
Reticular control of interpenetration in a complex metal–organic framework†
Trang T. M.
Nguyen
a,
Hung M.
Le
 a,
Yoshiyuki
Kawazoe
b and
Ha L.
Nguyen
*ac
a,
Yoshiyuki
Kawazoe
b and
Ha L.
Nguyen
*ac
aCenter for Innovative Materials and Architectures (INOMAR), Vietnam National University (VNUHCM), Ho Chi Minh City, 721337, Vietnam. E-mail: nlha@inomar.edu.vn
bNew Industry Creation Hatchery Center, Tohoku University, Sendai, 980-8579, Japan
cCenter for Research Excellence in Nanotechnology (CENT), King Fahd University of Petroleum and Minerals, Dhahran 34464, Saudi Arabia
First published on 11th September 2018
Abstract
Metal–organic frameworks (MOFs) are a new generation of crystalline porous materials covering a broad spectrum of research fields due to their high porosity and the feasibility of modification of their structure. Due to their large void spaces, MOF structures tend to be more stable when the crystals are grown to form interpenetrated frameworks. Control of interpenetrated frameworks in MOFs is not only useful for various kinds of practical application but also provides insight towards understanding the reaction process and catenated behavior in MOF chemistry. In this research, we sought to investigate and successfully achieve reticular control of the interpenetration of PCN-280, a complex MOF with a 2-fold interpenetrated framework containing heterogeneity building blocks (two kinds of polytopic organic linking unit based on carboxylate), to form a new isoreticular single-frame MOF, termed MOF-908 and isoreticular MOF-909. The crystal structure and underlying network of MOF-908 and MOF-909 are proven by X-ray diffraction analysis as well as other related characterizations. Indeed, the mechanism of reticular control of the interpenetration of PCN-280 is studied in detail using density functional theory (DFT) calculations.
Introduction
Reticular chemistry1 at its best opens the window toward the blue sky of metal–organic framework (MOF) chemistry,2 a highly-crystalline ordered material possessing permanent porosity and designable properties which can be potentially applied for gas/vapor uptake,3 catalysis,4,5 energy conversion6 and chemical sensing.7 Interpenetration behavior mainly comes from the huge void fraction of the large unit cells of MOFs, in which another framework can grow to occupy the vacancy resulting in the stabilization of the resulting structures.8 Given the fact that control of the reaction in which interpenetration occurs on increasing the lengths of organic linkers to obtain a single host framework is one of the most interesting studies9–11 for paving the way to understanding the mechanism of MOF growth, it has experienced a lack of full exploration in MOF research.It is noted that interpenetration reportedly occurs in MOFs generated through the reaction of single carboxylate-based linkers, which, in some cases, could be a mixture of carboxylate and bipyridine linking units or metal–oxo clusters.12 In order to entirely control the structures of interpenetrated frameworks,13 (i) synthetic conditions were varied, for example bulky molecules can be introduced into the reaction, which unpredictably produces a host single frame or another different structure;14 (ii) organic templates were utilized and somehow the interpenetration control is not clear;15 (iii) the organic linker was functionalized to modify the geometrical direction of the point of extension and the carboxylate moiety;12 (iv) the coordinated/uncoordinated solvent removal was additionally carried out after the structure formation.16 Here we report a strategy that utilizes the power of reticular chemistry in the control of the structure of a tertiary MOF. We define this methodology as “reticular control” and it involves independent 2-fold interpenetrated frameworks formed using triangular 4,4′,4′′-benzene-1,3,5-triyl-tris(benzoic acid) (H3BTB) and linear biphenyldicarboxylic acid (H2BPDC) linkers and a trigonal prism Fe3O(–CO2)6 cluster, using a heterogeneity MOF reported by Zhou and co-workers termed PCN-280.17 From two complex MOFs, namely MOF-908 and MOF-909 (Scheme 1), the isoreticular single net of PCN-280 is successfully produced by reticular control of the rational input linkers using a mixture of H2BPDC and 3,3′-azoBDC (azobenzene-3,3′-dicarboxylic acid). This is the first time the structure of a tertiary interpenetrated MOF has been controlled to achieve a non-interpenetrated framework. Indeed, the “deinterpenetration”, the process of removing the interpenetrated frameworks to obtain a single-frame structure, was not successful either using the reported methods, varying the synthetic conditions, using a template, using bulky organic solvents or in the absence of 3,3′-azoBDC. The role of 3,3′-azoBDC in the reticular control is subsequently demonstrated by adopting density functional theory (DFT) calculations to explore the process of the reaction. Both 3,3′-azoBDC2− and BPDC2− are competitive in the first reaction stage, but BPDC2− is more favorable for the substitution of 3,3′-azoBDC2− in a subsequent process, which leads to the formation of MOF-908. In terms of mechanical stability, we also verify that MOF-908 is much more stable than MOF-909. Not only does our finding provide new insight into interpenetration control in MOFs by reticular synthesis, but it also contributes to the understanding of reaction processes and mechanisms in MOF chemistry.
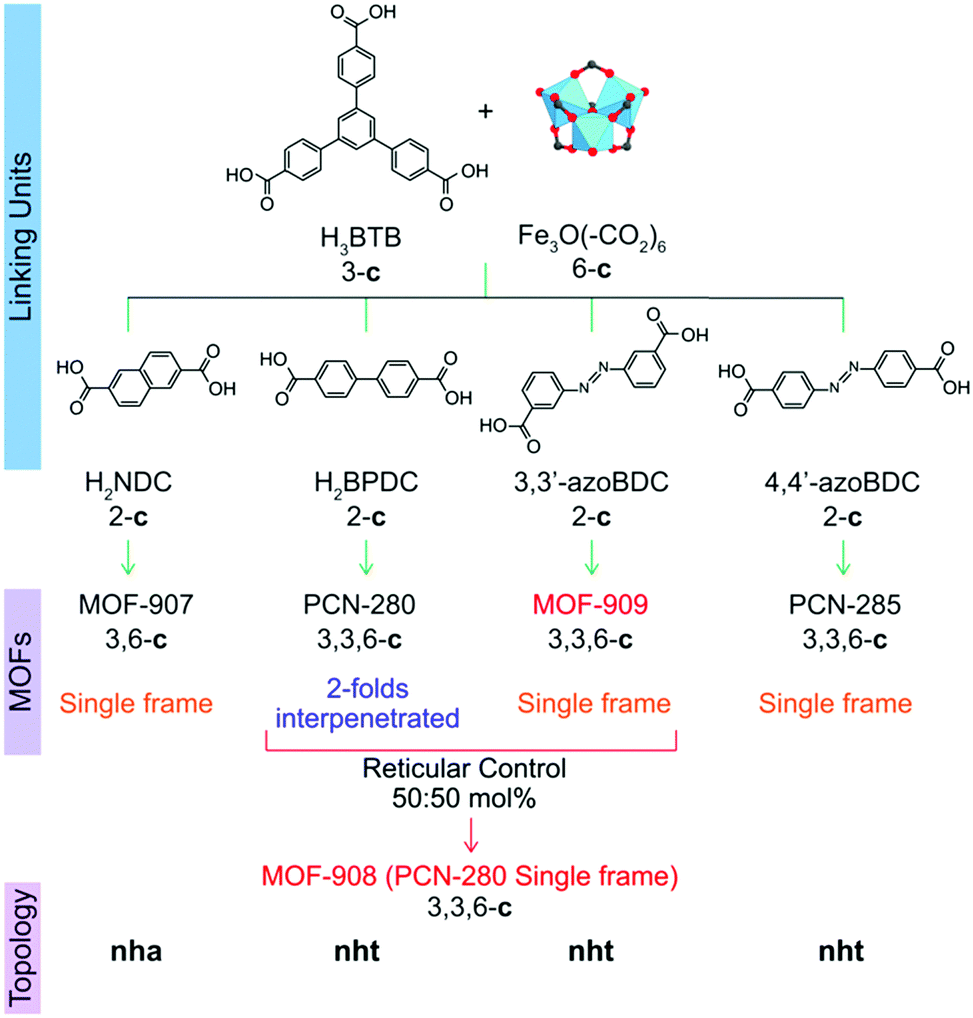 | ||
| Scheme 1 Synthetic strategy to produce heterogeneity MOFs based on a mixed linker system. From top to bottom: linking units containing the tritopic (3-c) and ditopic (2-c) organic linkers to react with the Fe-based cluster (6-c) to form complex MOFs including MOF-907,19 PCN-280,17 PCN-285,17 MOF-908, and MOF-909, whose topological networks are correspondingly presented. | ||
Results and discussion
To verify the role of the geometrical requirement of both organic linkers in a heterogeneity MOF,18 we previously reported the concept of “perfect fit” of the ratio lengths of the tritopic H3BTB linker over various ditopic linkers and the number of links surrounding the triangular Fe-based cluster, which is more convenient for non-interpenetrated structure formation.19 On increasing the length of the linear ditopic linker in the mixed linker system complex MOFs have shown a tendency to become interpenetrated structures; in particular, a series of MIL-142 (MIL-142A to E)20 and PCN-280 (a complex MOF structure comprising two kinds of link, BTB3− and BPDC2−, and trigonal prism Fe3O(–CO2)6 cluster) (Scheme 1) have shown this.17 However, by replacing BPDC2− in the PCN-280 structure by azobenzene-4,4′-dicarboxylate (4,4′-azoBDC2−), an isoreticular structure with a single net was formed that is termed PCN-285.17 This observation is still ambiguous and has not received any explanation from either experimental or calculation methodology.Before controlling the interpenetration behavior of PCN-280, we start by synthesizing the isoreticular structure of PCN-280 and PCN-285 in which the 3,3′-azoBDC linker, whose length is longer than H2BPDC and shorter than 4,4′-azoBDC, is used and a single frame MOF is expected to be obtained. MOF-909 microcrystalline powder was isoreticularly produced under synthetic conditions different to those used for PCN-280 and PCN-285 preparation, in line with our speculation. The powder X-ray diffraction (PXRD) patterns of the as-synthesized and activated samples are coincident with the pattern calculated from structural modeling (ESI,† Section 3). A single crystal of PCN-280 was then prepared by the same synthetic method utilized for MOF-909 manufacturing. The single-frame structure of PCN-280, MOF-908, cannot be obtained after exhausting efforts following the reported methodologies including synthetic modification, bulky organic solvent changing and use of templates. We hypothesize that there exists an energy barrier for the reaction when the mixed linker system, H3BTB and H2BPDC, interacts with the triangular Fe–oxo cluster, preventing the final structure from the single network of MOF-909- or PCN-285-type topology which is unseen in MOF chemistry and was not acknowledged in the original publication.17 In order to prove that postulation, we then move forward to rational investigation of the reaction in which the H3BTB linker was an independent reagent and was constrained to be unchanged in molar amount while the ratio of 3,3′-azoBDC to H2BPDC was proportionally controlled from 0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 mol% to 100:0 mol%. As depicted in Fig. 1, after 24 h of reaction the highly crystalline products were observed for all of the samples. The PXRD patterns clearly show that PCN-280 is only formed when the molar percentage of the 3,3′-azoBDC linker in the solution is in the range from 0% to 40%. Interestingly, increasing the molar ratio of 3,3′-azoBDC to H2BPDC to 60:40 mol% leads to the achievement of the non-interpenetrated structure of PCN-280, MOF-908 (a zoomed in image of Fig. 1 at low angles is found in Section 4 of the ESI†).
:100 mol% to 100:0 mol%. As depicted in Fig. 1, after 24 h of reaction the highly crystalline products were observed for all of the samples. The PXRD patterns clearly show that PCN-280 is only formed when the molar percentage of the 3,3′-azoBDC linker in the solution is in the range from 0% to 40%. Interestingly, increasing the molar ratio of 3,3′-azoBDC to H2BPDC to 60:40 mol% leads to the achievement of the non-interpenetrated structure of PCN-280, MOF-908 (a zoomed in image of Fig. 1 at low angles is found in Section 4 of the ESI†).
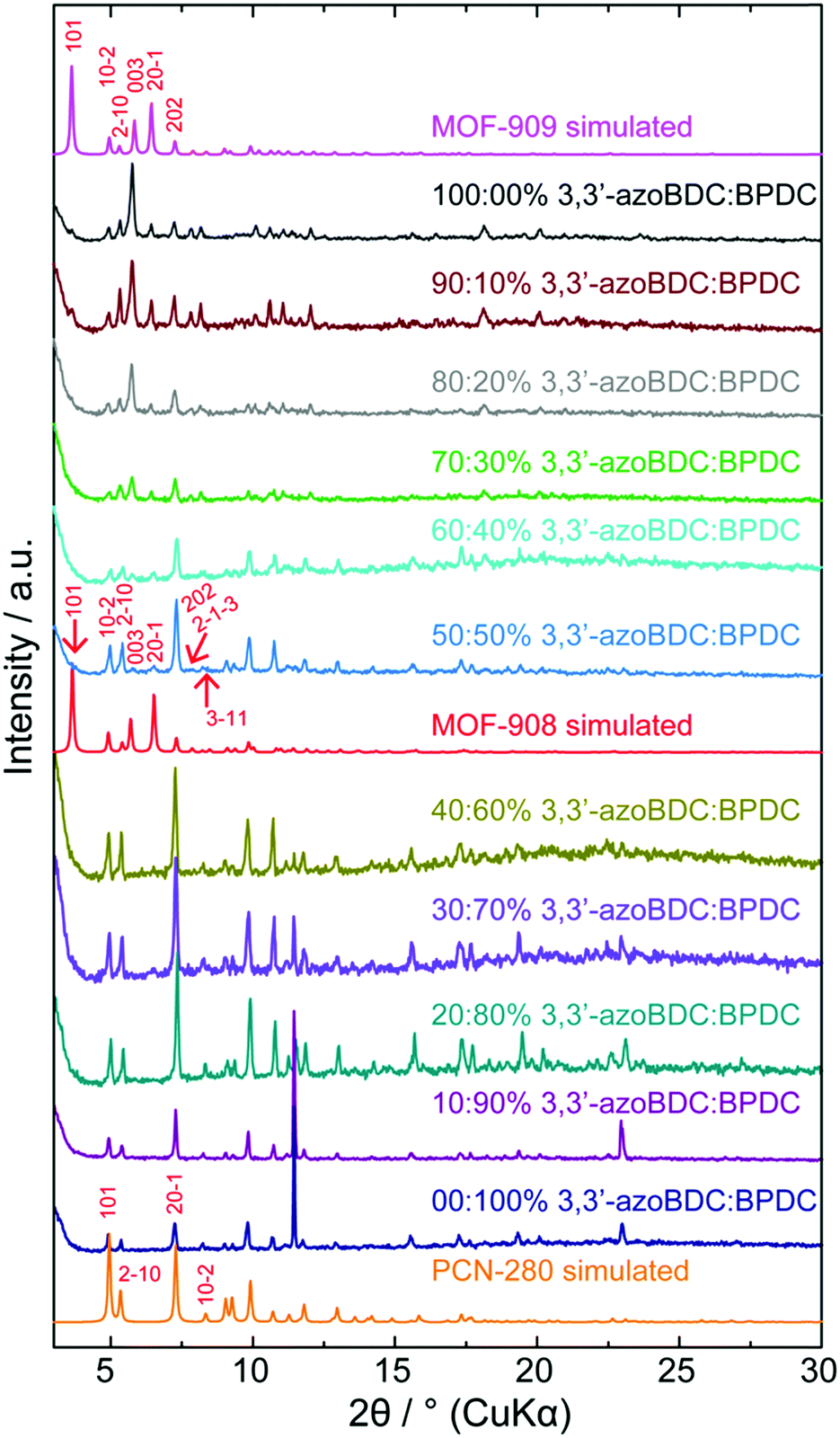 | ||
| Fig. 1 Powder X-ray diffraction analysis systematically demonstrates the control of interpenetration of 2-fold interpenetrated PCN-280 forming MOF-908 – a single frame version of PCN-280 – and an isoreticular structure of MOF-908, MOF-909. The reticular control is conducted via the rational design of input linkers (the ratio is based on molar proportions). | ||
The PXRD pattern of the 50:50 mol% as-synthesized sample exhibited 5 characteristic peaks coincident with the simulated pattern which belong to the Miller planes of (101), (003), (201), (2−1−3), and (3−11). It is noted that the (101) Bragg position of the 50:50 mol% sample obviously enhances in intensity after the solvent exchange and activation processes (ESI,† Section 4), confirming the observation of a single frame structure. PCN-280 appeared to be a single crystal represented by very thin hexagon shape while the single crystals of MOF-908 appeared in form of trigonal prisms due to crystal growth primarily along the c direction (ESI,† Section 2) resulting in a large unit cell in the crystal structure compared to PCN-280.
To further verify if the purely single phase of MOF-908 was obtained, the activated sample was digested for proton nuclear magnetic resonance spectroscopy (NMR). The post-digestion 1H-NMR spectrum displayed the presence of only the BPDC2− linking unit (ESI,† Section 4), undoubtedly proving that MOF-908 was purely produced by controlling the input ratio of the mixture of organic linkers. The phase transition from MOF-908 to MOF-909 is more distinctly displayed in those samples containing molar ratios of mixed linkers from 70:30 mol% to 100:0 mol% of 3,3′-azoBDC:H2BPDC (Fig. 1).
Due to the small size, large pore volume and nature of Fe-based MOFs, which leads to lowering of the resolution (a resolution of only 1.4 Å in the data collection was obtained by Mo X-ray radiation) of single crystal analysis, the MOF-908 crystal structure was finally elucidated by Rietveld refinement. This revealed that the structural single framework crystallized in a trigonal lattice with an R3 space group (no. 146) and unit cell parameters of a = b = 32.9145 Å and c = 46.0360 Å (ESI,† Section 3). MOF-909 is an isoreticular network of MOF-908 with similar unit cell parameters (a = b = 32.2658 Å and c = 45.8488 Å). The structure of MOF-908 is made up of two kinds of cage. The small primary cage with a pore size of 12 Å in diameter is a distorted octahedra constructed by six trigonal prism Fe–oxo clusters which link together through four BTB3− linkers connecting the vertices along opposing equatorial edges, and three BPDC2− moieties fill in the remaining equatorial edges composing the triangular window which is found to be 8 Å. The secondary cage is then built by the connection of eight primary cages with corners as nodes. In particular, those primary cages sharing four vertices, when translating a distance equal to the distance of two primary cages (approximately 23.2 Å), locate at the vertices of a cube and generate a cubic cage with a diameter of 23 Å. The square channel window of the secondary cage, which comprises four sharing vertices of the primary cage building units, was found to be 15.5 Å (Fig. 2b). Possessing the isoreticular structure of MOF-908, MOF-909, with the longer ditopic linking unit, had larger pore sizes but the triangular window size is smaller than that of MOF-908 due to the geometrical arrangement of 3,3′-azoBDC2−. In particular, the diameters of the distorted octahedra and cubic cage in MOF-909 are around 12.5 and 24 Å, respectively, while the triangular and square windows are calculated to be 7.5 and 16 Å, respectively. It is noted that all of the estimations are based on the model of the crystal structure verified by a Platon calculation, which is also applied to calculate the empty void space of approximately 82% for both MOF-908 and MOF-909.
 | ||
| Fig. 2 Representation of the single crystal structure and underlying net of PCN-280 and MOF-908. (a) Reaction of BTB3−, BPDC2−, and Fe3O(–CO2)6 to produce the 2-fold interpenetrated framework of PCN-280 whose the structure is looked down on from the c direction and the second frame is highlighted orange. (b) MOF-908 exhibits a single frame showing 2 kinds of pore, the distorted octahedral cage and the trigonal prism cage. (c) The natural tiling of the augmented nht-a topology is shown for MOF-908, MOF-909, and isoreticular PCN-285. It is noted that the network of PCN-280 belongs to the 2-fold nht network. Fe, blue polyhedral; C, black; and O, red. BTB3− linkers are shown in filled pink to aid in distinguishing them from the filled green BPDC2− linkers. The second framework of PCN-280 is orange. H atoms are omitted for clarity. | ||
The topological analysis of MOF-908 representing the underlying net of MOF-909, PCN-280, and PCN-285, which appeared to be isoreticular structures, was performed using the Topos Pro package.21 For the underlying net simplification, the Fe3O(–CO2)6 SBUs and the linkers containing BTB3− and BPDC2− were simplified as nodes which are 6 and 3 coordination points, corresponding to 6-c and 3-c, of the points of extension of the trigonal prismatic Fe–oxo cluster and the triangular node of the BTB moiety, respectively. The linear 2-c of the ditopic linker is reduced and no longer under consideration due to the formation of the 6-c point of extension for the Fe3O(–CO2)6 nodes. It is interesting that the structural deconstruction revealed an unseen network, termed the nht net, in MOF chemistry. The transitivity of the nht topology is described by the number of vertices p, the number of edges q, the number of faces r, and the type of tiling s. Indeed, the pqrs transitivity of nht is 3553 and the natural tiling of this kind of network is 3[4·72] + 3[43·53] + [3·53·76] (where the symbol […mn…] denotes n faces with m-membered rings in a topological network) (Fig. 2c).
The solvent-exchange was carried out using an anhydrous acetone solvent and the full activation procedure was subsequently followed by heating the material under low pressure at 100 °C for 24 h. The chemical formula of MOF-908 is Fe3O(OH)(BTB)4/3(BPDC), in which the mixed linker ratio of BTB:BPDC was found to be 4/3:1, as proven by 1H-NMR post-digestion (ESI,† Section 4). Thermal gravimetric analysis (TGA) measurements (ESI;† Section 4) further supported that formula because the observed residual mass (21.7%) perfectly matched with the expected mass of Fe2O3 (22.8%) after fully decomposing the structural framework at 600 °C. The elemental microanalysis (EA) continuously confirmed the formulation of MOF-908 due to the consistence of the CHN percentage between the experimental and theoretical model (ESI;† Section 2). Finally, the porosity of MOF-908 was proven by N2 adsorption isotherms at 77 K, which showed a permanent surface area based on the Brunauer–Emmett–Teller (Langmuir) method of 1850 (2315) m2 g−1 (ESI;† Section 4), which is in line with the geometrical surface area calculated from the model structure. This exceeds the BET value of 1500 m2 g−1 for PCN-280 and is lower than the value of 1945 m2 g−1 for MOF-909 (ESI;† Section 4), which were conducted under solvent-free conditions and under the same measurement conditions. The pore size distribution of PCN-280 and MOF-908 was analysed by DFT calculation based on the N2 adsorption isotherms, and the results showed the difference between the two: the pore size of MOF-908 (∼22 Å) is larger than that of PCN-280 (∼12 Å) (ESI,† Section 4).
We subsequently carried out first-principles density functional theory22,23 (DFT) calculations to unveil the mechanism of interpenetration control of PCN-280 to formulate MOF-908. We proposed to look at two different theoretical aspects which are believed to be helpful in explaining the overall scenario. First the reaction mechanisms between Fe3O(OH)(–CO2)6 and H2BPDC/3,3′-azoBDC were explored to clarify the energetic favorability of the two cluster-linker combinations. Second, by estimating elastic tensors of the abbreviated MOF-908 and MOF-909 structures, we were able to make conclusions on their relative stability in the condensed phase.
For the reaction mechanism investigation we employed the localized atomic-orbital-basis calculation method.24 The Perdew–Burke–Ernzerhof (PBE) functional25 was used to execute these calculations as well as the later condensed-phase calculations. The 6-31g* basis set26,27 was employed to construct the electronic wave-functions for C, H, O and N, while the LANL2dz basis set28,29 was utilized to describe the effective core potential for Fe. In both gas-phase and condensed-phase calculations we introduced a D3 correction to account for long-range van der Waals interactions.30 The quartet electronic spin state of the Fe3O(OH)(–CO2)6 cluster was revealed to be the most energetically-favored spin state; in other words, each Fe3+ site preferred to establish one unpaired electron. For the following investigation of reaction mechanisms we carried out all localized atomic-orbital-basis calculations at the quartet spin state.
As a linker (H2BPDC or 3,3′-azoBDC) attempts to substitute an acetate (CH3COO−) residue, one O atom from the linker approaches closely and makes a coordination bond to one Fe cation in the cluster. Because of this connection the Mulliken charge of the involved Fe3+ site decreased to 0.57, which is less positive than that of the other two Fe sites (0.74) in the cluster. The binding energy of the (H2BPDC)-cluster was 0.67 eV, while the binding energy of the (3,3′-azoBDC)-cluster was 0.59 eV, which showed the slight favorability of H2BPDC connection to the cluster. Such [Fe3O(OH)(–CO2)6]-linker complexes were regarded as initial reactants. The proton (H+) still remains linked with the linker molecule while attempting to make a weak bond with an O atom from acetate (Fig. 3a). To accomplish the cluster-linker connection, the system has to undergo two transformation stages: (i) an O atom from acetate first departs from Fe (a coordination bond is broken), and (ii) the O atom (from the –OH group of the linker) approaches and makes a connection with the other Fe cation linked to the same acetate residue. In the first process, H2BPDC or 3,3′-azoBDC actually play a role as a catalyst to loosen the Fe–acetate linkage. Actually, we performed a relaxed scan of the Fe–O(acetate) dissociation in each case and found a barrier of 0.76 eV (17.5 kcal mol−1) for both cases, as illustrated in Fig. 3b. Hence, there is competition between H2BPDC and 3,3′-azoBDC in the first binding process to the Fe3O cluster and the weakening of the Fe–acetate bond.
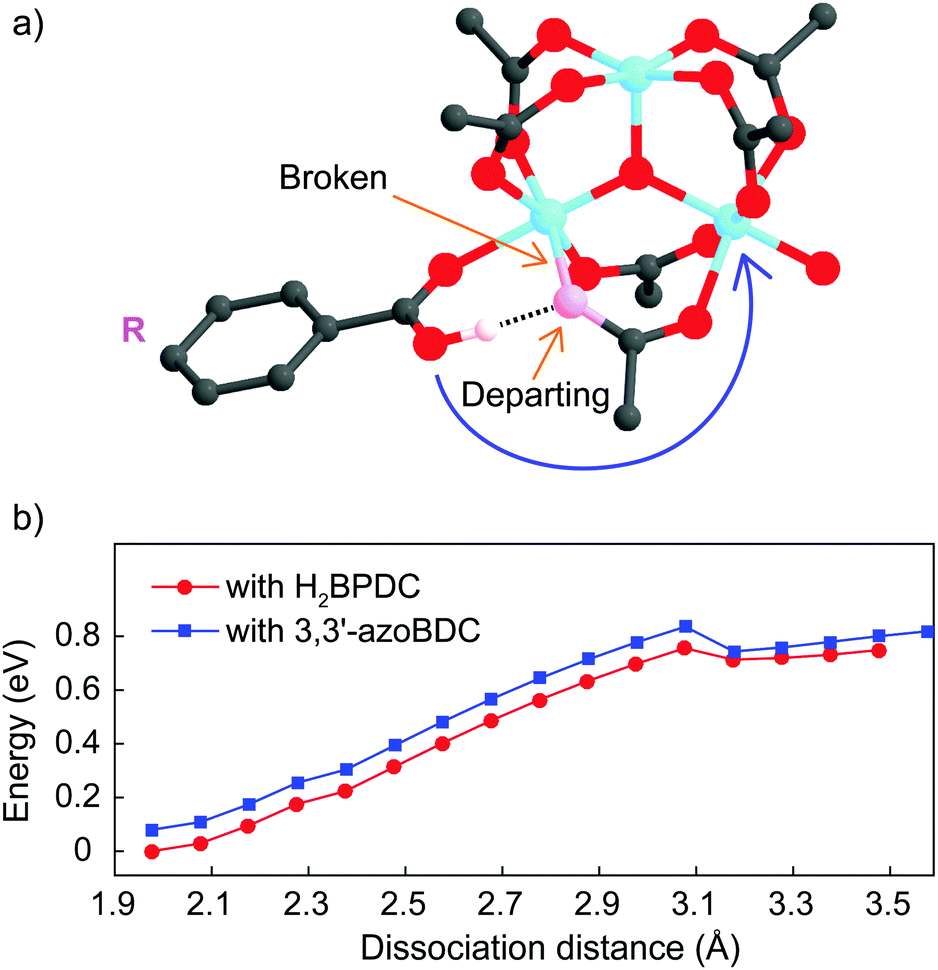 | ||
| Fig. 3 (a) The initial reaction in which one O atom from RCOOH (linker) attaches to one Fe site while the –OH group moves forward to donate its proton toward the pink O atom from CH3COO−. The pink O atom soon departs from Fe and gives RCOOH the chance to establish bonding to the second Fe site. (b) Relaxed scans of the Fe–O(acetate) bond in which the Fe–O bond is broken with the same energy barrier (0.76 eV). This indicates the competitiveness between H2BPDC or 3,3′-azoBDC in the first process of the substitution. The energy difference between the two curves comes from the difference in binding energies with the RCOOH-cluster (∼0.8 eV). | ||
Soon after the disruption of the Fe–O bond in the first step the linker is more motivated to attack the second Fe site. In the second process, we located the transition states when H2BPDC/3,3′-azoBDC attempt to replace acetate. In those transition states, the Fe–O(H2BPDC) distance was observed as 3.21 Å, while the Fe–O(3,3′-azoBDC) distance was similar (3.23 Å). However, the H2BPDC transition state was found to be 0.43 eV (9.9 kcal mol−1) above the initial reactant, while the 3,3′-azoBDC transition state was 0.76 eV (17.5 kcal mol−1) above the initial reactant. The energetic scheme (Fig. 4) shows that 3,3′-azoBDC is less competitive in forming the framework.
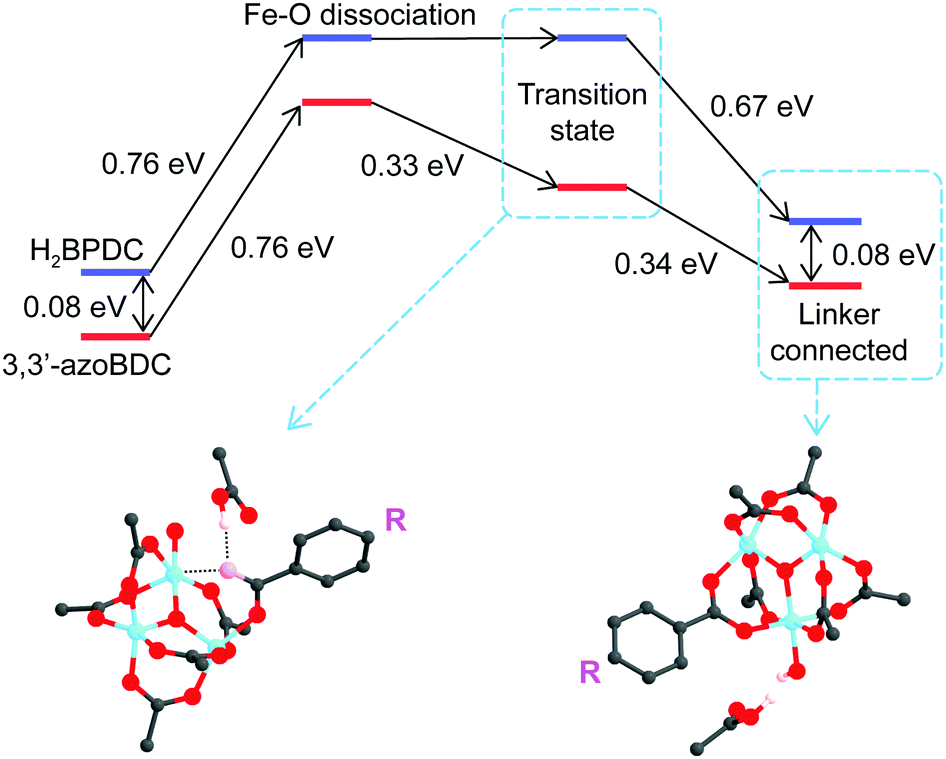 | ||
| Fig. 4 Energy profile of the two-step mechanism for RCOOH (linker) to substitute CH3COO−. In the second step, H+ is transferred to CH3OO−, while the second O from the linker approaches the second Fe site. H2BPDC is more favorable in the second step with the transition state located at 0.43 eV above the initial state, while the transition state of 3,3′-azoBDC is 0.76 eV above the initial state. | ||
Additionally, we also took into account a circumstance in which 3,3′-azoBDC successfully establishes connection to the cluster, and verified how H2BPDC could come in and replace the 3,3′-azoBDC linker. We believe that the substitution should be identical to the mechanism described above when H2BPDC (or 3,3′-azoBDC) substituted CH3COO−. As a matter of fact, the transition state of H2BPDC replacing 3,3′-azoBDC is 0.78 eV (18.0 kcal mol−1) above the initial state when 3,3′-azoBDC is connected to the cluster. The final state, in which H2BPDC makes a connection to the cluster and eliminates 3,3′-azoBDC, was found to be more stable than the initial state by 0.24 eV (5.5 kcal mol−1). At this stage, by conducting localized atomic-orbital-basis calculations, we found that H2BPDC and 3,3′-azoBDC are competitive to each other in the first stage of establishing bonding with one Fe cation. This first stage is very important because the participation of 3,3′-azoBDC is involved in the formulation of the single framework and avoided interpenetration. The second stage takes place rapidly after the first process, where H2BPDC was shown to be preferable in bonding with the iron cluster.
We also performed PBE calculations for the condensed phases of MOF-908 and MOF-909 to evaluate their relative stability using the projector-augmented wave method31 implemented in the Vienna Ab Initio Package.32–34 Those two structures were extremely large and sophisticated and therefore, we chose to simplify the model by substituting three acetylene residues into the position of three phenyls in BTB (the crystallographic data of the original MOF-908 and MOF-909 as well as the modified structures are available in the ESI†). Such substitution was believed to alter the unit cell parameters insignificantly. To afford computational feasibility, planewave expansion was only done around the Γ point with a cut-off energy of 350 eV. After a simultaneous relaxation of unit cells and atomic positions, we calculated the Cij elastic tensors for both systems by adopting the stress–strain relationship.35 It was observed that the obtained Cij tensors of MOF-908 and MOF-909 satisfied the four conditions of mechanical stability of hexagonal unit cells. Moreover, the Cij tensors of MOF-908 were much larger than those of MOF-909. To make a more quantitative justification, we then computed the shear (G), bulk (K), and Young's (E) moduli, as shown in Table 1 along with the Cij quantities. Remarkably, each of the moduli of MOF-908 was six times larger than the corresponding modulus computed for MOF-909. Moreover, all G, K, and E of MOF-909 were below 200 kbar, which is relatively low for a large unit cell and indicates that the framework of MOF-909 could suffer and collapse more easily. Such theoretical quantities clearly explain the experimental fact that MOF-909 actually partially collapsed after activation (the PXRD pattern shows the reduced intensity of the peaks located higher than 7.5° 2θ, ESI,† Section 4). Additionally, MOF-908 is more mechanically stable than MOF-909, as proven by the pellet pressing test which clearly shows that the MOF-908 framework still remains up to 11 tons cm−2 of pressure while the MOF-909 structure collapses at a pressure of 5 tons cm−2 (ESI,† Section 4).
| MOFs | C 11 | C 12 | C 13 | C 33 | C 44 | C 66 |
|---|---|---|---|---|---|---|
| MOF-908 | 1460 | 400 | 387 | 1263 | 510 | 530 |
| MOF-909 | 231 | 58 | 65 | 193 | 84 | 88 |
| MOFs | G | K | E |
|---|---|---|---|
| MOF-908 | 510 | 726 | 1241 |
| MOF-909 | 82 | 114 | 199 |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We extend our gratitude to Mr Long H. Ngo, Mr Bao N. Truong (KITECH, Korea), Mr Thanh T. Vu, Mr Dinh Q. Le, Ms Yen H. Ha, Mr Hieu C. Dong and Dr Tan L. H. Doan (INOMAR) for helpful discussions. H. L. N acknowledges financial support from VNU-HCM (no. B2017-50-01). We also acknowledge the supercomputing assistance from the Institute for Materials Research, Tohoku University.References
- H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- M. Li, D. Li, M. O'Keeffe and O. M. Yaghi, Chem. Rev., 2014, 114, 1343–1370 CrossRef PubMed.
- B. T. Nguyen, H. L. Nguyen, T. C. Nguyen, K. E. Cordova and H. Furukawa, Chem. Mater., 2016, 28, 6243–6249 CrossRef.
- L. Zhang, P. Cui, H. Yang, J. Chen, F. Xiao, Y. Guo, Y. Liu, W. Zhang, F. Huo and B. Liu, Adv. Sci., 2016, 3, 1500243 CrossRef PubMed.
- H. L. Nguyen, New J. Chem., 2017, 41, 14030–14043 RSC.
- C. A. Trickett, A. Helal, B. A. Al-Maythalony, Z. H. Yamani, K. E. Cordova and O. M. Yaghi, Nat. Rev. Mater., 2017, 2, 17045 CrossRef.
- M. G. Campbell, S. F. Liu, T. M. Swager and M. Dincă, J. Am. Chem. Soc., 2015, 137, 13780–13783 CrossRef PubMed.
- Y.-N. Bong, D.-C. Zhong and T.-B. Lu, CrystEngComm, 2016, 18, 2596–2606 RSC.
- A. Ferguson, L. Liu, S. J. Tapperwijn, D. Perl, F.-X. Coudert, S. Van Cleuvenbergen, T. Verbiest, M. A. van der Veen and S. G. Telfer, Nat. Chem., 2016, 8, 250–257 CrossRef PubMed.
- R. Haldar, N. Sikdar and T. K. Maji, Mater. Today, 2015, 18, 97–116 CrossRef.
- R. J. Marshall, C. T. Lennon, A. Tao, H. M. Senn, C. Wilson, D. Fairen-Jimenez and R. S. Forgan, J. Mater. Chem. A, 2018, 6, 1181–1187 RSC.
- O. K. Farha, C. D. Malliakas, M. G. Kanatzidis and J. T. Hupp, J. Am. Chem. Soc., 2010, 132, 950–952 CrossRef PubMed.
- H.-L. Jiang, T. A. Makal and H.-C. Zhou, Coord. Chem. Rev., 2013, 257, 2232–2249 CrossRef.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O’Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef PubMed.
- D. Sun, S. Ma, Y. Ke, D. J. Collins and H.-C. Zhou, J. Am. Chem. Soc., 2006, 128, 3896–3897 CrossRef PubMed.
- R. K. Deshpande, J. L. Minnaar and S. G. Telfer, Angew. Chem., Int. Ed., 2010, 49, 4598–4602 CrossRef PubMed.
- D. Feng, K. Wang, Z. Wei, Y.-P. Chen, C. M. Simon, R. K. Arvapally, R. L. Martin, M. Bosch, T.-F. Liu, S. Fordham, D. Yuan, M. A. Omary, M. Haranczyk, B. Smit and H.-C. Zhou, Nat. Commun., 2014, 5, 5723 CrossRef PubMed.
- H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. O. Yazaydin, R. Q. Snurr, M. O'Keeffe, J. Kim and O. M. Yaghi, Science, 2010, 239, 424–428 CrossRef PubMed.
- H. L. Nguyen, T. T. Vu, D.-K. Nguyen, C. A. Trickett, T. L. H. Doan, C. S. Diercks, V. Q. Nguyen and K. E. Cordova, Chem. Commun., 2018 Search PubMed , in press.
- H. Chevreau, T. Devic, F. Salles, G. Maurin, N. Stock and C. Serre, Angew. Chem., Int. Ed., 2013, 52, 5056–5060 CrossRef PubMed.
- V. A. Blatov, A. P. Shevchenko and D. M. Proserpio, Cryst. Growth Des., 2014, 14, 3576–3586 CrossRef.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864–B871 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef PubMed.
- V. A. Rassolov, J. A. Pople, M. A. Ratner and T. L. Windus, J. Chem. Phys., 1998, 109, 1223–1229 CrossRef.
- V. A. Rassolov, M. A. Ratner, J. A. Pople, P. C. Redfern and L. A. Curtiss, J. Comput. Chem., 2001, 22, 976–984 CrossRef.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284–298 CrossRef.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef.
- Y. Le Page and P. Saxe, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 104104 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Full synthesis and characterization of MOF-908, MOF-909, crystallographic data, and additional gas uptake measurement details. See DOI: 10.1039/c8qm00368h |
| This journal is © the Partner Organisations 2018 |