
A hierarchical porous N-doped carbon electrode with superior rate performance and cycling stability for flexible supercapacitors†
Meng
Cheng
ab,
Yuena
Meng
*a,
Qinghai
Meng
b,
Lijuan
Mao
b,
Miao
Zhang
b,
Kamran
Amin
b,
Aziz
Ahmad
b,
Sixin
Wu
*a and
Zhixiang
Wei
 *b
*b
aThe Key Laboratory for Special Functional Material, Henan University, Kaifeng, 474004, P. R. China. E-mail: mengyuena@henu.edu.cn; wusixin@henu.edu.cn
bNational Center for Nanoscience and Technology, No. 11 Beiyitiao Zhongguancun, Beijing 100190, P. R. China. E-mail: weizx@nanoctr.cn
First published on 7th March 2018
Abstract
A novel carbon electrode with high nitrogen content and a hierarchical porous structure has been prepared via a three-step carbonization of a three-dimensional graphene and polyaniline nanoarray composite film. The as-obtained nitrogen-doped electrodes show favorable features for flexible electrochemical energy storage, such as a hierarchical porous structure with interconnected three-dimensional (3D) pores, high flexibility and rich nitrogen-doping (9.71%). With respect to the multiple synergistic effects of these merits, a two-electrode-based supercapacitor has been fabricated. The assembled supercapacitor exhibited a high specific capacitance of 221 F g−1 at 5 mV s−1, and an excellent rate capability with a capacitance retention of 84% when the current density was varied from 3 A g−1 to 50 A g−1, and demonstrated no capacitance reduction even after 20 000 cycles of charging and discharging at 5 A g−1. Moreover, when the electrode was assembled into a flexible device, it exhibited good flexibility with 96% retention of its initial capacitance after 10 000 cycles at 5 A g−1 when the flexible device was bent to 180°. The strategy in this study represents a facile strategy for constructing well-defined 3D structures with a high nitrogen-doping content, which is promising for constructing a flexible energy storage system.
Introduction
Owing to their unique mechanical, electronic and chemical properties, heteroatom-doped materials especially nitrogen-doped carbon (NC) materials have recently been found to offer great potential in various applications such as solar cells,1,2 electrocatalysts,3,4 lithium batteries,5,6 and supercapacitors.7–10 Supercapacitors are appealing energy storage devices with high power density and long cycling life.11–13 When used as electrode materials for supercapacitors, NC materials exhibit an enhanced energy storage ability compared with their undoped carbon counterparts due to their better conductivity, better electrolyte wettability, and more importantly, the additional redox reactions induced by the nitrogen.14–16 Various approaches have been developed to prepare N-doped carbon materials, and these approaches include chemical vapor deposition (CVD) growth,8,17,18 post treatment with N-containing small molecules,19,20 and pyrolysis of N-containing precursors.12,21,22 The most superior materials with a high specific capacitance of 855 F g−1 and a good cycling stability in a three-electrode system are prepared using the CVD method.8 However, the application of the CVD method is limited by the high cost, low yield and presence of metal residuals in the final product. In the post-treatment route, an easier procedure is employed, although a generally lower nitrogen content is introduced compared with that in other routes.15 In the pyrolysis method, the nitrogen species, nitrogen content, and morphology of the NC material can be modulated by changing the initial state of the nitrogen precursor and the temperature.23 Moreover, the nanoscale architecture of NC materials provides continuous electron pathways, ensuring good electrical contact.24–26 These nanomaterials,2,25,27 however, normally require binders to produce electrodes, which normally reduce their electron transport properties. Therefore, to display high performance, NC electrode materials require a high N-doping level and interconnected porous structure to facilitate good electron and ion transport during a fast charge–discharge process.With the rapid development of portable electronic devices, flexible energy storage devices have shown great potential application as power sources.28–33 Carbon-based electrodes, especially graphene and carbon nanotube films with high flexibility and electrochemical stability, have been recently considered to be candicates for free standing flexible electrodes.34–36 However, their capacitances are limited by the energy storage mechanism of electrochemical double layer formation, which is determined by the available surface area of the electrode.37–39 Nano-structured pseudocapacitor materials, such as metal oxides and conducting polymers, are used to improve the energy storage ability of flexible carbon electrodes.30,40–43 In our previous work, polyaniline (PANI) nanoarrays were grown on a flexible porous graphene film, forming a hierarchical composite film, which demonstrates enhanced specific capacitance and rate performance.44 However, the above pseudocapacitor materials normally show poor cycling stability compared with electrochemical double-layer capacitors.45 A NC material, which combines the properties of both an electrical double-layer capacitor and a pseudocapacitor, is the best choice to balance the performance of good stability and high capacitance. Given the above, developing a flexible carbon freestanding film with high N-doping level and hierarchical structure is an ideal method to obtain electrodes displaying high performance and good stability.
In this manuscript, we report a hierarchical structured flexible carbon electrode with a high N-doping level. The free-standing flexible N-doped carbon materials with a nano-array structure and a hierarchical porous structure were fabricated through controllable carbonization of three-dimensional graphene and polyaniline composite films (RGO/PANI) via a three-step pyrolysis process. By contrast, materials obtained via a one-step or two-step pyrolysis process display a considerably decreased hierarchical structure. Owing to the synergistic effect of the hierarchical porous structure and N-doped carbon nano-arrays, the composite film exhibited a high specific capacitance of 221 F g−1 at 5 mV s−1, and an excellent rate capability with a capacitance retention of 84% as the current density was varied from 3 A g−1 to 50 A g−1, and demonstrated no capacitance reduction even after 20 000 cycles of charging and discharging at 5 A g−1. Moreover, a flexible device was assembled, and exhibited good flexibility with 96% retention of its initial capacitance after 10 000 cycles at 5 A g−1 when it was bent to 180°.
Experimental
Materials
Aniline was distilled before use. All other reagents were of analytical grade and used as received. GO was prepared from natural flaked graphite according to a modified Hummers method.46 The GO production process is described in the ESI.†Carbonization of three-dimensional RGO/PANI composite films
The preparation method for the three-dimensional RGO/PANI films was reported in our previous paper.44 The RGO/PANI composite films were carbonized in a horizontal furnace under a pure argon (Ar) gas atmosphere by using different annealing processes. The typical three-step process was performed as follows: After being dried in a vacuum oven at 60 °C for 12 h, the three-dimensional RGO/PANI film was placed in a tube furnace under flowing Ar, stored at 150 °C for 5 h at a heating rate of 1 °C min−1, further warmed up to 450 °C at a heating rate of 1 °C min−1, and heated up to 700 °C at a rate of 5 °C min−1, and the temperature was maintained at 700 °C for 3 h. The sample obtained via the three-stage pyrolysis was denoted as RGO/NC-3. For comparison, samples annealed via one-step (directly pyrolyzed at 700 °C for 3 h at a heating rate of 1 °C min−1) and two-step pyrolysis (heated up to 450 °C at 1 °C min−1, warmed up to 700 °C at 5 °C min−1, and carbonized at 700 °C for 3 h) were prepared and denoted as RGO/NC-1 and RGO/NC-2, respectively.Fabrication of supercapacitors
A symmetric supercapacitor of coin cell type was fabricated to evaluate the supercapacitive performance of the products. Briefly, the freestanding films were cut into round slices of electrode and tested in 2032-type coin cells at room temperature. Without the addition of a binding agent or conductive additive, such small pieces of film can maintain good mechanical strength. The electrodes were assembled in a stainless steel coin cell with a hydrophilic filter membrane as a separator, gold foils as a current collector and 0.5 mol L−1 H2SO4 as an electrolyte. The mass of each electrode was 0.8–0.9 mg. The thickness of the electrode film is estimated to be 90 μm by SEM.A symmetric flexible device was assembled to test the flexibility of the electrode. A gold-coated polyimide film was prepared by the vacuum evaporation method as both the current collector and encapsulating material. Two of the as-prepared N-doped carbon electrodes were assembled together with a separator (hydrophilic polytetrafluoroethylene membrance) sandwiched in between and 0.5 mol L−1 H2SO4 was used as the electrolyte. The edge of the flexible device was encapsulated by double-faced gum.
Characterization
The as-prepared films were characterized via scanning electron microscopy (SEM, Hitachi S-8220), Brunauer–Emmett–Teller (BET, Micromeritics Tristar II 3020), Thermogravimetric analysis (TGA, Diamond TG/DTA), X-ray photoelectron spectroscopy (XPS, ESCALAB250Xi), and Raman spectroscopy (Renishaw inVia plus, UK).Electrochemical measurements
The electrochemical performances of the electrodes were characterized via cyclic voltammetry (CV), galvanostatic charge–discharge (GCD) tests, and electrochemical impedance spectroscopy (EIS). The CV tests were performed at a potential range of 0 V to 1 V. The EIS tests were performed at a frequency range of 100 kHz to 0.01 Hz. The experiments were performed on an EG&G Princeton Applied Research VMP3 workstation. The cycle performance test was conducted on an Arbin STCS-5V1A at 5 A g−1.Results and discussion
Morphological aspects
The three-dimensional RGO/PANI composite was produced by the polymerization of PANI nanowire arrays in a 3D porous RGO film by a reported procedure.44 We propose a three-step process to maximally retain the array morphology and structure during the conversion of the PANI nanoarray into NC. Fig. 1 shows the typical SEM images of a pristine three-dimensional RGO/PANI composite material and carbonized RGO/PANI obtained via different pyrolysis processes. Fig. 1a and b show the SEM images of RGO/PANI at low and high magnifications, respectively. The vertically aligned nanoarrays were obtained on the outer and inner surfaces of the three-dimensional RGO. Fig. 1c and d show the SEM images of the RGO/NC-1 and RGO/NC-2 obtained via one- and two-step pyrolysis, respectively. Retaining the struture of the original nanoarrays via one-step pyrolysis was challenging, and the PANI nanoarrays were nearly completely decomposed (Fig. 1c). By contrast, the nanoarray morphology in RGO/NC-2 was retained to a certain extent after a two-step pyrolysis (Fig. 1d), although the nanoarrays are more sparse in RGO/NC-2 than in RGO/PANI. The morphology of the PANI nanoarrays was maintained in RGO/NC-3 after a three-step high-temperature carbonization process, although the size of the nanoarrays slightly decreased (Fig. 1f). The reason for this is that the three-step pyrolysis can guarantee enough time for the further cross-linking of PANI before it is converted into the conjugate structure. The RGO/NC-3 film showed a hierarchical structure while maintaining interconnected three-dimensional pores (Fig. 1e), which facilitated ion diffusion when the film was used as an electrode. The inset in Fig. 1e shows that the film displayed a good flexibility and was bent easily after the three-step carbonization. The SEM image in Fig. S8 (ESI†) shows the morphology of the electrode after the cycling performance test; there is not much difference compared to the morphology of the electrode before the test. The specific surface area of the RGO/NC-3 is 218 m2 g−1 as measured by the Brunauer–Emmett–Teller method (Fig. S4a, ESI†). The pore size distributions derived from the adsorption branches by using the BJH model indicate that RGO/NC-3 has a mesopore peak at 3.9 nm (Fig. S4b, ESI†).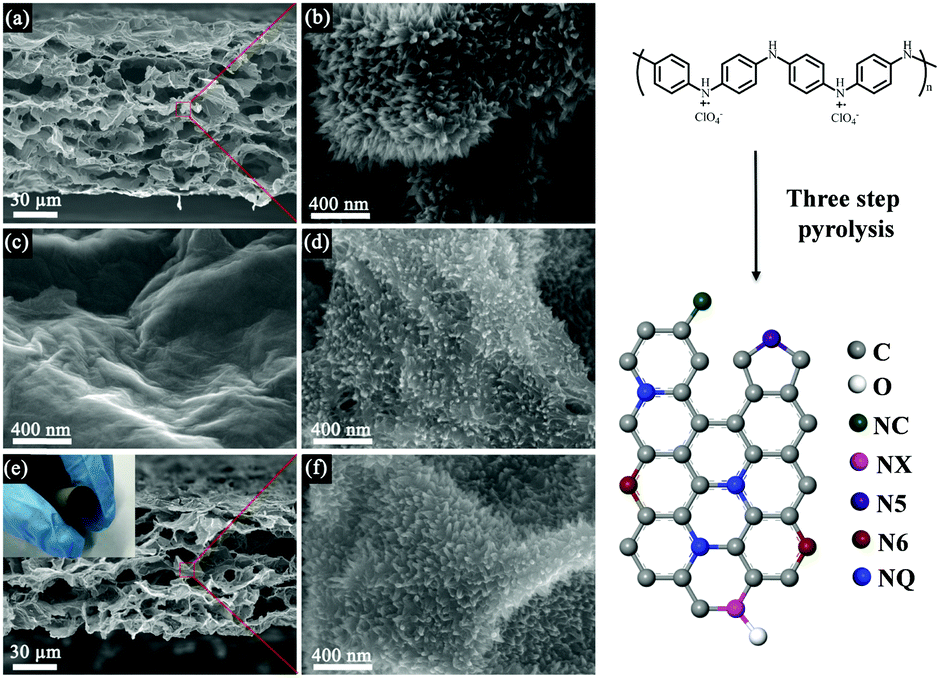 | ||
| Fig. 1 (a) and (b) Low and high magnification scanning electron microscopic (SEM) images of the three-dimensional graphene and polyaniline composite film (RGO/PANI). High-magnification SEM images of (c) RGO/PANI annealed via one-step pyrolysis (RGO/NC-1) and (d) RGO/PANI annealed via two-step pyrolysis (RGO/NC-2). (e) and (f) Low and high magnification SEM images of RGO/PANI annealed via three-step pyrolysis (RGO/NC-3); the inset in (e) illustrates the flexibility of the RGO/NC-3 film. | ||
Mechanism of multi-step pyrolysis
We performed thermogravimetric analysis (TGA) to understand the three-step pyrolysis mechanism (Fig. 2a). 3D-RGO/PANI showed four weight loss processes from room temperature to 1000 °C, and these processes can be attributed to the evolution of water, cross-linking of the PANI chains and the formation of phenazine heterocycles, breakdown of the polymer backbone resulting in the formation of the nitrogen doped carbon material, and its eventual transformation into amorphous carbon.47 The temperature of each pyrolysis section was chosen according to the TGA result in order to retain the array structure and simultaneously achieve the maximum N doping level. Fig. 2b shows the reaction processes in the three-step carbonization, and the samples collected in each reaction point were denoted as stages I, II, III, IV, V, and VI. The morphologies and chemical structures of these samples were characterized systematically. Their corresponding SEM images are shown in the ESI† (Fig. S3a–f).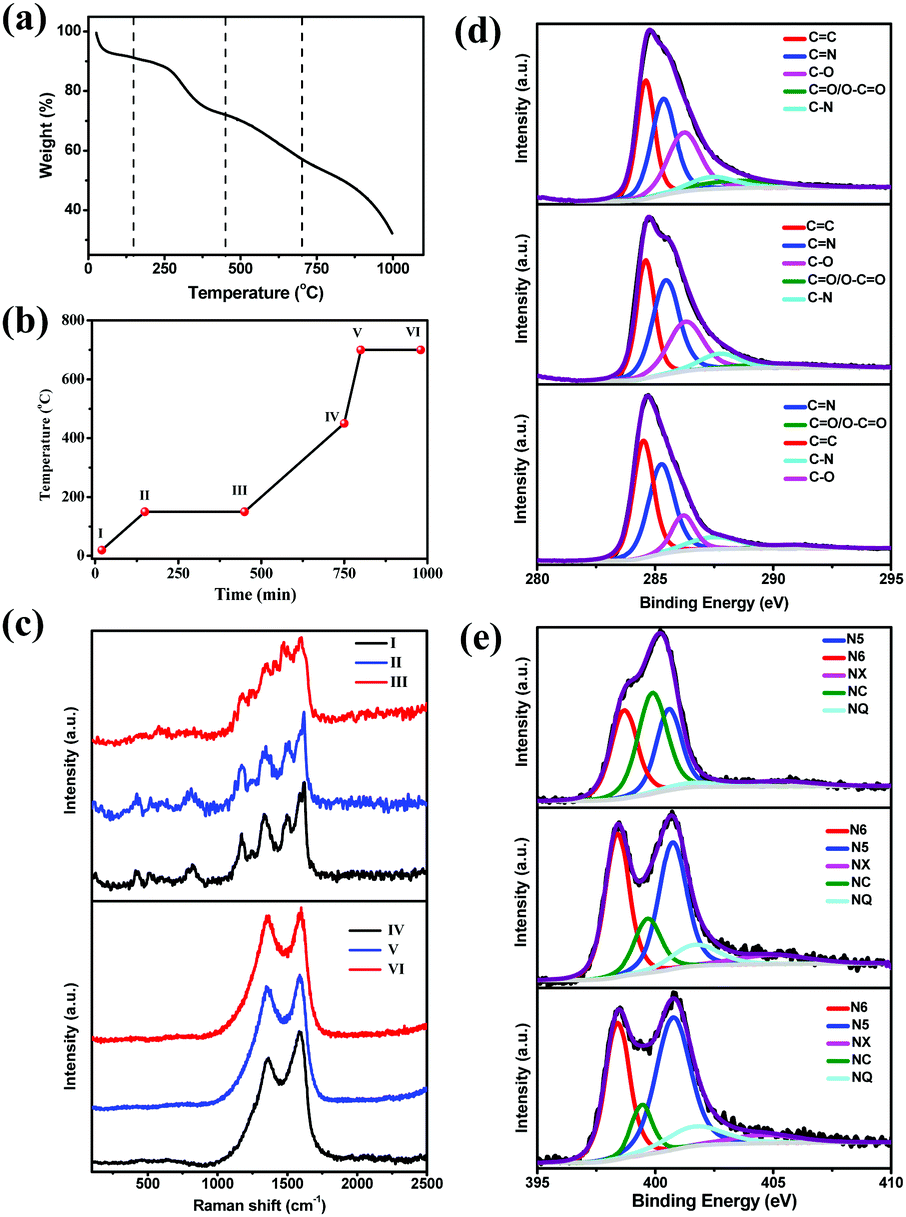 | ||
| Fig. 2 (a) Thermogravimetric analysis curve of the three-dimensional RGO/PANI. (b) Pyrolysis process in the three-step carbonization; reaction points were denoted as I, II, III, IV, V, and VI. (c) Raman spectra (at an excitation wavelength of 514 nm) of the different reaction periods during the three-step carbonization. (d) High-resolution C 1s spectra with peak deconvolution at I, II, and III. (e) High-resolution N 1s spectra with peak deconvolution at IV, V, and VI. | ||
The chemical bond structure of the composite film in different states as time progressed was studied via Raman spectroscopy (Fig. 2c). The Raman spectrum (obtained at 514 nm laser excitation) of the original RGO/PANI (see the spectrum of sample I in Fig. 2c) is similar to that reported in the literature.48,49 Table S2 (ESI†) shows the assignment of the characteristic Raman peaks of the three-dimensional RGO/PANI. Fig. 2c (top) shows the evolution of the Raman spectra of samples heated from stages I to III. As the reaction proceeded from stages I to III, the intensity of some of the characteristic peaks decreased, such as those assigned to N–H stretching vibration at 1514 cm−1, C–N+ vibration of delocalized polaronic structures at 1336 cm−1, and C–H bending vibration of the benzene ring and quinonoid ring at 1174 cm−1.48 By contrast, the intensity of other characteristic peaks increased, such as those peaks assigned to the phenazine-like or crosslinked structure at 1414 cm−1,49 attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching vibration of the quinonoid ring at 1498 cm−1 and the C–N stretching vibrations at 1239 cm−1.48 All of these changes support the concept that crosslinking is probably the most important reaction that modifies the molecular structure of PANI during ageing, wherein the PANI structure evolved into phenazine-like rings, which are beneficial in N doping.47 The spectrum changed dramatically after heat treatment at 450 °C (curve IV in Fig. 2c). The overall shape of the spectrum corresponds to carbonaceous material with two main bands derived from cross-linking and breakage of PANI chains. As shown in Fig. 2c (bottom), the Raman spectra of the samples treated from IV to VI consist of two broad bands centered at 1582 cm−1 (G peak) and 1358 cm−1 (D peak), which are specific to the graphite-like structure and to the defective sites of the graphite-like structure,50 respectively. Previous studies have shown that the intensity ratio of the D peak to G peak (ID/IG) can be used to estimate the degree of disorder in carbon samples.21,26,51 The calculated ID/IG values are 0.8, 0.91, and 0.94 for IV, V, and VI, respectively. The increased ID/IG ratio suggests that the defect sites in the graphite-like structure gradually increased.
N stretching vibration of the quinonoid ring at 1498 cm−1 and the C–N stretching vibrations at 1239 cm−1.48 All of these changes support the concept that crosslinking is probably the most important reaction that modifies the molecular structure of PANI during ageing, wherein the PANI structure evolved into phenazine-like rings, which are beneficial in N doping.47 The spectrum changed dramatically after heat treatment at 450 °C (curve IV in Fig. 2c). The overall shape of the spectrum corresponds to carbonaceous material with two main bands derived from cross-linking and breakage of PANI chains. As shown in Fig. 2c (bottom), the Raman spectra of the samples treated from IV to VI consist of two broad bands centered at 1582 cm−1 (G peak) and 1358 cm−1 (D peak), which are specific to the graphite-like structure and to the defective sites of the graphite-like structure,50 respectively. Previous studies have shown that the intensity ratio of the D peak to G peak (ID/IG) can be used to estimate the degree of disorder in carbon samples.21,26,51 The calculated ID/IG values are 0.8, 0.91, and 0.94 for IV, V, and VI, respectively. The increased ID/IG ratio suggests that the defect sites in the graphite-like structure gradually increased.
Furthermore, X-ray photoelectron spectroscopy (XPS) was employed to collect information on the chemical composition and estimate the bonding states of the samples obtained through different pyrolysis processes. Fig. 2d depicts the comparative deconvoluted C 1s spectra of the samples I, II and III; after deconvolution, the five separated peaks clearly corresponded to graphitic sp2-hybridized C (CC, 284.5 ± 0.1 eV), carbon–nitrogen double bonds (CN, 285.4 ± 0.1 eV),52 hydroxyl and epoxy linkages (C–O, 286.2 ± 0.1 eV), carbon–nitrogen single bonds (C–N, 287.6 ± 0.2 eV), and carbonyl and carboxylic groups (CO/O–CO, 288.5–291.0 eV).53Table 1 shows the relative amount of carbon atoms in different states. As the reaction process proceeds from I to III, the amounts of CC, CN, and C–N increased, whereas those of C–O and CO/O–CO decreased greatly. This result obviously suggests that the PANI structure evolved into a phenazine-like or cross-linked structure, which is beneficial for N doping. Moreover, prolonging the holding time in processes II to III is conducive for sufficient formation of crosslinking structures for the PANI molecular chains, and this approach is beneficial for the conversion of the cross-linked structure into a conjugated structure in the subsequent reaction processes. As the reaction process proceeds from IV to VI, the N 1s core level spectra of the three samples were deconvoluted into five components (Fig. 2e), namely, pyridinic–N (N6, 398.5 ± 0.2 eV), pyrrolic–N (N5, 400.7 ± 0.1 eV), quaternary–N (NQ, 401.6 ± 0.2 eV), amides/amines or nitrile N (NC, 399.6 ± 0.2 eV), and N-oxides (NX, 403–405 eV).11,54 The relative atomic ratios of each type of N species are summarized in Table 2 to further understand the N doping mode in these hybrids. Fig. 2e and Table 2 show that as the reaction proceeds from IV to VI, the N5 and N6 contents significantly increased, enhancing the surface polarity, electric conductivity, and electron-donor tendency of the carbon materials.8,12
| Samples | CC (%) |
CN (%) |
C–O (%) | C–N (%) | CO/O–CO (%) |
|---|---|---|---|---|---|
| I | 28.17 | 30.90 | 25.4 | 7.23 | 8.31 |
| II | 29.60 | 34.45 | 23.47 | 8.17 | 4.32 |
| III | 38.22 | 36.69 | 12.86 | 9.82 | 2.41 |
| Samples | N6 (%) | NC (%) | N5 (%) | NQ (%) | NX (%) |
|---|---|---|---|---|---|
| IV | 29.46 | 36.8 | 25.87 | 3.36 | 4.5 |
| V | 31.11 | 18.66 | 36.3 | 9.25 | 4.68 |
| VI | 31.52 | 10.22 | 42.6 | 10.6 | 5.06 |
Fig. S5a (ESI†) shows the wide-scan XPS survey spectra of the RGO/NC samples. The peaks at approximately 285.1, 399.9, and 532.3 eV correspond to the characteristic peaks of C 1s, N 1s, and O 1s.26 The existence of N 1s indicates successful N doping in all three samples during pyrolysis. Quantitative analysis indicates that the N content of RGO/NC-3 was 9.70%, significantly higher than that of RGO/NC-2 (6.66%) and RGO/NC-1 (3.17%); this result suggests that segmented heat treatment improves N content, which is expected to exert a positive effect on energy storage. Table S1 (ESI†) shows the relative content of N atoms in different states. As for RGO/NC-3, the percentage of N atoms is much higher on the edge of the graphite plane (N5, N6, and NX) than in the middle of the graphite plane (NQ). NQ with the center and valley position of the defective graphene lattice can effectively improve electronic conductivity, electron transfer and capacitance especially at high rates.2 The N6 bonding configuration has two carbon atoms at the defect site and contributes one P electron to the π system and N5 contributes two P electrons to the π system,25 and these negatively charged nitrogen groups can provide abundantly accessible defects for vast charge storage and active sites for generating additional surface intercalation capacitance14,55 and increasing the cycling stability for supercapacitors. Owing to its porous, N-doped, and conductive network structure, the three-dimensional RGO/NC-3 (sample VI) is expected to display an excellent electrochemical performance in supercapacitors.
Electrochemical characteristics
To evaluate the supercapacitive performance of the different samples, we fabricated two-electrode coin-type supercapacitors with 0.5 mol L−1 H2SO4 aqueous solution as an electrolyte. The performance was evaluated via cyclic voltammetry (CV), galvanostatic charge–discharge, electrochemical impedance spectroscopy (EIS), and cycle life tests. Specific capacitance can be calculated according to the equation: C = 2(I × t)/(m × V), where C is specific capacitance (F g−1), I and t are charge–discharge current and time, respectively, V is the charge voltage range, which is 1 V in our measurement, and m is the mass of one piece of the whole film electrode. Fig. 3 shows the typical electrochemical performance of the three-dimensional RGO/NC-based supercapacitors. Fig. 3a shows the nearly triangular charge–discharge curves with a slight bend, a result of the pseudocapacitive effect. Moreover, RGO/NC-3 displays the longest discharging time, implying that RGO/NC-3 possesses the highest specific capacitance. The specific capacitance calculated based on the galvanostatic charge–discharge curves under different current densities is shown in Fig. 3b. For RGO/NC-3, the specific capacitance at 3 A g−1 was 202 F g−1. When the current density was increased to 50 A g−1, the specific capacitance was 170 F g−1, indicating a capacitance retention ratio of 84%. Both the specific capacitance and the rate performance of the RGO/NC-3 film electrode are higher than those of the reported NC materials.2,21,25 By contrast, the specific capacitance of RGO/NC-1 and RGO/NC-2 decreased more rapidly with the increase in current density, suggesting that bushy nanoarrays combined with a hierarchical porous structure resulted in a good rate performance. Fig. S6a (ESI†) shows the CV curves of RGO/NC-1, RGO/NC-2, and RGO/NC-3 at a scan rate of 20 mV s−1. The CV test results similarly showed that RGO/NC-3 displays a considerably high specific capacitance and an enhanced rate capability. The superior capacitance and rate performance of RGO/NC-3 is attributed to its high N-doped content with dominant N6, N5, and NQ configurations. The Nyquist plots in Fig. 3c show the electrochemical resistance of the three film electrodes. All the curves of the NC materials show a rapid upward trend in the low-frequency region, indicating a good capacitive behavior.40 The equivalent series resistances (Res), where the resistance of the electrolyte is usually predominant, obtained from the x-intercept of the Nyquist plots are 0.29, 0.93, and 1.02 Ω, implying that RGO/NC-3 displays the smallest electrical resistance. The much smaller charge transfer resistance (Rct: the diameter of the semicircle in the high-frequency region) of RGO/NC-3 than the other electrodes originates from the larger ion-accessible surface area and the faster ion diffusion due to the much better wettability endowed by the much higher nitrogen content. The corresponding equivalent circuit diagram (Fig. 3c inset; R1 corresponding to Res, R2 corresponding to Rct51) also indicates that the Res and Rct (Table S3, ESI†) of the RGO/NC-3 electrode could be smaller than those of the other NC electrodes. Compared with the carbon materials carbonized via a one-step or two-step pyrolysis process, wherein these materials do not show a significant hierarchical structure, the sample annealed through a three-step carbonization demonstrates an improved electrochemical impedance property and enhanced electrochemical energy storage properties when used as a supercapacitor electrode material.56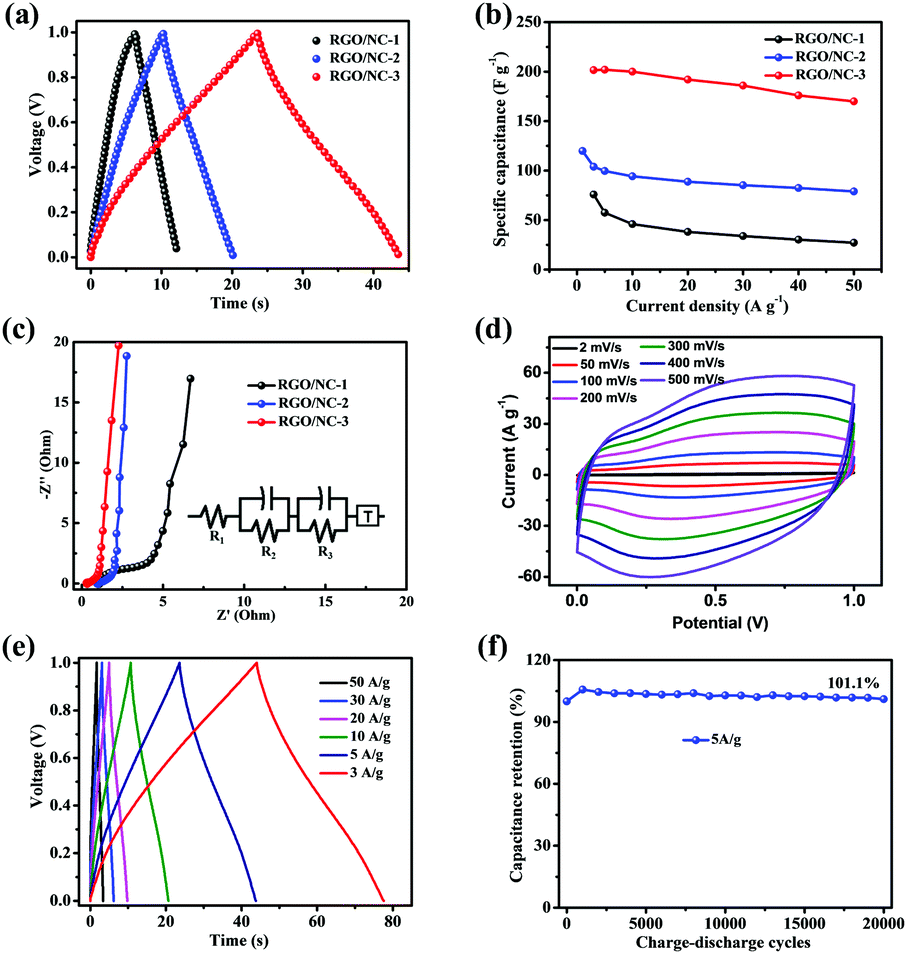 | ||
| Fig. 3 (a) Galvanostatic charge–discharge curves of RGO/NC-1, RGO/NC-2, and RGO/NC-3 at a current density of 5 A g−1. (b) Plot of specific capacitance calculated from the discharge curves at various current densities. (c) Nyquist plots of RGO/NC-1, RGO/NC-2, and RGO/NC-3 (inset: the corresponding equivalent circuit diagram). (d) Cyclic voltammetry curves of RGO/NC-3 at various scan rates. (e) Galvanostatic charge–discharge curves of RGO/NC-3 at various current densities. (f) Capacitance retention of RGO/NC-3 after 20 000 cycles at 5 A g−1. | ||
The electrochemical performances of three-dimensional RGO/NC-3 are presented systematically in Fig. 3d–f. The CV curves of RGO/NC-3 at a scan rate of 2–500 mV s−1 are present in Fig. 3d. All the CV curves exhibit shapes with the quasi-rectangular feature even at a high scan rate of 500 mV s−1, again demonstrating an excellent rate performance. The appearance of humps in the CV curves can be ascribed to extra pseudocapacitance due to redox reactions of active N atoms (N5/N6) in the electrodes.8,12Fig. 3e shows the galvanostatic charge–discharge curves of RGO/NC-3 at different current densities. The charge curves of RGO/NC-3 are nearly linear and symmetrical to its corresponding discharge counterpart, indicating the excellent reversibility of RGO/NC-3. The voltage drop during the measurement is negligible, given that it is too slight to affect measurement accuracy. To further evaluate the cycle life of RGO/NC-3, we performed a galvanostatic charge–discharge measurement at a current density of 5 A g−1 for 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles, RGO/NC-3 demonstrates an excellent stability, maintaining 101.1% of its initial capacitance (Fig. 3f). Interestingly, the specific capacitance for the two electrodes gradually increases during the first 1000 cycles, which may be attributed to the gradual activation process of the N-doped carbon materials.51 Moreover, energy density (E) and power density (P) are important parameters used to evaluate the electrochemical performance of supercapacitors.11,27 Table S2 (ESI†) compares the E and P of the RGO/NC-3 device with those of the recently reported NC material-based supercapacitors. Our RGO/NC-3 electrode in an aqueous electrolyte can maintain 84.3% of max enengy density under a high power density of 25000 W kg−1. The comparison suggests that the energy and power densities of RGO/NC-3 electrodes are comparable or even superior to those of the reported advanced NC materials.
000 cycles, RGO/NC-3 demonstrates an excellent stability, maintaining 101.1% of its initial capacitance (Fig. 3f). Interestingly, the specific capacitance for the two electrodes gradually increases during the first 1000 cycles, which may be attributed to the gradual activation process of the N-doped carbon materials.51 Moreover, energy density (E) and power density (P) are important parameters used to evaluate the electrochemical performance of supercapacitors.11,27 Table S2 (ESI†) compares the E and P of the RGO/NC-3 device with those of the recently reported NC material-based supercapacitors. Our RGO/NC-3 electrode in an aqueous electrolyte can maintain 84.3% of max enengy density under a high power density of 25000 W kg−1. The comparison suggests that the energy and power densities of RGO/NC-3 electrodes are comparable or even superior to those of the reported advanced NC materials.
Flexible characteristics
To test the flexibility of the RGO/NC-3 and the influence of this flexibility on electrical performance, the RGO/NC-3 film was assembled into a flexible film supercapacitor. Fig. 4 shows the pictures and schematics of the film supercapacitor in different bending states. The CV tests were performed at 20 mV s−1 when the supercapacitor was bent into structures with different radii (Fig. 4a, right), namely, 1.17, 0.83, 0.67, and 0.58 cm (the calculation57 was detailed in ESI†); no obvious changes were observed in the CV curves (Fig. 4b) suggesting the stable performance of the RGO/NC-3-based film supercapacitors at different bending states. The bending-induced strain was also calculated using the method reported in the related literature.58,59 And the bending-induced strain as a function of the radius of curvature is shown in Fig. S10 (ESI†). Moreover, the electrochemical performance during the bending process is important in the practical application of flexible supercapacitors. We performed a dynamic bending test to further measure the flexible performance of the prepared flexible film supercapacitor. Fig. 4a (left) shows the states of the film supercapacitor during the dynamic bending test. During the dynamic bending test, the film supercapacitor exhibited excellent cycle stability with 93% retention of its initial capacitance after 2000 cycles at 5 A g−1 (Fig. S8b, ESI†). When the flexible device is bent to 180°, it still can keep 96% of its original capacitance after 10000 cycles at 5 A g−1 (Fig. 4c). Therefore, this material displays great potential for application in the development of a flexible energy storage system.
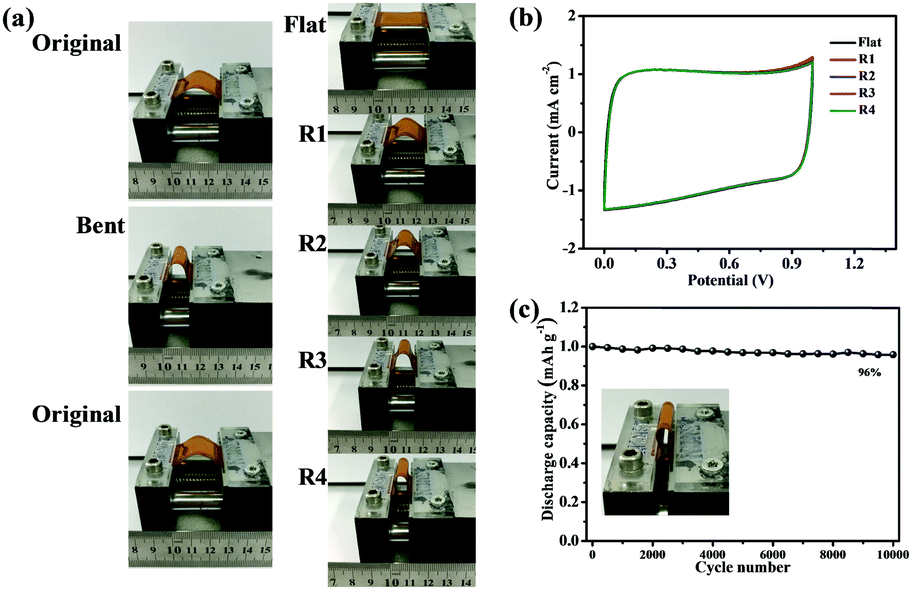 | ||
| Fig. 4 (a) Images of a flexible film supercapacitor during the dynamic bending test (left) and under different bending states (right: the corresponding bending radii from top to bottom were 1.17, 0.83, 0.67, and 0.58 cm). (b) Cyclic voltammetry curves of the film supercapacitor at a scan rate of 20 mV s−1 under different bending states. (c) Capacitance retention of the RGO/NC-3 film supercapacitor after 10000 cycles at 5 A g−1 when the device is bent to 180°. | ||
Conclusions
In summary, we developed a novel carbon electrode with high nitrogen content and hierarchical porous structure via a multi-step pyrolysis process; this electrode displays an excellent electrochemical performance in both coin-type batteries and a flexible device. When the coin-type battery was tested, the electrode exhibited excellent rate capability with a capacitance retention of 84% as the current density was varied from 3 A g−1 to 50 A g−1. Moreover, it demonstrated no capacitance reduction even after 20 000 cycles of charging and discharging at 5 A g−1. This superior rate performance of the electrode is attributed to its hierarchical porous structure, which allowed the electrolyte ions to quickly pass through during the rapid charge–discharge process, and its high nitrogen content, which provides additional pseudo-capacitance with cycling stability for supercapacitors. Furthermore, the electrode was assembled into a flexible device, which also exhibited good flexibility with 96% retention of its initial capacitance after 10 000 cycles at 5 A g−1 when the device is bent to 180°. With freestanding, flexible and high conductive features, this unique structure of three-demensional RGO/NC-3 represents great potential in the development of a flexible energy storage system.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support extended by the Ministry of Science and Technology of China (No. 2016YFA0200700), the National Natural Science Foundation of China (Grant No. 21534003), and the Chinese Academy of Sciences.Notes and references
- Z. P. Wang, D. P. McMeekin, N. Sakai, S. van Reenen, K. Wojciechowski, J. B. Patel, M. B. Johnston and H. J. Snaith, Adv. Mater., 2017, 29, 1604186 CrossRef PubMed.
- W. Q. Tian, Q. M. Gao, L. M. Zhang, C. X. Yang, Z. Y. Li, Y. L. Tan, W. W. Qian and H. Zhang, J. Mater. Chem. A, 2016, 4, 8690–8699 CAS.
- H. Zhu, L. Gu, D. Yu, Y. Sun, M. Wan, M. Zhang, L. Wang, L. Wang, W. Wu, J. Yao, M. Du and S. Guo, Energy Environ. Sci., 2017, 10, 321–330 CAS.
- Z. Pei, H. Li, Y. Huang, Q. Xue, Y. Huang, M. Zhu, Z. Wang and C. Zhi, Energy Environ. Sci., 2017, 10, 742–749 CAS.
- L. P. Wang, Y. Leconte, Z. Feng, C. Wei, Y. Zhao, Q. Ma, W. Xu, S. Bourrioux, P. Azais, M. Srinivasan and Z. J. Xu, Adv. Mater., 2017, 29, 1603286 CrossRef PubMed.
- J. Dong, Y. Xue, C. Zhang, Q. Weng, P. Dai, Y. Yang, M. Zhou, C. Li, Q. Cui, X. Kang, C. Tang, Y. Bando, D. Golberg and X. Wang, Adv. Mater., 2017, 29, 1603692 CrossRef PubMed.
- B. Li, F. Dai, Q. Xiao, L. Yang, J. Shen, C. Zhang and M. Cai, Energy Environ. Sci., 2016, 9, 102–106 CAS.
- T. Q. Lin, I. W. Chen, F. X. Liu, C. Y. Yang, H. Bi, F. F. Xu and F. Q. Huang, Science, 2015, 350, 1508–1513 CrossRef CAS PubMed.
- Q. Xu, X. Yu, Q. Liang, Y. Bai, Z.-H. Huang and F. Kang, J. Electroanal. Chem., 2015, 739, 84–88 CrossRef CAS.
- K. Yuan, T. Hu, Y. Xu, R. Graf, L. Shi, M. Forster, T. Pichler, T. Riedl, Y. Chen and U. Scherf, Mater. Chem. Front., 2017, 1, 278–285 RSC.
- J. Zhao, H. W. Lai, Z. Y. Lyu, Y. F. Jiang, K. Xie, X. Z. Wang, Q. Wu, L. J. Yang, Z. Jin, Y. W. Ma, J. Liu and Z. Hu, Adv. Mater., 2015, 27, 3541–3545 CrossRef CAS PubMed.
- K. Xiao, L. X. Ding, G. X. Liu, H. B. Chen, S. Q. Wang and H. H. Wang, Adv. Mater., 2016, 28, 5997–6002 CrossRef CAS PubMed.
- K. Zhu, Y. Wang, J. A. Tang, S. Guo, Z. Gao, Y. Wei, G. Chen and Y. Gao, Mater. Chem. Front., 2017, 1, 958–966 RSC.
- Y. Cheng, L. Huang, X. Xiao, B. Yao, L. Yuan, T. Li, Z. Hu, B. Wang, J. Wan and J. Zhou, Nano Energy, 2015, 15, 66–74 CrossRef CAS.
- Y. Deng, Y. Xie, K. Zou and X. Ji, J. Mater. Chem. A, 2016, 4, 1144–1173 CAS.
- X. J. Wei, S. G. Wan and S. Y. Gao, Nano Energy, 2016, 28, 206–215 CrossRef CAS.
- J. L. Shi, C. Tang, H. J. Peng, L. Zhu, X. B. Cheng, J. Q. Huang, W. C. Zhu and Q. Zhang, Small, 2015, 11, 5243–5252 CrossRef CAS PubMed.
- Y. Ito, C. Christodoulou, M. V. Nardi, N. Koch, H. Sachdev and K. Muellen, ACS Nano, 2014, 8, 3337–3346 CrossRef CAS PubMed.
- X. Du, H.-Y. Liu and Y.-W. Mai, ACS Nano, 2016, 10, 453–462 CrossRef CAS PubMed.
- H.-L. Guo, P. Su, X. Kang and S.-K. Ning, J. Mater. Chem. A, 2013, 1, 2248–2255 CAS.
- Y. Li, J. Dong, J. Zhang, X. Zhao, P. Yu, L. Jin and Q. Zhang, Small, 2015, 11, 3476–3484 CrossRef CAS PubMed.
- T. Mondal, A. K. Bhowmick and R. Krishnamoorti, Chem. Mater., 2015, 27, 716–725 CrossRef CAS.
- M. Fan, Z.-Q. Feng, C. Zhu, X. Chen, C. Chen, J. Yang and D. Sun, J. Mater. Sci., 2016, 51, 10323–10349 CrossRef CAS.
- Z. Xu, X. Zhuang, C. Yang, J. Cao, Z. Yao, Y. Tang, J. Jiang, D. Wu and X. Feng, Adv. Mater., 2016, 28, 1981–1987 CrossRef CAS PubMed.
- M. Kota, X. Yu, S.-H. Yeon, H.-W. Cheong and H. S. Park, J. Power Sources, 2016, 303, 372–378 CrossRef CAS.
- B. Duan, X. Gao, X. Yao, Y. Fang, L. Huang, J. Zhou and L. N. Zhang, Nano Energy, 2016, 27, 482–491 CrossRef CAS.
- B. Duan, X. Gao, X. Yao, Y. Fang, L. Huang, J. Zhou and L. Zhang, Nano Energy, 2016, 27, 482–491 CrossRef CAS.
- H. Sun, S. L. Xie, Y. M. Li, Y. S. Jiang, X. M. Sun, B. J. Wang and H. S. Peng, Adv. Mater., 2016, 28, 8431–8438 CrossRef CAS PubMed.
- L. B. Dong, C. J. Xu, Y. Li, Z. Z. Pan, G. M. Liang, E. L. Zhou, F. Y. Kang and Q. H. Yang, Adv. Mater., 2016, 28, 9313–9319 CrossRef CAS PubMed.
- S. Z. Wang, C. L. Sun, Y. L. Shao, Y. Z. Wu, L. Zhang and X. P. Hao, Small, 2017, 13, 1603330 CrossRef PubMed.
- Y. Yang, Q. Huang, L. Niu, D. Wang, C. Yan, Y. She and Z. Zheng, Adv. Mater., 2017, 29, 1606679 CrossRef PubMed.
- Y. Lin, Y. Gao and Z. Fan, Adv. Mater., 2017, 29, 1701736 CrossRef PubMed.
- Q. Huang, D. Wang and Z. Zheng, Adv. Energy Mater., 2016, 6, 1600783 CrossRef.
- H.-K. Kim, A. R. Kamali, K. C. Roh, K.-B. Kim and D. J. Fray, Energy Environ. Sci., 2016, 9, 2249–2256 CAS.
- X. Xiao, X. Peng, H. Jin, T. Li, C. Zhang, B. Gao, B. Hu, K. Huo and J. Zhou, Adv. Mater., 2013, 25, 5091–5097 CrossRef CAS PubMed.
- M. Zhang, C. Hou, A. Halder, H. Wang and Q. Chi, Mater. Chem. Front., 2017, 1, 37–60 RSC.
- L. Zhang, Y. Jiang, L. Wang, C. Zhang and S. Liu, Electrochim. Acta, 2016, 196, 189–196 CrossRef CAS.
- B. G. Hyun, H. J. Son, S. Ji, J. Jang, S.-H. Hur and J.-U. Park, J. Electroceram., 2016, 38, 43–50 CrossRef.
- A. Eftekhari and Z. Fan, Mater. Chem. Front., 2017, 1, 1001–1027 RSC.
- Y. Wang, X. Yang, A. G. Pandolfo, J. Ding and D. Li, Adv. Energy Mater., 2016, 6, 1600185 CrossRef.
- G. Qu, J. Cheng, X. Li, D. Yuan, P. Chen, X. Chen, B. Wang and H. Peng, Adv. Mater., 2016, 28, 3646–3652 CrossRef CAS PubMed.
- L. Liu, Y. Yu, C. Yan, K. Li and Z. Zheng, Nat. Commun., 2015, 6, 7260 CrossRef CAS PubMed.
- J. Yang, Y. Liu, S. Liu, L. Li, C. Zhang and T. Liu, Mater. Chem. Front., 2017, 1, 251–268 RSC.
- Y. N. Meng, K. Wang, Y. J. Zhang and Z. X. Wei, Adv. Mater., 2013, 25, 6985–6990 CrossRef CAS PubMed.
- A. Saha, A. Mondal, S. Maiti, S. C. Ghosh, S. Mahanty and A. B. Panda, Mater. Chem. Front., 2017, 1, 1585–1593 RSC.
- K. Krishnamoorthy, M. Veerapandian, K. Yun and S. J. Kim, Carbon, 2013, 53, 38–49 CrossRef CAS.
- M. Trchová, P. Matějka, J. Brodinová, A. Kalendová, J. Prokes and J. Stejskal, Polym. Degrad. Stab., 2006, 91, 114–121 CrossRef.
- Q. Wang, Q. Yao, J. Chang and L. D. Chen, J. Mater. Chem., 2012, 22, 17612–17618 RSC.
- Z. Rozlívková, M. Trchová, M. Exnerová and J. Stejskal, Synth. Met., 2011, 161, 1122–1129 CrossRef.
- L. S. Panchokarla, K. S. Subrahmanyam, S. K. Saha, A. Govindaraj, H. R. Krishnamurthy, U. V. Waghmare and C. N. R. Rao, Adv. Mater., 2009, 21, 4726–4730 Search PubMed.
- K. Xiao, L. X. Ding, H. B. Chen, S. Q. Wang, X. H. Lu and H. H. Wang, J. Mater. Chem. A, 2016, 4, 372–378 CAS.
- Y. Shang, H. Xu, M. Li and G. Zhang, NANO, 2017, 12, 1750018 CrossRef CAS.
- A. Śliwak, B. Grzyb, N. Díez and G. Gryglewicz, Appl. Surf. Sci., 2017, 399, 265–271 CrossRef.
- T. Palaniselvam, M. O. Valappil, R. Illathvalappil and S. Kurungot, Energy Environ. Sci., 2014, 7, 1059–1067 CAS.
- H. M. Jeong, J. W. Lee, W. H. Shin, Y. J. Choi, H. J. Shin, J. K. Kang and J. W. Choi, Nano Lett., 2011, 11, 2472–2477 CrossRef CAS PubMed.
- Y. Gao, Y. Lin, Z. Peng, Q. Zhou and Z. Fan, Nanoscale, 2017, 9, 18311–18317 RSC.
- H. P. Wu, Q. H. Meng, Q. Yang, M. Zhang, K. Lu and Z. X. Wei, Adv. Mater., 2015, 27, 6504–6510 CrossRef CAS PubMed.
- J. Jang, B. G. Hyun, S. Ji, E. Cho, B. W. An, W. H. Cheong and J.-U. Park, NPG Asia Mater., 2017, 9, 172–181 Search PubMed.
- S. Ji, J. Jang, E. Cho, S. H. Kim, E. S. Kang, J. Kim, H. K. Kim, H. Kong, S. K. Kim, J. Y. Kim and J. U. Park, Adv. Mater., 2017, 29, 1700538 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8qm00023a |
| This journal is © the Partner Organisations 2018 |