
Driving π-plane to π-bowl through lateral coordination at room temperature†
Ruili
Geng
 ,
Xueqing
Hou
,
Yantao
Sun
,
Chaoxian
Yan
,
Yuewei
Wu
,
Hao-Li
Zhang
and
Xiangfeng
Shao
*
,
Xueqing
Hou
,
Yantao
Sun
,
Chaoxian
Yan
,
Yuewei
Wu
,
Hao-Li
Zhang
and
Xiangfeng
Shao
*
State Key Laboratory of Applied Organic Chemistry, Lanzhou University, Tianshui Southern Road 222, Lanzhou 730000, Gansu Province, China. E-mail: shaoxf@lzu.edu.cn; Fax: +86 0931 8915557; Tel: +86 0931 8912500
First published on 18th May 2018
Abstract
A series of hetero polycycles (5a-6b) are created through the hybridization of the bowl-shaped trichalcogenasumanenes and planar triazacoronene. Among them, 6a and 6b bear the 2,2′-bipyridine (bpy) ligand in the lateral direction. The crystallographic analyses show that 6a and 6b possess planar cores, whereas they are driven to bowl-shaped (bowl depth, 0.38–0.50 Å) via chelation of Zn2+ by the bpy unit at room temperature (RT). This is caused by the formation of a five-membered ring upon coordination with Zn2+. The coordination of Zn2+ also brings distinct variation of the optical properties, i.e., the crystals of 6a and 6a[ZnCl2] respectively show green and red fluorescence. Moreover, 5a-6b exhibit a reversible color change and a fluorescence OFF/ON response upon acidification and neutralization. Owing to the multiple protonation process, 6a and 6b display a gradient color change from yellow to magenta, violet, and blue as the amount of acid increases. These new hetero polycycles (5a-6b) have potential applications as chemosensors and/or switches.
Introduction
Exploration of polycyclic aromatic hydrocarbons (PAHs) with diverse functionalities is of fundamental importance in synthetic chemistry and materials science.1 It has been well-documented that the optical/electrical properties and assembly nature of PAHs can be finely tuned via molecular engineering, and introducing heteroatoms onto PAHs is an efficient method toward this purpose.2,3 Coronene is a representative PAH showing multiple applications.4 The molecular engineering of coronene is mainly performed by peripheral modification5 and/or substitution of the outer benzenes with different rings.6–8 For example, triazacoronene is derived from coronene by replacing the three outer benzenes with pyridines (Scheme 1).6a–c Meanwhile, the C3v-symmetric buckybowl sumanene9 and the heterasumanenes10–15 might also be regarded as coronene analogues by replacing the outer benzenes with five-membered rings. | ||
| Scheme 1 The chemical structures of the representative compounds and the molecular design rationale of this work. | ||
The bowl-shaped PAHs, including buckybowls16 and “fold-in” bowls,17 are of growing interest.18 Bowl-shaped PAHs are mainly formed by embedding the five-membered rings into hexagonal sheets, and sometimes harsh conditions such as pyrolysis are required to overcome the ring strain energy.16c We have reported the non-pyrolytic and gram-scale synthesis of bowl-shaped trichalcogenasuamenes,19 and their regioselective functionalization.20 Recently, we found that the molecular geometry could be tuned from planar to bowl-shaped by tailoring the size of the side amidine rings.20b One may be intrigued as to whether it is possible to transform the planar PAHs into bowl-shaped ones by embedding the five-membered rings formed by the dative covalent bonds, for example, the metal–ligand bonding along the lateral direction.
In this regard, a new type of hetera PAHs is designed by virtue of hybridizing trichalcogensumanenes and triazacoronene as shown in Scheme 1. The molecular structure optimization reveals that the conjugated cores of these hetera PAHs are almost planar. Further, the 2,2′-bipyridine (bpy) ligand is built up to chelate metal ions along the lateral direction.21 Herein, we report the synthesis of these hetera PAHs and the successive coordination of metal ions. This work proves that the transformation of the π-plane into a π-bowl can be achieved via a lateral dative bond at room temperature (RT). This transformation also results in distinct modulation of the optical properties. Moreover, the absorbance and emission of these PAHs show a reversible response upon acidification and neutralization that could be applied in a chemosensor and/or switch.
Results and discussion
The synthetic route to the new hetera PAHs is shown in Scheme 2. 2,3,6,7,10,11-hexabutoxytriphenylene (HBT) was tetralithiated by n-BuLi (4 equiv.), then sulfur powder was inserted to construct one thiophene ring and one 1,2-dithiin ring at the bay regions of HBT, say, compound 1a (yield 90%). 1a was desulfurized by heating at 200 °C in the presence of copper nano powder (80–100 nm mesh) to give 2a in the yield of 78%, and 2a was then converted to 3a (yield 89%) by reaction with tert-butyl nitrite (TBN). It should be noted that the nitration of 2a by fuming HNO3 mainly resulted in the oxidation of the thiophene ring. 3a was reduced under the conditions of Zn/AcOH to afford 4a in a yield of 90%. In the final step, 5a (yield 71%) and 6a (yield 80%) were respectively synthesized via the Pictet–Spengler reaction of 4a with benzaldehyde and pyridine-2-aldehyde.22 The synthesis of 5b/6b was conducted using the same procedure. To gain insight into the influence on the structural and electronic features caused by hybridization, the hexabutoxy substituted trithiasumanene (3S),19a triselenasumanene (3Se),19a and triazacoronene (3N)6a were also synthesized according to the literature reports. The chemical structures of 3S, 3Se, and 3N are shown in the ESI,† Scheme S1. Compounds 5a-6b are soluble in most of the common organic solvents such as dicloromethane, chloroform, tetrahydrofuran, toluene, and so on. The thermogravimetric analyses (TGA) indicate that 5a-6b show good thermal stability with the decomposed temperature higher than 288 °C (see Table S1 and Fig. S1 in the ESI†). This is beneficial for fabricating 5a-6b onto molecular electronics via the solution process and/or vacuum deposition.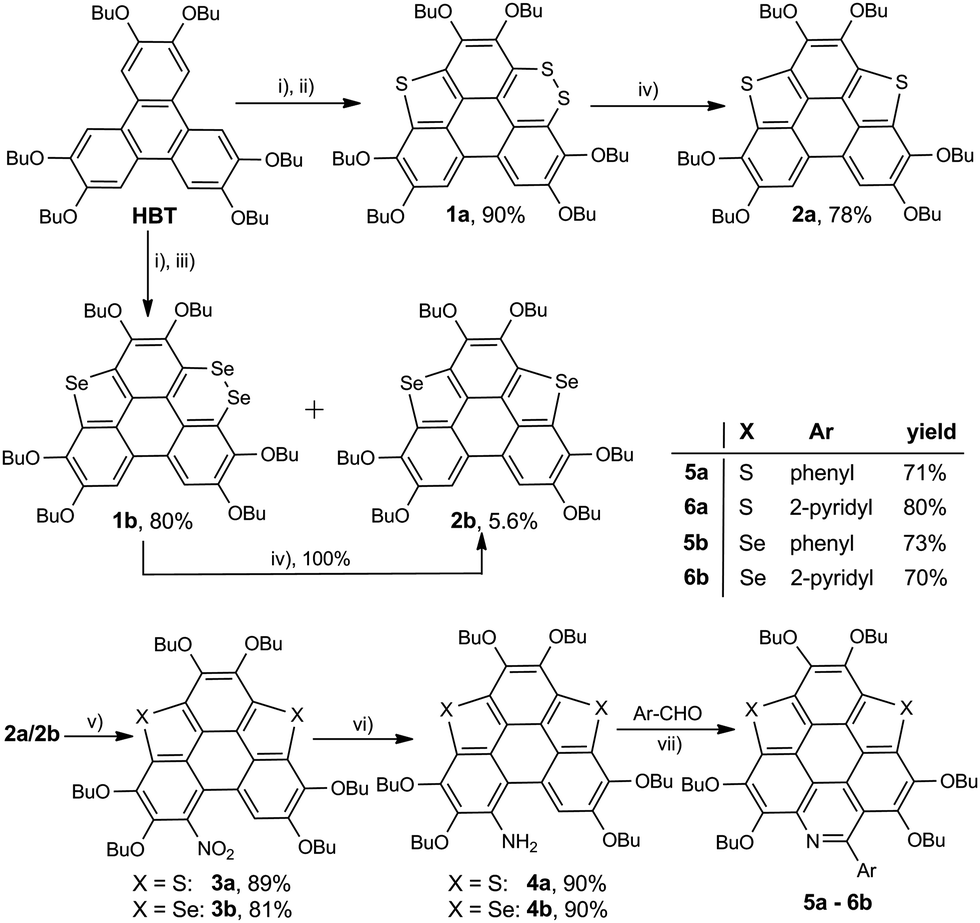 | ||
| Scheme 2 Synthetic route to 5a-6b: (i) HBT (0.01 mol), TMEDA (6 mL), n-BuLi (0.04 mol), 60 °C, 3 h; (ii) S powder (0.08 mol), −78 °C to RT; (iii) Se powder (0.08 mol), −78 °C to RT; (iv) Cu powder (80–100 nm, 10 equiv.), 200 °C, 2 h; (v) 2a/2b (0.01 mol), TBN (0.03 mol), CH2Cl2 (100 mL), RT, 4 h; (vi) 3a/3b (1 mmol), Zn (5 mmol), AcOH (0.1 mL), EtOH (7 mL), 0 °C to RT, 3 h; (vii) 4a/4b (1 mmol), ArCHO (2 mmol), and TFA (3 mL) in sealed Schlenk tube, 120 °C, 12 h. | ||
3S 11 and 3Se16a adopt bowl-shaped conformations with bowl depths of 0.70 Å and 0.47 Å, respectively. It is also reported that 3N is a shallow bowl with a bowl depth of 0.17 Å.6a However, the molecular structure optimization by DFT at the B3LYP/6-31G(d,p) level reveals that the central cores of 5a-6b are planar. To further elucidate the molecular geometries, the crystal structures of 6a and 6b were solved.23Fig. 1 depicts the structure of 6a. The central core of 6a is planar, which is consistent with structure optimization. The two pyridine rings on the bpy unit are almost perpendicular to each other. The 6a molecules stack in a head-to-tail manner as shown in Fig. S2 (ESI†). The structure of 6b is very similar to that of 6a, i.e., planar π-framework and head-to-tail type stack (Fig. S3, ESI†).
 | ||
| Fig. 1 Crystal structure of 6a: (a) top view and (b) side view of molecule 6a. The butyl groups and hydrogen atoms in (b) are omitted for clarity. | ||
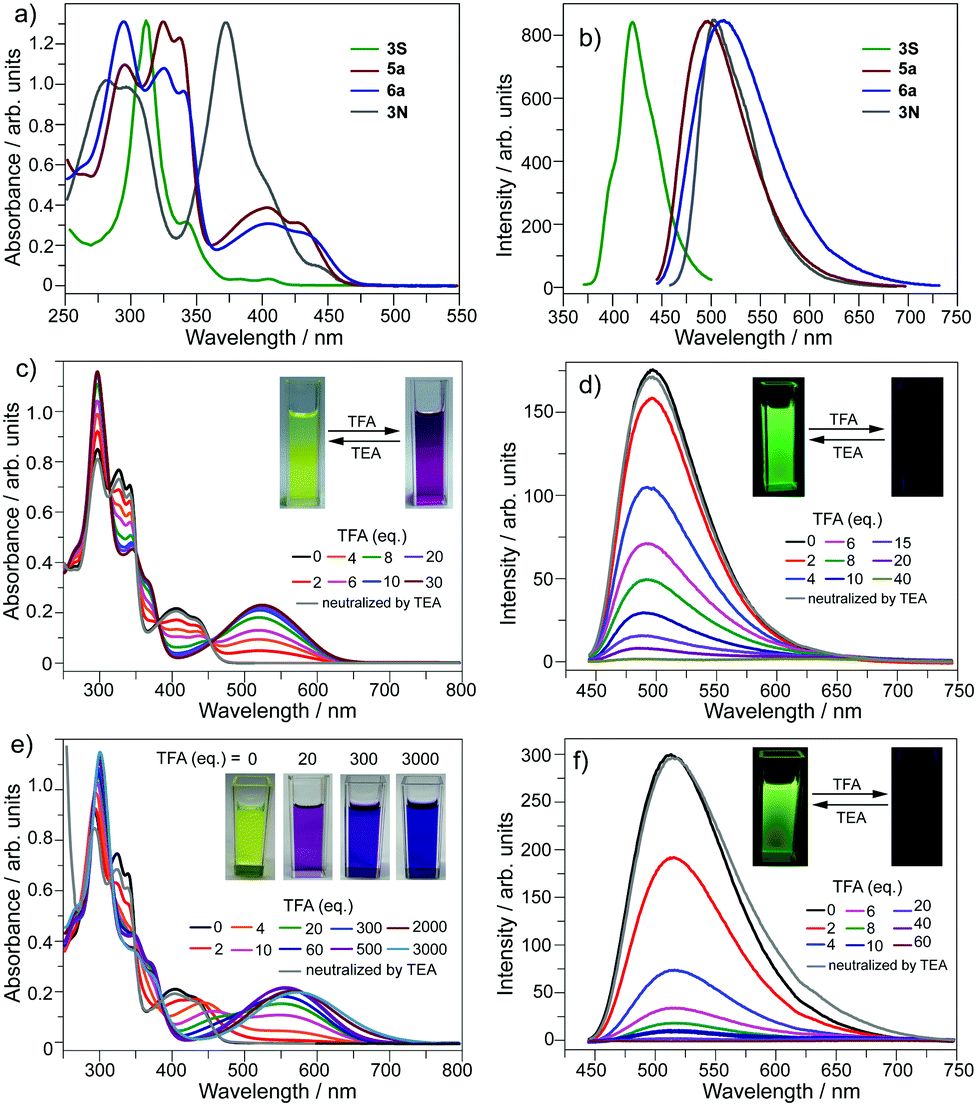
The optical properties of 5a and 6a are almost identical to those of 5b and 6b, respectively. The optical properties of 5a/6a are herein reported and those of 5b/6b are shown in Fig. S15–S19 (ESI†). The UV-Vis absorption spectra of 5a/6a are distinct from those of 3S and 3N (Fig. 2a). 3S shows a sharp absorption peak at 311 nm and a weak broad band at 370–420 nm, and 3N exhibits the strongest absorption at 370 nm. 5a/6a have three strong absorption peaks at 295, 325, and 337 nm, and a broad band at 360–475 nm with moderate absorbance. In comparison with 3S, the lowest energy band of 5a/6a is red-shifted, since they have both electron-rich (thiophene, butoxy) and electron-deficient (pyridine) units on the main skeleton, resulting in an intramolecular charge-transfer (ICT) transition. The theoretical investigation indicates that the HOMO–LUMO energy gaps of 3S and 6a are 3.96 eV and 3.46 eV respectively, which is consistent with the absorption spectra. The theoretical calculation also reveals that the peripheral aryls on 5a/6a show big contribution on their LUMO orbitals (see Fig. 3 for 6a). This is consistent with the discrepancy between the absorption spectra of 5a and 6a, that is, the lowest energy band of 6a is red-shifted about 10 nm as compared with 5a. 5a and 6a respectively display green (λex, 439 nm; λem, 493 nm; ΦF, 4.2%) and yellowish green (λex, 439 nm; λem, 512 nm; ΦF, 3.5%) emission in CH2Cl2 solution (Fig. 2b), which is clearly red-shifted as compared with 3S (λem, 420 nm). The emission of 3N is similar to that of 5a/6a, but the half-band width is narrower than that of 5a/6a.
 | ||
| Fig. 2 (a) UV-Vis and (b) emission spectra of 5a, 6a, 3S, and 3N; (c) UV-Vis and (d) emission spectra of 5a upon titration with TFA; (e) UV-Vis and (f) emission spectra of 6a upon titration with TFA. Measurement conditions: 20 °C, CH2Cl2 solution with c = 10−5 mol L−1. | ||
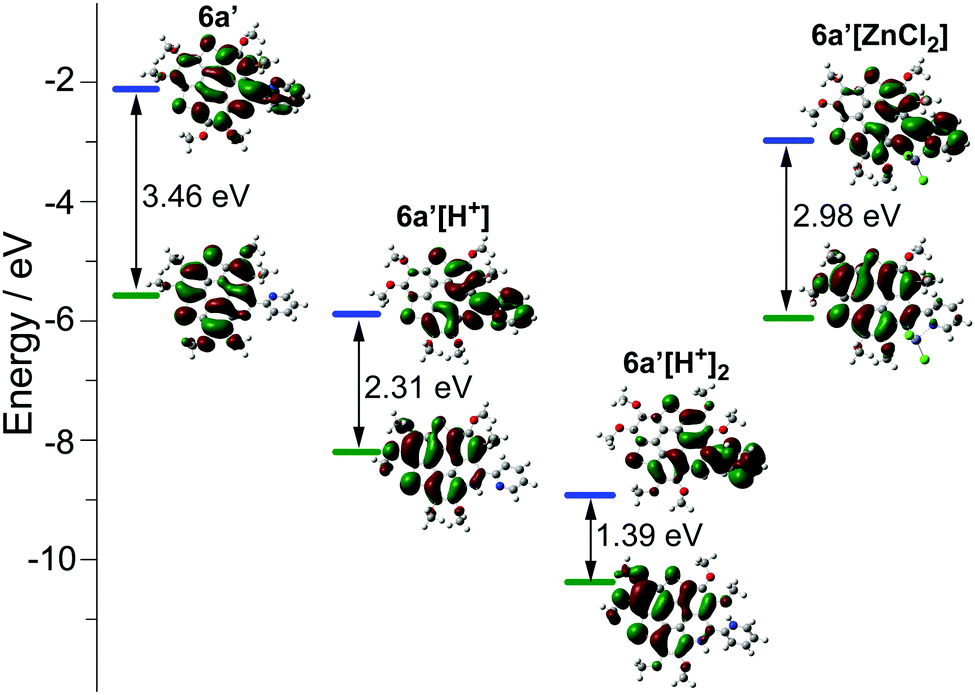 | ||
| Fig. 3 Calculated molecular orbitals and energy levels for 6a′, 6a′[H+], 6a′[H+]2, and 6a′[ZnCl2] at the B3LYP/6-31G(d,p) level of theory. 6a′ is an analogue of 6a by replacing the butyl groups with methyl groups. | ||
The optical properties of 5a/6a can be modulated in the presence of acid. Referring to Fig. 2c, by titrating 5a with trifluoroacetic acid (TFA), the band at 360–475 nm diminishes and a new band appears at 400–650 nm (λmax, 524 nm), associated with the color change of the solution from yellow to magenta. There are four isosbestic points at 313, 350, 380, and 451 nm, indicating that the protonation of 5a solely occurs on the pyridine ring. The protonation on the pyridine ring would profoundly lower the LUMO energy level of 5a to shrink the HOMO–LUMO energy gap.
However, titrating 6a with TFA shows a different phenomenon (Fig. 2e). The band at 360–475 nm is gradually red-shifted and vanishes in the presence of excess TFA, and a new band at a lower energy region appears and extends to 750 nm. The color of the solution turns from yellow to magenta (TFA, 20 eq.), violet (TFA, 300 eq.), and blue (TFA, 3000 eq.). There is no isosbestic point between the new band and the original one, therefore this process would include multiple steps. Regarding two pyridine rings on 6a, the protonation of 6a would mainly involve two states (Scheme 3), 6a[H+] and 6a[H+]2. Theoretical calculation shows that the two pyridine rings on the bpy unit are almost co-planar when one H+ is added onto 6a (Fig. 3), agreeing with the proposed structure of 6a[H+] in Scheme 3. 6a, 6a[H+], and 6a[H+]2 respectively have HOMO–LUMO gaps of 3.46, 2.31, and 1.39 eV (Fig. 3), consistent with the absorption spectra. The fluorescence of 5a/6a is quenched upon adding TFA (Fig. 2d–f). As aforementioned, 5a/6a show an ICT transition, and the protonation of 5a/6a would enhance the ICT transition to give rise to the diminishing of the fluorescence. On the other hand, the original absorption and emission of 5a/6a can be restored upon neutralization with triethylamine (TEA), indicating that it is a reversible process that could be applied in a chemosensor or switch.24
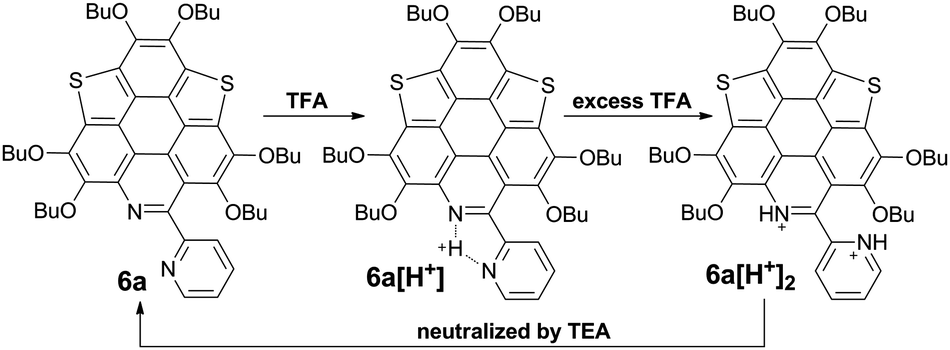 | ||
| Scheme 3 Protonation and neutralization of 6a by TFA and TEA. | ||
The bpy unit on 6a/6b could chelate metal ions;21 herein we show the coordination of 6a with Zn2+. By titrating 6a with ZnCl2, the band at 360–475 nm vanishes, and a new band appears with λmax = 494 nm and extends to 575 nm (Fig. 4a). Four isosbestic points are observed at 315, 349, 381, and 444 nm, indicating that a single product is formed. According to the Job-plot (Fig. 4b), the combination ratio of 6a and ZnCl2 is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The calculated binding constant is 4684 M−1. The reaction between ZnCl2 and 6a is shown in Scheme 4, i.e., the bpy unit chelates Zn2+ to form 6a[ZnCl2]. The HOMO–LUMO gap of 6a[ZnCl2] is 2.98 eV, smaller than that of 6a but larger than that of 6a[H+] and 6a[H+]2 (Fig. 3). This is consistent with the UV-Vis absorption spectra, i.e., the lowest energy bands of 6a, 6a[ZnCl2], 6a[H+], and 6a[H+]2 respectively extend to 475 nm, 575 nm, 650 nm, and 750 nm. Similar to the protonation of 6a by TFA, the fluorescence of 6a decreases upon titration with ZnCl2 and the color of the fluorescence changes from yellowish green to red (Fig. 4c). This is because the 6a[ZnCl2] displays red emission (λex, 492 nm; λem, 640 nm; ΦF, 1.4%) as shown in Fig. S20 (ESI†). The formation of 6a[ZnCl2] can also be detected by 1H NMR. The chemical shifts of the protons on the terminal pyridine ring show clear downfield shifts and split (Fig. 5). 6b exhibits similar behavior to 6a upon titration with ZnCl2, whereas 5a and 5b show no response to ZnCl2.
:1. The calculated binding constant is 4684 M−1. The reaction between ZnCl2 and 6a is shown in Scheme 4, i.e., the bpy unit chelates Zn2+ to form 6a[ZnCl2]. The HOMO–LUMO gap of 6a[ZnCl2] is 2.98 eV, smaller than that of 6a but larger than that of 6a[H+] and 6a[H+]2 (Fig. 3). This is consistent with the UV-Vis absorption spectra, i.e., the lowest energy bands of 6a, 6a[ZnCl2], 6a[H+], and 6a[H+]2 respectively extend to 475 nm, 575 nm, 650 nm, and 750 nm. Similar to the protonation of 6a by TFA, the fluorescence of 6a decreases upon titration with ZnCl2 and the color of the fluorescence changes from yellowish green to red (Fig. 4c). This is because the 6a[ZnCl2] displays red emission (λex, 492 nm; λem, 640 nm; ΦF, 1.4%) as shown in Fig. S20 (ESI†). The formation of 6a[ZnCl2] can also be detected by 1H NMR. The chemical shifts of the protons on the terminal pyridine ring show clear downfield shifts and split (Fig. 5). 6b exhibits similar behavior to 6a upon titration with ZnCl2, whereas 5a and 5b show no response to ZnCl2.
 | ||
| Fig. 4 (a) UV-Vis spectra, (b) UV-Vis Job-plot, and (c) emission spectra upon titration of 6a with ZnCl2 in CH2Cl2 (c = 10−5 mol L−1); (d) emission spectra of 6a and 6a[ZnCl2] in the solid state. | ||
 | ||
| Scheme 4 Formation of 6a[ZnCl2]via the coordination of 6a and ZnCl2. | ||
 | ||
| Fig. 5 1H NMR for the protons of the terminal pyridine rings on 6a and 6a[ZnCl2] in CDCl3. The 1H NMR for the protons on the butoxy groups is shown in Fig. S21 (ESI†). | ||
By evaporating the CH2Cl2 solution of the mixture of ZnCl2 and 6a (molar ratio, 1:1), needle-like red crystals were formed. Single crystal structure analysis shows that it has the same structure as that proposed in Scheme 4, namely, 6a[ZnCl2]. As shown in Fig. 4d, the crystal of 6a[ZnCl2] displays red fluorescence (λex, 590 nm; λem, 661 nm; ΦF, 0.4%), distinct from the crystal of 6a which shows green fluorescence. Single crystals of 6b[ZnCl2] were prepared in the same way.
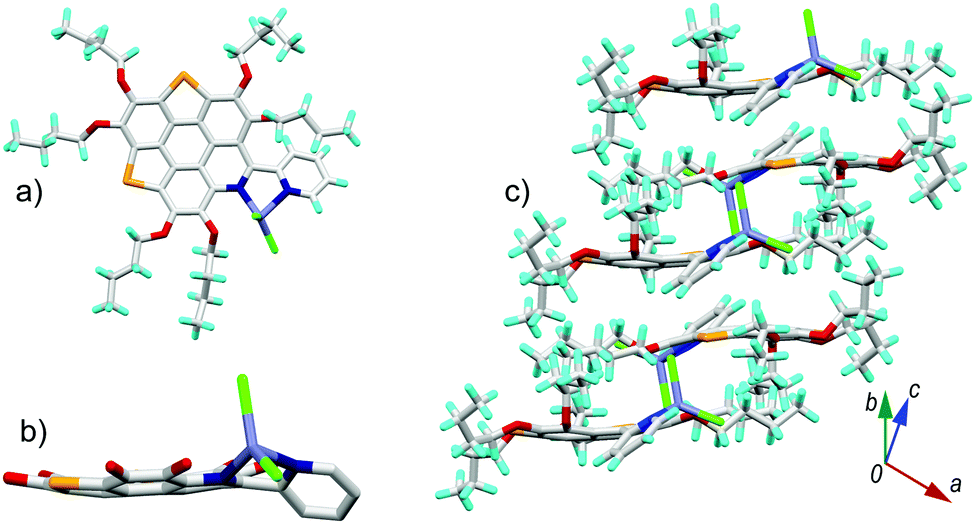
6a[ZnCl2] crystallizes in the P21/c space group with one crystallographically unique molecule. As shown in Fig. 6a, the Zn2+ is chelated by the bpy unit to form a five membered ring with Zn–N bond lengths of 2.09 Å and 2.12 Å. The fusion of the five-membered ring in the lateral direction of 6a has a big influence on the molecular geometry. The C–S bond lengths on 6a[ZnCl2] are 1.78–1.79 Å (Fig. S4, ESI†), almost identical to those in the bowl-shaped trithiasumanene11 but shorter than those of the planar 6a (1.79–1.81 Å). The bond lengths of the three outer C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds on the edged benzene rings are 1.40, 1.41, and 1.42 Å (Fig. S4, ESI†), which are shorter than the corresponding ones in 6a (1.42, 1.42, 1.45 Å). The pyridine ring on the central conjugated core of 6a[ZnCl2] is contorted; this could be due to the stereo hindrance between the butoxy group and terminal pyridine ring, and the non-planar five-membered ring involving a Zn–N bond as well. Consequently, the central core of 6a in 6a[ZnCl2] is bowl-shaped (Fig. 6b), in good agreement with our hypothesis. The bowl depth is 0.50 Å, slightly deeper than that of 3Se (0.47 Å).19a This is the first transformation of a π-plane (6a) into a π-bowl (6a[ZnCl2]) by means of lateral coordination at room temperature. The 6a[ZnCl2] stacks in a “-convex-concave-” manner (Fig. 6c), but there is no crucial π–π interaction between the conjugated cores of the neighboring molecules.
C double bonds on the edged benzene rings are 1.40, 1.41, and 1.42 Å (Fig. S4, ESI†), which are shorter than the corresponding ones in 6a (1.42, 1.42, 1.45 Å). The pyridine ring on the central conjugated core of 6a[ZnCl2] is contorted; this could be due to the stereo hindrance between the butoxy group and terminal pyridine ring, and the non-planar five-membered ring involving a Zn–N bond as well. Consequently, the central core of 6a in 6a[ZnCl2] is bowl-shaped (Fig. 6b), in good agreement with our hypothesis. The bowl depth is 0.50 Å, slightly deeper than that of 3Se (0.47 Å).19a This is the first transformation of a π-plane (6a) into a π-bowl (6a[ZnCl2]) by means of lateral coordination at room temperature. The 6a[ZnCl2] stacks in a “-convex-concave-” manner (Fig. 6c), but there is no crucial π–π interaction between the conjugated cores of the neighboring molecules.
 | ||
| Fig. 6 Crystal structure of 6a[ZnCl2]: (a) top and (b) side views of the molecule; (c) packing structure. The butyl groups and hydrogen atoms in (b) are omitted for clarity. The bond lengths for 6a and 6a[ZnCl2] are shown in Fig. S4 (ESI†). | ||
The molecular geometry variation from planar 6a to bowl-shaped 6a[ZnCl2] has a big influence on the electronic structure. Theoretical calculation reveals that the nucleus-independent chemical shifts (NICS) of two thiophenes on 6a[ZnCl2] show large positive-shifts as compared with those on 6a, and the NICS values of the other rings remain almost unchanged (Table S3, ESI†). The 6a[ZnCl2] is thus of low aromaticity as compared with 6a. Meanwhile, 6a[ZnCl2] possesses HOMO and LUMO energy levels much higher than those of the planar 6a[H+] (Fig. 3).
6b[ZnCl2] also crystallizes in the P21/c space group with one unique molecule. The crystal structure of 6b[ZnCl2] is shown in Fig. 7. The overall feature for the crystal structure of 6b[ZnCl2] is very similar to that of 6a[ZnCl2]. The central conjugated core of 6b in 6b[ZnCl2] is bowl shaped with a bowl depth of 0.38 Å, slightly shallower than that of 3Se.16a The transformation of the 6b π-plane into a 6b[ZnCl2] π-bowl is also caused by the shrinkage of the bond lengths of the outer CC and C–Se bonds (Fig. S5, ESI†), as well as the contortion of the pyridine ring on the central skeleton.
 | ||
| Fig. 7 Crystal structure of 6b[ZnCl2]: (a) top and (b) side views of the molecule; (c) packing structure. The butyl groups and hydrogen atoms in (b) are omitted for clarity. The bond lengths for 6b and 6b[ZnCl2] are shown in Fig. S5 (ESI†). | ||
Conclusions
In summary, the transformation of a π-plane to a π-bowl has been successfully performed by virtue of lateral coordination at room temperature. The work is carried out by rational design of polycycles bearing a bpy unit (6a/6b). The conjugated core of 6a/6b is derived through hybridization of bowl-shaped trichalcogenasumanenes and triazacoronene to show a planar π-framework. By chelating Zn2+, the conjugated cores of 6a and 6b are changed to be bowl-shaped with bowl depths of 0.50 Å and 0.38 Å, respectively. The coordination of 6a/6b with Zn2+ also shows a significant influence on the optical properties. 6a and 6a[ZnCl2] respectively display green and red fluorescence in the solid state. Moreover, the absorbance and emission of 5a-6b show a reversible response upon acidification and neutralization that could be applied in a chemosensor and/or switch. While π-bowls are mainly built up by covalent bonds, this work demonstrates that a π-bowl with a moderate bowl depth can be created through lateral coordination. This would be promising to construct curved π-systems. Further investigation on 6a/6b with transition metals to generate photocatalysts is underway in our group.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the grant from the National Natural Science Foundation of China (21522203), the National Key R&D Program of China (2017YFA0204903), and the Fundamental Research Funds for the Central Universities (lzujbky-2016-ct06).Notes and references
-
(a) H. Dong, H. Zhu, Q. Meng, X. Gong and W. Hu, Chem. Soc. Rev., 2012, 41, 1754 RSC
; (b) C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208 CrossRef PubMed
- J. Mei, Y. Diao, A. L. Appleton, L. Fang and Z. Bao, J. Am. Chem. Soc., 2013, 135, 6724 CrossRef PubMed
- W. Jiang, Y. Li and Z. Wang, Chem. Soc. Rev., 2013, 42, 6113 RSC
-
(a) L. Chen, Y. Hernandez, X. Feng and K. Müllen, Angew. Chem., Int. Ed., 2012, 51, 7640 CrossRef PubMed
-
(a) J. Holzwarth, K. Y. Amsharov, D. I. Sharapa, D. Reger, K. Roshchyna, D. Lungerich, N. Jux, F. Hauke, T. Clark and A. Hirsch, Angew. Chem., Int. Ed., 2017, 56, 12184 CrossRef PubMed
-
(a) J. Wei, B. Han, Q. Guo, X. Shi, W. Wang and N. Wei, Angew. Chem., Int. Ed., 2010, 49, 8209 CrossRef PubMed
-
(a) X.-Y. Wang, F.-D. Zhuang, R.-B. Wang, X.-C. Wang, X.-Y. Cao, J.-Y. Wang and J. Pei, J. Am. Chem. Soc., 2014, 136, 3764 CrossRef PubMed
- Q. Zhang, H. Peng, G. Zhang, Q. Lu, J. Chang, Y. Dong, X. Shi and J. Wei, J. Am. Chem. Soc., 2014, 136, 5057 CrossRef PubMed
- H. Sakurai, T. Daiko and T. Hirao, Science, 2003, 301, 1878 CrossRef PubMed
- For recent reviews, see:
(a) X. Li and X. Shao, Synlett, 2014, 1795 Search PubMed
-
(a) K. Imamura, K. Takimiya, Y. Aso and T. Otsubo, Chem. Commun., 1999, 1859 RSC
- S. Furukawa, Y. Suda, J. Kobayashi, T. Kawashima, T. Tada, S. Fujii, M. Kiguchi and M. Saito, J. Am. Chem. Soc., 2017, 139, 5787 CrossRef PubMed
-
(a) S. Furukawa, J. Kobayashi and T. Kawashima, J. Am. Chem. Soc., 2009, 131, 14192 CrossRef PubMed
-
(a) Q. Tan, S. Higashibayashi, S. Karanjit and H. Sakurai, Nat. Commun., 2012, 3, 891 CrossRef PubMed
- For comprehensive reviews, see:
(a) P. W. Rabideau and A. Sygula, Acc. Chem. Res., 1996, 29, 235 CrossRef
- For comprehensive reviews, see:
(a) P. W. Rabideau and A. Sygula, Acc. Chem. Res., 1996, 29, 235 CrossRef
-
(a) D. Myśliwiec and M. Stępień, Angew. Chem., Int. Ed., 2013, 52, 1713 CrossRef PubMed
- M. Saito, H. Shinokubo and H. Sakurai, Mater. Chem. Front., 2018, 2, 635 RSC
-
(a) X. Li, Y. Zhu, J. Shao, B. Wang, S. Zhang, Y. Shao, X. Jin, X. Yao, R. Fang and X. Shao, Angew. Chem., Int. Ed., 2014, 53, 535 CrossRef PubMed
-
(a) X. Li, Y. Zhu, J. Shao, L. Chen, S. Zhao, B. Wang, S. Zhang, Y. Shao, H.-L. Zhang and X. Shao, Angew. Chem., Int. Ed., 2015, 54, 267 CrossRef PubMed
-
(a) H.-L. Kwong, K.-M. Lau, W.-S. Lee and W.-T. Wong, New J. Chem., 1999, 23, 629 RSC
-
(a) E. D. Cox and J. M. Cook, Chem. Rev., 1995, 95, 1797 CrossRef
- CCDC 1830627, 1830628, 1830629, and 1830630 respectively contain crystallographic data of 6a, 6b, 6a[ZnCl2], and 6b[ZnCl2]†.
-
(a) H. Jeon, J. Lee, M. H. Kim and J. Yoon, Macromol. Rapid Commun., 2012, 33, 972 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details, characterization of the compounds, the theoretical calculations, additional optical spectra, and the crystallographic data in CIF format. CCDC 1830627–1830630. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8qm00168e |
| This journal is © the Partner Organisations 2018 |