DOI:
10.1039/C8QM00171E
(Research Article)
Mater. Chem. Front., 2018,
2, 1618-1625
Controlling the canonical/zwitterionic balance through intramolecular proton transfer: a strategy for vapochromism†
Received
16th April 2018
, Accepted 17th May 2018
First published on 18th May 2018
Abstract
A series of N-substituted diamino-benzoquinone diimines featuring tunable aryl electron-withdrawing functions were synthesized. For the first time, the subtle variation of electronic richness allows controlling the canonical vs. zwitterionic equilibrium through the tuning of an intramolecular proton transfer. The establishment of a ground-state zwitterionic electronic structure, in lieu of the canonical one, within a monocyclic architecture is evidenced by the X-ray structure of one compound (4) and the telltale absorption spectra of the whole series that display an unexpected signature at low energy (ca. 700 nm). The evolution of this peculiar band is concomitant with the electron-withdrawing strength of the aryl substituents and theoretical calculations highlight that this band can be attributed to a charge transfer transition from the anionic cyanine towards the cationic cyanine. Finally, such a color change is advantageously used for the rapid and naked-eye detection of N,N-dimethylformamide using 3a as a vapochromic material. This latter experiment shows the first case of intramolecular solid-state proton transfer occurring in a polar environment.
Introduction
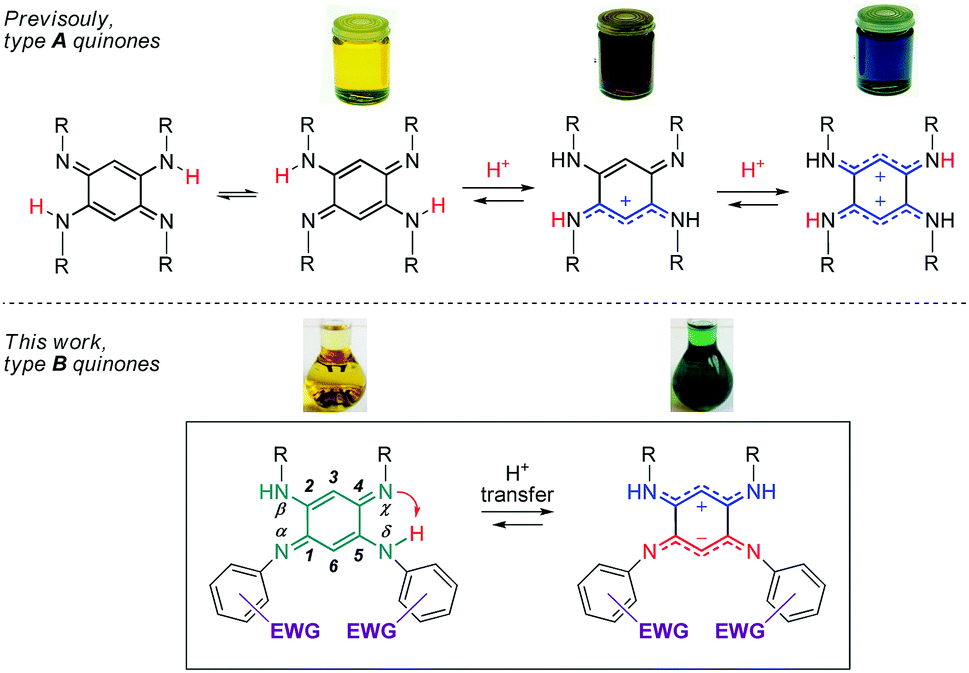
Quinoidal molecules have attracted extraordinary attention for decades because of their remarkable fundamental features and their diverse applications in many fields ranging from organic and coordination chemistry to biology and physics.1 Among them, 2,5-diamino-1,4-benzoquinonediimines (QDIs) of type A (Fig. 1) are particularly interesting because of two major features inherent to their specific molecular structure. First, the presence of a fast intramolecular double proton transfer leads to two degenerate tautomers in equilibrium in solution (Fig. 1).2 This phenomenon could be explained by the proximity of two H-donor/acceptor sites (amine/imine) and similar acidity (i.e., same N-substituent) of the NH proton that allows symmetrical H transfer. Second, type A quinones revealed an unusual distribution of their overall 12π electron system, which can be described as two nearly-independent 6π electron subunits chemically connected through two C–C single bonds. This unique division, the so-called “coupling principle”, first suggested 50 years ago based on theoretical studies,3 was confirmed experimentally in 2003, giving rise to fascinating optical properties depending on the degree of protonation (Fig. 1).4 The observed drastic colour change could be explained by the different distribution of the π electrons which are delocalized – only upon protonation of the imine function(s) – to stabilize the charge(s) between the two nitrogen atoms. These features prompted us to study dissymmetrical QDIs of type B (Fig. 1) since the presence of two different substituents on each 6π electron subunit should (i) induce only one intramolecular proton transfer by tuning the acid–base properties of the H-donor/acceptor sites, and (ii) allow the formation of ground state zwitterions accompanied by a remarkable colour change because of the two charges stabilized by intramolecular delocalization (by analogy with the di-protonated A-based species having a blue colour).
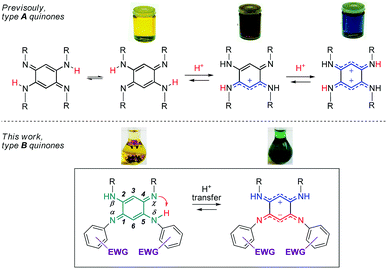 |
| | Fig. 1 [N,N,N,N] quinones under canonical and zwitterionic forms (EWG = electron-withdrawing group). | |
Importantly, the control of the canonical (uncharged) vs π-zwitterionic‡ balance induced by the intramolecular proton transfer, i.e., by chemical engineering, would then become attainable whereas this feature is hitherto unknown. In this context, 12π electron quinones B appear as unique candidates because the H-donor and H-acceptor sites are in optimal proximity to avoid issues related to spatial and conformational effects that might plague the H-transfer. Consequently, this process can be controlled by exclusively modulating the electron density on the neighbouring sites.§![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 5 In the course of this study, we could isolate a new class of ground-state π-zwitterions which were scarcely reported in the literature6–9 whereas they are very valuable because of the presence of both a positive and a negative charge in a conjugated molecule, which paves the way to several applications in the fields of linear and nonlinear optics and organic electronics.10
5 In the course of this study, we could isolate a new class of ground-state π-zwitterions which were scarcely reported in the literature6–9 whereas they are very valuable because of the presence of both a positive and a negative charge in a conjugated molecule, which paves the way to several applications in the fields of linear and nonlinear optics and organic electronics.10
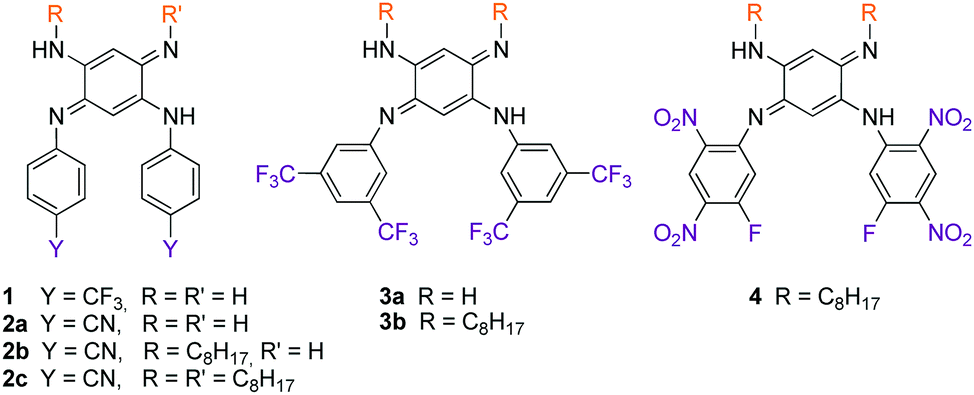
We therefore develop herein quinones of type B in which tuneable N-aryl substituents modulate the basicity/acidity of the imine/amine sites and, consequently, control the intramolecular H-transfer (Fig. 1 and Chart 1). We evidence the above-mentioned phenomenon through the isolation of the zwitterionic electronic structure of QDIs. A combined experimental-theoretical study evidences that the subtle variations of the electron-withdrawing aryl substituents tune the zwitterionic/canonical balance in both solution and solid states, an unprecedented achievement. The controlled intramolecular H-transfer within these systems is used as a new strategy to develop vapochromic materials featuring an unequivocal visible coloration change in response to N,N-dimethylformamide (DMF) or dimethylsulfoxide (DMSO) vapours.
 |
| | Chart 1 Scope of the study (only the canonical form is represented). | |
Results and discussion
Synthesis
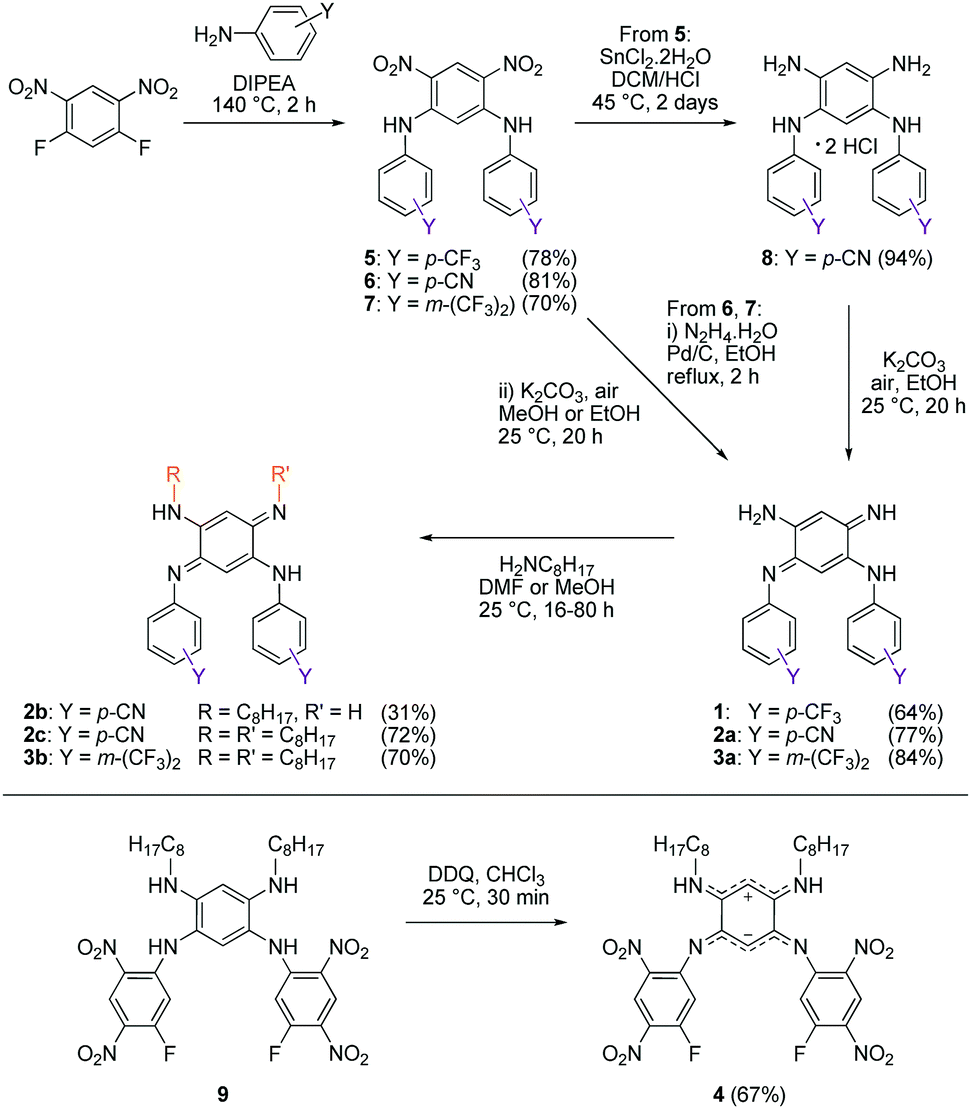
We first envisage the synthesis of di-substituted quinones 1, 2a and 3a11 following a classical three-step strategy (Scheme 1).1g Aniline derivatives were introduced on commercial 1,5-difluoro-2,4-dinitrobenzene using a very efficient aromatic nucleophilic substitution (ca. 70–80% yield for 5–7). Then, the nitro functions were reduced in the presence of SnCl2 in acidic medium or Pd/C with hydrazine, and the tetraaminobenzene intermediates of type 8 were finally oxidized to the corresponding quinones under aerobic conditions.
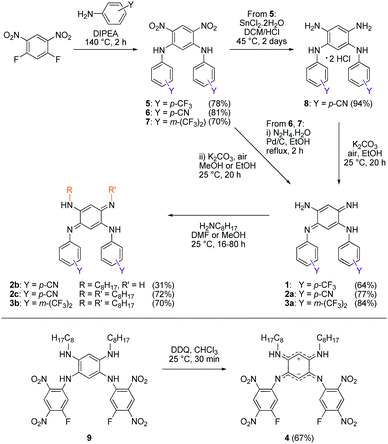 |
| | Scheme 1 Synthetic routes to compounds 1–4. | |
To modulate the electronic density on the upper part of the molecule, we then decided to prepare the corresponding tri- and tetra-substituted quinones. The introduction of alkyl chains to form compounds 2b–c and 3b was performed by transamination reactions on 2a and 3a in the presence of n-octylamine.¶12 Compound 4 was obtained by instantaneous oxidation of the corresponding aromatic precursor 913 using one equivalent of 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) in chloroform (67% yield). Unlike the other compounds that were isolated as orange or red solids, molecule 4 was isolated as a green powder. All the final compounds were fully characterized (see the ESI†), including by X-ray analysis for 3a and 4 (vide infra). Remarkably, 1H NMR data revealed the different chemical shifts for 3a depending on the solvent (Fig. 2). First, going from chloroform to DMSO, a dramatic upfield shift of the broad NH signal is observed from 8.44 to 6.95 ppm. Second, the signal corresponding to the H atom borne by C(6) is progressively shielded from 5.84 to 5.22 ppm upon increasing the solvent polarity. These observations are consistent with the establishment of a zwitterionic structure in 3a in the most polar environment (DMSO), with the cationic trimethine (resp. anionic trimethine) progressively lowering (resp. increasing) the electronic density around the NH (resp. C6(H)) nuclei.
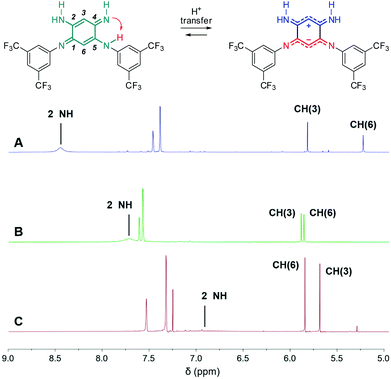 |
| | Fig. 2
1H NMR (298 K, 400 MHz) of 3a in DMSO-d6 (A), acetone-d6 (B) and CDCl3 (C) revealing progressive establishment of a zwitterionic form upon increasing solvent polarity. | |
Structural analysis
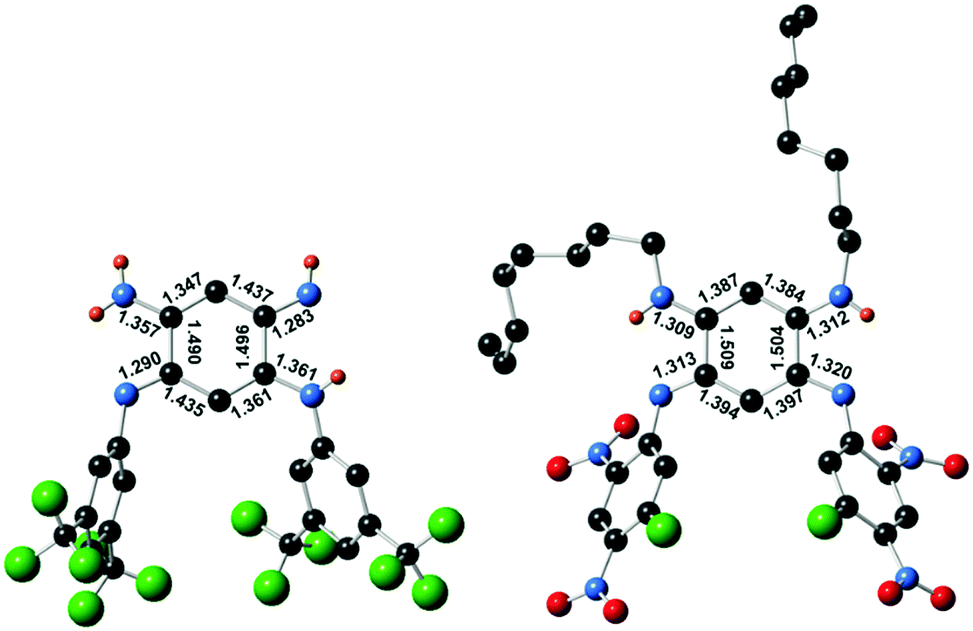
Single crystals suitable for diffraction analysis were obtained by slow evaporation of methanol and acetonitrile solutions for 3a and 4, respectively. The bond lengths determined for the central cycle of 3a unambiguously establish its quinone structure, with single and double bond alternation within each 6π subunit, linked by longer single C(1)–C(2) and C(4)–C(5) bonds (ca. 1.49 Å, Fig. 3). As such, 3a parallels all previous cases of [N,N,N,N] quinoidal ligands in the literature exhibiting a canonical structure in the ground-state.1g In contrast, the structure of 4 revealed two subunits with a perfectly vanishing bond length alternation (BLA = 0) in the N(1)–C(1)–C(6)–C(5)–N(3) and N(2)–C(2)–C(3)–C(4)–N(3) moieties, connected through two single C–C bonds (ca. 1.50 Å distance in C(1)–C(2) and C(4)–C(5)) and H atoms being borne by N(β) and N(γ).4b This observation proves the first appearance of a ground-state zwitterion in monocyclic [N,N,N,N] quinones, constituted by separated and highly delocalized anionic (ArN⋯C⋯CH⋯C⋯NAr) and cationic (RHN⋯C⋯CH⋯C⋯NHR) trimethine cyanines. This structure presumably results from the highly electron-withdrawing character of the fluoro-dinitrophenyl rings that increases the acidity of the H atom and favour its transfer from N(δ) to N(γ) (Fig. 1). The resulting negative charge is then stabilized by intramolecular delocalization between N(α) and N(δ) atoms. The canonical or zwitterionic character for 3a and 4 was also assessed by the experimental determination of the protonation for both compounds (Section S-I in the ESI†).
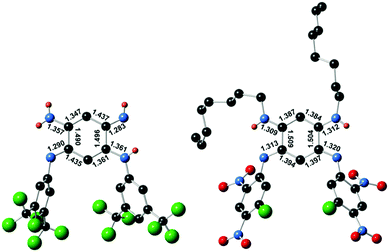 |
| | Fig. 3 Single crystal structures of canonical 3a (left) and zwitterionic 4 (right) and selected bond lengths (Å). Hydrogen atoms only shown when borne by nitrogen atoms. | |
Electronic absorption
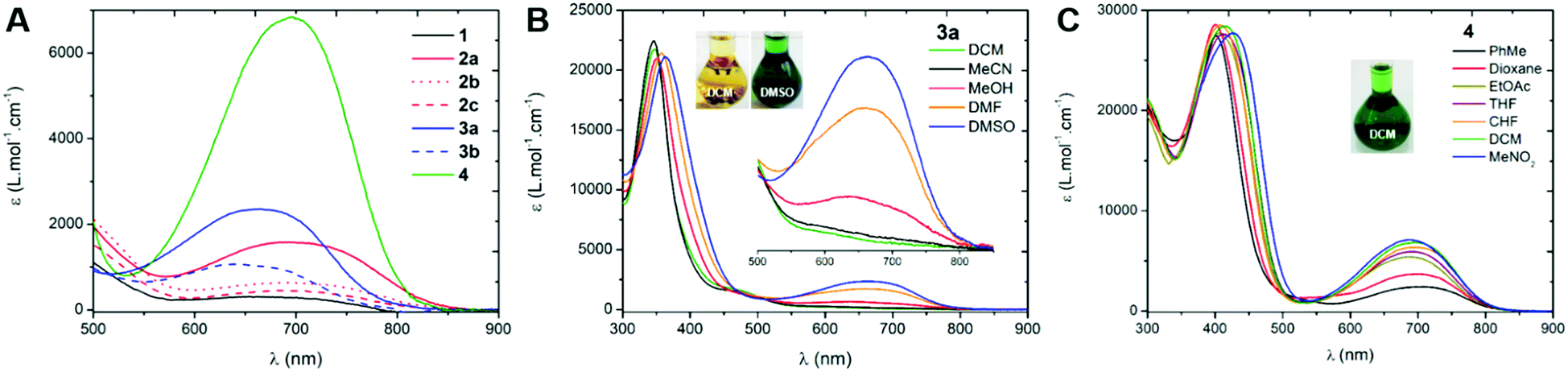
We first investigated the UV-Vis-NIR absorption spectra of the compounds in solution (ca. 2 × 10−5 M) using solvents of various polarities to demonstrate the canonical/zwitterionic equilibrium. The spectra are presented in Fig. 4 and Table 1 (see also in Section S-V of the ESI†). In the most polar solvent (DMSO), all the chromophores exhibit a weak to intense absorption band in the orange-red region, between 600–800 nm (Fig. 4A), this transition being the fingerprint of the zwitterionic form in solution. In the case of di-substituted quinone 1, presenting p-trifluoromethyl moieties, the molar extinction coefficient is particularly low (ε = 310 M−1 cm−1) which indicates a predominant canonical ground-state. However in 2a and 3a, featuring p-cyanophenyl and m-bis(trifluoromethyl)phenyl substituents, the molar extinction coefficient increases together with the electron-withdrawing strength of the aryl substituents (ε = 1580 and 2360 M−1 cm−1, respectively), indicating that the zwitterionic form is relatively more favoured. The introduction of one or two n-octyl chains on tri- and tetra-substituted compounds 2b and 2c results in a decrease of the transition intensity, with ε = 640 and 450 M−1 cm−1, respectively. A similar, though less pronounced trend is found in di-substituted 3a and tetra-substituted 3b probably due to stronger electron-withdrawing aryl moieties. Compound 4, featuring highly electron-withdrawing aryl moieties, shows the most intense transition around 700 nm with an extinction coefficient reaching ca. 7000 M−1 cm−1 (Fig. 4C). Interestingly, solvent polarity has a strong effect on the stabilization of the zwitterionic structure, as shown by the solvatochromism of 3a which reveals an exclusive canonical form in dichloromethane or acetonitrile, and a sharp enhancement of the 665 nm band when going to methanol, DMF and DMSO (Fig. 4B), accompanied by a green coloration of the solution due to the presence of the zwitterionic form. This trend was also evidenced for compounds 1, 2a–c and 3b, with however a stronger stabilization of the canonical form in polar solvents as the compounds are electronically enriched. Compound 4 exhibits the most pronounced zwitterionic character as the low energy band is still observable even in the less polar solvents. For this compound, a noticeable milestone is reached in dichloromethane since the intensity of the band remains quasi-constant upon increasing the polarity of the medium.||
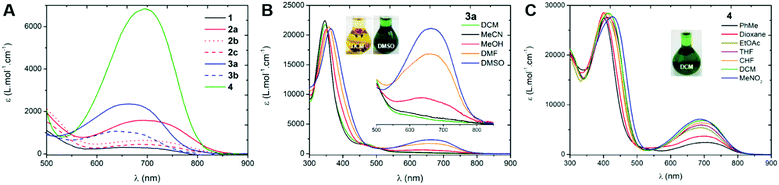 |
| | Fig. 4 Electronic absorption spectra of the compounds in DMSO (in DCM for 4, A) and UV-Vis-NIR absorption solvatochromism of compounds 3a (B) and 4 (C). | |
Table 1 Longest wavelength of maximal absorption and related molar extinction coefficients
| Compound |
Solvent |
λ
max (nm) |
ε (M−1 cm−1) |
|
1
|
DCM |
350 |
20100 |
| DMSO |
653 |
310 |
|
2a
|
DCM |
356 |
23500 |
| DMSO |
693 |
1580 |
|
2b
|
DCM |
364 |
25500 |
| DMSO |
697 |
640 |
|
2c
|
DCM |
371 |
24400 |
| DMSO |
707 |
450 |
|
3a
|
DCM |
348 |
21700 |
| DMSO |
665 |
2360 |
|
3b
|
DCM |
361 |
26500 |
| DMSO |
641 |
1100 |
|
4
|
DCM |
697 |
6800 |
Influence of the pH
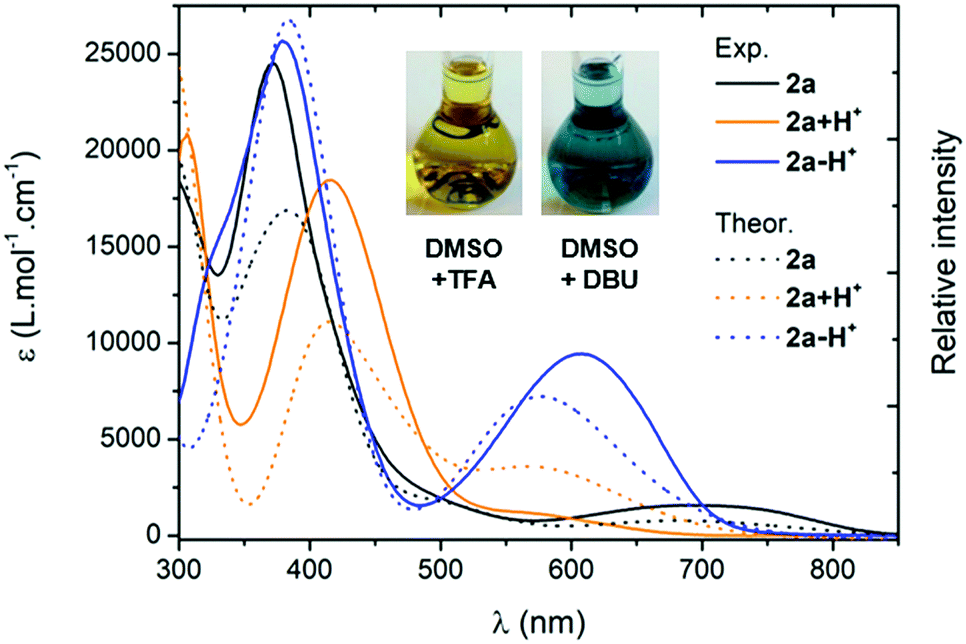
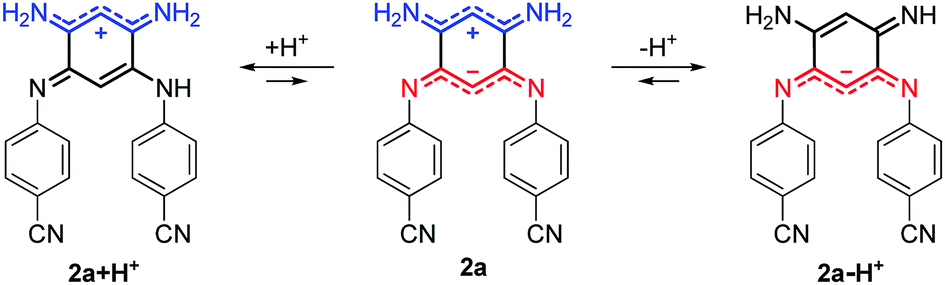
For all the compounds, the presence of acid induces the disappearance of the low energy absorption maximum in favour of a weak absorption band at ca. 550 nm and an intense transition in the UV-blue region (ca. 400–450 nm, see Fig. 5). These changes were attributed by theory to the mono-protonated species, as exemplified with molecule 2a + H+ (Scheme 2 and below). Curiously, although this protonation is a well-known phenomenon in the chemistry of quinones and “coupled polymethines”,4b,9 it was not possible to reach the di-protonated species 2a + 2H+ upon addition of excess trifluoroacetic acid (TFA), presumably due to the particularly electron-poor character of the second 6π subunit.
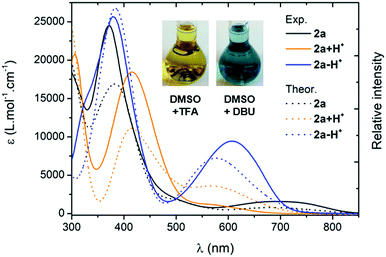 |
| | Fig. 5 Electronic absorption spectra of the protonated (TFA 0.1 M) and deprotonated (DBU 0.1 M) species of 2a in DMSO (plain lines) and corresponding TD-DFT theoretical spectra obtained through Boltzmann averaging of the different rotamers/tautomers (half-width at half-maximum, HWHM: 2000 cm−1, dotted lines). | |
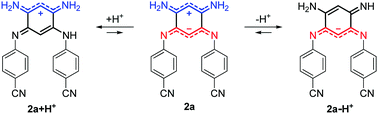 |
| | Scheme 2 Structures generated upon protonation or deprotonation of zwitterionic 2a. | |
Upon addition of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) to a DMSO solution of 2a, the solution instantaneously turned blue and the absorption revealed a broad band centred at 600 nm. Theoretical calculations indicated that the mono-deprotonated species 2a − H+ was generated in solution, the Gaussian-shaped transition being attributed to a charge-transfer from the anionic 6π subunit towards the electron-withdrawing aryl substituents (vide infra). It is noteworthy that, while the cationic derivatives can be generated from both canonical and zwitterionic forms – i.e., in solvents of any polarity – the anionic derivative is only accessible from zwitterionic tautomers (see Fig. S20–S26, ESI†).
Theoretical calculations
DFT and TD-DFT calculations have been performed on selected systems (see the ESI† for details). For 3a modelled in the gas phase, theory finds that the canonical form is more stable than the zwitterionic structure by 5.5 kcal mol−1, which is consistent with the X-ray analysis result. In contrast for 4, the DFT gas-phase free energies computed for the two tautomers starting from the structure of Fig. 3 (octyl chains substituted by methyl) show virtually no energy difference between the canonical and zwitterionic structures (<0.1 kcal mol−1), which is in line with the fact that strong electron-withdrawing groups favour the presence of the charge-separated structure. For 2a, we have performed a detailed investigation of the various possible tautomers that could be envisaged, including a conformational analysis (see Section S-VII in the ESI†). It turns out that the canonical structure remains the most stable form irrespective of the considered medium (gas, DCM and DMSO). However, increasing the polarity of the medium stabilizes the zwitterionic form, which is 5.5 kcal mol−1 above the canonical structure in the gas phase, but only 0.8 kcal mol−1 higher in DMSO (Table S2, ESI†). The Boltzmann distribution hints at the presence of ca. 14% of zwitterions for 2a in DMSO. This is well in line with the experimental absorption spectrum that shows the emergence of a weak long-wavelength absorption in that solvent. Indeed, TD-DFT foresees that only the zwitterionic structures develop an absorption band above 500 nm (Fig. S35, ESI†), with an absorption peaking at 684 nm in DMSO, in excellent match with experiment (693 nm). As we compare here experimental λmax to vertical transition energies, this agreement is of course partly due to error compensations.14 However, calculations of 0–0 energies were impossible in the present case. In contrast, in the canonical tautomers, this long-wavelength absorption is absent and the first maximum appears at 383 nm according to the calculations (Fig. S36, ESI†), which is again in line with the data of Table 1. For a given tautomer, theory returns almost the same transition energies and oscillator strengths in both DCM and DMSO, so that the experimentally observed differences in the UV-Vis absorption spectra are indeed signatures of the changes in the relative population of the various forms, rather than a direct solvent effect.
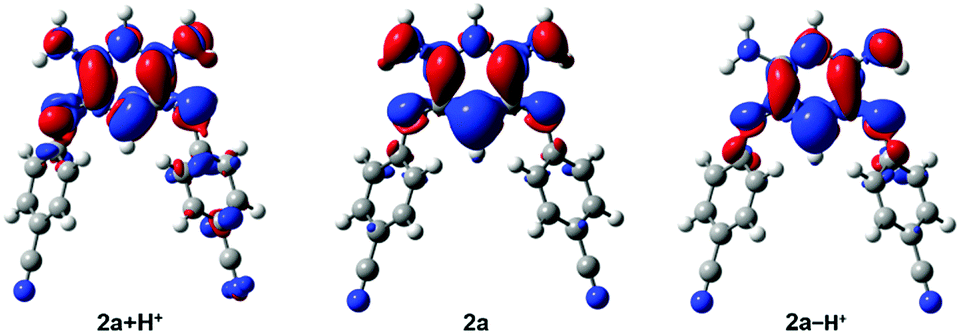
Interestingly, the density difference plot corresponding to the typical zwitterionic absorption band (Fig. 6, centre) can be interpreted in two complementary ways. On the one hand, one notices a strong increase of the density on the central σ bonds (mostly in red) indicating that in the excited state, the conjugation between the two halves of the molecule is restored. This interpretation is in line with a previous work dedicated to tetraphenylhexaazaanthracenes.15 On the other hand, one can also notice that there is a clear transfer of electron density from the negatively charged cyanine fragment (mostly in blue) to the positively charged cyanine fragment (mostly in red). This stands as an interesting occurrence of cyanine-to-cyanine charge-transfer excitation. Such nature of the low-energy transition in the zwitterions might explain why the maxima of the long-wavelength absorption band are poorly impacted among the various compounds (Fig. 4A).
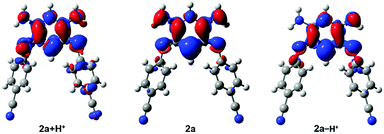 |
| | Fig. 6 Density difference plots corresponding to the long-wavelength band of 2a + H+ (left), 2a (centre) and 2a − H+ (right). In red (blue) are shown the zones corresponding to the increase (decrease) of the electron density upon absorption. | |
For 2a, the computed aromaticity of the three six-member cycles is essentially unaffected by the proton transfer (Fig. S38, ESI†), the two p-cyanophenyl rings being strongly aromatic, whereas the central tetra-amino cycle is only slightly more anti-aromatic in the zwitterionic form than in the canonical one. The strong charge separation in 2a is confirmed by an analysis of the partial atomic charges (Fig. S39, ESI†) as well as by the very large ground-state dipole moment (23.1 D). The amplitude of the dipole significantly decreases in the excited state (18.3 D).
We further used theoretical calculations to ascertain the nature of the protonated and deprotonated species. The results displayed in Fig. 5 and in the ESI† unambiguously demonstrate that: (i) only mono-protonated and mono-deprotonated species are present in solution, as the di-ionic species would lead to absorption spectra not matching experiment; (ii) the most stable tautomers for the two species are the one displayed in Scheme 2. The density difference plots corresponding to the longest-wavelength absorption of 2a + H+ and 2a − H+ are respectively displayed on the left-hand-side and right-hand-side of Fig. 6. They correspond to the weakly-intense shoulder at ca. 550 nm for the protonated form and the intense peak at 608 nm for the deprotonated form (see Fig. 5). As can be seen, the nature of the excited states is similar to the one of 2a (Fig. 6, centre), but, counter-intuitively, the changes are mostly found on one nitrogen atom that is not protonated/deprotonated, i.e., nitrogen atom borne by C(1) and C(2) for 2a + H+ and 2a − H+, respectively.
Cyclic voltammetry
Care was taken to evaluate the electronic richness of the series of compounds by recording their redox properties in DCM and DMF solutions (Table S1 and Fig. S27–S33 in the ESI†). Quinone 1 in DCM presents a reduction process at −1.41 V vs. Fc/Fc+ and its oxidation occurs at 0.32 V. Upon introduction of stronger electron-withdrawing cyano moieties (2a), the reduction becomes easier (−1.33 V) while the oxidation is found at exactly the same potential (0.32 V). A similar tendency is found for the reduction processes of compounds 3a and 4 which are facilitated and found at −1.29 and −0.83 V, respectively. However their oxidations are observed at more anodic potentials (0.44 and 0.65 V, respectively). We highlight that a distinct second reduction wave is present for compounds 3a, 3b, and 4, featuring the strongest electron-withdrawing substituents. Interestingly, the introduction of octyl chains on 2a or 3a does not tune the oxidation potential, while a strong effect is evidenced on the reduction processes which are found at −1.65 and −1.40 V for 2c and 3b, respectively. Globally, changing the solvent to DMF leads to a slight cathodic shift of the oxidations and anodic shift of the reductions, thus lowering the electrochemical gap (ΔE). This evolution is consistent with the establishment of zwitterions in polar solvents (vide supra).
Vapochromism
The detection of volatile organic compounds (VOC) is of particular interest for protection against harmful chemical species. In the context of optical gas detection, vapochromic materials are mainly represented by organometallic and coordination complexes,16 including quinone-based metal complexes.17 For such metallic systems, instances of DMF-responsive vapochromes are few and rarely instantaneous and specific.18 Aside, vapochromism was recently observed in the instance of organic dyes through an intermolecular proton shift,19 or modification of intermolecular π-stacking interactions.20 To the best of our knowledge, vapochromism induced at the molecular level by intramolecular proton transfer has never been reported so far.
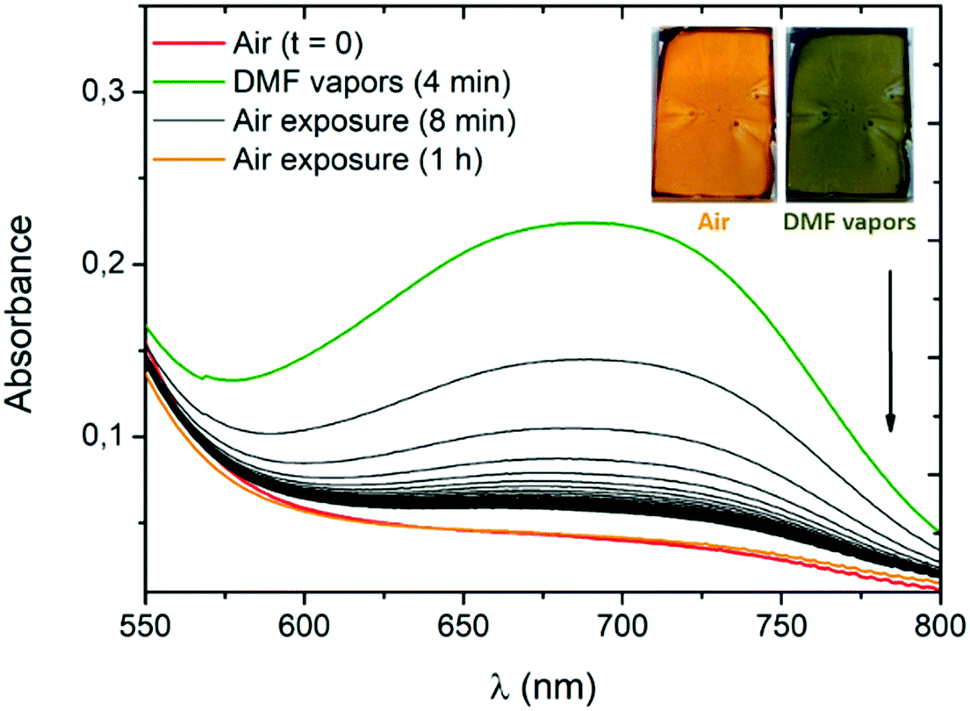
To evaluate the potential of our systems, we use compound 3a as its conversion to the zwitterionic form is accompanied by a pronounced visible colour change (Fig. 4B) occurring exclusively in the presence of the most polar solvents (MeOH, DMF and DMSO). High-concentration thin films of 3a are prepared by spin-coating deposition of the dye in dichloromethane (166 mg mL−1) with polyvinylpyrrolidone (PVP). The use of the PVP polymer and ITO substrate was necessary to obtain uniform thin films of 3a. The amount of PVP was kept low (10 wt%) to avoid any spectroscopic change of 3a due to the polar polymer environment. In a closed receptacle, the originally orange film turns green within ca. 2 minutes in the presence of DMF vapours (Fig. 7 and Fig. S44, ESI†). The vapours of this highly polar solvent induce a solid-state proton shift to generate the zwitterionic structure of 3a, responsible for the visible colour change. Upon exposure to air, the film recovers its initial coloration within a few seconds. This phenomenon can be monitored by absorption spectra recorded after 4 minutes of exposure to DMF vapours (Fig. 7). As expected, the band at ca. 700 nm starts to rapidly decrease, the initial absorption profile being fully recovered after one hour of exposure to air.
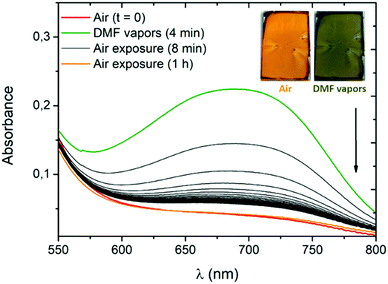 |
| | Fig. 7 Absorption spectra of a 3a/PVP film (9:1, wt./wt.) on an indium tin oxide (ITO) substrate in the presence of air or DMF vapours (24 second interval between every black curve). The inset shows pictures of the film used for the measurement, before and just after exposure to DMF vapours. | |
The same experiment carried out in the presence of DMSO vapours shows the need for a longer time to observe a colour change (50 minutes) and of several hours of air exposure to recover the initial form, presumably due to the lower vapour pressure of this solvent (Fig. S45, ESI†).**19 The spectroscopic changes observed in the presence of DMF or DMSO vapours suggest an unprecedented intramolecular proton transfer mechanism in the solid-state that enables efficient and unambiguous vapochromism without any need of specific organization of the solid phase or presence of additional proton-donating molecules.
Conclusions
To conclude, the occurrence of zwitterionic ground state in monocyclic diaminoquinone diimines has been evidenced for the first time. The establishment of the charge-separated structure occurs through an intramolecular proton transfer that is successfully controlled via the engineering of the electron-withdrawing groups. The colourful zwitterions are characterized by a low-energy transition relative to a cyanine-to-cyanine charge-transfer, which is strongly influenced by the polarity of the medium (solution or vapour). Moreover, it was highlighted that the corresponding cationic and anionic species were readily and quantitatively generated upon acidic and basic treatment, respectively. With its well-balanced zwitterionic/canonical character, molecule 3a constitutes, to the best of our knowledge, the first example of a fully organic DMF-responsive vapochromic dye and it features clear naked-eye, rapid and reversible detection of DMF and, to a lesser extent, DMSO vapours. From a broader perspective, these results let foresee multiple molecular engineering possibilities (heteroatoms, cycle size, acceptor nature…) to extend the scope this new family of zwitterions for further applications in the field of organic optics and electronics and the development of a functional device for the quantitative detection of DMF vapours.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors thank the ANR for support within the framework of the EMA grant. C. A. and D. J. thank A. Varlot (Nantes) for his help with the modelling compound 4. This research used resources of (i) the GENCI-CINES/IDRIS; (ii) Centre de Calcul Intensif des Pays de Loire; (iii) a local Troy cluster; and (iv) HPC resources from ArronaxPlus (Grant No. ANR-11-EQPX-0004 funded by the French National Agency for Research).
Notes and references
-
(a)
E. R. Brown, in The Quinonoid Compounds (1988), John Wiley & Sons, Inc., 2010, pp. 1231–1292 Search PubMed;
(b) S. Hünig, Pure Appl. Chem., 1990, 62, 395–406 CrossRef;
(c) J. Moussa and H. Amouri, Angew. Chem., Int. Ed., 2008, 47, 1372–1380 CrossRef PubMed;
(d) J. Casado, R. Ponce Ortiz and J. T. Lopez Navarrete, Chem. Soc. Rev., 2012, 41, 5672–5686 RSC;
(e) C. Asche, Mini-Rev. Med. Chem., 2005, 5, 449–467 CrossRef PubMed;
(f) J. López, F. de la Cruz, Y. Alcaraz, F. Delgado and M. A. Vázquez, Med. Chem. Res., 2015, 24, 3599–3620 CrossRef;
(g) S. Pascal and O. Siri, Coord. Chem. Rev., 2017, 350, 178–195 CrossRef.
- H. Rumpel and H. H. Limbach, J. Am. Chem. Soc., 1989, 111, 5429–5441 CrossRef.
- S. Dähne and D. Leupold, Angew. Chem., Int. Ed., 1966, 5, 984–993 CrossRef.
-
(a) K. Elbl, C. Krieger and H. A. Staab, Angew. Chem., Int. Ed., 1986, 25, 1023–1024 CrossRef;
(b) O. Siri, P. Braunstein, M.-M. Rohmer, M. Bénard and R. Welter, J. Am. Chem. Soc., 2003, 125, 13793–13803 CrossRef PubMed.
-
(a) F. B. Tamboura, C. S. J. Cazin, R. Pattacini and P. Braunstein, Eur. J. Org. Chem., 2009, 3340–3350 CrossRef;
(b) A. D. Bailey, B. P. Murphy and H. Guan, J. Phys. Chem. A, 2016, 120, 8512–8520 CrossRef PubMed.
- Bis-dithiazole π-zwitterions:
(a) L. Beer, R. T. Oakley, J. R. Mingie, K. E. Preuss, N. J. Taylor and A. W. Cordes, J. Am. Chem. Soc., 2000, 122, 7602–7603 CrossRef;
(b) S. M. Winter, R. J. Roberts, A. Mailman, K. Cvrkalj, A. Assoud and R. T. Oakley, Chem. Commun., 2010, 46, 4496–4498 RSC;
(c) A. Mailman, A. A. Leitch, W. Yong, E. Steven, S. M. Winter, R. C. M. Claridge, A. Assoud, J. S. Tse, S. Desgreniers, R. A. Secco and R. T. Oakley, J. Am. Chem. Soc., 2017, 139, 2180–2183 CrossRef PubMed.
- Tetraphenylhexaazaanthracene π-zwitterions:
(a) K. Hutchison, G. Srdanov, R. Hicks, H. Yu, F. Wudl, T. Strassner, M. Nendel and K. N. Houk, J. Am. Chem. Soc., 1998, 120, 2989–2990 CrossRef;
(b) P. Langer, A. Bodtke, N. N. R. Saleh, H. Görls and P. R. Schreiner, Angew. Chem., Int. Ed., 2005, 44, 5255–5259 CrossRef PubMed;
(c) P. Langer, S. Amiri, A. Bodtke, N. N. R. Saleh, K. Weisz, H. Görls and P. R. Schreiner, J. Org. Chem., 2008, 73, 5048–5063 CrossRef PubMed;
(d) T. A. Ioannou, P. A. Koutentis, H. Krassos, G. Loizou and D. L. Re, Org. Biomol. Chem., 2012, 10, 1339–1348 RSC;
(e) C. P. Constantinides, G. A. Zissimou, A. A. Berezin, T. A. Ioannou, M. Manoli, D. Tsokkou, E. Theodorou, S. C. Hayes and P. A. Koutentis, Org. Lett., 2015, 17, 4026–4029 CrossRef PubMed;
(f) G. A. Zissimou, C. P. Constantinides, M. Manoli, G. K. Pieridou, S. C. Hayes and P. A. Koutentis, Org. Lett., 2016, 18, 1116–1119 CrossRef PubMed;
(g) G. A. Zissimou, A. Kourtellaris and P. A. Koutentis, Org. Lett., 2018, 20, 844–847 CrossRef PubMed;
(h) G. A. Zissimou, A. Kourtellaris and P. A. Koutentis, J. Org. Chem., 2018, 83, 4754–4761 CrossRef PubMed.
- Quinoxalinophenzaine π-zwitterions:
(a) F. Wudl, P. A. Koutentis, A. Weitz, B. Ma, T. Strassner, K. N. Houk and S. I. Khan, Pure Appl. Chem., 1999, 71, 295–302 CrossRef;
(b) C. P. Constantinides, P. A. Koutentis and J. Schatz, J. Am. Chem. Soc., 2004, 126, 16232–16241 CrossRef PubMed.
- Zwitterionic [N,O,O,N] quinones:
(a) O. Siri and P. Braunstein, Chem. Commun., 2002, 208–209 RSC;
(b) P. Braunstein, O. Siri, J.-P. Taquet, M.-M. Rohmer, M. Bénard and R. Welter, J. Am. Chem. Soc., 2003, 125, 12246–12256 CrossRef PubMed;
(c) Y. Haas and S. Zilberg, J. Am. Chem. Soc., 2004, 126, 8991–8998 CrossRef PubMed;
(d) Q.-Z. Yang, O. Siri and P. Braunstein, Chem. Commun., 2005, 2660–2662 RSC;
(e) A. Muranaka, S. Ohira, D. Hashizume, H. Koshino, F. Kyotani, M. Hirayama and M. Uchiyama, J. Am. Chem. Soc., 2012, 134, 190–193 CrossRef PubMed;
(f) N. Toriumi, A. Muranaka, K. Hirano, K. Yoshida, D. Hashizume and M. Uchiyama, Angew. Chem., Int. Ed., 2014, 53, 7814–7818 CrossRef PubMed;
(g) N. Bonneau, G. Chen, D. Lachkar, A. Boufridi, J.-F. Gallard, P. Retailleau, S. Petek, C. Debitus, L. Evanno, M. A. Beniddir and E. Poupon, Chem. – Eur. J., 2017, 23, 14454–14461 CrossRef PubMed.
- Selected reviews and recent examples:
(a) A. Ajayaghosh, Acc. Chem. Res., 2005, 38, 449–459 CrossRef PubMed;
(b) L. Beverina and G. A. Pagani, Acc. Chem. Res., 2014, 47, 319–329 CrossRef PubMed;
(c) Y. Zheng and F. Wudl, J. Mater. Chem. A, 2014, 2, 48–57 RSC;
(d) Y. Zheng, M.-S. Miao, Y. Zhang, T.-Q. Nguyen and F. Wudl, J. Am. Chem. Soc., 2014, 136, 11614–11617 CrossRef PubMed;
(e) Y. Shi, A. J. T. Lou, G. S. He, A. Baev, M. T. Swihart, P. N. Prasad and T. J. Marks, J. Am. Chem. Soc., 2015, 137, 4622–4625 CrossRef PubMed;
(f) Z. A. Li, P. Zhao, S. Tofighi, R. Sharma, T. R. Ensley, S.-H. Jang, D. J. Hagan, E. W. Van Stryland and A. K. Y. Jen, J. Phys. Chem. C, 2016, 120, 15378–15384 CrossRef.
- L. Lavaud, Z. Chen, M. Elhabiri, D. Jacquemin, G. Canard and O. Siri, Dalton Trans., 2017, 46, 12794–12803 RSC.
- F. Santoro and D. Jacquemin, WIREs Comput. Mol. Sci., 2016, 6, 460–486 CrossRef.
- J. Andeme Edzang, Z. Chen, H. Audi, G. Canard and O. Siri, Org. Lett., 2016, 18, 5340–5343 CrossRef PubMed.
- Z. Chen, R. Haddoub, J. Mahé, G. Marchand, D. Jacquemin, J. Andeme Edzang, G. Canard, D. Ferry, O. Grauby, A. Ranguis and O. Siri, Chem. – Eur. J., 2016, 22, 17820–17832 CrossRef PubMed.
-
(a) O. S. Wenger, Chem. Rev., 2013, 113, 3686–3733 CrossRef PubMed;
(b) A. Kobayashi and M. Kato, Eur. J. Inorg. Chem., 2014, 4469–4483 CrossRef.
-
(a) A. Kobayashi, M.-A. Dosen, M. Chang, K. Nakajima, S.-I. Noro and M. Kato, J. Am. Chem. Soc., 2010, 132, 15286–15298 CrossRef PubMed;
(b) M. Chang, A. Kobayashi, K. Nakajima, H.-C. Chang and M. Kato, Inorg. Chem., 2011, 50, 8308–8317 CrossRef PubMed;
(c) P. Kar, M. Yoshida, Y. Shigeta, A. Usui, A. Kobayashi, T. Minamidate, N. Matsunaga and M. Kato, Angew. Chem., Int. Ed., 2017, 56, 2345–2349 CrossRef PubMed.
-
(a) A. Kobayashi, Y. Fukuzawa, H.-C. Chang and M. Kato, Inorg. Chem., 2012, 51, 7508–7519 CrossRef PubMed;
(b) Y. Li, S. Zhang and D. Song, Angew. Chem., Int. Ed., 2013, 52, 710–713 CrossRef PubMed;
(c) L.-Y. Zhang, L.-J. Xu, X. Zhang, J.-Y. Wang, J. Li and Z.-N. Chen, Inorg. Chem., 2013, 52, 5167–5175 CrossRef PubMed;
(d) J. S. Ovens and D. B. Leznoff, Chem. Mater., 2015, 27, 1465–1478 CrossRef;
(e) A. S. Sergeenko, J. S. Ovens and D. B. Leznoff, Inorg. Chem., 2017, 56, 7870–7881 CrossRef PubMed.
- A. Sakon, A. Sekine and H. Uekusa, Cryst. Growth Des., 2016, 16, 4635–4645 Search PubMed.
- T. Ogoshi, Y. Shimada, Y. Sakata, S. Akine and T.-A. Yamagishi, J. Am. Chem. Soc., 2017, 139, 5664–5667 CrossRef PubMed.
-
C. Reichardt, Solvents and Solvent Effects in Organic Chemistry, Wiley-VCH, 3rd edn, 2003 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental protocols, 1H and 13C NMR spectra, additional solvatochromism and pH-dependent absorption spectra, cyclic voltammograms, theoretical calculations details and additional vapochromism details. CCDC 1831850 and 1831851 contain the X-ray crystallographic data for 3a and 4. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8qm00171e |
| ‡ For clarity reasons, the term canonical describes the uncharged localized form, even if, formally, canonical zwitterions can be drawn. |
| § Derivatization of N-substituent needs electron-rich primary amines (see ref. 9d). Only three N-aryl BQMI are reported with Ar = Ph, p-MeOPh or p-NH2Ph; see ref. 5. |
| ¶ Noteworthily, the use of a catalytic amount of acid was not necessary to perform transamination since the solvent used for the reaction was polar enough to ensure the presence of zwitterionic structures in the starting reactants. |
| || In DMF, and to a lesser extent in DMSO, the absorption spectra of 4 are noticeably modified. Comparisons of the data show that these absorption shifts correspond to the formation of the deprotonated species 4 − H+ in solution (see Fig. S26, ESI†). |
| ** Vapour pressures for DMF and DMSO are 3.5 hPa (20 °C) and 0.61 hPa (25 °C), respectively (from ref. 20). Exposure to water vapor did not induce any colour change of the thin film. Toluene, tetrahydrofuran and methanol vapours led to a partial degradation of the thin film, probably due to the higher vapour pressure of these solvents, without inducing any visible colour change. Moreover, we have carried out a few additional experiments using toluene–DMF mixtures (see ESI† Section S-VIII). |
|
| This journal is © the Partner Organisations 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a,
Lucien
Lavaud
*a,
Lucien
Lavaud