by the substitution on the aromatic ring, either by electron-withdrawing or electron-donating groups, to acheive unique optical or electronic properties.8,9 Porphyrin systems, as energy acceptors, have been covalently linked to donor units, such as organic compounds that have fluorescence properties, to mimic photosynthetic devices and to develop new efficient molecular systems.10 The porphyrin core easily allows the attachment of donor units near the periphery, directly or via spacers, and such a light-harvesting system has great potential as a suitable system for the functioning of antennae.11 Among the well-known fluorophores, compounds based on the BODIPY core are preferred because they show significant characteristics, such as a high excitation coefficient, high quantum yield, good solubility in most organic solvents, and excellent stability.12 Small modifications to BODIPY structures facilitate the tuning of their fluorescence properties, and these types of structures are widely used to label proteins or energy transfer cassettes.13 From the literature studies, it can be observed that dyads composed of BODIPY and porphyrin units exploit BODIPY's ability to act as an antenna molecule.14–22
Additionally, o-carborane (1,2-dicarba-closo-dodecaborane), a subclass of boron cluster compounds, has unique features based on its icosahedral structure and highly polarisable sigma-aromatic character.23–25 This cluster has a significantly unsymmetrical electron density distribution, which shows electron-withdrawing characteristic when it is bonded through C vertices.26–29 It has been reported that structural parameters obtained from X-ray diffraction studies of numerous carboranes and metallacarboranes indicate that the C1–C2 distances vary considerably if substituents with lone pairs are exo-cluster bonded to the C1 or C2.30,31 Computational methods31 suggested that the cause of elongation is the transfer of the electron density from the available lone pairs on the substituents to the antibonding orbitals on carbon, producing a decrease in the C1–C2 bond order and, thereby, an increase in the C1–C2 distance.32
In the last decade, several research groups were interested in combining organic π-conjugated systems with carborane derivatives, to modulate either one or more of their electronic, optical, and photophysical properties.33–35o-Carborane derivatives linked to the π-conjugated groups at the C1 (and/or C2) position have shown aggregation-induced emission (AIE) or aggregation-enhanced emission (AEE) properties. The intramolecular charge transfer from the π-conjugated groups to the C1–C2 bond of o-carborane quenches the emission in solution, but the reduction in emission can be reversed by freezing the C1–C2 bond vibration in the aggregates.36–38
Despite numerous studies on the synthesis and energy transfer properties of porphyrin–BODIPY dyads14,17,19,39,40 and carborane–BODIPY dyads,25 to the best of our knowledge, no prior publication has documented a triad that includes these three units. Here, we report the synthesis, characterisation and energy transfer process of novel porphyrin–BODIPY dyad and porphyrin–o-carborane–BODIPY triad systems, where the BODIPY moiety acts as the energy donor (D), the porphyrin core serves as the energy acceptor (A) and o-carborane functions as the photo-induced charge transfer compound or the quencher.41 The selective excitation of the donor moiety in such a D–A type dyad system causes an efficient singlet energy-transfer to the acceptor core, while the energy transfer efficiency is quenched by o-carborane for the triad system.
Results and discussion
Scheme 1 outlines the synthesis procedures of the dyad (3) and triad (4). Precursor porphyrin (1) and BODIPY (2) were prepared as described in the literature.42–45 The detailed procedures for 3 and 4 are given in the Experimental section. Briefly, the copper-free palladium-catalysed Sonogashira coupling reaction of porphyrin 1 and BODIPY 2 yielded dyad 3. The formation of 3 was identified by the observation of a red spot by TLC (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v hexane/dichloromethane). After purification by column chromatography over SiO2, the desired compound was obtained, with a yield of 55%. The formation of the triad was followed, by the insertion of decaborane to the ethynyl unit of the dyad (3). Treatment of 3 with decaborane in the presence of N,N-dimethylaniline in dry toluene generated compound 4, in 46% yield. Compound 4 appeared more polar compared to the starting compound 3. Hence, it was readily identified and easy to handle, using the same chromatographic conditions as 3. The resulting compounds were readily soluble in common organic solvents, such as THF, CH2Cl2, CH3Cl, and toluene but insoluble in methanol and ethanol. The chemical purities and structures were fully characterised by FT-IR, 1H NMR, 13C NMR, 11B NMR, MALDI-TOF-MS and UV-Vis spectroscopy.
:1 v/v hexane/dichloromethane). After purification by column chromatography over SiO2, the desired compound was obtained, with a yield of 55%. The formation of the triad was followed, by the insertion of decaborane to the ethynyl unit of the dyad (3). Treatment of 3 with decaborane in the presence of N,N-dimethylaniline in dry toluene generated compound 4, in 46% yield. Compound 4 appeared more polar compared to the starting compound 3. Hence, it was readily identified and easy to handle, using the same chromatographic conditions as 3. The resulting compounds were readily soluble in common organic solvents, such as THF, CH2Cl2, CH3Cl, and toluene but insoluble in methanol and ethanol. The chemical purities and structures were fully characterised by FT-IR, 1H NMR, 13C NMR, 11B NMR, MALDI-TOF-MS and UV-Vis spectroscopy.
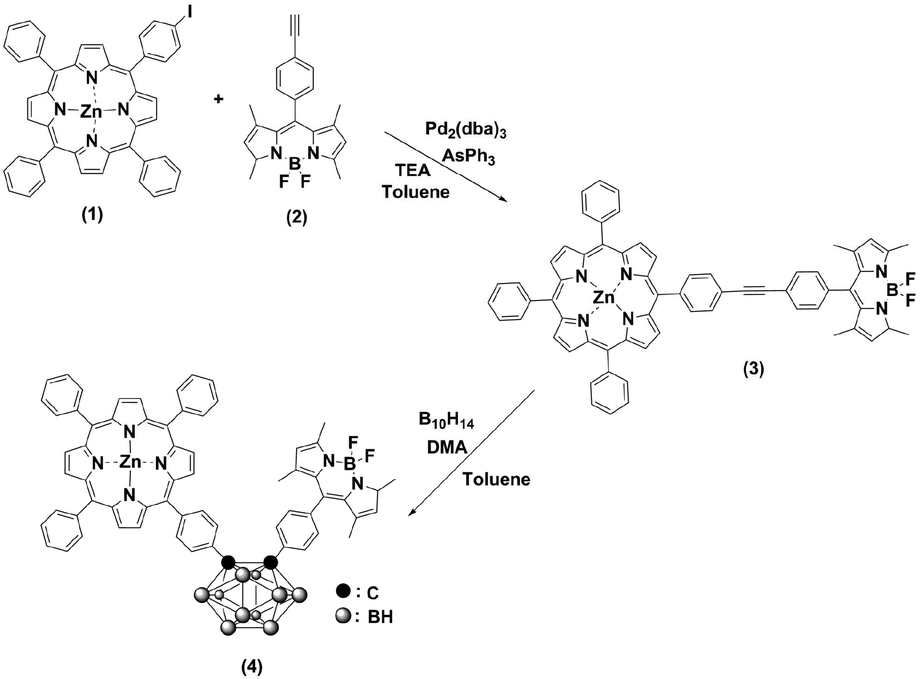 | ||
| Scheme 1 Synthetic routes towards 3 and 4. | ||
In the FT-IR spectra of 4, the disappearance of the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C absorption band at 2214 cm−1 and the formation of B–H stretching vibrations at 2572 cm−1 verified an immediate conversion of 3 to 4 (see ESI, Fig. S1 and S2†).
C absorption band at 2214 cm−1 and the formation of B–H stretching vibrations at 2572 cm−1 verified an immediate conversion of 3 to 4 (see ESI, Fig. S1 and S2†).
The 1H and 13C NMR chemical shifts of 3 and 4 were assigned, by using homonuclear and heteronuclear 2D NMR techniques. The 1H and 13C NMR spectra of compounds 3 and 4 agreed with their structures. The NMR spectra of the compounds (Fig. 1) evidenced a combination of the porphyrin, carborane and BODIPY signals. The insertion of decaborane to compound 3 brings the BODIPY unit spatially close to the porphyrin ring. Consequently, the 1H chemical shift changes observed for the BODIPY protons, due to the porphyrin ring, are dependent on the orientation and proximity relative to the porphyrin macrocycle. The BODIPY methyl groups at 2.59 and 1.52 ppm shifted to 2.52 and 1.13 ppm, respectively. Similarly, the pyrrolic protons of the BODIPY unit moved from 6.03 to 5.65 ppm. Conversely, the BH protons were observed as a broad signal between 1 and 3 ppm for 4.
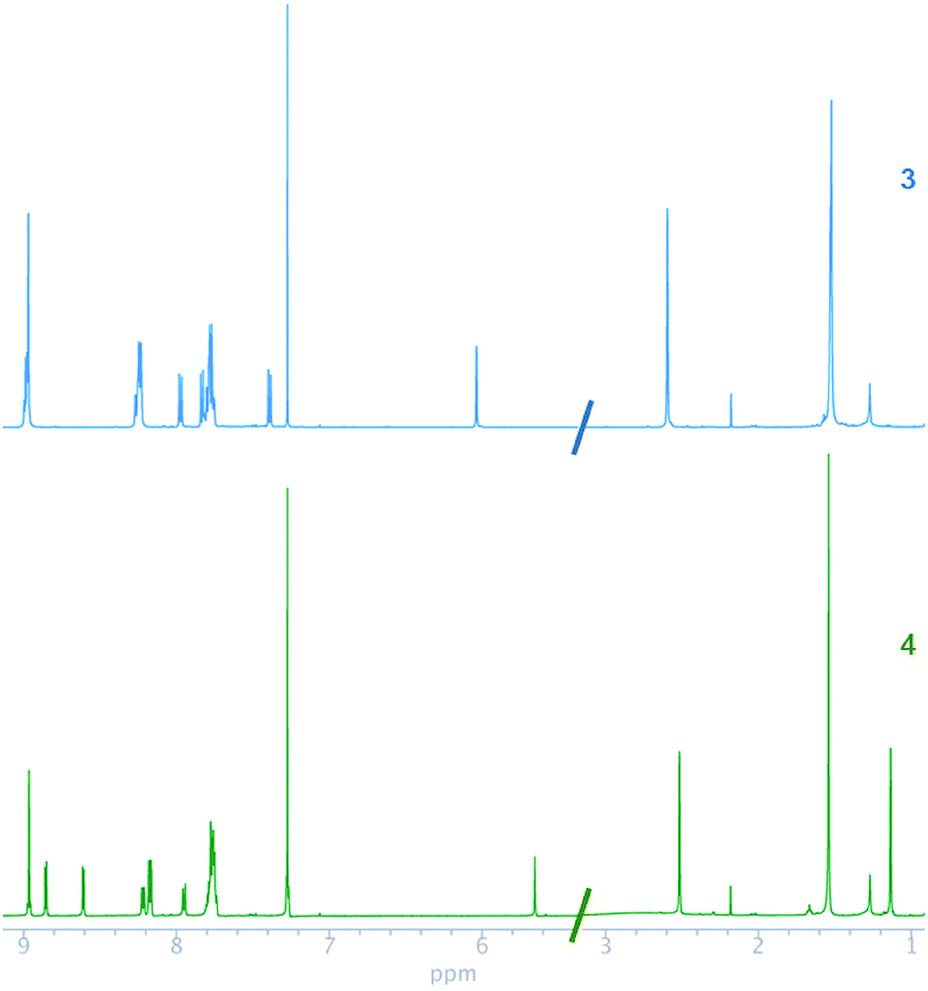 | ||
| Fig. 1 1H NMR spectra of 3 and 4 in CDCl3. | ||
In the 13C NMR spectra of compounds 3 and 4, the chemical shifts of ethynyl carbons at 89.66 and 90.90 ppm changed to 84.21 and 85.17 ppm, respectively, after the insertion of decaborane. This alteration in the position of the ethynyl carbons confirmed that the insertion reaction occurred. BODIPY complexes show 19F NMR resonances at approximately 147 ppm as a quartet, which originates from the coupling between 11B and 19F.46,47 The 19F chemical shift of the BODIPY fluorine atoms for 3 and 4 was observed as quartets at 146.04 and 146.15 ppm, respectively. The 11B NMR chemical shift of compound 3 was seen as a triplet at 0.65 ppm, and the 11B chemical shift of 4 had an additional two broad signals at −2.05 and −9.89 ppm, respectively, which indicate carborane formation.25
The MALDI-TOF mass spectra showed the molecular ion peaks at m/z = 1022.88 [M+]+ for 3 and m/z = 1142.13 [M]+ for 4, together with the separated fragmentation at 1005.27 and 1122.84 m/z, respectively, of one fluorine atom. The structure of 4 was confirmed by X-ray crystallography. In order to obtain suitable crystals, methanol was slowly diffused into a DCM solution of 4. However, immediate deterioration occurred in air. Thus, it was not possible to obtain diffraction data from these crystals. As a second approach, crystals were grown by using the same method but with the addition of pyridine. Even if the resulting crystal indicated very weak diffraction, it was satisfactory to determine the molecular structure via X-ray analysis [Fig. 2] (CCDC 1575485†). Compound 4 crystallised in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) .
.
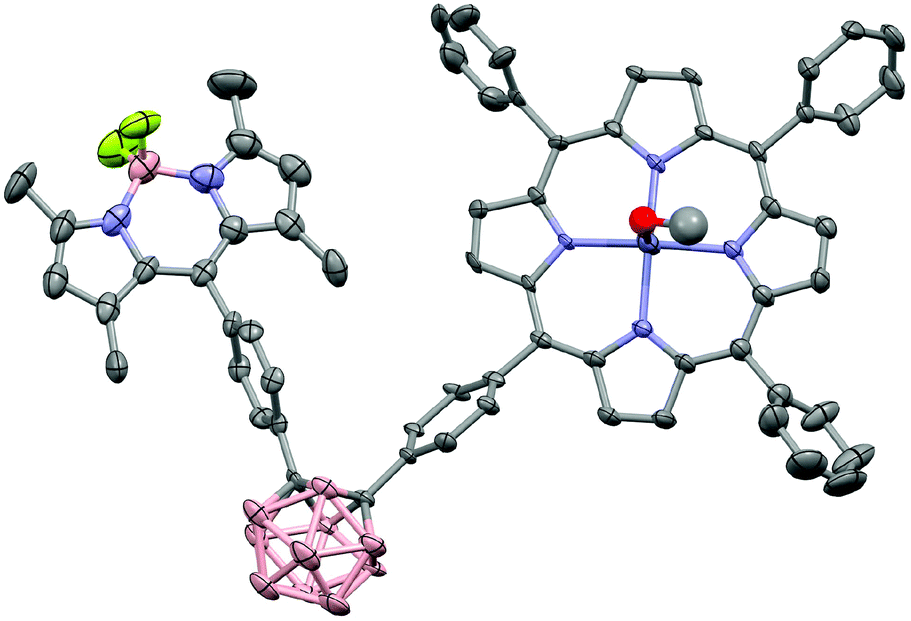 | ||
| Fig. 2 X-ray molecular structure of 4. Thermal ellipsoids are shown at 50% probability level. The hydrogen atoms and disordered solvent molecules were omitted for clarity. | ||
The UV-Vis spectra of the compounds (1a, 2–4) in toluene are shown in Fig. 3. The peaks at 424 and 506 nm correspond to the absorption bands of 1a (ZnTPP) (see ESI, Scheme S1†) and 2, respectively. As shown in Fig. 3, the spectra of 3 and 4 are the superposition of the individual spectra of 1a and 2, and there is not a considerable ground state interaction between the molecules in the compounds.12
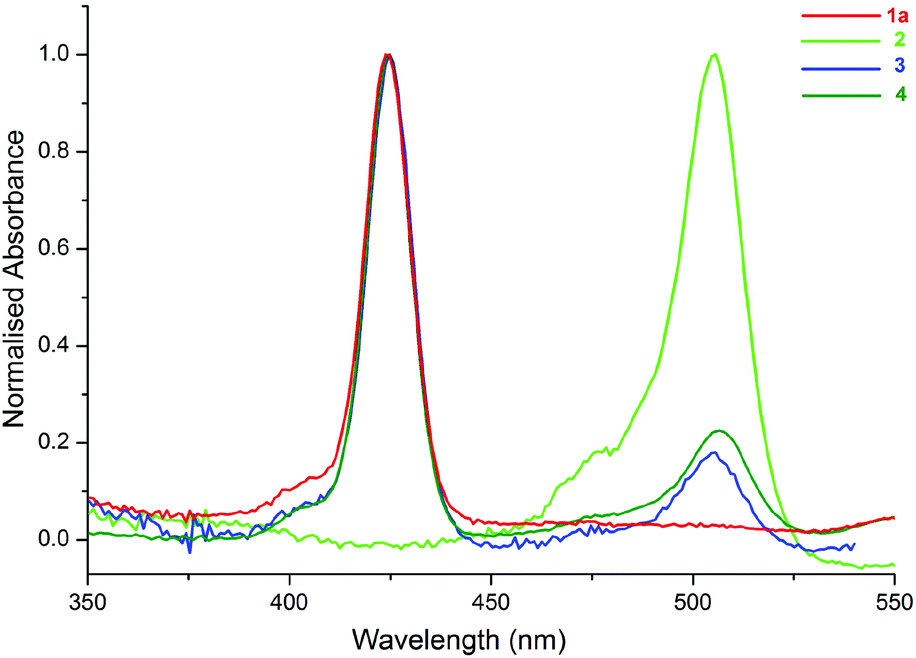 | ||
| Fig. 3 UV-Vis spectra of 1a, 2, 3 and 4 in toluene. | ||
The fluorescence emission spectra of 3, 4 and the 1:1 mixture of 1a and 2 are given in Fig. 4. Fig. 4(a) and (b) show the emission spectra of the molecules selectively excited at 424 and 506 nm, respectively. In Fig. 4(a), the emission intensity of 3 is stronger than the others, and the emission intensity of 4 is the weakest. These conflicting results, with respect to the free forms of 1a, are related to the quantum yields of the compounds and the quenching effect of o-carborane in 4. The quantum yield of 3 was calculated as 0.041, according to eqn (1), and it is larger than the quantum yield of 4, determined as 0.028 and the free form of 1a, 0.033.48o-Carborane is an electron-withdrawing unit when it is attached to the C atom. Therefore, when 1a or 2 is selectively excited, an efficient photo-induced electron transfer (PET) occurs through the antibonding orbitals of the C–C bonds of o-carborane and the fluorescence of 4 is quenched considerably.14,15,25,49,50 In Fig. 4(b), the emission intensity of 4 is the weakest, as expected, due to the quenching effect of o-carborane but the emission intensity of 3 is less than that of the free form of 2, which contrasts with the results observed in Fig. 4(a). These data give some clues about the RET from BODIPY to ZnTPP in 3.
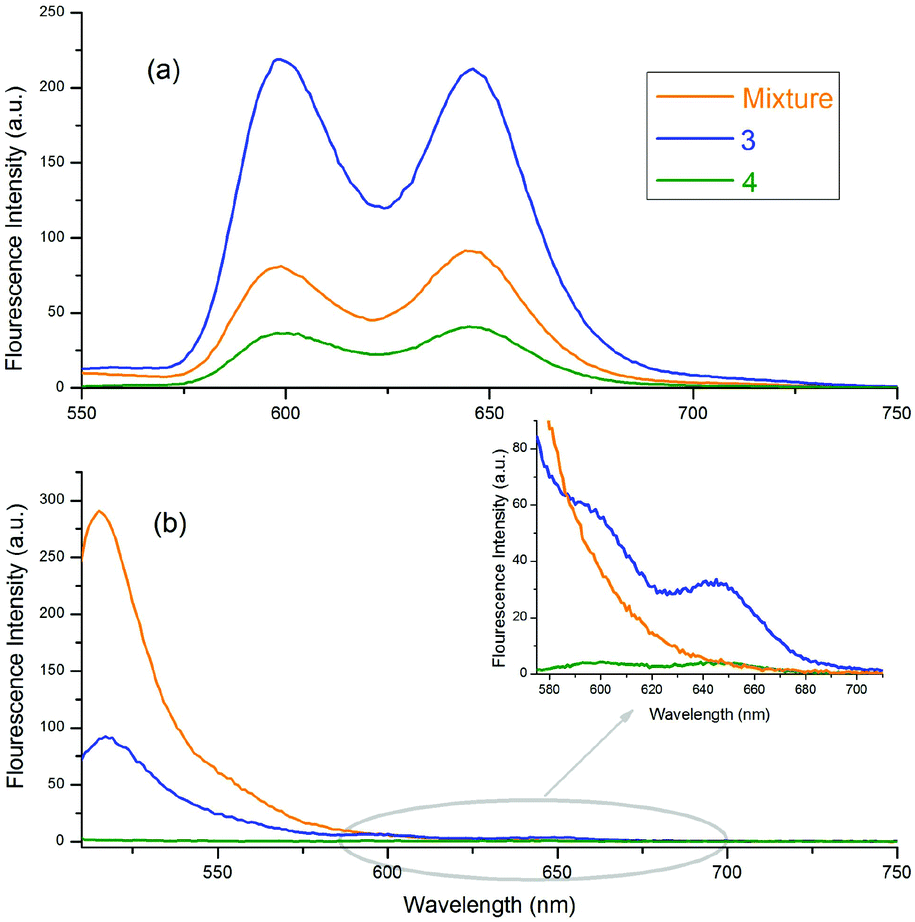 | ||
| Fig. 4 Emission spectra of 3, 4 and 1:1 mixture of 1a and 2 in toluene. The excitation wavelengths of the compounds shown in (a) and (b) are 424 nm and 506 nm, respectively. | ||
When the BODIPY unit in 3 is selectively excited, its emission intensity decreased compared to that of the free form of BODIPY. This behaviour occurred because the energies of some of the excited BODIPY molecules were transferred to ZnTPP in 3 and, thus, the intensity of the selectively excited BODIPY molecules decreased. Also, some peaks with low intensity, belonging to ZnTPP in the dyad and triad, could be seen, due to the RET [Fig. 4(b) inset]. When these two selectively excited emission spectra are compared, it can be concluded that the emission of 2 is quenched more than that of 1a. Indeed, upon excitation of the compounds at 506 nm, the excitation wavelength of 2, almost no emission was observed from BODIPY, whereas there was considerable emission intensity when the compounds were excited at 424 nm, the excitation wavelength of 1a.
Similar results were observed in the excitation spectra of 2 in free and bound forms of the compounds, as shown in Fig. 5. Here, the typical excitation peak of 2 at 506 nm was noted for both 3 and the mixture of 1a and 2, but it disappeared for 4. The peak intensity was strongest for the mix, i.e., the free form of 2. When the BODIPY is bound to porphyrin through a CC bridge, the intensity decreases due to the transfer of the excited energy from some of the BODIPY units to the ZnTPP moiety in 3, as mentioned above. Thus, the data demonstrated an energy transfer from the BODIPY unit to the ZnTPP moiety in 3, which is consistent with the literature.14,15,17,20–22,25 In 4, the intensity of the peak can be assumed to be zero because of the efficient quenching properties of carborane.
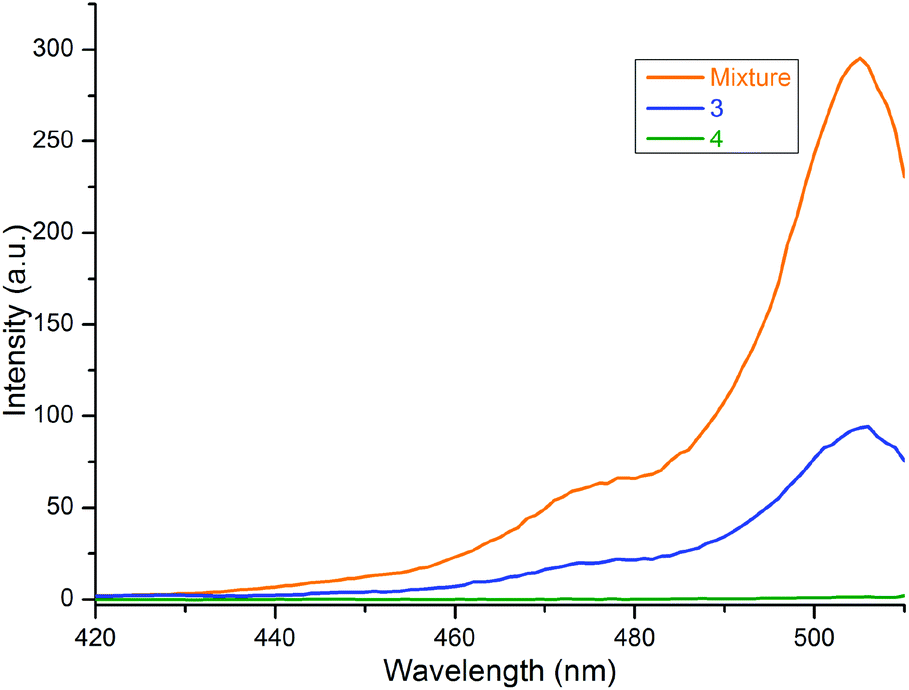 | ||
| Fig. 5 Excitation spectra of 3, 4 and 1:1 mixture of 1a and 2 in toluene. The monitored emission wavelength is 515 nm. | ||
Fig. 6 displays the normalized excitation spectrum of 1a and the normalized emission spectrum of 2 in toluene. A considerable overlap existed between the emission and the excitation spectra. Collectively, the results indicate that the excited energy is transferred from the BODIPY moiety to the porphyrin in 3 and 4. Considerable energy transfer efficiency was calculated as 0.90 for the dyad. However, the energy transfer efficiency could not be calculated for 4 as the fluorescence of the donor, 2, was almost completely quenched, due to the PET process in the presence of o-carborane,14,15,25 as seen in Fig. 4(b). Moreover, the decrease in the emission intensity of the free form of BODIPY [Fig. 3(b)] to near zero upon binding to 1a compared to that of o-carborane to form 4, may suggest an efficient energy transfer from 2 to 1a. If this energy transfer occurred, substantial emission of 1a in 4 should be observed in Fig. 4(b). Nevertheless, 1a produced a very low emission intensity. Therefore, it can be concluded that most of the selectively excited BODIPY units in 4 were quenched by o-carborane, which prevents correct energy transfer efficiency for 4.
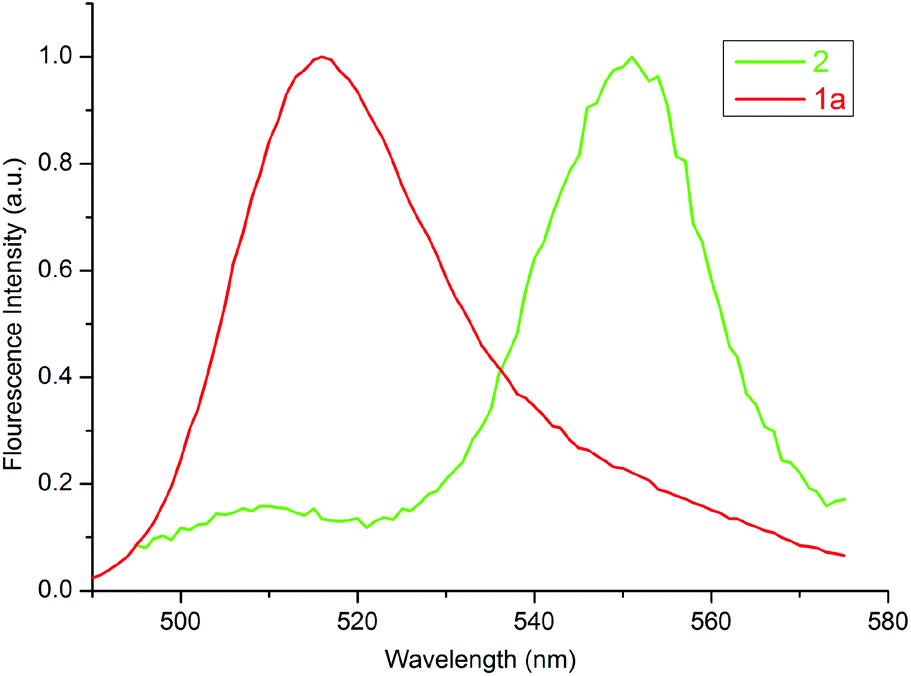 | ||
| Fig. 6 Normalized excitation and emission spectra of 1a and 2 in toluene, respectively. The concentration of each compound was adjusted to 1 × 10−6 M. | ||
It is known in the literature that in the presence of an o-carborane unit in a compound, the fluorescence emission intensity of the compound increases as the molecules aggregate, referred to as AIE or AEE.36,38 In solution, o-carborane, which is bound to the peripheral units through single C bonds, acts as an electron-withdrawing unit26,27 and triggers the PET process in the system.50 In addition, the aryl units rotate around the single bonds which connect the peripheral units.51 These two mechanisms, PET and rotation, increase the nonradiative decay probability and thus quench the fluorescence emission. When the molecules start to aggregate in a solution in which the solubility of the molecule is small, the efficiency of PET and rotation around the single bonds decreases and the fluorescence emission intensity starts to appear in AIE or increases in AEE. In our compound 4, the fluorescence emission intensity is observable in pure THF, whereas, with the addition of a small amount of water it decreases, followed by an increase as the amount of water is increased (Fig. 7 and Table 1).
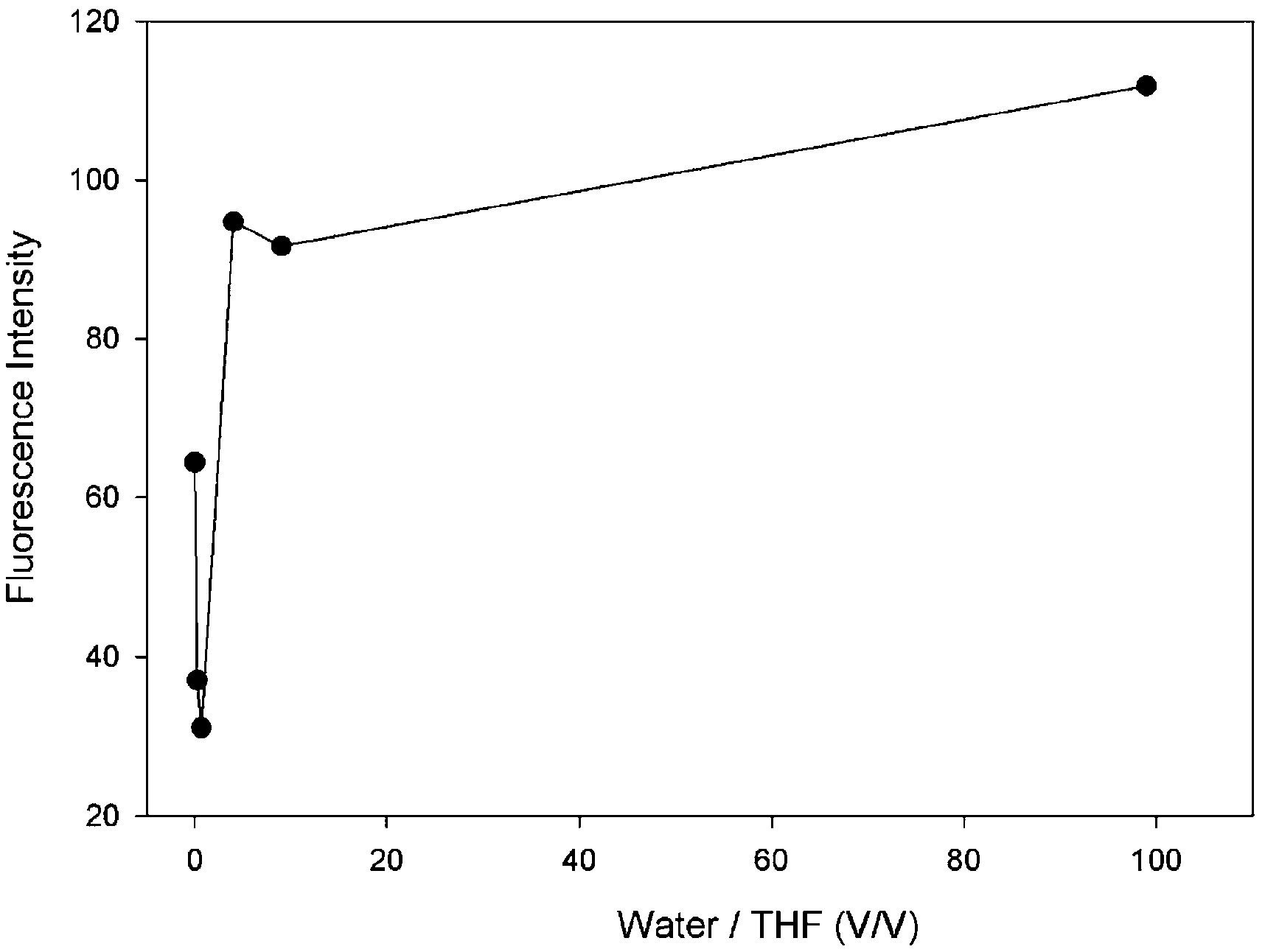 | ||
| Fig. 7 Intensity at 653 nm of the emission spectra of the triad versus water/THF ratio. | ||
| Water (ml) | THF (ml) | Water/THF (v/v) | If |
|---|---|---|---|
| 0.00 | 5.00 | 0.00 | 64.4 |
| 1.00 | 4.00 | 0.25 | 37.1 |
| 2.00 | 3.00 | 0.67 | 31.1 |
| 4.00 | 1.00 | 4.00 | 94.7 |
| 4.50 | 0.50 | 9.00 | 91.7 |
| 4.95 | 0.05 | 99.00 | 111.8 |
As the amount of water is increased, the amount of the aggregated compound 4 increases. Due to the interactions between the molecules and water, and the low mobility of 4 in water, which is the result of a low solubility in water, a decrease in the rotational modes of 4 is observed. This results in a decreasing probability of nonradiative decay and an increase in the intensity of the fluorescence emission. Hence, the triad molecule (4) synthesized in this study exhibits AEE properties in the mixtures of THF/water.
The redox properties of the dyad (3), triad (4), ethynyl-substituted porphyrin reference (1b)52 (see ESI, Scheme S2†), and BODIPY reference (2)45 were determined by cyclic voltammetry (CV) and square wave voltammetry (SWV). These measurements were performed in anhydrous DCM, using tetra-n-butylammonium perchlorate (TBAP) as the supporting electrolyte. The half-peak potential E1/2 was determined as (Epa + Epc)/2, where Epa and Epc are the respective anodic and cathodic peak potentials, determined from the CV measurements. Fig. 8 shows the CV data for 3, 4, and the reference compounds, and the corresponding redox potentials are summarised in Table 2.
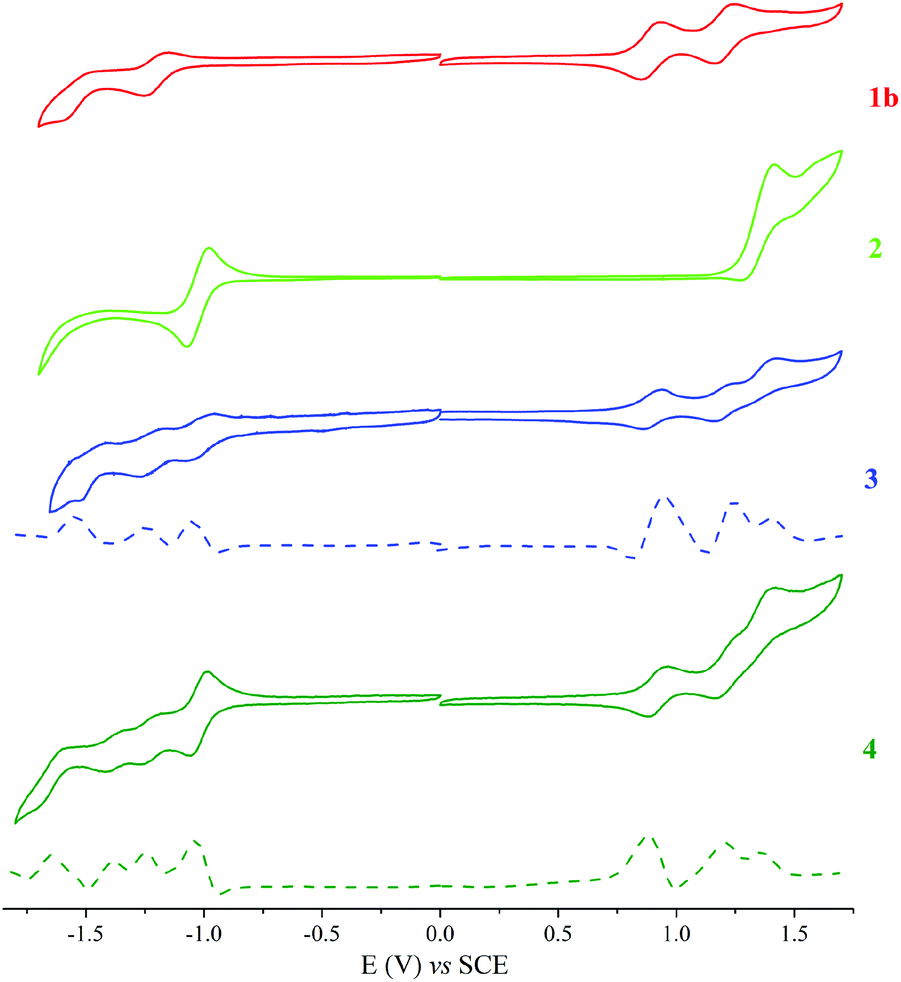 | ||
| Fig. 8 Cyclic and square voltammograms for a 0.1 mM CH2Cl2 solution of 1b, 2, 3 and 4 containing 0.1 M TBAP obtained at a scan rate of 0.025 V s−1. | ||
| Compound | R4 | R3 | R2 | R1 | Ox1 | Ox2 | Ox3 |
|---|---|---|---|---|---|---|---|
| R: Reduction, Ox: Oxidation. | |||||||
| 1b | −1.6 | −1.25 | 0.85 | 1.16 | |||
| 2 | −1.04 | 1.39 | |||||
| 3 | −1.58 | −1.21 | −1.03 | 0.94 | 1.24 | 1.4 | |
| 4 | −1.65 | −1.35 | −1.25 | −1.01 | 0.88 | 1.19 | 1.36 |
With the aid of reference compounds 1b and 2, it was possible to assign the potentials corresponding to the different entities of the dyad 3 and triad 4. For 3, there were three oxidation peaks at 0.94, 1.24 and 1.4 V vs. SCE. The first and second oxidation peaks can be assigned to be the oxidation of the porphyrin group and the third peak can be assigned to be the oxidation of the BODIPY moiety. Also, three reduction peaks at −1.03, −1.21 and −1.58 V vs. SCE were apparent. The first reduction peak can be attributed to that of the BODIPY group. The second and third reduction peaks can be assigned to that of the porphyrin moiety. For 4, the oxidation potentials are comparable to that of 3, with an additional reduction process at −1.35 V vs. SCE that can be ascribed to the o-carborane group reduction.25,38 Due to the electron-withdrawing characteristics of carborane, the oxidation and reduction potentials of 4 shifted from the anodic to the cathodic site.
Experimental
Materials, instruments and methods
Compounds 1 and 2 were synthesized according to the literature procedure.42–45,53 All NMR spectra were recorded on an Agilent VNMRS 500 MHz at 25 °C and chemical shifts were referenced internally using the residual solvent resonances. Matrix-assisted laser desorption time of flight mass spectrometry (MALDI-TOF-MS) was carried out on a BRUKER Microflex LT using 2,5-dihydroxybenzoic acid as the matrix. Electrochemical measurements were carried out using a Gamry 600 Potentiostat/Galvanostat. The cell comprised inlets for a glassy carbon working electrode, a platinum wire counter electrode and a saturated calomel electrode (SCE) reference electrode. Typically, 0.1 M solution of TBAP in CH2Cl2 containing the sample was purged with nitrogen for 20 min, and then the voltammograms were recorded at room temperature. The UV-Vis spectra were obtained on a Scinco S-3100 spectrophotometer using 1 cm path length cuvettes at room temperature. The steady state fluorescence spectra of the samples were collected by using a Varian Carry Eclipse fluorescence spectrometer at room temperature. The excitation wavelengths were adjusted depending on the absorption and the excitation spectra of the molecules used in this study.Quantum yields of the compounds were calculated by using the comparative method as given in eqn (1).54,55
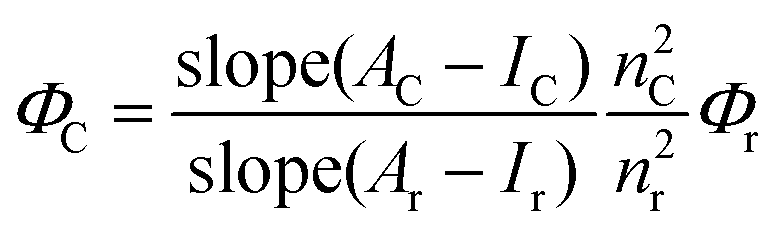 | (1) |
ZnTPP (1a) was used as the reference (R), which has excitation and emission peaks at 424 nm and 603 nm, respectively, and has quantum yield, ΦF = 0.033,56 in toluene which are the same as the excitation and emission wavelengths of 3 and 4. 5 to 7 different concentrations of 3, 4 and R ranging between 1 × 10−7 M and 7 × 10−7 M were used and all measurements were performed under the same conditions.
The resonance energy transfer (RET) occurred from the BODIPY unit to the ZnTPP part of 3 and 4, i.e. BODIPY and ZnTPP are called the donor and acceptor, respectively. The energy transfer efficiency, εT, from the donor to acceptor is given by eqn (2):
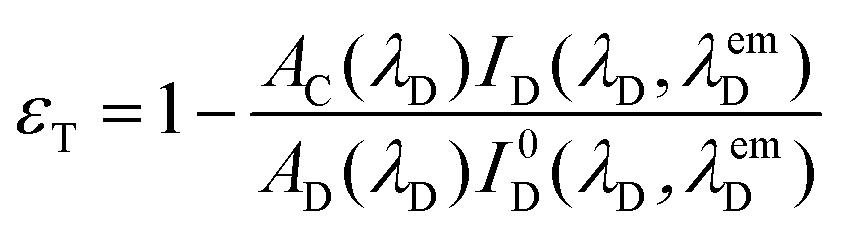 | (2) |
For AEE measurements, 6 mixtures of water and THF with different water/THF ratios, where the water/THF ratio was changed from 0 to 99 (V/V), were prepared (Table 1). The total volume of each sample was 5 ml and the concentration of compound 4 in each mixture was kept fixed at 10−5 M. The excitation wavelength for the fluorescence measurements of the mixtures was adjusted at 424 nm and the fluorescence emission intensity at 654 nm was monitored.
Single crystals of 4 were mounted on a MicroMount (MiTeGen). The crystallinity of 4 deteriorated quickly in air. The crystal of 4 was immediately cooled to prevent crystal degradation. The crystallographic data of the compound was obtained from a Bruker D8 VENTURE single crystal X-ray diffractometer equipped with a PHOTON 100 CMOS detector and KryoFlex II low-temperature apparatus operating at 120 K, using graphite monochromatized MoKα radiation (λ = 0.71073 Å). All of the data were corrected for absorption effects using the multiscan technique. The structures were solved by direct methods and refined on F2 by full-matrix least-squares using SHELXL 2014.58 All the hydrogen atoms were added to their geometrically ideal positions. All non-hydrogen atoms were refined with anisotropic displacement parameters.
Synthesis and characterization
:1 mixture of dry toluene/triethylamine (20 mL). The solution was degassed by bubbling nitrogen for 15 min, then Pd2(dba)3 (23 mg, 0.025 mmol), and AsPh3 (76.2 mg, 0.248 mmol) were added to the mixture and the reaction mixture was stirred at 80 °C for 12 h. The formation of the desired compound was identified by the observation of a red colour spot by TLC (hexane/DCM (2:1 v/v)). After cooling to room temperature, the organic layer was extracted with CH2Cl2 (3 × 50 mL) and dried over Na2SO4. The solvent was removed by evaporation under high vacuum and the residue was purified by column chromatography on silica gel, using hexane/DCM (2:1 v/v) as the eluent. A reddish solid was obtained as the product (140 mg, 55%).
FT-IR νmax (cm−1): 3051.58, 3027.77, 2920.18, 2849.20, 2214.28, 1596.99, 1540.54, 1508.32, 1467.75, 1440.49, 1407.04, 1371.73, 1339.11, 1305.66, 1190.13, 1155.26, 1119.84, 1070.43, 1020.36, 979.38, 834.00, 796.61, 763.71, 718.70, 700.31, 660.70. UV/Vis (toluene): λmax/nm = 424, 506, 551 nm. 1H NMR (CDCl3): δ 9.0–8.97 (m, 8H), 8.27–8.25 (m, 8H), 7.97 (d, 2H), 7.84–7.77 (m, 11H), 7.40 (d, 2H), 6.03 (s, 2H), 2.59 (s, 6H), 1.52 (s, 6H). 11B NMR (CDCl3): δ 0.65. 13C NMR (CDCl3): δ 155.80, 150.33, 150.22, 149.82, 143.38, 143.04, 142.71, 140.84, 135.12, 134.54, 134.41, 132.45, 132.23, 12.08, 131.62, 131.24, 129.88, 128.38, 127.96, 126.57, 124.21, 122.04, 121.43, 121.31, 120.01, 90.90, 89.66, 31.59, 22.66. 19F NMR (CDCl3): δ −146.04 ppm. MALDI-TOF: calcd for C65H46BF2N6Zn: 1023.31 [M+], found 1022.88, 1005.27 [M − 19 + H]+.
:1 v/v)), where 4 appeared more polar when compared to the starting compound 3. After cooling to room temperature, the organic layer was extracted with CH2Cl2 (3 × 50 mL) and dried over Na2SO4. Solvents were removed by evaporation under high vacuum and the residue was purified by column chromatography on silica gel, using hexane/dichloromethane (2:1 v/v) as the eluent. A reddish solid was obtained as the product (65 mg, 46%).
FT-IR νmax (cm−1): 3006.31, 2989.79, 2921.69, 2850.91, 2572.00, 1597.74, 1546.00, 1511.70, 1467.73, 1440.13, 1404.29, 1372.82, 1339.41, 1307.88, 1275.61, 1261.00, 1189.00, 1155.66, 1119.95, 1084.48, 1021.67, 979.68, 840.32, 797.37, 750.20, 701.00, 660.54. UV/Vis (toluene): λmax/nm = 688, 621, 356, 260 nm. 1H NMR (CDCl3): δ 8.96 (m, 4H), 8.85 (m, 2H), 8.60 (d, 2H), 8.23 (d, 2H), 8.16 (d, 4H), 7.94 (d, 2H), 7.75 (m, 13H), 7.26 (d, 2H), 5.65 (s, 2H), 3.1–2.4 (b, 10H), 2.52 (s, 6H), 1.13 (s, 6H). 11B NMR (CDCl3): δ 0.65, −2.05, −9.89. 13C NMR (CDCl3): δ 155.98, 150.42, 150.35, 150.12, 149.20, 145.25, 142.58, 142.48, 139.35, 137.52, 134.39, 132.29, 132.19, 131.46, 131.16, 130.92, 129.87, 128.82, 128.40, 127.61, 126.62, 121.71, 121.46, 121.36, 118.32, 85.17, 84.21, 14.85, 14.60. 19F NMR (CDCl3): δ −146.15 ppm. MALDI-TOF: calcd for C65H56B11F2N6Zn: 1143.48 [M+], found 1142.13, 1122.84 [M − 19]+.
Conclusions
In summary, we have synthesised and characterised a novel porphyrin–BODIPY dyad (3) and a porphyrin–o-carborane-BODIPY triad (4), where the BODIPY moiety acts as the energy donor, the porphyrin core serves as the energy acceptor and o-carborane functions as the photo-induced charge transfer compound. The redox properties of the dyad and triad were determined by CV and SWV. It was concluded that the quantum yield of ZnTPP increases upon binding to BODIPY to form the dyad (3), whereas it decreases in the triad (4) due to the quenching effect of o-carborane. When the BODIPY unit in dyad 3 and triad 4, respectively, was selectively excited, the RET from the BODIPY group to the corresponding ZnTPP unit in dyad 3 and triad 4 was observed. The RET efficiency was calculated as 0.90 for the dyad, but it could not be calculated for the triad (4), due to the quenching effect of o-carborane. We concluded that the calculation of the energy transfer efficiency for the triad (4) would not be physically meaningful, due to the lack of information about the fluorescence of the donor/acceptor pair, which is quenched considerably by o-carborane. Furthermore, it was noted that the emission intensity of the triad in THF was increased by the increasing aggregation probability upon adding water to THF. This result shows that triad (4) exhibits AEE properties in aqueous THF. Thus, the synthesised dyad (3) and triad (4) may have potency for use in energy conversion devices or the design of new AEE materials. Their electrochemical properties revealed that o-carborane insertion extended the reduction potentials together with a cathodic shift.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Research Fund of the Istanbul Technical University. The authors acknowledge the valuable help of Prof. Vickie Mckee for crystal structure solutions.References
- S. Das, H. R. Bhat, N. Balsukuri, P. C. Jha, Y. Hisamune, M. Ishida, H. Furuta, S. Mori and I. Gupta, Inorg. Chem. Front., 2017, 4, 618–638 RSC.
- A. Langlois, H.-J. Xu, P.-L. Karsenti, C. P. Gros and P. D. Harvey, Chem. – Eur. J., 2017, 23, 5010–5022 CrossRef CAS PubMed.
- C.-B. Huang, L. Xu, J.-L. Zhu, Y.-X. Wang, B. Sun, X. Li and H.-B. Yang, J. Am. Chem. Soc., 2017, 139, 9459–9462 CrossRef CAS PubMed.
- A. Concellón, M. Marcos, P. Romero, J. L. Serrano, R. Termine and A. Golemme, Angew. Chem., Int. Ed., 2017, 56, 1259–1263 CrossRef PubMed.
- M. Özçeşmeci, İ. Özçeşmeci and E. Hamuryudan, Polyhedron, 2010, 29, 2710–2715 CrossRef.
- S. Xuan, N. Zhao, Z. Zhou, F. R. Fronczek and M. G. H. Vicente, J. Med. Chem., 2016, 59, 2109–2117 CrossRef CAS PubMed.
- M. Koepf, A. Trabolsi, M. Elhabiri, J. A. Wytko, D. Paul, A. M. Albrecht-Gary and J. Weiss, Org. Lett., 2005, 7, 1279–1282 CrossRef CAS PubMed.
- Y. Liu, J. Jin, H. Deng, K. Li, Y. Zheng, C. Yu and Y. Zhou, Angew. Chem., Int. Ed., 2016, 128, 8084–8089 CrossRef.
- L. Zhang, L. Hou, X. Zhao, Z. Zhang, Y. Wang and J. Li, Inorg. Chem. Front., 2017, 4, 360–367 RSC.
- D. Baskaran, J. W. Mays, X. P. Zhang and M. S. Bratcher, J. Am. Chem. Soc., 2005, 127, 6916–6917 CrossRef CAS PubMed.
- J. Xu, L.-J. Xue, J.-L. Hou, Z.-N. Yin, X. Zhang, Q.-Y. Zhu and J. Dai, Inorg. Chem., 2017, 56, 8036–8044 CrossRef CAS PubMed.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed.
- J. D. Spiegel, I. Lyskov, M. Kleinschmidt and C. M. Marian, Chem. Phys., 2017, 482, 265–276 CrossRef CAS.
- D. Gao, S. M. Aly, P.-L. Karsenti, G. Brisard and P. D. Harvey, Dalton Trans., 2017, 46, 6278–6290 RSC.
- C. Y. Lee, J. K. Jang, C. H. Kim, J. Jung, B. K. Park, J. Park, W. Choi, Y.-K. Han, T. Joo and J. T. Park, Chem. – Eur. J., 2010, 16, 5586–5599 CrossRef CAS PubMed.
- E. Maligaspe, T. Kumpulainen, N. K. Subbaiyan, M. E. Zandler, H. Lemmetyinen, N. V. Tkachenko and F. D'Souza, Phys. Chem. Chem. Phys., 2010, 12, 7434–7444 RSC.
- F. D'Souza, P. M. Smith, M. E. Zandler, A. L. McCarty, M. Itou, Y. Araki and O. Ito, J. Am. Chem. Soc., 2004, 126, 7898–7907 CrossRef PubMed.
- G. N. Lim, E. Maligaspe, M. E. Zandler and F. D'Souza, Chem. – Eur. J., 2014, 20, 17089–17099 CrossRef CAS PubMed.
- T. K. Khan, M. Bröring, S. Mathur and M. Ravikanth, Coord. Chem. Rev., 2013, 257, 2348–2387 CrossRef CAS.
- A. Bagaki, H. B. Gobeze, G. Charalambidis, A. Charisiadis, C. Stangel, V. Nikolaou, A. Stergiou, N. Tagmatarchis, F. D'Souza and A. G. Coutsolelos, Inorg. Chem., 2017, 56, 10268–10280 CrossRef CAS PubMed.
- M. E. El-Khouly, C. A. Wijesinghe, V. N. Nesterov, M. E. Zandler, S. Fukuzumi and F. D'Souza, Chem. – Eur. J., 2012, 18, 13844–13853 CrossRef CAS PubMed.
- M. J. Leonardi, M. R. Topka and P. H. Dinolfo, Inorg. Chem., 2012, 51, 13114–13122 CrossRef CAS PubMed.
- D. Zhao, Functionalization of Carborane via Carboryne Intermediates, Springer Nature, Singapore, 2016 Search PubMed.
- S. Mukherjee and P. Thilagar, Chem. Commun., 2016, 52, 1070–1093 RSC.
- G. F. Jin, Y.-J. Cho, K.-R. Wee, S. A. Hong, I.-H. Suh, H.-J. Son, J.-D. Lee, W.-S. Han, D. W. Cho and S. O. Kang, Dalton Trans., 2015, 44, 2780–2787 RSC.
- A. M. Spokoyny, C. W. Machan, D. J. Clingerman, M. S. Rosen, M. J. Wiester, R. D. Kennedy, C. L. Stern, A. A. Sarjeant and C. A. Mirkin, Nat. Chem., 2011, 3, 590 CrossRef CAS PubMed.
- R. N. Grimes, in Carboranes (Third Edition), Academic Press, Boston, 2016, pp. 7–18 Search PubMed.
- R. Núñez, P. Farràs, F. Teixidor, C. Viñas, R. Sillanpää and R. Kivekäs, Angew. Chem., Int. Ed., 2006, 45, 1270–1272 CrossRef PubMed.
- F. Teixidor, R. Núñez, C. Viñas, R. Sillanpää and R. Kivekäs, Angew. Chem., Int. Ed., 2000, 39, 4290–4292 CrossRef CAS.
- J. Llop, C. Viñas, J. M. Oliva, F. Teixidor, M. A. Flores, R. Kivekas and R. Sillanpää, J. Organomet. Chem., 2002, 657, 232–238 CrossRef CAS.
- J. Llop, C. Viñas, F. Teixidor, L. Victori, R. Kivekäs and R. Sillanpää, Organometallics, 2001, 20, 4024–4030 CrossRef CAS.
- J. M. Oliva, N. L. Allan, P. v. R. Schleyer, C. Viñas and F. Teixidor, J. Am. Chem. Soc., 2005, 127, 13538–13547 CrossRef CAS PubMed.
- I. Nar, A. Gül, I. B. Sivaev and E. Hamuryudan, Synth. Met., 2015, 210, 376–385 CrossRef CAS.
- R. Núñez, M. Tarrés, A. Ferrer-Ugalde, F. F. de Biani and F. Teixidor, Chem. Rev., 2016, 116, 14307–14378 CrossRef PubMed.
- R. N. Grimes, in Carboranes (Third Edition), Academic Press, Boston, 2016, pp. 283–502 Search PubMed.
- H. Naito, K. Nishino, Y. Morisaki, K. Tanaka and Y. Chujo, Angew. Chem., Int. Ed., 2017, 56, 254–259 CrossRef CAS PubMed.
- H. Naito, K. Nishino, Y. Morisaki, K. Tanaka and Y. Chujo, Chem. – Asian J., 2017, 12, 2134–2138 CrossRef CAS PubMed.
- K. Hosoi, S. Inagi, T. Kubo and T. Fuchigami, Chem. Commun., 2011, 47, 8632–8634 RSC.
- S. Pascal, L. Bucher, N. Desbois, C. Bucher, C. Andraud and C. P. Gros, Chem. – Eur. J., 2016, 22, 4971–4979 CrossRef CAS PubMed.
- B. Brizet, A. Eggenspiller, C. P. Gros, J.-M. Barbe, C. Goze, F. Denat and P. D. Harvey, J. Org. Chem., 2012, 77, 3646–3650 CrossRef CAS PubMed.
- A. Ferrer-Ugalde, E. J. Juárez-Pérez, F. Teixidor, C. Viñas, R. Sillanpää, E. Pérez-Inestrosa and R. Núñez, Chem. – Eur. J., 2012, 18, 544–553 CrossRef CAS PubMed.
- A. D. Adler, F. R. Longo, J. D. Finarelli, J. Goldmacher, J. Assour and L. Korsakoff, J. Org. Chem., 1967, 32, 476–476 CrossRef CAS.
- R. Luguya, L. Jaquinod, F. R. Fronczek, M. G. H. Vicente and K. M. Smith, Tetrahedron, 2004, 60, 2757–2763 CrossRef CAS.
- J. Rochford and E. Galoppini, Langmuir, 2008, 24, 5366–5374 CrossRef CAS PubMed.
- X. Wu, W. Wu, X. Cui, J. Zhao and M. Wu, J. Mater. Chem. C, 2016, 4, 2843–2853 RSC.
- T. W. Hudnall and F. P. Gabbai, Chem. Commun., 2008, 4596–4597 RSC.
- J. A. Jacobsen, J. R. Stork, D. Magde and S. M. Cohen, Dalton Trans., 2010, 39, 957–962 RSC.
- F. Li, S. I. Yang, Y. Ciringh, J. Seth, C. H. Martin, D. L. Singh, D. Kim, R. R. Birge, D. F. Bocian, D. Holten and J. S. Lindsey, J. Am. Chem. Soc., 1998, 120, 10001–10017 CrossRef CAS.
- S. Inagi, K. Hosoi, T. Kubo, N. Shida and T. Fuchigami, Electrochemistry, 2013, 81, 368–370 CrossRef CAS.
- A. Ferrer-Ugalde, A. González-Campo, C. Viñas, J. Rodríguez-Romero, R. Santillan, N. Farfán, R. Sillanpää, A. Sousa-Pedrares, R. Núñez and F. Teixidor, Chem. – Eur. J., 2014, 20, 9940–9951 CrossRef CAS PubMed.
- K. Kokado and Y. Chujo, J. Org. Chem., 2011, 76, 316–319 CrossRef CAS PubMed.
- J. R. Burns, K. Göpfrich, J. W. Wood, V. V. Thacker, E. Stulz, U. F. Keyser and S. Howorka, Angew. Chem., Int. Ed., 2013, 52, 12069–12072 CrossRef CAS PubMed.
- C. He, Q. He, C. Deng, L. Shi, D. Zhu, Y. Fu, H. Cao and J. Cheng, Chem. Commun., 2010, 46, 7536–7538 RSC.
- B. Valeur, Molecular fluorescence principles and applications, Wiley-VCH, Germany: Weinheim, 2002 Search PubMed.
- İ. Özceşmeci, A. Gelir and A. Gül, Dyes Pigm., 2012, 92, 954–960 CrossRef.
- J.-P. Strachan, S. Gentemann, J. Seth, W. A. Kalsbeck, J. S. Lindsey, D. Holten and D. F. Bocian, J. Am. Chem. Soc., 1997, 119, 11191–11201 CrossRef CAS.
- İ. Özceşmeci, A. Gelir and A. Gül, J. Lumin., 2014, 147, 141–146 CrossRef.
- G. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic data and additional experimental procedures. CCDC 1575485. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7qi00608j |
| This journal is © the Partner Organisations 2018 |