DOI:
10.1039/C8QM00035B
(Research Article)
Mater. Chem. Front., 2018,
2, 1112-1118
Design and synthesis of a novel azobenzene-containing polymer both in the main- and side-chain toward unique photocontrolled isomerization properties†
Received
20th January 2018
, Accepted 21st March 2018
First published on 21st March 2018
Abstract
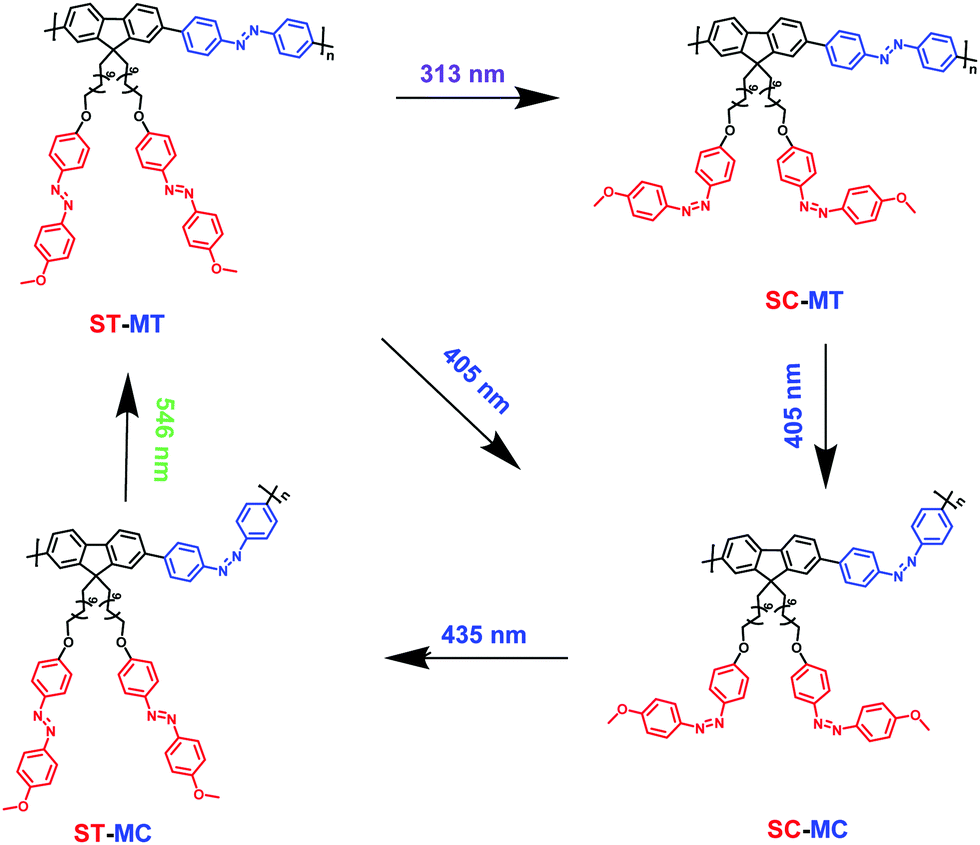
Azobenzene (Azo)-containing polymers have been widely exploited due to their unique reversible trans–cis photoisomerization properties driven by UV/Vis or all visible light. Herein, the copolymer AzoF8-alt-Azo was designed and synthesized, which contained Azo units both in the main- and side-chain. The π–π* band maximum absorption of the side-chain Azo (s-Azo) trans-isomer locates at 360 nm (UV region), and the n–π* band maximum absorption is near 450 nm (Vis region). The π–π* band maximum absorption of the main-chain Azo (m-Azo) trans-isomer locates at 425 nm (Vis region), and the n–π* band maximum absorption is near 520 nm (Vis region). The separation of absorptions of the π–π* bands of the trans-isomers and the n–π* bands of the cis-isomer of the s- and m-Azo units endows the possibility to control the photoisomerization behaviour of different Azo units in the main- and side-chain. The structural changes were achieved upon alternating photoirradiation in the order of 365 nm light (cis-form of s-Azo and trans-form of m-Azo, SC-MT), 435 nm light (trans-form of s-Azo and cis-form of m-Azo, ST-MC), and 546 nm light (trans-form of s- and m-Azo, ST-MT), which were monitored by UV-Vis spectroscopy. This indicates that three forms, SC-MT, ST-MC and ST-MT in same polymer chain can be successfully obtained by the above strategy. Furthermore, upon the irradiation in the order of 313 nm, 405 nm, 435 nm and 536 nm light, four forms, ST-MT, SC-MT, SC-MC and ST-MC, were successfully achieved in the same AzoF8-alt-Azo chain. These interesting findings will not only expand the photoisomerization behaviours of Azo-containing polymers, but also give cues for the design of novel optical materials.
Introduction
The photoisomerization of azobenzene (Azo) chromophore, discovered in 1937, is one of the most striking features of novel Azo-containing materials, since it can be reversibly switched only by versatile light to trigger the change of the molecules’ shape, size and dipole moment.1–6 To date, remarkable examples in the context of photochemical switching applications have been extensively reported including photoactuators, photocontrolled supramolecular chirality, optically switchable electronic devices as well as photoregulated liquid crystal polymers.7–16 Generally, the reversible trans–cis isomerization can be efficiently conducted by alternating irradiation with ultraviolet (UV) and visible (Vis) light, which induces the trans–cis and cis–trans transition through Azo π–π* excitation and n–π* excitation, respectively. For instance, Ikeda et al.9 reported that the incorporation of Azo chromophore into polymer films gave rise to the shape change of materials in response to light, and they found that the bending and unbending of the ferroelectric liquid-crystalline elastomer were successfully constructed upon alternating irradiation with UV and Vis light. In most examples, UV light is necessary to isomerize the switch in at least one way. However, UV light is not suitable for application in biological systems because it is strongly scattered, making the penetration of organisms difficult and severely damaging cells and tissues.17–20
To address these limitations in vivo, an Azo photoswitch completely addressable in the visible region has emerged.17,18,21–27 Thus far, several strategies, such as extended π-conjugation,10,21 push–pull systems,17,19,22 energy transfer27 and electron transfer,26 have been employed to achieve visible-light-activated (VLA) Azo molecules. In the group of Woolley and Hecht,19,22 they followed a related strategy with Azo functionalized in the ortho positions with either the methoxy electron-donating group or the fluorine electron-withdrawing group and prepared the all-VLA Azo molecules according to the electronic effects originating from the ortho substituents. Yu et al.21,27 described that the use of the upconversion nanophosphors or a longer π-conjugated azotolane in response to near-IR light or sunlight stimuli can drive the photoinduced deformation of Azo polymer films along with trans-to-cis isomerization. In the past few decades, most studies were limited to small molecule systems, wherein the examples of VLA Azo polymers were very rare. Zhu et al.28 reported that the photoisomerization process driven by visible light can be attributed to the incorporation of an electron-donating amino substituent (dimethylamino) onto the side-chain Azo group. In our previous studies,10,29,30 larger π-conjugated units (fluorenes) were employed to link with the Azo unit in the main chain, which lowered the HOMO–LUMO gap and caused the red-shift of absorption. As we know, the structure of Azo polymers switched by UV-visible or only visible light is completely different. It will be very intriguing to introduce two kinds of photocontrolled isomerization Azo units to the same polymer chain.
Toward this end, a novel copolymer poly[(9,9-di(4-methoxyazobenzene-4-oxy)octylfluorenyl-2,7-diyl)-alt-4,4′-azobenzene] (AzoF8-alt-Azo) was designed and synthesized by utilizing the Pd-catalyzed Suzuki coupling reaction, which contained the VLA main-chain Azo (m-Azo) and the UV-visible activated side-chain Azo (s-Azo). The π–π* bands of the trans-isomers and the n–π* bands of the cis-isomer of the s- and m-Azo units were effectively separated, which contributed to the controlled photoisomerization behaviour of different Azo units in the main- and side-chain. The controlled photoisomerization changes of s- and m-Azo were easily achieved upon alternating photoirradiation with different wavelengths (313 nm, 365 nm, 405 nm, 435 nm and 536 nm). The current research provides an innovative way to design novel Azo-containing polymer relative optical materials.
Experimental section
Materials
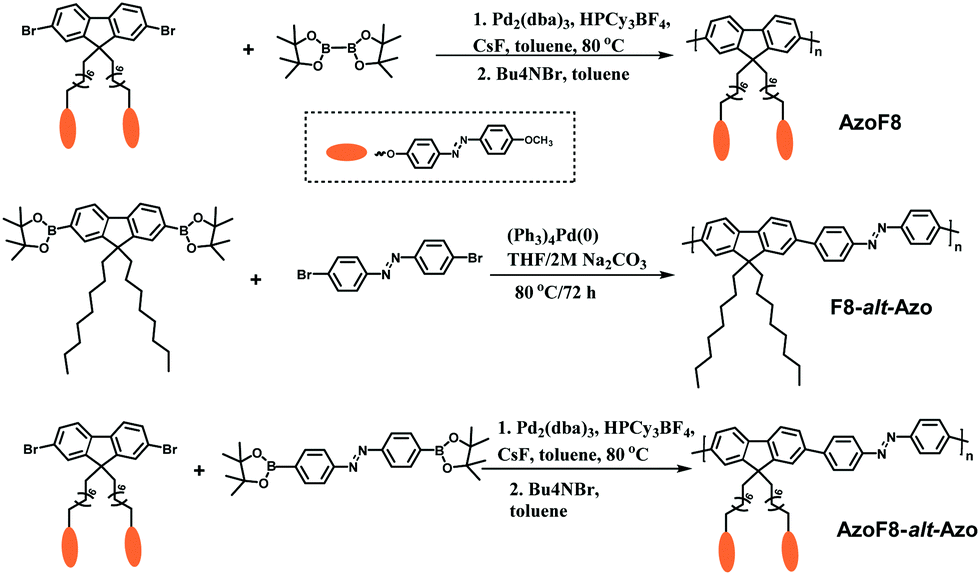
Tetrakis(triphenylphosphine)-palladium (0) (97.0%, TCI (Shanghai) Development Co., Ltd), 4,4,4′,4′,5,5,5′,5′-octamethyl-2,2′-bi-1,3,2-dioxaborolane (98.0%, J&K Scientific Ltd Shanghai), 2,7-dibromo-9H-fluorene (98.0%, TCI (Shanghai) Development Co., Ltd), and 1,8-dibromooctane (99.0%, TCI) were used as received. Dioxane was obtained from TCI (TCI (Shanghai) Development Co., Ltd) and purified by distillation under reduced pressure prior to use. Spectroscopic grade chloroform from TCI Development Co., Ltd was used to prepare the polymer solution for measurements. Anhydrous toluene was purchased from Wako and generally used without further purification. Tetrahydrofuran (THF) (99.9%), acetone (99.9%), dichloromethane (99.9%), methanol, dimethyl sulfoxide, hydrochloric acid (36.0%), potassium carbonate (99.9%), sodium carbonate (85.0%), potassium acetate, sodium hydroxide (98.0%) and other chemicals were obtained from Shanghai Chemical Reagents Co., Ltd and used as received. The monomers 9,9′-dioctyl-2,7-dibromofluorene, azobenzene-4,4′-diboronic ester and the copolymer poly[(9,9-di-n-octylfluorenyl-2,7-diyl)-alt-4,4′-azobenzene] were synthesized according to the previously reported procedures.10 Synthetic routes for the new copolymers are shown in Scheme 1.
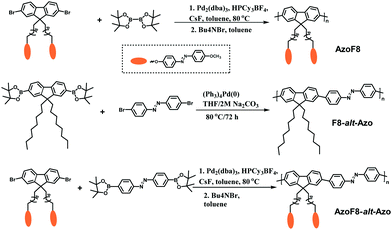 |
| | Scheme 1 Illustration of synthetic routes to AzoF8, F8-alt-Azo and AzoF8-alt-Azo. | |
Synthesis of 9,9-di(4-methoxyazobenzene-4′-oxy)octyl-2,7-dibromofluorene
To a stirred solution of 1-bromo-8-(4-methoxyazobenzene-4′-oxy)octane (2.09 g, 5.0 mmol) and 2,7-dibromo-9H-fluorene (0.74 g, 2.9 mmol) in dimethyl sulfoxide (40 mL) was added sodium hydroxide powder (0.91 g, 22.7 mmol) rapidly at room temperature and then the temperature was raised to 56 °C. The solution was stirred for 36 h at 56 °C. The product was diluted in 200 mL of ethyl acetate. After filtration, the product was extracted with salt brine and washed with water, dried over anhydrous MgSO4, and filtered and concentrated under reduced pressure. The product was recrystallized from a mixture of dichloromethane and methanol to give the desired product as a yellow solid (2.97 g, 86.0%). 1H NMR (300 MHz, CDCl3): δ 7.92–7.82 (8H, m), 7.57–7.41 (6H, m), 7.04–6.91 (8H, m), 4.05–3.93 (4H, m), 3.91–3.86 (6H, s), 1.97–1.86 (4H, m), 1.80–1.66 (4H, m), 1.48–1.00 (16H, m), 0.67–0.52 (4H, s). 13C NMR (300 MHz, CDCl3): δ 161.53, 161.21, 152.49, 147.10, 146.89, 139.07, 130.22, 126.17, 124.34, 121.52, 121.20, 114.68, 114.17, 68.27, 55.69, 55.56, 40.14, 31.61, 29.74, 29.21, 29.13, 29.08, 25.89, 25.61, 22.68.
Preparation of poly[(9,9-di(4-methoxyazobenzene-4-oxy)octylfluorenyl-2,7-diyl)] (AzoF8)
To a 25 mL Schlenk tube containing a magnetic stir bar was added 9,9-di(4-methoxyazobenzene-4′-oxy)octyl-2,7-dibromofluorene (227.5 mg, 0.228 mmol), bis(pinacolato)diboron (57.9 mg, 0.228 mmol), Pd2(dba)3 (4.97 mg, 0.0045 mmol), tricyclohexylphosphonium tetrafluoroborate (4.72 mg, 0.0135 mmol), and cesium fluoride (235 mg, 1.55 mmol). The flask was evacuated and backfilled with argon three times, after which degassed toluene (9 mL) was transferred to the mixture via a cannula. The reaction was immediately immersed in an oil bath at 80 °C and stirred for 24 h at that temperature. Tetrabutylammonium bromide (14.7 mg, 0.046 mmol) dissolved in degassed toluene (1 mL) was added to the reaction via a syringe, and the reaction was stirred for an additional 24 h. The reaction was cooled to room temperature, the solvent was removed in vacuo, and the product was diluted in 100 mL of THF and filtered through neutral alumina and concentrated under reduced pressure, reconstituted in a minimal amount of hot THF (1 mL) and precipitated into a mixture of methanol and water (50 mL, CH3OH/H2O = 10/1 (v/v)). The precipitate was recovered by filtration and washed with a large amount of water. The resulting precipitate was washed for 72 h in a Soxhlet extracting apparatus using acetone to remove oligomers and catalyst residues. The resulting polymer produced a yellow solid (123.6 mg, 41.2%). Mn = 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 g mol−1, Mw/Mn = 1.46. 1H NMR (300 MHz, CDCl3): δ 7.92–7.41 (14H, m), 7.04–6.91 (8H, m), 4.05–3.86 (10H, m), 1.97–0.52 (28H, m).
800 g mol−1, Mw/Mn = 1.46. 1H NMR (300 MHz, CDCl3): δ 7.92–7.41 (14H, m), 7.04–6.91 (8H, m), 4.05–3.86 (10H, m), 1.97–0.52 (28H, m).
Preparation of poly[(9,9-di(4-methoxyazobenzene-4-oxy)octylfluorenyl-2,7-diyl)-alt-4,4′-azobenzene] (AzoF8-alt-Azo)
To a 25 mL Schlenk tube containing a magnetic stir bar was added 9,9-di(4-methoxyazobenzene-4′-oxy)octyl-2,7-dibromofluorene (227.5 mg, 0.228 mmol), azo-4,4′-diboronic ester (99 mg, 0.228 mmol), Pd2(dba)3 (4.97 mg, 0.0045 mmol), tricyclohexylphosphonium tetrafluoroborate (4.72 mg, 0.0135 mmol), and cesium fluoride (235 mg, 1.55 mmol). The flask was evacuated and backfilled with argon three times, after which degassed toluene (9 mL) was transferred to the mixture via a cannula. The reaction was immediately immersed in an oil bath at 80 °C and stirred for 24 h at that temperature. Tetrabutylammonium bromide (14.7 mg, 0.046 mmol) dissolved in degassed toluene (1 mL) was added to the reaction via a syringe, and the reaction was stirred for an additional 24 h. The reaction was cooled to room temperature, the solvent was removed in vacuo, and the product was diluted in 100 mL of THF and filtered through neutral alumina and concentrated under reduced pressure, reconstituted in a minimal amount of hot THF (1 mL) and precipitated into a mixture of methanol and water (50 mL, CH3OH/H2O = 10/1 (v/v)). The precipitate was recovered by filtration and washed with a large amount of water. The resulting precipitate was washed for 72 h in a Soxhlet extracting apparatus using acetone to remove oligomers and catalyst residues. The resulting polymer produced a yellow solid (110.3 mg, 33.8%). Mn = 20200 g mol−1, Mw/Mn = 1.67. 1H NMR (300 MHz, CDCl3): δ 8.20–7.30 (22H, d), 7.04–6.80 (8H, d), 4.05–3.71 (10H, m), 2.20–0.50 (28H, m).
Characterization
1H and 13C NMR spectra were recorded on a Bruker 300 MHz nuclear magnetic resonance instrument using CDCl3 as the solvent and tetramethylsilane (TMS) as an internal standard at 25 °C. The number-average molecular mass (Mn) and polydispersity (PDI = Mw/Mn) of the polymers were determined by using a HLC-8320 size exclusion chromatograph (SEC, TOSOH) equipped with a refractive index detector, using two TSKgel Super Multipore HZ-N columns (4.6 × 150 mm, 3 μm particle size) in series with molecular weights ranging from 7 × 102 to 2 × 105 g mol−1. THF was used as the eluent at a flow rate of 0.35 mL min−1 and 40 °C. These samples were calibrated with PS standards purchased from TOSOH. Ultraviolet-visible (UV-Vis) absorption spectra of the samples were recorded on a Shimadzu UV-2600 spectrophotometer at room temperature. Photoisomerization of the Azo polymer solution was conducted using a 500 W high-pressure mercury lamp (Ushio (Tokyo, Japan), Optiplex SX-UID and 502HUV), and light of different wavelengths (313 nm, 365 nm, 405 nm, 435 nm and 546 nm) were obtained through narrow bandpass filters (Sigma, Koki, Shanghai, China). The light intensities were 190 μW cm−2 at 313 nm, 135.1 μW cm−2 at 365 nm, 159 μW cm−2 at 405 nm, 169 μW cm−2 at 435 nm and 328 μW cm−2 at 546 nm. The thermal behaviors of polymers were measured by using a Q200 DSC instrument.
Results and discussion
Synthesis and characterization of AzoF8, F8-alt-Azo and AzoF8-alt-Azo polymers
Suzuki cross-coupling polymerization is one of the most powerful methods for the synthesis of π-conjugated polymers, which allows straightforward formation of carbon–carbon bonds under the condition of palladium as the catalyst.10Scheme 1 illustrates the synthetic routes of AzoF8, F8-alt-Azo and AzoF8-alt-Azo by Suzuki polycondensation reaction. Firstly, the monomer (9,9-di(4-methoxyazobenzene-4′-oxy)octyl-2,7-dibromofluorene) containing the Azo unit in the side chain was successfully synthesized according to a series of organic reactions, as shown in Scheme S1 and Fig. S1, S2 (ESI†). The homopolymer AzoF8 was achieved by Suzuki coupling polycondensation occurring between the dibromofluorene monomer and bis(pinacolato)diboron. Also, F8-alt-Azo was obtained in a similar way, using Pb(0) as the catalyst. In order to prepare the AzoF8-alt-Azo containing Azo units both in the main- and side-chain, we designed and synthesized the Azo monomer (azobenzene-4,4′-diboronic ester), which is displayed in the ESI.†
Similarly, the Suzuki polycondensation reaction occurred between dibromofluorene and the Azo monomer, and the AzoF8-alt-Azo polymer was successfully obtained with the reversible trans–cis photoisomerization driven by UV/Vis in the side chain and all visible light in the main chain. SEC and NMR analyses were used to characterize the molecular mass and the structure of the π-conjugated polymers. The SEC results showed that the number-average molecular mass and polydispersity indices of AzoF8, F8-alt-Azo and AzoF8-alt-Azo were 18800 g mol−1 and 1.46, 22000 g mol−1 and 1.58, and 20200 g mol−1 and 1.67, respectively.
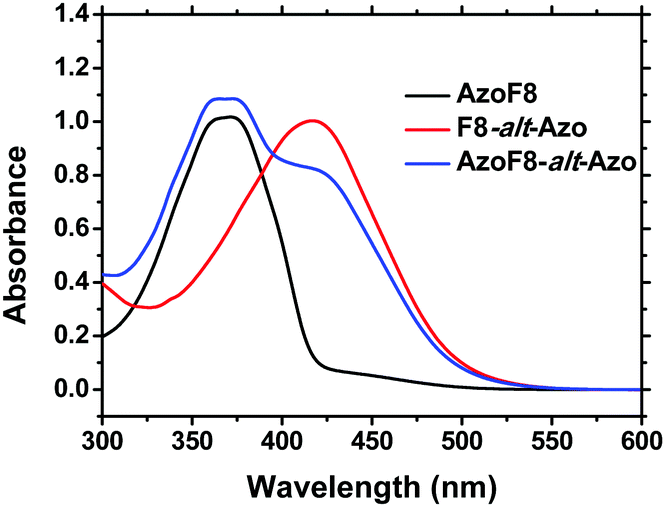
Their structures were satisfactorily identified by the corresponding 1H NMR spectra (Fig. S3 and S4, ESI†). The assignments of each proton and carbon agree well with the proposed molecular structures. The UV-vis spectra (Fig. 1) indicated that the absorption maxima of the π–π* and n–π* band of s-Azo located at 360 nm and 450 nm, while the absorption maxima of m-Azo are near 425 nm and 520 nm. The absorption spectra of AzoF8-alt-Azo demonstrated the complete separation of absorptions of the π–π* bands (∼65 nm) of the trans-isomers and the n–π* bands (∼70 nm) of the cis-isomer of the s- and m-Azo units, providing the possibility to control the photoisomerization behaviors of different Azo units in the main- and side-chain. Additionally, Azo-containing homopolymers31 or block polymers32 often show unique liquid-crystalline phase transition behaviors due to the rigid and rod-like structure of Azo chromophores. DSC results reveal that no obvious liquid-crystalline phases of the synthesized polymers were observed in the range of 20–200 °C (Fig. S5, ESI†).
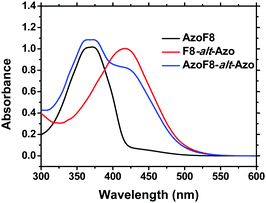 |
| | Fig. 1 UV-Vis absorption spectra of AzoF8 ([repeating unit]0 = 2.38 × 10−5 mol L−1), F8-alt-Azo ([repeating unit]0 = 3.87 × 10−5 mol L−1) and AzoF8-alt-Azo ([repeating unit]0 = 2.45 × 10−5 mol L−1) in CHCl3. | |
Photoisomerization behaviour of AzoF8
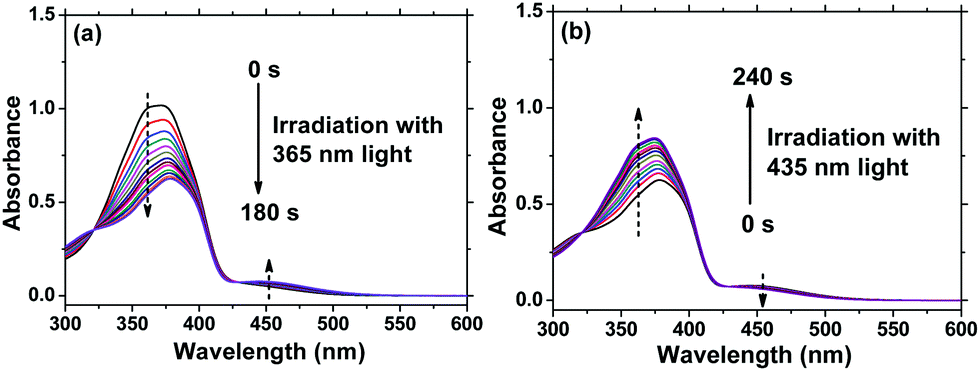
It is well-known that reversible photoisomerization is one of the most fundamental and important properties of Azo polymers under alternative irradiation with UV/Vis light.33 Generally, the absorption spectra show a high-intensity π–π* transition band of the Azo chromophore located in the UV region and a relatively low-intensity n–π* transition band located in the visible region. Here, the position of the π–π* band maximum absorption of AzoF8 is around 360 nm (UV region), and the n–π* band maximum absorption is near 450 nm (visible region). To characterize the procedure of AzoF8 photoisomerization, we dissolved the polymer in chloroform (CHCl3) and selected UV light (365 nm) and Vis light (435 nm) as the stimulus. Upon irradiation with UV light at 365 nm, the UV-Vis spectra displayed that the π–π* absorption band (λmax = 360 nm) gradually decreases and the n–π* transition (λmax = 450 nm) gradually increases (Fig. 2a). The photostationary state of the photoisomerization of AzoF8 was reached after 180 s of 365 nm light irradiation. The reversibility of the photoisomerization process was achieved when the AzoF8 solution was irradiated with 435 nm light, during which the π–π* absorption band gradually increased and the n–π* transition gradually decreased (Fig. 2b). The photostationary state of the isomerization of AzoF8 was reached after 240 s of 435 nm light irradiation. The reversible photoisomerization process can be conducted many times by alternative 365/435 nm light irradiation (Fig. S6, ESI†). To eliminate the effect of 546 nm light on the side-chain Azo isomerization, we irradiated the above polymer solution with 546 nm light and found that no distinct trans–cis transition was detected (Fig. S7, ESI†).
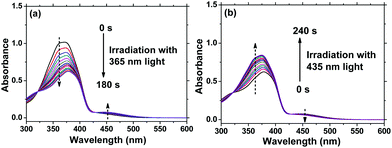 |
| | Fig. 2 Changes in UV-Vis spectra of AzoF8 in CHCl3 under different irradiation times with 365 nm and 435 nm light at 25 °C. (a) 365 nm light, (b) 435 nm light. [repeating unit]0 = 2.38 × 10−5 mol L−1. | |
Photoisomerization behaviour of F8-alt-Azo
Due to the extended π-conjugated system in F8-alt-Azo, the absorption of Azo units is red-shifted to the visible region.10 The position of the π–π* band maximum absorption of F8-alt-Azo is around 425 nm (visible region), and the n–π* band maximum absorption is near 520 nm (visible region). The polymer dissolved in CHCl3 was irradiated with visible light (435 nm and 546 nm). UV-Vis spectra indicated that the π–π* absorption band (λmax = 425 nm) gradually decreased and the n–π* transition (λmax = 520 nm) gradually increased (Fig. S8, ESI†). The photostationary state of the photoisomerization of F8-alt-Azo was reached after 140 s of 435 nm light irradiation. Compared to AzoF8, F8-alt-Azo showed a relatively poorer photoswitching ability. The possible reason is that the relatively rigid main chain of F8-alt-Azo hinders the photoisomerization of Azo units by the rotation mechanism.6 The cis-to-trans photoisomerization was achieved when the AzoF8 solution was irradiated with 546 nm light. The photostationary state of the isomerization of F8-alt-Azo was reached after 40 min of 546 nm light irradiation.
Photoisomerization behaviour of AzoF8-alt-Azo upon alternating irradiation with 365 nm, 435 nm and 546 nm light
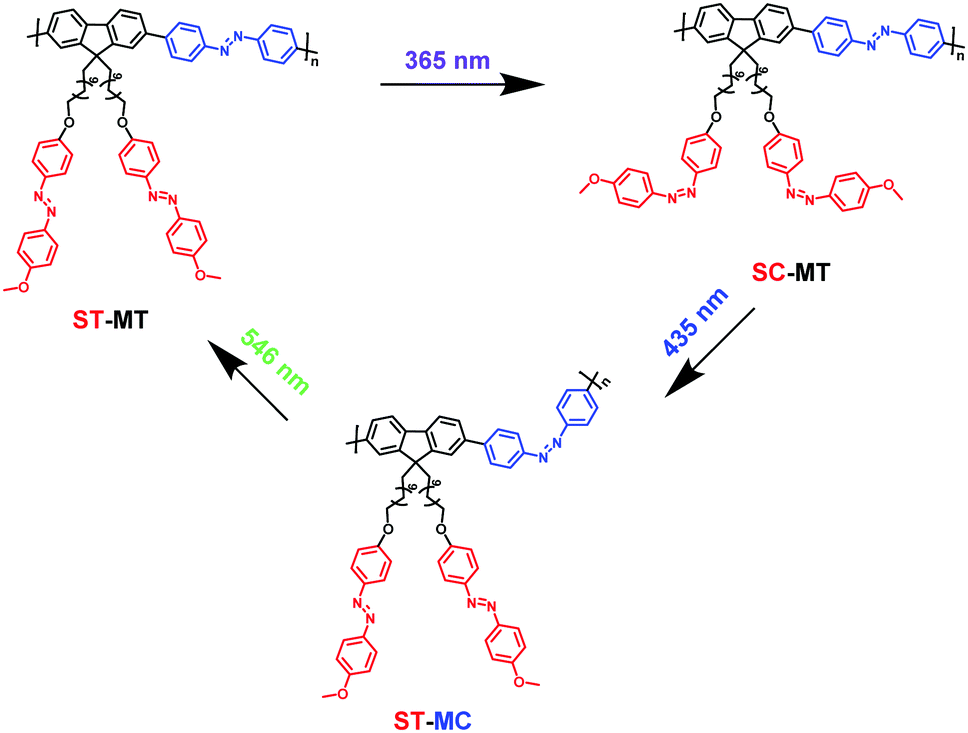
AzoF8-alt-Azo containing UV-visible activated s-Azo and VLA m-Azo units was firstly dissolved in CHCl3 and irradiated by UV light (365 nm) for 100 s. The π–π* absorption band (λmax = 360 nm) of the trans-form of s-Azo gradually decreased, and the n–π* absorption band (λmax = 450 nm) of the cis-form of s-Azo gradually increased (Fig. 3a). However, the π–π* absorption band (λmax = 425 nm) of the trans-form of m-Azo remained almost stable. The spectral variation evidenced that the successful trans-to-cis transition of s-Azo was triggered by UV light while the trans-form of m-Azo was unchanged. Upon irradiation with visible light (435 nm), the π–π* absorption band (λmax = 360 nm) of the trans-form of s-Azo gradually increased, along with the decrease of the π–π* absorption band (λmax = 425 nm) of the trans-form of m-Azo (Fig. 3b). Those changes in the UV-Vis spectra under 435 nm light irradiation confirmed the cis-to-trans isomerization of s-Azo and the trans-to-cis transition of m-Azo simultaneously occurring in the same polymer chain. Further, cis-to-trans isomerization of m-Azo was observed by 546 nm light irradiation while the trans-form of s-Azo remained unchanged, as shown in Fig. 3c and d. Three forms, the trans-form of s- and m-Azo (ST-MT), the cis-form of s-Azo and the trans-form of m-Azo (SC-MT), and the trans-form of s-Azo and the cis-form of m-Azo (ST-MC) in the same polymer chain can be conveniently obtained by alternating photoirradiation in the order of 365 nm, 435 nm and 546 nm light. Based on these results, the photoisomerization process was proposed in Scheme 2.
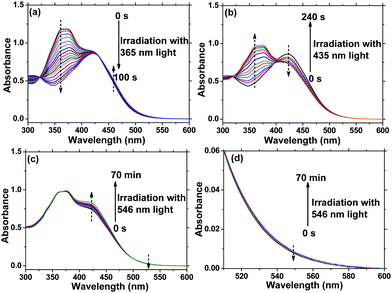 |
| | Fig. 3 Changes in UV-Vis spectra of AzoF8-alt-Azo in CHCl3 under different irradiation times with 365 nm, 435 nm and 546 nm light at 25 °C. (a) 365 nm, (b) 435 nm, (c) 546 nm light, (d) magnified parts of (c). [repeating unit]0 = 2.45 × 10−5 mol L−1. | |
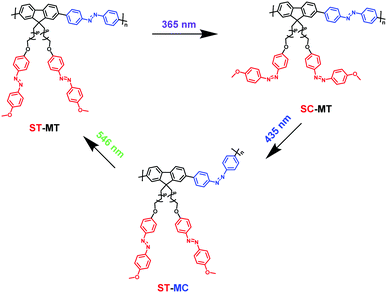 |
| | Scheme 2 Illustration of photoisomerization process of AzoF8-alt-Azo upon irradiation in the order of 365 nm (UV), 435 nm (visible) and 536 nm (visible) light. | |
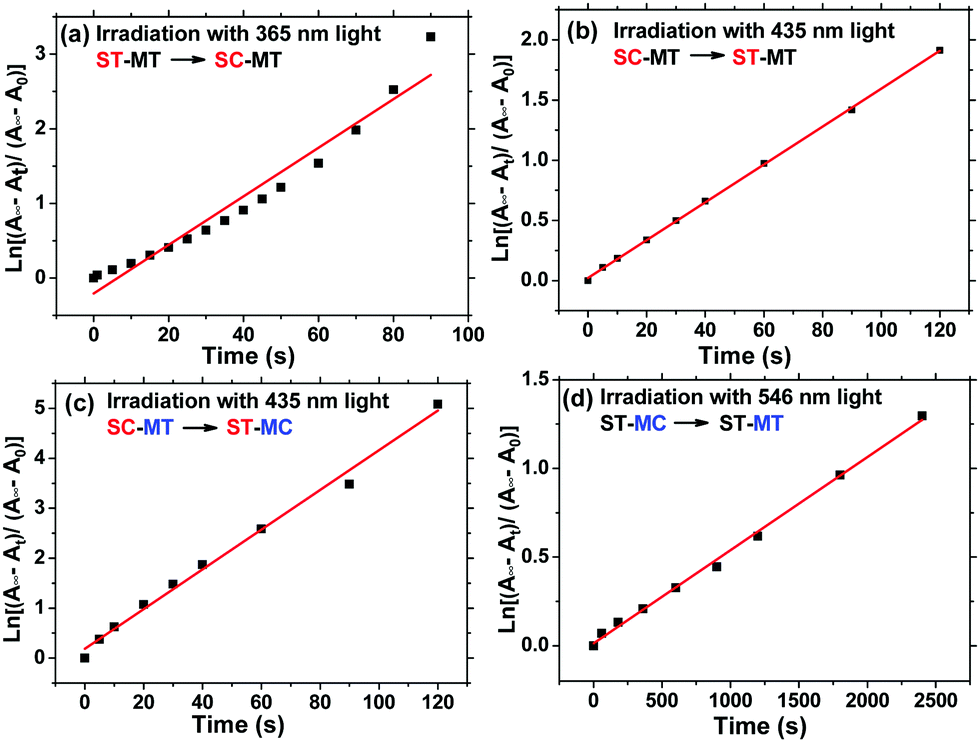
To further investigate the difference of photoisomerization rate of the s- and m-chain Azo units, we calculated the photoisomerization rate constants of trans-to-cis (Kes) and cis-to-trans (KHs) of the Azo unit. Fig. 4 indicates the first-order kinetic plots of the trans-to-cis or cis-to-trans isomerization of the s- and m-Azo units. The Kes and KHs values, calculated in the ESI,† of s-Azo triggered by 365 (UV) and 435 nm (visible) light are 0.0325 and 0.0157 s−1, respectively. The total photoisomerization rate constant of both the s- and m-Azo unit (λmax = 425 nm) via 435 nm light irradiation is 0.0398 s−1. Therefore, the Kes and KHs values of m-Azo triggered by 435 (visible) and 546 nm (visible) light are 0.0241 and 5.31 × 10−4 s−1 (Table 1), respectively. Both the Kes and KHs values of s-Azo are distinctly higher than those of m-Azo, which is attributed to the flexible side-chain structure of s-Azo.
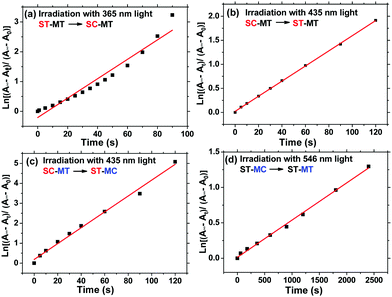 |
| | Fig. 4 First-order kinetics for reversible isomerization of AzoF8-alt-Azo. (a) The trans–cis photoisomerization of s-Azo (ST-SC) via 365 nm light irradiation, (b) the cis–trans photoisomerization of s-Azo (SC-ST) via 435 nm light irradiation, (c) the total photoisomerization of both s- and m-Azo (SC-MT to ST-MC) via 435 nm light irradiation, and (d) the cis–trans photoisomerization of m-Azo (MC-MT) via 546 nm light irradiation. | |
Table 1 Photoisomerization rate of AzoF8-alt-Azo
| Polymer |
λ
max/nm |
λ
irradiation/nm |
K
e/s−1 |
K
H/s−1 |
| AzoF8-alt-Azo |
360 |
365/435 |
0.0325 |
0.0157 |
| 425 |
435/546 |
0.0241 |
5.31 × 10−4 |
Photoisomerization behaviour of AzoF8-alt-Azo upon alternating irradiation with 313 nm, 405 nm, 435 nm and 546 nm light
In order to enrich the structural changes of AzoF8-alt-Azo isomer, we used 313 nm and 405 nm light to substitute 365 nm light, both of which can induce the trans-to-cis isomerization of the s-Azo unit (AzoF8), as displayed in Fig. S9 (ESI†). The m-Azo unit (F8-alt-Azo) can be effectively switched from the trans-form to cis-form under 405 nm (visible) light irradiation, while having no response to 313 nm light (Fig. S10, ESI†). Upon irradiation in the order of 313 nm, 405 nm, 435 nm and 536 nm light, four forms were successfully obtained in the same AzoF8-alt-Azo chain, including the cis-form of s-Azo and the trans-form of m-Azo (ST-MT), the cis-form of s-Azo and the cis-form of m-Azo (SC-MT), the trans-form of s-Azo and the cis-form of m-Azo (SC-MC) and the trans-form of s-Azo and the trans-form of m-Azo (ST-MC). The absorption changes of the s- and m-Azo units were recorded by the UV-Vis spectra (Fig. 5). The first-order kinetic plots, and Kes and KHs values were deduced from Fig. 5, and they are shown in Fig. S11 and Table S1 (ESI†). Based on these results, the four structural forms of the AzoF8-alt-Azo isomer are proposed in Scheme 3 upon the alternating irradiation with 313 nm, 405 nm, 435 nm and 546 nm light.
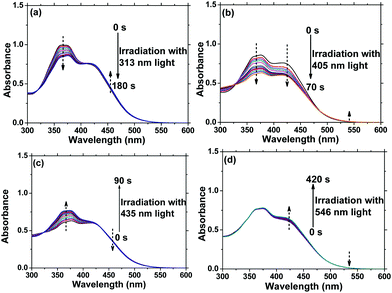 |
| | Fig. 5 Changes in UV-Vis spectra of AzoF8-alt-Azo in CHCl3 under different irradiation times with 313 nm, 405 nm, 435 nm and 546 nm light at 25 °C. (a) 313 nm, (b) 405 nm, (c) 435 nm, (d) 546 nm. [repeating unit]0 = 2.04 × 10−5 mol L−1. | |
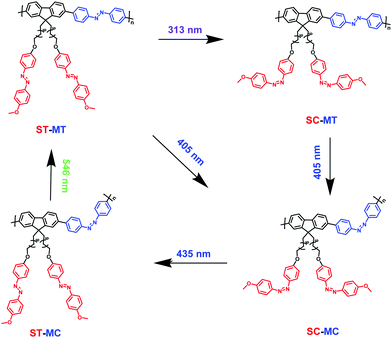 |
| | Scheme 3 Illustration of photoisomerization process of AzoF8-alt-Azo upon irradiation in the order of 313 nm (UV), 405 nm (visible), 435 nm (visible) and 536 nm (visible) light. | |
Back thermal photoisomerization of AzoF8-alt-Azo
In general, the cis-azobenzene isomers can relax very quickly back to the trans one for some small azobenzene molecules and polymers. In order to evaluate the stability of the cis-isomer in the solution, we kept the polymer solution of the cis-photostationary state at 40 °C. As shown in Fig. S12 and S13 (ESI†), the cis-isomer of both the side-chain and main-chain Azo moieties displayed excellent stability, and the relaxation times of returning back to the trans-photostationary state are both more than 250 min at 40 °C. This behaviour might be attributed to the rigid conjugated structure of AzoF8-alt-Azo, which suppresses the photoisomerization transition of Azo moieties.
Conclusions
In summary, a novel copolymer AzoF8-alt-Azo was rationally designed and successfully synthesized by utilizing the Pd-catalyzed Suzuki coupling reaction, containing VLA main-chain Azo (m-Azo) and UV-Vis light activated side-chain Azo (s-Azo). The extended π-conjugated structure of m-Azo causes the effective red-shift of the π–π* absorption band of the m-Azo unit (λmax = 425 nm) and provides the possibility to flexibly control the photoisomerization process of s- and m-Azo irradiated by different wavelength light. Three structural forms can be achieved upon alternating photoirradiation with 365 nm (cis-form of s-Azo and trans-form of m-Azo), 435 nm (trans-form of s-Azo and cis-form of m-Azo), and 546 nm (trans-form of s- and m-Azo) light. Upon irradiation in the order of 313 nm, 405 nm, 435 nm and 536 nm, four forms can be also successfully obtained in the same AzoF8-alt-Azo chain, including the cis-form of s-Azo and trans-form of m-Azo (313 nm), the cis-form of s-Azo and cis-form of m-Azo (405 nm), the trans-form of s-Azo and cis-form of m-Azo (435 nm) and the trans-form of s-Azo and trans-form of m-Azo (546 nm). These interesting findings will provide new cues for the design of novel optical materials.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors are grateful for the financial support from the National Nature Science Foundation of China (21574089), the Priority Academic Program Development (PAPD) of Jiangsu Higher Education Institutions and the Program of Innovative Research Team of Soochow University.
Notes and references
- G. S. Hartley, Nature, 1937, 140, 281 CrossRef CAS
 .
.
- A. A. Beharry and G. A. Woolley, Chem. Soc. Rev., 2011, 40, 4422–4437 RSC .
- H. Koshima, N. Ojima and H. Uchimoto, J. Am. Chem. Soc., 2009, 131, 6890–6891 CrossRef CAS PubMed .
- H. M. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC .
- A. N. Tamai and H. Miyasaka, Chem. Rev., 1999, 100, 1875–1890 CrossRef .
- D. Bléger and S. Hecht, Angew. Chem., Int. Ed., 2015, 54, 11338–11349 CrossRef PubMed .
- J.-a. Lv, Y. Liu, J. Wei, E. Chen, L. Qin and Y. Yu, Nature, 2016, 537, 179–184 CrossRef CAS PubMed .
- X. Lu, S. Guo, X. Tong, H. Xia and Y. Zhao, Adv. Mater., 2017, 29, 1606467 CrossRef PubMed .
- Y. Yu, T. Maeda, J. Mamiya and T. Ikeda, Angew. Chem., 2007, 119, 899–901 CrossRef .
- W. Zhang, K. Yoshida, M. Fujiki and X. Zhu, Macromolecules, 2011, 44, 5105–5111 CrossRef CAS .
- L. Wang, Y. Lu, W. Zhang, X. Zhu and M. Fujiki, J. Am. Chem. Soc., 2017, 139, 13218–13226 CrossRef CAS PubMed .
- G. Zou, H. Jiang, H. Kohn, T. Manaka and M. Iwamoto, Chem. Commun., 2009, 5627–5629 RSC .
- E. Orgiu, N. Crivillers, M. Herder, L. Grubert, M. Pätzel, J. Frisch, E. Pavlica, D. T. Duong, G. Bratina and A. Salleo, Nat. Chem., 2012, 4, 675–679 CrossRef CAS PubMed .
- M. E. Gemayel, K. Börjesson, M. Herder, D. T. Duong, J. A. Hutchison, C. Ruzié, G. Schweicher, A. Salleo, Y. Geerts and S. Hecht, Nat. Chem., 2015, 6, 6330 Search PubMed .
- S. Kurihara, A. Sakamoto, D. Yoneyama and T. Nonaka, Macromolecules, 1999, 31, 6493–6498 CrossRef .
- Z. Cheng, S. Ma, Y. Zhang, S. Huang, Y. Chen and H. Yu, Macromolecules, 2017, 50, 8317–8324 CrossRef CAS .
- A. A. Beharry, O. Sadovski and G. A. Woolley, J. Am. Chem. Soc., 2011, 133, 19684–19687 CrossRef CAS PubMed .
- S. Samanta, A. A. Beharry, O. Sadovski, T. M. McCormick, A. Babalhavaeji, V. Tropepe and G. A. Woolley, J. Am. Chem. Soc., 2013, 135, 9777–9784 CrossRef CAS PubMed .
- S. Samanta, T. M. McCormick, S. K. Schmidt, D. S. Seferos and G. A. Woolley, Chem. Commun., 2013, 49, 10314–10316 RSC .
- M. Dong, A. Babalhavaeji, S. Samanta, A. A. Beharry and G. A. Woolley, Acc. Chem. Res., 2015, 48, 2662–2670 CrossRef CAS PubMed .
- R. Yin, W. Xu, M. Kondo, C.-C. Yen, J.-i. Mamiya, T. Ikeda and Y. Yu, J. Mater. Chem., 2009, 19, 3141–3143 RSC .
- D. Bléger, J. Schwarz, A. M. Brouwer and S. Hecht, J. Am. Chem. Soc., 2012, 134, 20597–20600 CrossRef PubMed .
- C. Knie, M. Utecht, F. Zhao, H. Kulla, S. Kovalenko, A. M. Brouwer, P. Saalfrank, S. Hecht and D. Bléger, Chem. – Eur. J., 2014, 20, 16492–16501 CrossRef CAS PubMed .
- S. Samanta, C. Qin, A. J. Lough and G. A. Woolley, Angew. Chem., Int. Ed., 2012, 51, 6452–6455 CrossRef CAS PubMed .
- R. Siewertsen, H. Neumann, B. Buchheim-Stehn, R. Herges, C. Näther, F. Renth and F. Temps, J. Am. Chem. Soc., 2009, 131, 15594–15595 CrossRef CAS PubMed .
- M. Kurihara, A. Hirooka, S. Kume, M. Sugimoto and H. Nishihara, J. Am. Chem. Soc., 2002, 124, 8800–8801 CrossRef CAS PubMed .
- W. Wu, L. Yao, T. Yang, R. Yin, F. Li and Y. Yu, J. Am. Chem. Soc., 2011, 133, 15810–15813 CrossRef CAS PubMed .
- R. Dong, B. Zhu, Y. Zhou, D. Yan and X. Zhu, Polym. Chem., 2013, 4, 912–915 RSC .
- L. Wang, X. Pan, Y. Zhao, Y. Chen, W. Zhang, Y. Tu, Z. Zhang, J. Zhu, N. Zhou and X. Zhu, Macromolecules, 2015, 48, 1289–1295 CrossRef CAS .
- L. Wang, Y. Chen, L. Yin, S. Zhang, N. Zhou, W. Zhang and X. Zhu, Polym. Chem., 2016, 7, 5407–5413 RSC .
- R. Tang, Z. Liu, D. Xu, J. Liu, L. Yu and H. Yu, ACS Appl. Mater. Interfaces, 2015, 7, 8393–8397 CAS .
- T. Wang, X. Li, Z. Dong, S. Huang and H. Yu, ACS Appl. Mater. Interfaces, 2017, 9, 24864–24872 CAS .
- Y. Zhao and J. He, Soft Matter, 2009, 5, 2686–2693 RSC .
Footnotes |
| † Electronic supplementary information (ESI) available: The UV-Vis, 1H, 13C NMR. See DOI: 10.1039/c8qm00035b |
| ‡ Kai Wang and Lu Yin contributed equally to this work. |
|
| This journal is © the Partner Organisations 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 * and
Xiulin
Zhu
* and
Xiulin
Zhu