
Ultralong needle-like N-doped Co(OH)F on carbon fiber paper with abundant oxygen vacancies as an efficient oxygen evolution reaction catalyst†
Jiaqi
Lv
,
Xiaoxuan
Yang
,
Hong-Ying
Zang
 *,
Yong-Hui
Wang
and
Yang-Guang
Li
*
*,
Yong-Hui
Wang
and
Yang-Guang
Li
*
Key Lab of Polyoxometalate, Science of Ministry of Education, Key Laboratory of Nanobiosensing and Nanobioanalysis at Universities of Jilin Province, Institute of Functional Material Chemistry, Faculty of Chemistry, Northeast Normal University, Changchun, 130024, Jilin, P. R. China. E-mail: zanghy100@nenu.edu.cn; liyg658@nenu.edu.cn
First published on 10th September 2018
Abstract
Although significant efforts have been devoted to designing efficient electrocatalysts for the oxygen evolution reaction, further progress is still needed regarding modulating the microstructures of electrocatalysts to further improve catalytic activity. Herein, utilizing ionic compound NaF and heteroatom-N-rich urea as morphology directing reagents, ultralong needle-like N-doped Co(OH)F supported on carbon fiber paper has been synthesized through a one-step hydrothermal method. Specifically, the hydrothermal kettle provides a stable environment for the formation of ultra-long needle-like structures. Meanwhile, with the addition of halogen anion F− and heteroatom N, a wealth of oxygen vacancies are obtained, and the OER activity is enhanced. The as-prepared N-doped Co(OH)F-CFP (N:Co(OH)F-CFP) has excellent hydrophilicity, which facilitates its contact with the electrolyte and then accelerates the passing of electrons back and forth between the surface of the material and the electrolyte. Under alkaline conditions, N:Co(OH)F-CFP's OER activity is superior to that of IrO2. Its potential can reach 1.540 V (vs. RHE) at 10 mA cm−2 with a Tafel slope of 69.75 mV dec−1 in 1 M KOH solution.
Introduction
The greenhouse effect and smog problems have plagued people. Seeking an effective pollution-free energy source to replace traditional fossil fuels or to explore a new way of energy conversion is a major challenge in the twenty-first century.1,2 Electrochemical water splitting has been the focus of recent research on energy conversion and storage, including the oxygen evolution reaction and hydrogen evolution reaction (OER and HER).3–5 Currently, the bottleneck of these two half-reactions is the too high overpotential,6 especially for the OER, whose complex reaction process involves multi-electron transfer.7–9 Therefore, the OER plays a vital role in the process of water splitting.10 At present, RuO2 and IrO2 are the most effective catalysts for the OER.11 But RuO2 and IrO2 are expensive and cannot be used on a large scale because of their limited output and relatively poor stability.12 Therefore, it is urgent to develop a new type of low cost catalytic material instead. Among a wide range of new OER catalysts, one of the best choices is based on earth-abundant metals, such as Fe, Co, Ni, etc.,13,14 which are rich in output and have a lower price. Recently, some research also showed that their oxides,15 sulfides,16,17 carbides,18 hydroxides19,20 and nitrides21,22 are effective OER catalyst replacements.Co-based materials have been studied for application in electrochemical water splitting due to their high intrinsic reactivity.23,24 Besides, cobalt-based materials have exceptional 3d electronic configurations.25 Under the influence of some electron-withdrawing atoms, defects are easily generated in the hybrid materials. The unsaturated vacancies due to the lack of oxygen atoms are not easily stable under alkaline conditions and the OH− groups in the solution will be filled into the defect. Meanwhile, the overpotential of H2O dissociation can be reduced, which in turn enhanced the affinity for the reactant with intermediates.26,27 The defects on the electrocatalyst can provide accessible active sites to activate H2O molecules and accelerate the rate-determining step of the OER reaction so as to increase the kinetics of the reaction.28–31 The cobalt-based materials (especially cobalt (oxy)hydroxides) have attracted intensive attention in the field of energy conversion.32 It was discovered to have special activities in catalyzing oxygen and hydrogen evolution reactions in alkaline solution.33 However, the conventional cobalt-based (oxy)hydroxides are easy to gather25 and induce magnetism, which will absorb the electrons and make them difficult to leave.19 So we propose to employ a substrate (carbon cloth or carbon fiber paper with good conductivity and higher supportive areas)34,35 to reduce the aggregation of substances, and meanwhile, this design will improve its conductivity and the speed of electronic transmission. Furthermore, the material loaded on the substrate is conducive to large-scale promotion and application.
To some extent, the amount of halogen elements and heteroatoms in the catalyst are also important factors that influence the electrocatalytic activity.23 When the anion F− is introduced into the cobalt hydroxide, it has some positive effect on catalysis. Zhu et al. reported the concept of Co(OH)F for the first time.36 This is also a design strategy for fluoride-assisted synthesis. Sun's group used Co(OH)F as a precursor, phosphating it as an OER electrochemical catalyst in neutral media.37 Then Co(OH)F was further sulfided and loaded on carbon cloth as an efficient catalyst for the HER.38 Wan et al. synthesized a high-dimensional (3D) Co(OH)F architecture used for water oxidation.39 The above description shows that the introduction of F− not only improves the catalytic activity of cobalt hydroxide, but also expands the application field of cobalt hydroxide. For nitrogen, the experimental results of Wu et al. demonstrate that the nitrogen-doped material can improve the catalytic properties of the ORR and OER.13 Wang et al. also proved that the nitrogen-doped material is beneficial for the oxygen catalytic performance of the material.40,41 In the hydrothermal reaction, urea is often chosen as a supplier of heteroatom nitrogen, thereby improving the properties of the material.42 Hydrothermal reactions have been extensively studied, because in a confined container, we can ensure that all the reactants have a relatively stable reaction environment. By controlling the reaction time and temperature, the material's morphology and structure can be adjusted.
Herein, we introduced the heteroatom nitrogen into the Co(OH)F material through a hydrothermal process. We chose a one-step hydrothermal synthetic method to obtain a catalyst that contains heteroatoms N and F.43 It is called N-doped Co(OH)F, which is supported on the substrate of carbon fiber paper (N:Co(OH)F-CFP). In the N:Co(OH)F-CFP, the electrical conductivity is improved and the morphology is kept, and meanwhile, there are a lot of oxygen vacancies in the N:Co(OH)F-CFP material which guarantees excellent OER activity.
Experimental section
Chemicals and materials
All chemical reagents were AR grade, with no further treatment: Co(NO3)2·6H2O (≥98.5%, Sinopharm Chemical Reagent Co., Ltd), CH4N2O (≥99.0%, Sinopharm Chemical Reagent Co., Ltd), NaF (≥98.5%, Tianjin Fuchen chemical reagents factory), and C6H12O6 (≥98.5%, Tianjin Fuchen chemical reagents factory).Preparation of carbon fiber paper
Cut-sheet CFPs were cleaned with 6 mol L−1 HNO3 solution for 2 h, then ultrasonicated for about 30 minutes. After that, the CFPs were dipped in 0.1 M KOH solution for another 30 minutes with heating, then rinsed with deionized water several times and dried at 60 °C overnight in a vacuum oven.The surface of the CFP treated with 6 mol L−1 HNO3 solution produces some oxygen functional groups, –COO– and –OH, and these groups can act as anchors to receive a variety of ions to facilitate the growth of materials on the top of the CFP after hydrothermal synthesis; meanwhile, the adhesion of them increases.44
Synthesis of N:Co(OH)F-CFP
The Co(OH)F was synthesised by using a one-step hydrothermal method. It was prepared by mixing Co(NO3)2·6H2O (0.291 g), NaF (0.226 g) and urea (0.610 g) in 40 ml deionized water, and uniformly stirring for about 20 min to generate a homogeneous ink. The CFP (2 cm × 3 cm × 0.01 cm (thickness)) was put into a Teflon-lined stainless steel autoclave. Then the mixture solution was transferred into the Teflon-lined stainless steel autoclave. The reactor was heated at 120 °C for 6 h in an oven. Teflon-lined stainless steel was cooled to room temperature naturally. The carbon fiber paper was rinsed with ethanol and deionized water several times. Then the product was put in a vacuum drying oven overnight.Synthesis of N:Co(OH)x-CFP
The method for the synthesis of N:Co(OH)x-CFP was the same as above. Co(NO3)2·6H2O (0.291 g) and urea (0.610 g) were added in 40 ml deionized water, and uniformly stirred for about 20 min to generate a homogeneous ink. The CFP (2 cm × 3 cm × 0.01 cm (thickness)) was put into a Teflon-lined stainless steel autoclave. The reactor was placed in an oven at 120 °C for 6 hours, then naturally dropped to room temperature. The CFP was then removed from the kettle, washed with ethanol and water, and dried in a vacuum drying oven.Synthesis of Co(OH)F-CFP
For the synthesis of Co(OH)F-CFP. Co(NO3)2·6H2O (0.291 g), C6H12O6 (1.80 g) and NaF (0.226 g) were added in 40 ml deionized water, and uniformly stirred for about 20 min to generate a homogeneous ink. The CFP (2 cm × 3 cm × 0.01 cm (thickness)) was put into a Teflon-lined stainless steel autoclave. The reactor was placed in an oven at 120 °C for 6 hours, then naturally dropped to room temperature.Synthesis of Co(OH)x-CFP
The method for the synthesis of Co(OH)x-CFP was the same as above. After getting N:Co(OH)F-CFP, it was immersed in 1 M KOH solution. Then the solution was transferred to the reactor again. The reactor was baked at 120 °C for 2 h and cooled to room temperature. The kettle was then opened to remove the CFP, which was washed with ethanol and deionized water several times, and then Co(OH)x-CFP was obtained.Characterization
Scanning electron microscopy (SEM) and energy dispersive X-ray spectroscopy (EDS) were performed with a Hitachi SU-8010A scanning electron microscope. Transmission electron microscopy (TEM) and selected area electron diffraction (SAED) were carried out on a JEM-2100F. A Siemens D5005 diffractometer with Cu-Kα radiation was used as an instrument to test the Powder X-ray diffraction (PXRD). X-ray photoelectron spectroscopy (XPS) was conducted with a VG ESCALAB MKII using a monochromic Al-Kα achromatic X-ray source. IR was tested by Nicolet 6700-FTIR; the test range is 400–4000 cm−1, and the resolution of it is 0.125 cm−1.Electrochemical measurements
All the electrochemical measurements were performed on an electrochemical analysis station (CHI 760 E, CH Instruments, China) at 25 °C using a standard three-electrode cell. The N:Co(OH)F-CFP was the working electrode, and a carbon rod and a Hg/HgO electrode in saturated KOH served as the counter and reference electrodes, respectively. After the reaction, we measured the mass of each CFP compared to pre-reaction, and their loading was same. IrO2 with the same amount coated on the CFP was also used as the working electrode for comparison. OER polarization curves were obtained by linear sweep voltammetry scanning from 0 V to 1 V at a scan rate of 10 mV s−1 in N2 saturated in 1 M KOH solution. Cyclic voltammograms (CVs) were obtained by scanning between 0.1 V and 0.35 V at different scan rates in N2 saturated solution.All the potentials versus the Hg/HgO reference electrode were converted to the reversible hydrogen electrode (RHE) scale via the Nernst equation:
| ERHE = EHg/HgO + 0.0592 pH + EθHg/HgO |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logj + a, where η is the overpotential, b is the Tafel slope, j is the current density and a is a constant. An amperometric i–t curve was tested at a constant potential to evaluate the stability of the materials. Electrochemical impedance spectroscopy (EIS) was tested at an amplitude of 5 mV, and the test range was 1–100000 Hz. An electrochemically active surface area (ECSA) test was performed by measuring the CV at different sweep speeds of the sample from 0.1 V to 0.35 V vs. RHE and then plotting the CV curve. According to different CV cycles, the sweep rate was taken as the abscissa and half of the difference between the up and down values was taken as the ordinate to get the electric double layer capacitance diagram. The double-layer capacitance Cdl is the slope of the corresponding pattern, which can also be said to be one of the means of ECSA characterization.45,46
logj + a, where η is the overpotential, b is the Tafel slope, j is the current density and a is a constant. An amperometric i–t curve was tested at a constant potential to evaluate the stability of the materials. Electrochemical impedance spectroscopy (EIS) was tested at an amplitude of 5 mV, and the test range was 1–100000 Hz. An electrochemically active surface area (ECSA) test was performed by measuring the CV at different sweep speeds of the sample from 0.1 V to 0.35 V vs. RHE and then plotting the CV curve. According to different CV cycles, the sweep rate was taken as the abscissa and half of the difference between the up and down values was taken as the ordinate to get the electric double layer capacitance diagram. The double-layer capacitance Cdl is the slope of the corresponding pattern, which can also be said to be one of the means of ECSA characterization.45,46
Results and discussion
Structure and morphology of materials
In this work, we used a one-step hydrothermal method to synthesize a class of defective OER catalytic material N-doped Co(OH)F on carbon fiber paper (N:Co(OH)F-CFP). Fig. 1 schematically describes the growth process of the 3D needle-like N:Co(OH)F arrays on the CFP. First, small thorn strips of N:Co(OH)F were formed, which were intertwined with each other. When there was no CFP, they stacked into spiked spheres over time, as process (i) showed. With the addition of CFP, it began to form large spikes and uniformly grew on the surface of CFP. The overall structure was like a needle (ii). During the hydrothermal reaction, urea acts as a nitrogen source together with NaF to act as a structure-directing agent. They control the formation of the needle-like structure. On the other hand, the treated CFP surface has many oxygen-containing functional groups, such as –O–H, –O–OH etc., providing nucleation sites for the growth of this N:Co(OH)F thorn and avoiding agglomeration.44 The vertical staggered growth of sharp thorns provides an open space with rich edge activity.47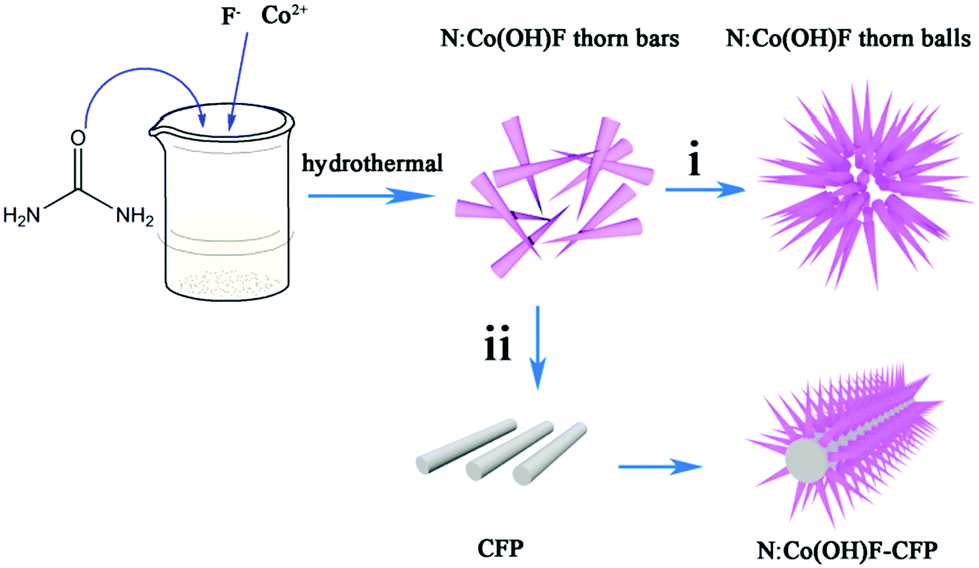 | ||
| Fig. 1 Schematic representation of the growth process of the 3D needle-like N:Co(OH)F array structure on CFP. | ||
Fig. 2a displays the PXRD pattern of N:Co(OH)F-CFP. The diffraction peaks at 26.42° and 54.53° correspond to carbon's standard PDF card (PDF#41-1487).48 This comes from the substrate carbon fiber paper. The other peaks at 33.62° and 39.22° are Co(OH)F (27753-ICSD).39 The substrate of carbon fiber paper exhibited strong carbon peaks in the PXRD patterns, which covered other substances’ peaks, leading to the weak peaks of Co(OH)F. In order to further highlight the peaks of the synthetic material, we scraped the material N:Co(OH)F off the surface of the CFP and compared it with the standard XRD card of Co(OH)F (ICSD-27753), as shown in Fig. 2a. Then Fig. 2b shows the XRD diffraction of N:Co(OH)F-CFP before and after the 10 h IT stability test, and we can see that the position of the main peaks showed almost no change and revealed the stability of the material's skeleton.
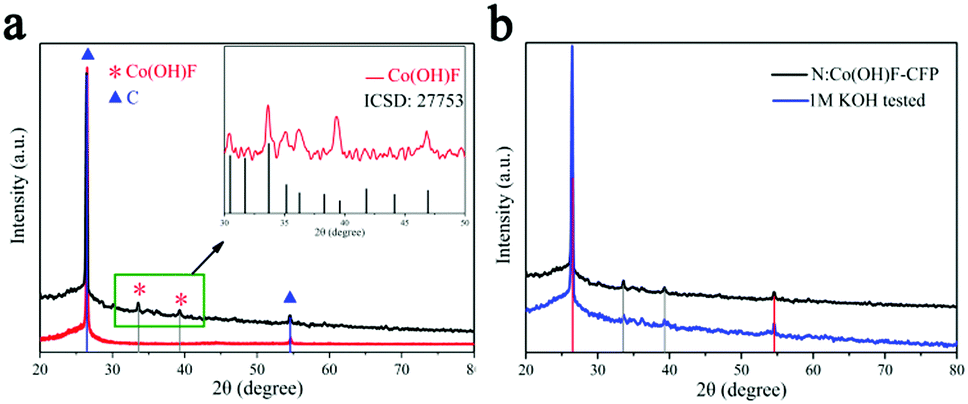 | ||
| Fig. 2 (a) PXRD patterns of the CFP without and with N:Co(OH)F. Red for CFP, black for N:Co(OH)F-CFP. Inset “a” is the XRD of N:Co(OH)F and standard Co(OH)F XRD card. (b) PXRD patterns of N:Co(OH)F-CFP before and after the IT stability test. | ||
The scanning electron microscopy (SEM) image of N:Co(OH)F-CFP (Fig. 3a) shows that the ultralong thorns of N:Co(OH)F are vertically aligned on the CFP substrate, forming a uniform nanowire array. Fig. S1a–e (ESI†) exhibit SEM images of different sizes of N:Co(OH)F-CFP, respectively. Fig. S1f (ESI†) shows a side sectional view of N:Co(OH)F on the surface of CFP. Different patterns of thorns close to the CFP surface can be seen. Fig. 3b is a partial TEM of N:Co(OH)F, which is clearly seen to be consistent with SEM, while Fig. 3c shows that the thorn is about 3–4 μm long and 50 nm in the radial direction (wide).39 Ultralong thorns facilitate contact with the electrolyte and accelerate conduction. In order to further explore the significance of NaF for the formation of the material, we did not add NaF when the original ratio conditions were the same, and the non-added SEM images are shown in Fig. S2 (ESI†). It's obvious that when there is no NaF involved in the reaction, it does not grow vertically on the surface of CFP. It's just a simple cross-like composition of network-like strips, which are attached to the CFP surface. Simultaneously, this structure cannot have enough contact with the electrolyte solution, and its OER activity is worse than N:Co(OH)F-CFP. This result is shown in the electrochemical test section of this article. Meanwhile, F− can form a strong Co–F bond with Co2+; this will be proved in the following XPS. The formation of the bond makes the overall structure of N:Co(OH)F more stable, thereby facilitating the formation of the arrays on the surface of CFP. This also confirms the important role of F− in this reaction. Next, for the importance of CFP, we did an experiment under the same conditions without adding CFP to collect the powder product. As shown in Fig. 3d, it reunited on its own into a ball-spike structure, which also showed that CFP could prevent the reunion of N:Co(OH)F to avoid unnecessary electrostatic resistance. And the catalytic activity of N:Co(OH)F was worse than that of N:Co(OH)F-CFP (Fig. S3, ESI†). Besides we also tested the SEM images of IrO2-CFP and the results are shown in Fig. S4 (ESI†). The mapping images (Fig. S5, ESI†) of the material prove that N, O, Co and F in the sample are uniformly distributed. Since the substrate is CFP, mapping of C is not considered. The SAED pattern (inset in Fig. 3e) indicates the crystalline nature of needle-like N:Co(OH)F-CFP. Sharp peaks in the XRD also show that these particles are crystallised. The lattice fringes with spacing of 0.26 nm are indexed to the (201) facets of N:Co(OH)F (Fig. 3e).39 After the 10 hours’ IT stability test, we tested the SEM of N:Co(OH)F-CFP. The result is shown in Fig. S6 (ESI†). As can be seen from Fig. S6a and b (ESI†), there are some interwoven thorns on the surface of the arrays of CFP. This is because a few arrays are caused to drop under the high current of the stability test. However, Fig. 3c and d show that the ultralong thorns of N:Co(OH)F-CFP after the IT test are unchanged. This indicates the stability of the array structure.
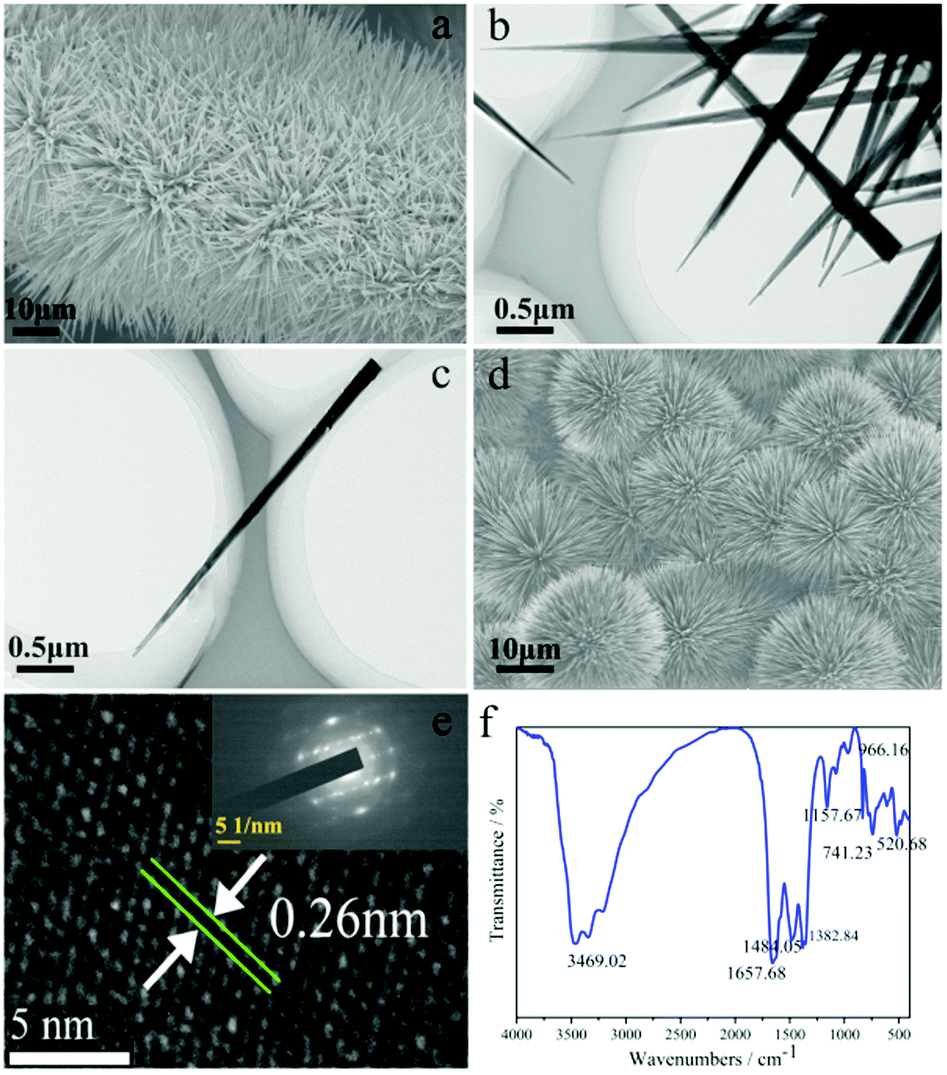 | ||
| Fig. 3 (a) SEM of N:Co(OH)F-CFP. (b and c) TEM of N:Co(OH)F-CFP. (d) The SEM of the N:Co(OH)F-CFP not grown on the CFP. (e) HRTEM; the inset in “e” is the SAED pattern of the sample. (f) IR. | ||
In order to further illustrate the types of functional groups contained in the N:Co(OH)F-CFP, IR testing was performed (Fig. 3f). IR analysis showed a broad peak at 3469.02 cm−1 was attributed to the stretching vibrations of the O–H bonds. This reveals the presence of hydroxyl functional groups on its surface, which further explains why it has hydrophilicity.49 The peak at 1657.68 cm−1 originates from the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bonds.50 This shows that the N:Co(OH)F has a close combination with the CFP surface, further illustrating the binding mode between N:Co(OH)F and CFP. The peaks of 1484.05 and 1382.84 cm−1 are stretching vibrations ν(O–C–O–O) and ν(CO3), respectively. The peak at low wavenumber (741.23 cm−1) is from the O–C–O bending mode.51 The peaks at 966.16 and 520.68 cm−1 correspond to δ(Co–OH) and ρw(Co–OH) bending modes.37,51
N double bonds.50 This shows that the N:Co(OH)F has a close combination with the CFP surface, further illustrating the binding mode between N:Co(OH)F and CFP. The peaks of 1484.05 and 1382.84 cm−1 are stretching vibrations ν(O–C–O–O) and ν(CO3), respectively. The peak at low wavenumber (741.23 cm−1) is from the O–C–O bending mode.51 The peaks at 966.16 and 520.68 cm−1 correspond to δ(Co–OH) and ρw(Co–OH) bending modes.37,51
N2 adsorption can be used to study the microstructures for needle-like N:Co(OH)F. BET measurement displays that the surface area of needle-like N:Co(OH)F is 37.14 m2 g−1 (Fig. S7, ESI†). We think this is the result of the pyrolysis and release of NH3 and H2O during the hydrothermal reaction.37 Meanwhile, under the action of water, urea decomposed to produce NH3 and CO2, and then NH3 combined with water forming into NH4+ and OH−.
| Co2+ + F− → CoF+ |
| Co(NO3)2·H2O → Co2+ + NO3− |
| CO(NH2)2 + H2O → CO2 + 2NH3 |
| NH3 + H2O → NH3·H2O → NH4+ + OH− |
In the solution, Co2+ combines with F− to produce CoF+, and urea decomposes into OH− and NH4+, which co-produce Co(OH)F and NH3·H2O, and then turn into N:Co(OH)F as follows:
| CoF+ + OH− + NH4+ → Co(OH)F + NH3·H2O → N:Co(OH)F. | (1) |
| Co2+ + NH3·H2O → [Co(NH3)]2+ + H2O |
| [Co(NH3)]2+ + OH− → [Co(OH)]+ + NH3 |
| Co(OH)+ + OH− ↔ CoOOH |
| [Co(OH)]+ + F− → Co(OH)F + NH3·H2O → N:Co(OH)F | (2) |
The contact angle measurements of bare CFP and N:Co(OH)F-CFP are shown in Fig. S9 (ESI†). The water contact angle of bare CFP was 98°, showing obvious hydrophobicity. While the contact angles of N:Co(OH)F and N:Co(OH)F-CFP were both 0°, indicating that they had special affinity for water, namely hydrophilicity. At the same time, it is also demonstrated that there is a clear open coordination site on the surface of N:Co(OH)F-CFP to promote the rate of oxygen evolution, favoring the departure of electrons in the OER reaction.52 The surface effect is an important factor affecting a material's properties.26 So we conducted an XPS test to analyze the surface situations of N:Co(OH)F-CFP.
The XPS test can be used to characterize the existence of oxygen vacancies and the valence of cations. The X-ray photoelectron spectroscopy (XPS) full-spectrum analysis is shown in Fig. 4a.53 XPS full spectrum analysis showed that the sample contained elements Co, N, O, C and F, where Co/N was 3.54, indicating that the product was a non-stoichiometric compound. But this has shown that N successfully incorporates into Co(OH)F.54 The C is mainly provided by the base CFP. Table S1 of the ESI† shows the XPS full-spectrum analysis of the comparative samples Co(OH)F-CFP, N:Co(OH)x-CFP and Co(OH)x-CFP. This further confirms the elemental composition of the comparative samples.
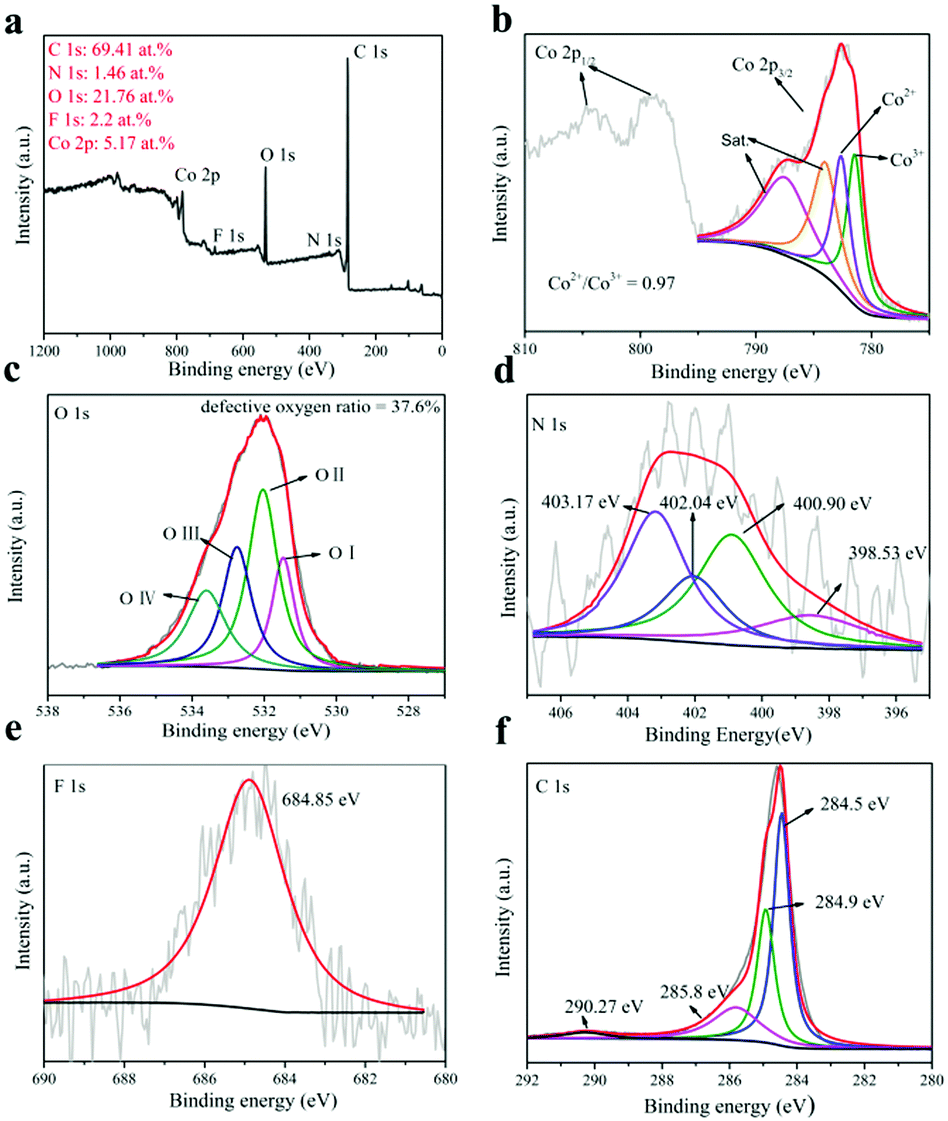 | ||
| Fig. 4 (a) XPS spectrum of the N:Co(OH)F-CFP catalyst, (b) Co 2p XPS spectra, (c) O 1s XPS spectra, (d) N 1s XPS spectra, (e) F 1s XPS spectra and (f) C 1s XPS spectra. | ||
For cobalt, there are four peaks in its XPS (Fig. 4b) and we find the Co3+ peak in its spectrum. When we washed the synthesized N:Co(OH)F-CFP and dried it at 60 °C, due to the defects of N:Co(OH)F, some Co2+ on the surface was oxidized to Co3+. The XPS was based on the in situ analysis, so that Co3+ was present in the N:Co(OH)F-CFP. The positions of the two main peaks and the broad nature of the satellite peak agree with the Co 2p3/2 signals from inorganic Co(II) species with unpaired 3d electrons as reported in the literature. The Co 2p3/2 787.47 eV and 784 eV peaks are fitted with the shake-up (satellites) peaks of Co2+, respectively. The two fitted peaks for Co 2p3/2 are respectively Co3+ (CoOOH) (≈781.44 eV) and Co2+ (≈782.6 eV) (Co–F).53 It should be noted that the area ratio of Co2+/Co3+ to N:Co(OH)F is 0.97. However, in the comparative samples N:Co(OH)x-CFP and Co(OH)x-CFP (Fig. S10, ESI†), we can observe that the area ratio of Co2+/Co3+ is 0.51 and 0.43, which is smaller than that in N:Co(OH)F-CFP. But in the Co(OH)F-CFP, there is only Co2+. The higher atomic ratio of Co2+/Co3+ can be indicative of relatively more oxygen vacancies,53,56 thereby increasing OER activity. In this case, the sample N:Co(OH)F-CFP contains more oxygen vacancies. The presence of vacancies leads to the exposure of the coordination sites of cobalt. The unsaturated sites lacking oxygen easily absorb the OH− in the solution, resulting in the formation of –O–O–H groups. Therefore, oxygen vacancies act as the active sites of OER and promote the reaction activity.26,27 According to previous studies, the valence control can also significantly determine the electrocatalytic performance.57–60
The Co atom with low coordination numbers near the surface could offer unsaturated coordinating sites for oxygen species chemisorption, which can be characterized by the O 1s from XPS spectra. The O 1s spectrum can be divided into four regions (Fig. 4c) which represent the bonds of cobalt metal with oxygen (Co–O) at 531.47 eV (O-I); the lower oxygen coordination defect position (532.02 eV) (O-II); oxygen single bonds in hydroxyl groups (532.75 eV) (O-III); and –OOH or adsorbed water molecules (533.59 eV) (O-IV).55,61,62 Under the influence of fluorine, the O element loses electrons, and its binding energy shifts towards the high field direction. Here we can see this through the O1s XPS spectra of N:Co(OH)F-CFP and Co(OH)x-CFP. Meanwhile, defective oxygen (O-II) accounts for 37.6% of the total proportion. But in the N:Co(OH)x-CFP and Co(OH)x-CFP samples, oxygen which represents defects is respectively 26.5% and 22.4% (Fig. S10, ESI†). The higher ratio of oxygen represents defects in the N:Co(OH)F-CFP, indicating its higher oxygen hole density. In the Co(OH)F-CFP sample, there is about 30.1% defect oxygen. However, since only Co2+ is present in the Co(OH)F-CFP sample, this indicates that the addition of F− can produce defective oxygen, but without the addition of N, it is difficult to cause Co to change its valence, which is particularly important for the OER reaction. This further shows that the synergy between halogen F and heteroatom N enhances the hole electron transfer ability of the N:Co(OH)F-CFP. In order to further determine the presence of oxygen vacancies, we performed photoluminescence spectroscopy. The results showed that there was a clear absorption peak around 391 nm with photoelectron reorganization occurrence. N:Co(OH)F-CFP has higher luminescence intensity, which further proves its higher oxygen vacancy content63,64 (Fig. S11, ESI†).
For N, the peaks at 398.53, 400.90 and 403.17 eV are ascribed to pyridinic nitrogen, pyridonic/pyrrolic nitrogen or N–F, pyridine-N-oxide (N–O), respectively. This indicates that N is incorporated into the base carbon fiber paper and material Co(OH)F. We did an EDX on the array itself, proving the Co(OH)F material contains the nitrogen element (Fig. S12, ESI†). The peak of 402.04 eV is due to N-species adsorbed on the CFP surface such as NO2 or N2 (Fig. 4d).65Fig. 4e displays that the peak of 684.85 eV is identical to the position of a single F 1s of metal hydroxyl fluoro.39 Finally, Fig. 4f is the XPS spectrum of C. The main peak of C at 284.5 eV is the sp2 C–C species,48 which is attributed to the base of carbon fiber paper. The peaks with binding energies of 284.9 and 285.8 eV are the C–N and C–O bonds, respectively.66,67 However, 290.27 eV is the binding energy of COOH. This corresponds to the mechanistic process above.68
The role of NaF was discussed when analyzing SEM in the above. Experiments with different amounts of NH4F have been carried out in Cao et al.'s work to verify its effect on the morphology of Co(OH)F.39 Here we changed the amount of NaF and urea under the same experimental conditions of ours and found that when the amount of halogen ion F− and urea was changed to different amounts, the morphologies of N-containing Co(OH)F will be different (Fig. S13, ESI†) (see the ESI† for more details). But according to the LSV of the OER, it shows no enhancement. When the ratios of urea and NaF are 2:1, 1:2, 4:1, 1:1 and 10:1, respectively, the potentials at 10 mA cm−2 are 1.540 V, 1.594 V, 1.580 V, 1.604 V and 1.589 V vs. RHE, respectively. This shows that the thickness and flexibility of the synthesized material N:Co(OH)F are most suitable for the OER reaction only when the ratio of urea and NaF is 2:1. (Fig. S14, ESI†).
The hydrothermal reaction temperature is another important factor influencing the morphology and activity.69 In our experiment, the temperature of 120 °C was selected because at this temperature sufficient oxygen vacancies can be produced.70,71 Furthermore, we took reaction time as a variable and did a series of experiments at 120 °C, the results showing that there were some differences in physical appearance, as shown in Fig. S15 (ESI†). However, LSV found that the best reaction time was 6 h, proving that the right temperature and reaction time were important for the formation of the right morphology (Fig. S16, ESI†). This further confirms the common structure-oriented effect of NaF and urea in the morphology.
Oxygen evolution activity
iR-corrected linear sweep voltammogram (LSV) curves were carried out in 1 M KOH in the potential range of 1.0–2.0 V (vs. RHE) with a standard three-electrode system; a carbon rod and a Hg/HgO electrode in saturated KOH served as the counter and reference electrodes, respectively.46 The N:Co(OH)F-CFP as a working electrode without polymerization adhesive was tested directly. Bare CFP, N:Co(OH)x-CFP, Co(OH)F-CFP, Co(OH)x-CFP and the IrO2-coated CFP were tested in the same way and the results are shown in Fig. 5a. For the loading of catalysts on the CFP, we ensure its consistence by controlling the amount of reactants. We found that the catalyst loading was 2 mg by measuring the mass of CFP before and after the reaction. This further ensures that the amount of catalysts involved in the reaction is consistent. Bare CFP showed negligible OER activity. The overpotential of our material was found to be 310 mA cm−2 in 1 M KOH solution, better than that of commercial IrO2-CFP. Table S2 (ESI†) shows the performance comparison of the OER at 10 mA cm−2 for some other cobalt-based catalysts on different substrates recently reported in alkaline media. In addition, to confirm the important role of halogen anion F− and heteroatom N, we tested the OER activity of N:Co(OH)x-CFP, Co(OH)F-CFP and Co(OH)x-CFP. When F− and N are not present at the same time, their OER catalytic activity greatly reduced, further illustrating the synergy effect between F− and N on the catalytic activity. In Fig. 5b, we can see that the Tafel slope of bare CFP is 92.53 mV dec−1, N:Co(OH)x-CFP (86.5 mV dec−1), Co(OH)F-CFP (110 mV dec−1), Co(OH)x-CFP (122 mV dec−1), and IrO2-coated CFP (178 mV dec−1), which is bigger than that of N:Co(OH)F-CFP (69.75 mV dec−1), demonstrating N:Co(OH)F-CFP's good OER kinetic activity. We also find that the Tafel slope of CFP was smaller than that of IrO2-CFP, however, that is not contradictory because the Tafel slope is related to the specific surface area of the material. CFP itself is a cross-porous structure. When IrO2 is spin-coated on the surface of CFP, it will fill the holes of CFP, resulting in a decrease in specific surface area, and thus the Tafel slope will increase. However, the onset potential of IrO2 is earlier than that of CFP, and at 10 mA cm−2, 20 mA cm−2, 30 mA cm−2, etc., the overpotential of IrO2 is smaller than that of CFP. Obviously IrO2's OER performance is better than that of CFP. At the same time the Tafel slopes of all these materials are higher. We know that the OER is a highly oxidative process during the reaction, and so the catalyst is enduring a relatively high current state. There are two simultaneous competitions that exist in the whole system: the process of converting Co2+ into Co3+ with electron transfer and some of the electrons being involved in the oxygen evolution process, resulting in a relatively high Tafel slope.45 The occurrence of this competitive process is consistent with the actual kinetic reaction.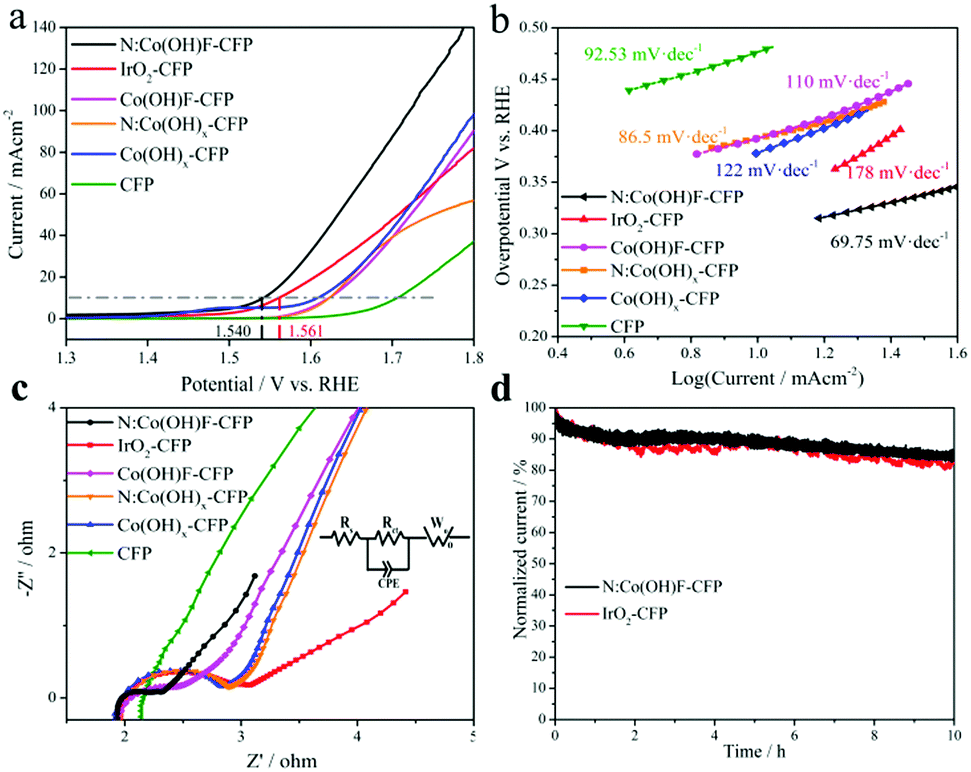 | ||
| Fig. 5 (a) LSV with iR-correction, (b) Tafel slope and (c) electrochemical impedance spectroscopy (EIS) (Nyquist plots) in 1 M KOH solution. The inset in (c) is the equivalent electric circuit of the cell. (d) IT test at a potential of 10 mA cm−2 for 10 h in 1 M KOH. | ||
The N:Co(OH)F-CFP material with a thorn bar structure showed a lower interfacial charge transfer resistance, as shown in Fig. 5c by the Nyquist plots derived from the EIS studies.34 In the Nyquist plots, the impedance spectrum of bare CFP is not a standard half-circle form. Obviously it has a larger internal resistance; its own structure leads to a greater contact resistance with the solution, which hinders the OER reaction with the electrolyte solution. But for IrO2-CFP, N:Co(OH)x-CFP, Co(OH)F-CFP, Co(OH)x-CFP and N:Co(OH)F-CFP, there are two parts in the Nyquist plots, including polarization resistance (Rct) and liquid resistance (Rs). The equivalent circuit models of the typical Nyquist plots were fitted by the Zview Tools (inset in Fig. 5c). CPE represents the constant phase element. Rct, Rs and Wo (Warburg-Open Circuit Terminus) are connected in series, as the solution impedance.72 The semicircular diameter of the mid-high frequency region of the EIS plots is related to the electron transfer process.73 A smaller diameter represents better electrochemical kinetics, indicating the fastest charge transport among all the studied counterparts. This also proves that it is a non-ideal nanostructured surface that produces an electric double layer capacitor.45
The stability of the sample has always been an important indicator evaluating its electrochemical properties. For this, we conducted a 10 h IT test at the corresponding potential of 10 mA cm−2. The results show that after 10 h, the current of N:Co(OH)F-CFP has decreased, because in the stability test, the surface of the material always receives strong oxidizing current, which will inevitably cause Co2+ oxidation on the surface of the material to become Co3+. The decrease in the ratio of Co2+/Co3+ causes changes in the catalytic activity of the material. On the other hand, some of the oxygen vacancies of the material will also be filled with the generated O2 gas to cause a certain change, resulting in a decrease in the number of active sites and a corresponding decrease in current. However, the decreased rate of IrO2-CFP was higher than that of N:Co(OH)F-CFP after 10 hours, which was consistent with our other electrochemical test data (Fig. 5d). This further demonstrates that the addition of F and N can improve the stability of the N:Co(OH)F material in the OER reaction.39 Meanwhile, from the stability test image, we can see the stability curve of IrO2 fluctuates greater than that of N:Co(OH)F-CFP, which is due to the fact that O2 accumulated on the surface during the OER reaction and cannot leave in time. In order to solve the problem of gas accumulation in the test device and try to reduce the damage of the material by high oxidation current, we passed nitrogen gas to the device for half an hour before the LSV test after finishing the stability test. LSV comparison of OER activity before and after the IT test further showed the stability N:Co(OH)F-CFP (Fig. S17, ESI†). Meanwhile, XRD analysis of the catalyst before and after the reaction also proved that the main peaks were unchanged, with only a slight change of intensity. All of these are due to the force in the process of highly oxidized electrons transporting back and forth. In order to evaluate the active area of the catalyst, the current density at different sweep rates was tested in a non-Faradaic region, whose voltage range is from 0.12 to 0.32 V vs. RHE by cyclic voltammograms (Fig. S18a, ESI†). After analyzing the double-layer capacitance (Cdl) of the electrodes, we found it linearly proportional to the active surface area which played a significant role in the reaction (Fig. S18b, ESI†).45 Although we cannot extract the exact values due to the unknown behavior of the catalysts, we can easily estimate the relative surface areas for all electrodes. It can be calculated that the Cdl value of N:Co(OH)F-CFP is 14.68 mF cm−2, but 12.82 mF cm−2 for IrO2-CFP. The results show that the Cdl of N:Co(OH)F-CFP is higher, meaning that N:Co(OH)F-CFP possesses more active sites. At the same time, the ECSA values are also consistent with the results of previous electrochemical tests such as LSV etc.
The above results indicate that the OER catalytic activity of N:Co(OH)F-CFP is superior to that of the commercial IrO2, which may be attributed to the following aspects: (1) the hydrothermal synthesis of N:Co(OH)F-CFP has a unique ultralong needle-like structure. On the one hand, more active sites can be exposed for oxygen absorption and release. On the other hand, it increases the contact area with the electrolyte to accelerate the departure of the gas. (2) The 3D needle-like N:Co(OH)F directly grown on the carbon fiber paper substrate improves the conductivity, and it is conducive to electron transport of N:Co(OH)F-CFP, being in favor of the catalytic reaction. (3) XPS (shown in Fig. 4b and c) shows that Co and O exist around different environments in N:Co(OH)F-CFP, revealing the process of material changes during the reaction. In addition, the direct bonds between Co and F, and N are in favor of catalysis.
After analysis, we find that there are mainly three parts playing roles in the whole reaction system: (1) surface oxygen holes, (2) active sites of oxygen vacancies and (3) incorporation of heteroatom N and F. The three parts work together to promote oxygen evolution. Meanwhile, it reveals that the existence of oxygen vacancies leads to the formation of new gap states where the electrons previously associated with the Co–O bonds tend to be delocalized, resulting in much higher electrical conductivity and electrocatalytic activity.74
Conclusions
We have successfully prepared an ultralong needle-like catalyst N:Co(OH)F on carbon fiber paper via hydrothermal synthesis. NaF and urea as co-morphology directing reagents, promote the formation of ultra-long needle-like structures on the surface of CFP. Our investigation has proved that heteroatomic N and halogen element F played vital roles in producing oxygen vacancies in N-doped Co(OH)F-CFP, which can improve the speed of actual OER in water splitting. The ultralong needle-like catalyst N:Co(OH)F-CFP has a wealth of oxygen vacancies, which is beneficial for the OER. The overpotential of N:Co(OH)F-CFP at 10 mA cm−2 in 1 M KOH is 310 mV, showing that the activity of OER is better than that of IrO2-CFP in alkaline solution, which can potentially be used as the catalyst in electrocatalytic water oxidation. This can provide a new strategy for the design of cobalt-based efficient electrocatalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (No. 21471028, 21671036, 21871042), the National Key Basic Research Program of China (No. 2013CB834802), Changbai Mountain Scholarship, the Natural Science Foundation of Jilin Province (No. 20150101064JC) and the Fundamental Research Funds for the Central Universities (No. 2412015KJ012, 2412017BJ004). We would like to acknowledge the support from the Jilin Provincial Department of Education.Notes and references
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. Shao-Horn, Nat. Chem., 2011, 3, 546–550 CrossRef PubMed.
- P. Chen, K. Xu, S. Tao, T. Zhou, Y. Tong, H. Ding, L. Zhang, W. Chu, C. Wu and Y. Xie, Adv. Mater., 2016, 28, 7527–7532 CrossRef PubMed.
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 0003 CrossRef.
- A. P. Wu, Y. Xie, H. Ma, C. G. Tian, Y. Gu, H. J. Yan, X. M. Zhang, G. Y. Yang and H. G. Fu, Nano Energy, 2018, 44, 353–363 CrossRef.
- M. J. Chen, L. Wang, H. P. Yang, S. Zhao, H. Xu and G. Wu, J. Power Sources, 2018, 375, 277–290 CrossRef.
- J. Wang, Y. Gao, D. Chen, J. Liu, Z. Zhang, Z. Shao and F. Ciucci, ACS Catal., 2018, 8, 364–371 CrossRef.
- W. Zhou, X. J. Wu, X. Cao, X. Huang, C. Tan, J. Tian, H. Liu, J. Wang and H. Zhang, Energy Environ. Sci., 2013, 6, 2921–2924 RSC.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef PubMed.
- P. Z. Chen, K. Xu, T. P. Zhou, Y. Tong, J. C. Wu, H. Cheng, X. L. Lu, H. Ding, C. Z. Wu and Y. Xie, Angew. Chem., Int. Ed., 2016, 55, 2488–2492 CrossRef PubMed.
- B. Q. Li, C. Tang, H. F. Wang, X. L. Zhu and Q. Zhang, Sci. Adv., 2016, 2, 2375–2548 Search PubMed.
- X. Kong, K. Xu, C. Zhang, J. Dai, S. Norooz Oliaee, L. Li, X. Zeng, C. Wu and Z. Peng, ACS Catal., 2016, 6, 1487–1492 CrossRef.
- F. Cheng and J. Chen, Chem. Soc. Rev., 2012, 41, 2172–2192 RSC.
- G. Wu, A. Santandreu, W. Kellogg, S. Gupta, O. Ogoke, H. G. Zhang, H.-L. Wang and L. M. Dai, Nano Energy, 2016, 29, 83–110 CrossRef.
- S. S. Zheng, X. R. Li, B. Y. Yan, Q. Hu, Y. X. Xu, X. Xiao, H. G. Xue and H. Pang, Adv. Energy Mater., 2017, 7, 1602733 CrossRef.
- S. Dou, X. Y. Li, L. Tao, J. Huo and S. Y. Wang, Chem. Commun., 2016, 52, 9727–9730 RSC.
- J. Zhang, T. Wang, D. Pohl, B. Rellinghaus, R. Dong, S. Liu, X. Zhuang and X. Feng, Angew. Chem., Int. Ed., 2016, 55, 6702–6707 CrossRef PubMed.
- B. Li, P. Gu, G. X. Zhang, Y. Lu, K. S. Huang, H. G. Xue and H. Pang, Small, 2018, 14, 1702184 CrossRef PubMed.
- C. Tang, H. F. Wang, X. Chen, B. Q. Li, T. Z. Hou, B. Zhang, Q. Zhang, M. M. Titirici and F. Wei, Adv. Mater., 2016, 28, 6845–6851 CrossRef PubMed.
- Y. Zhan, G. Du, S. Yang, C. Xu, M. Lu, Z. Liu and J. Y. Lee, ACS Appl. Mater. Interfaces, 2015, 7, 12930–12936 CrossRef PubMed.
- X. X. Zou, A. Goswami and T. Asefa, J. Am. Chem. Soc., 2013, 135, 17242–17245 CrossRef PubMed.
- Y. Liu, S. B. Yin and P. K. Shen, ACS Appl. Mater. Interfaces, 2018, 10, 23131–23139 CrossRef PubMed.
- K. Xu, P. Chen, X. Li, Y. Tong, H. Ding, X. Wu, W. Chu, Z. Peng, C. Wu and Y. Xie, J. Am. Chem. Soc., 2015, 137, 4119–4125 CrossRef PubMed.
- J. Masa, W. Xia, I. Sinev, A. Zhao, Z. Sun, S. Grutzke, P. Weide, M. Muhler and W. Schuhmann, Angew. Chem., Int. Ed., 2014, 53, 8508–8512 CrossRef PubMed.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef PubMed.
- Y. Tong, P. Chen, T. Zhou, K. Xu, W. Chu, C. Wu and Y. Xie, Angew. Chem., Int. Ed., 2017, 56, 7121–7125 CrossRef PubMed.
- J. Q. Zhu, Z. Y. Ren, S. C. Du, Y. Xie, J. Wu, H. Y. Meng, Y. Z. Xue and H. G. Fu, Nano Res., 2017, 10, 1819–1831 CrossRef.
- M.-Q. Yang, J. Wang, H. Wu and G. W. Ho, Small, 2018, 14, 1703323 CrossRef PubMed.
- N. Tymińska, G. Wu and M. Dupuis, J. Phys. Chem. C, 2017, 121, 8378–8389 CrossRef.
- D. Yan, Y. Li, J. Huo, R. Chen, L. Dai and S. Wang, Adv. Mater., 2017, 29, 1606459 CrossRef PubMed.
- H. Li, C. Tsai, A. L. Koh, L. L. Cai, A. W. Contryman, A. H. Fragapane, J. H. Zhao, H. S. Han, H. C. Manoharan, F. Abild-Pedersen, J. K. Nørskov and X. L. Zheng, Nat. Mater., 2016, 15, 48–53 CrossRef PubMed.
- D. Chen, J. Wang, Z. Zhang, Z. Shao and F. Ciucci, Chem. Commun., 2016, 52, 10739–10742 RSC.
- M. S. Burke, M. G. Kast, L. Trotochaud, A. M. Smith and S. W. Boettcher, J. Am. Chem. Soc., 2015, 137, 3638–3648 CrossRef PubMed.
- Z. Xiao, Y. Wang, Y. C. Huang, Z. Wei, C. L. Dong, J. Ma, S. Shen, Y. Li and S. Wang, Energy Environ. Sci., 2017, 10, 2563–2569 RSC.
- P. Wang, F. Song, R. Amal, Y. H. Ng and X. Hu, ChemSusChem, 2016, 9, 472–477 CrossRef PubMed.
- X. Li, Y. Fang, X. Lin, M. Tian, X. An, Y. Fu, R. Li, J. Jin and J. Ma, J. Mater. Chem. A, 2015, 3, 17392–17402 RSC.
- L. P. Zhu, Z. Wen, W. M. Mei, Y. G. Li and Z. Z. Ye, J. Phys. Chem. C, 2013, 117, 20465–20473 CrossRef.
- L. S. Xie, R. Zhang, L. Cui, D. N. Liu, S. Hao, Y. J. Ma, G. Du, A. M. Asiri and X. P. Sun, Angew. Chem., 2017, 129, 1084–1088 CrossRef.
- L. L. Chen, J. Y. Zhang, X. Ren, R. X. Ge, W. Q. Teng, X. P. Sun and X. M. Li, Nanoscale, 2017, 9, 16632–16637 RSC.
- S. Wan, J. Qi, W. Zhang, W. Wang, S. Zhang, K. Liu, H. Zheng, J. Sun, S. Wang and R. Cao, Adv. Mater., 2017, 29, 1700286 CrossRef PubMed.
- J. Wang and F. Ciucci, Small, 2017, 13, 1604103 CrossRef PubMed.
- J. Wang, H. Zhao, Y. Gao, D. Chen, C. Chen, M. Saccoccio and F. Ciucci, Int. J. Hydrogen Energy, 2016, 41, 10744–10754 CrossRef.
- L. Sun, L. Wang, C. Tian, T. Tan, Y. Xie, K. Shi, M. Li and H. Fu, RSC Adv., 2012, 2, 4498–4506 RSC.
- U. Y. Qazi, C. Z. Yuan, N. Ullah, Y. F. Jiang, M. Imran, A. Zeb, S. J. Zhao, R. Javaid and A. W. Xu, ACS Appl. Mater. Interfaces, 2017, 9, 28627–28634 CrossRef PubMed.
- G. Zhang, S. Sun, D. Yang, J. P. Dodelet and E. Sacher, Carbon, 2008, 46, 196–205 CrossRef.
- M. Blasco-Ahicart, J. Soriano-Lopez, J. J. Carbo, J. M. Poblet and J. R. Galan-Mascaros, Nat. Chem., 2018, 10, 24–30 CrossRef PubMed.
- J. W. Li, W. M. Xu, J. X. Luo, D. Zhou, D. W. Zhang, L. C. Wei, P. M. Xu and D. S. Yuan, Nano-Micro Lett., 2018, 10, 2150–5551 Search PubMed.
- Z. J. Liu, Z. H. Zhao, Y. Y. Wang, S. Dou, D. F. Yan, D. D. Liu, Z. H. Xia and S. Y. Wang, Adv. Mater., 2017, 29, 1606207 CrossRef PubMed.
- G. Yan, C. X. Wu, H. Q. Tan, X. J. Feng, L. K. Yan, H. Y. Zang and Y. G. Li, J. Mater. Chem. A, 2017, 5, 765–772 RSC.
- D. Qu, M. Zheng, P. Du, Y. Zhou, L. G. Zhang, D. Li, H. Q. Tan, Z. Zhao, Z. G. Xie and Z. C. Sun, Nanoscale, 2013, 5, 12272–12277 RSC.
- D. GiGmes, A. Gaudel-Siri, S. R. A. Marque, D. Bertin, P. Tordo, P. Astolfi, L. Greci and C. Rizzoli, Helv. Chim. Acta, 2006, 89, 2312–2326 CrossRef.
- R. Xu and H. C. Zeng, J. Phys. Chem. B, 2003, 107, 12643–12649 CrossRef.
- J. L. Meng, J. Q. Fu, M. J. Wei, S. Liang, H. Y. Zang, H. Q. Tan, Y. H. Wang and Y. G. Li, Inorg. Chem. Front., 2017, 4, 1791–1797 RSC.
- T. Y. Ma, S. Dai, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2014, 136, 13925–13931 CrossRef PubMed.
- J. G. Yu, Q. J. Xiang and M. H. Zhou, Appl. Catal., B, 2009, 90, 595–602 CrossRef.
- L. Z. Zhuang, L. Ge, Y. S. Yang, M. R. Li, Y. Jia, X. D. Yao and Z. H. Zhu, Adv. Mater., 2017, 29, 1606793 CrossRef PubMed.
- Y. Li, P. Hasin and Y. Wu, Adv. Mater., 2010, 22, 1926–1929 CrossRef PubMed.
- W. Q. Song, Z. Ren, S.-Y. Chen, Y. T. Meng, S. Biswas, P. Nandi, H. A. Elsen, P.-X. Gao and S. L. Suib, ACS Appl. Mater. Interfaces, 2016, 8, 20802–20813 CrossRef PubMed.
- B. S. Yeo and A. T. Bell, J. Am. Chem. Soc., 2011, 133, 5587–5593 CrossRef PubMed.
- Y. Y. Liang, H. L. Wang, J. G. Zhou, Y. G. Li, J. Wang, T. Regier and H. J. Dai, J. Am. Chem. Soc., 2012, 134, 3517–3523 CrossRef PubMed.
- Y. X. Zhang, F. Ding, C. Deng, S. Y. Zhen, X. Y. Li, Y. F. Xue, Y. M. Yan and K. N. Sun, Catal. Commun., 2015, 67, 78–82 CrossRef.
- Y. Jia, L. Zhang, A. Du, G. Gao, J. Chen, X. Yan, C. L. Brown and X. Yao, Adv. Mater., 2016, 28, 9532–9538 CrossRef PubMed.
- J. Kim, X. Yin, K. C. Tsao, S. Fang and H. Yang, J. Am. Chem. Soc., 2014, 136, 14646–14649 CrossRef PubMed.
- J. Bao, X. D. Zhang, B. Fan, J. J. Zhang, M. Zhou, W. L. Yang, X. Hu, H. Wang, B. C. Pan and Y. Xie, Angew. Chem., Int. Ed., 2015, 54, 7399–7404 CrossRef PubMed.
- V. M. Jiménez, A. Fernández, J. P. Espinós and A. R. González-Elipe, J. Electron Spectrosc. Relat. Phenom., 1995, 71, 61–71 CrossRef.
- J. A. Rengifo-Herrera, E. Mielczarski, J. Mielczarski, N. C. Castillo, J. Kiwi and C. Pulgarin, Appl. Catal., B, 2008, 8, 448–456 CrossRef.
- C. Ronning, H. Feldermann, R. Merk and H. Hofsäss, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 2207–2215 CrossRef.
- G. M. Burke, D. E. Wurster, M. J. Berg, P. Veng-Pedersen and D. D. Schottelius, Pharm. Res., 1992, 9, 126–130 CrossRef.
- Y. Taki and O. Takai, Thin Solid Films, 1998, 316, 45–50 CrossRef.
- I. Roger, R. Moca, H. N. Miras, K. G. Grawford, D. A. J. Moran, A. Y. Ganin and M. D. Symes, J. Mater. Chem. A, 2017, 5, 1472–1480 RSC.
- X. Y. Pan, M. Q. Yang, X. Z. Fu, N. Zhang and Y. J. Xu, Nanoscale, 2013, 5, 3601–3614 RSC.
- Y. R. Qi, L. Q. Mu, J. M. Zhao, Y. S. Hu, H. Z. Liu and S. Dai, J. Mater. Chem. A, 2016, 4, 7178–7184 RSC.
- H. Zhong, Z. H. Fu, J. M. Taylor, G. Xu and R. H. Wang, Adv. Funct. Mater., 2017, 27, 1701465 CrossRef.
- K. L. Yan, J. F. Qin, Z. Z. Liu, B. Dong, J. Q. Chi, W. K. Gao, J. H. Lin, Y. M. Chai and C. G. Liu, Chem. Eng. J., 2018, 334, 922–931 CrossRef.
- W. Z. Lai, R. Cao, G. Dong, S. Shaik, J. N. Yao and H. Chen, J. Phys. Chem. Lett., 2012, 3, 2315–2319 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8qm00405f |
| This journal is © the Partner Organisations 2018 |