
Cross-coupling of sulfonic acid derivatives via aryl-radical transfer (ART) using TTMSS or photoredox†
Eric D.
Nacsa
and
Tristan H.
Lambert
 *
*
Department of Chemistry, Columbia University, New York, New York 10027, USA. E-mail: tl2240@columbia.edu
First published on 20th September 2017
Abstract
The intramolecular cross-coupling of sulfonic acid derivatives occurs in the presence of tris(trimethylsilyl)silane (TTMSS) at room temperature and in air to form biaryl compounds. A photoredox-catalyzed procedure is also described. These protocols provide mild and convenient alternatives to standard tin-mediated reactions. Combined with the trivial preparation of the substrates from activated sulfonic acids and 2-halophenols or anilines, this work presents a useful means to employ sulfonic acid derivatives in cross-coupling transformations. A modified linker to realize high regioselectivity is also presented. Finally, a one-pot cross-coupling procedure is demonstrated.
Cross-coupling reactions are a cornerstone of modern organic synthesis (Fig. 1a). Among the numerous functionalities that can participate in such transformations, boronic acids1 have proven to be one of the most useful, and many derivatives are now commercially available. Nevertheless, expanding the range of functional groups that can be reliably engaged in cross-couplings remains of high interest, and notable recent efforts have demonstrated the use of ubiquitous functionalities such as carboxylic acids2 and alcohols,3 among others.4,5 However, one notable class of substrates that remains poorly represented in cross-coupling efforts are the sulfonic acids and their derivatives.6 This paucity stands in spite of the commercial availability of hundreds of such compounds and the substantial synthetic utility of the sulfonate group.7 The expansion of technologies that allow for the engagement of sulfonic acid derivatives in cross-coupling reactions would thus offer useful new capabilities for synthetic chemistry. In this communication, we report two approaches for intramolecular cross-coupling of aryl sulfonates using the mechanistic blueprint of aryl-radical transfer (ART).8
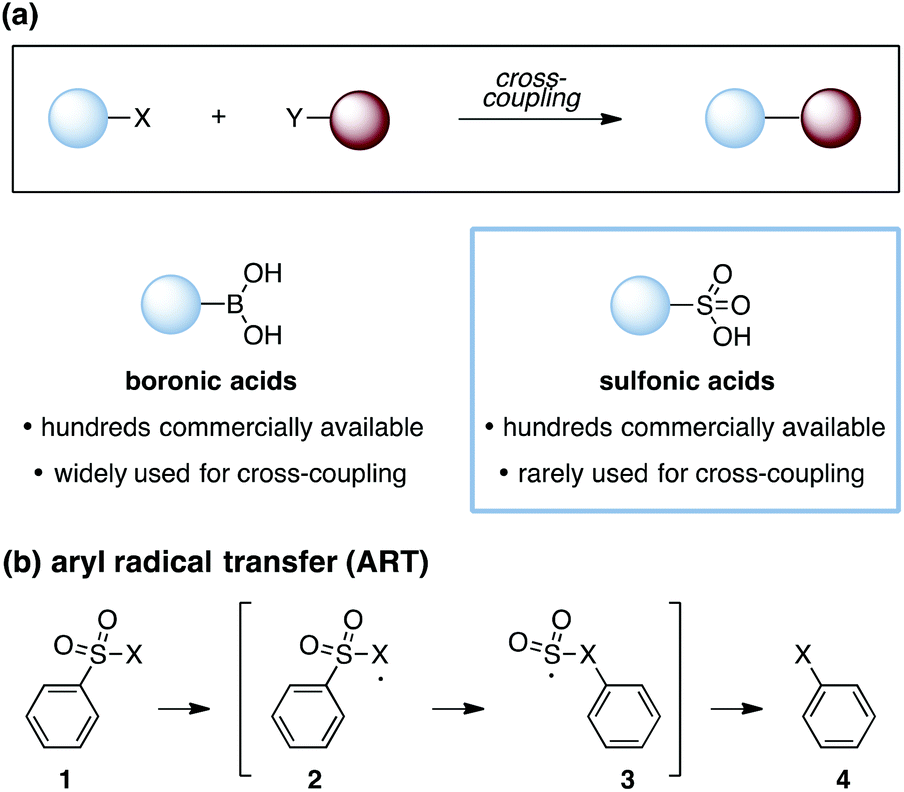 | ||
| Fig. 1 (a) Cross-coupling reactions, boronic and sulfonic acids as cross-coupling substrates. (b) Aryl radical transfer (ART). X = generic substituent. | ||
As first reported by Speckamp,9 aryl sulfonate derivatives 1, when converted to a radical species 2, can undergo a transfer of the aryl group from sulfur to the radical-bearing atom (Fig. 1b). Following collapse of the resulting intermediate 3, a biaryl cross-coupled product 4 is thus produced. While such transformations offer a mechanistically unique approach to biaryl bond formation,10,11 the utility of ART reactions has remained limited by their general reliance on stoichiometric quantities of organotin reagents and the often-competitive formation of sultone (e.g., 7) or other byproducts (eqn (1)). We reasoned that the application of more modern radical-forming strategies would offer major practical improvements to ART reactions and therefore enable chemists to exploit the synthetic opportunities these transformations provide. Specifically, we have identified that tris(trimethylsilyl)silane (TTMSS)12 enables the efficient synthesis of biaryl compounds from sulfonic acid derivatives (e.g., 8) under exceptionally mild conditions (eqn (2)).
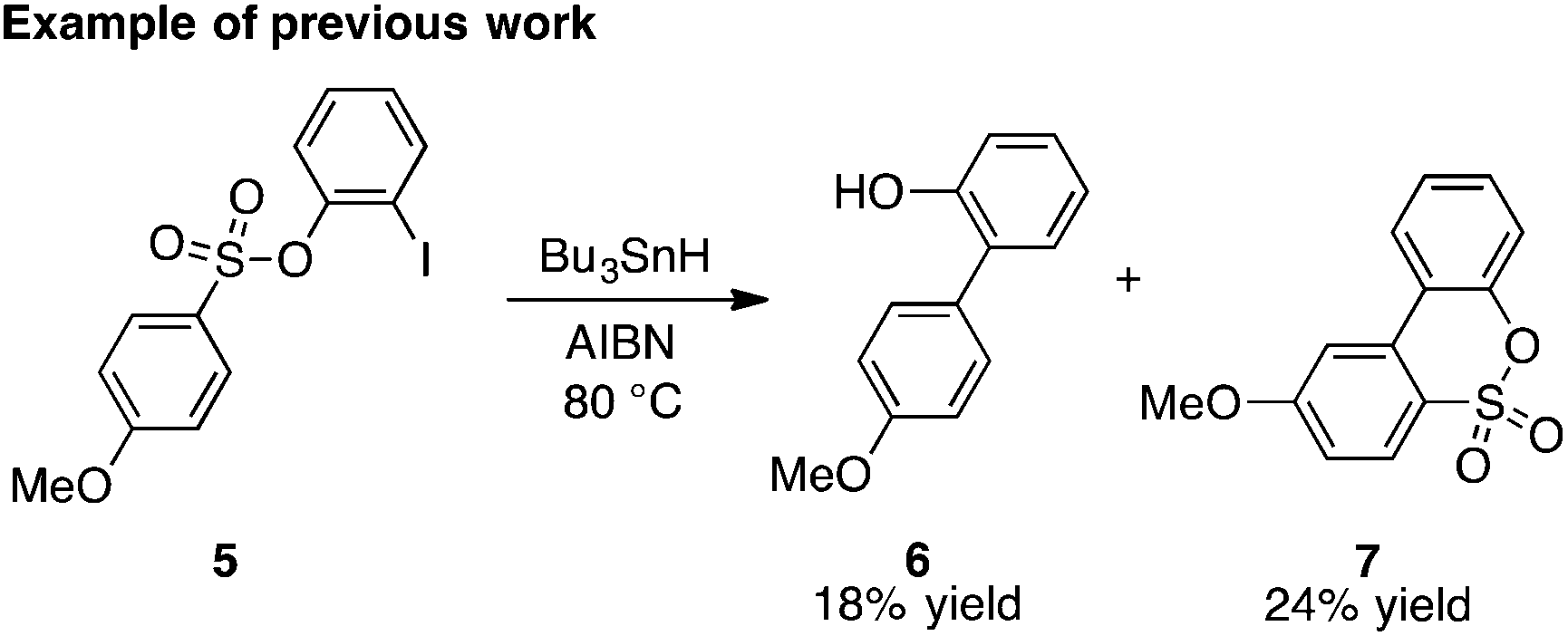 | (1) |
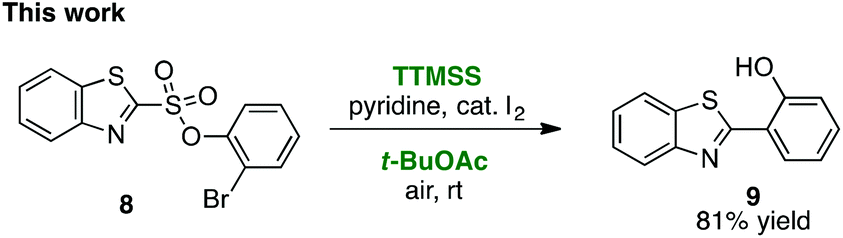 | (2) |
As originally reported by Curran,12 TTMSS generates aryl radicals from aryl iodides at room temperature in the presence of pyridine and catalytic amounts of iodine, and is promoted by air. Although we screened a variety of other radical generation methods13 to replace the traditional organotin reagents such as Bu3SnH, Curran's protocol proved uniquely capable of effecting this transformation.
Some important highlights of our reaction development studies are shown in Table 1. Optimally, we found that when sulfonate 8 was treated with TTMSS (2 equiv.), pyridine, and a catalytic amount of I2 in t-BuOAc under air at room temperature, biaryl 9 was isolated in 81% yield after 20 h (entry 1). The reaction was slightly less efficient when 1.5 equiv. of TTMSS was used (entry 2). Under an argon atmosphere, the conversion appears to have been limited by the amount of I2 added (entry 3), although an atmosphere of pure O2 was also inferior to air (entry 4). Concentration (0.01 M) was important, as 9 was obtained in only 18% yield at 0.1 M (entry 5). Inorganic bases such as Na2CO3 inhibited the reaction completely (entry 6), and temperatures deviating from room temperature also lowered the yield of 9 (entries 7 and 8). Especially critical was the solvent medium. Thus while in addition to t-BuOAc the reaction could be performed with reasonable efficiency in acetic acid (entry 9), most other common solvents lowered yields more dramatically (see ESI†). We suggest that many of these solvents fail due to their capacity to donate hydrogen atoms to the radical intermediates. Finally, the corresponding aryl iodide substrate converted with comparable efficiency to the bromide 8 (entry 10).
|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | % yielda |
| a Substrate 8 was treated with TTMSS (2 equiv.), pyridine (6 equiv.) and I2 (∼10 mol%) in t-BuOAc (0.01 M) under air for 24 h. Yields are based on 1H NMR analysis. b Isolated yield. TTMSS = tris(trimethylsilyl)silane. | ||
| 1 | None | 81b |
| 2 | 1.5 equiv. TTMSS | 70 |
| 3 | Argon atmosphere | 15 |
| 4 | O2 atmosphere | 50 |
| 5 | 0.1 M | 18 |
| 6 | Na2CO3 base | 0 |
| 7 | 0 °C | 55 |
| 8 | 50 °C | 28 |
| 9 | AcOH solvent | 69 |
| 10 | Iodide substrate | 84b |
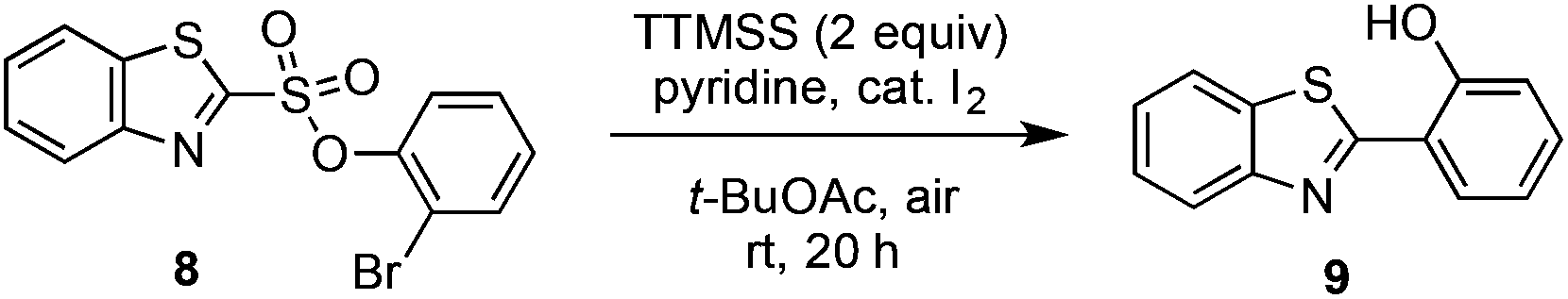
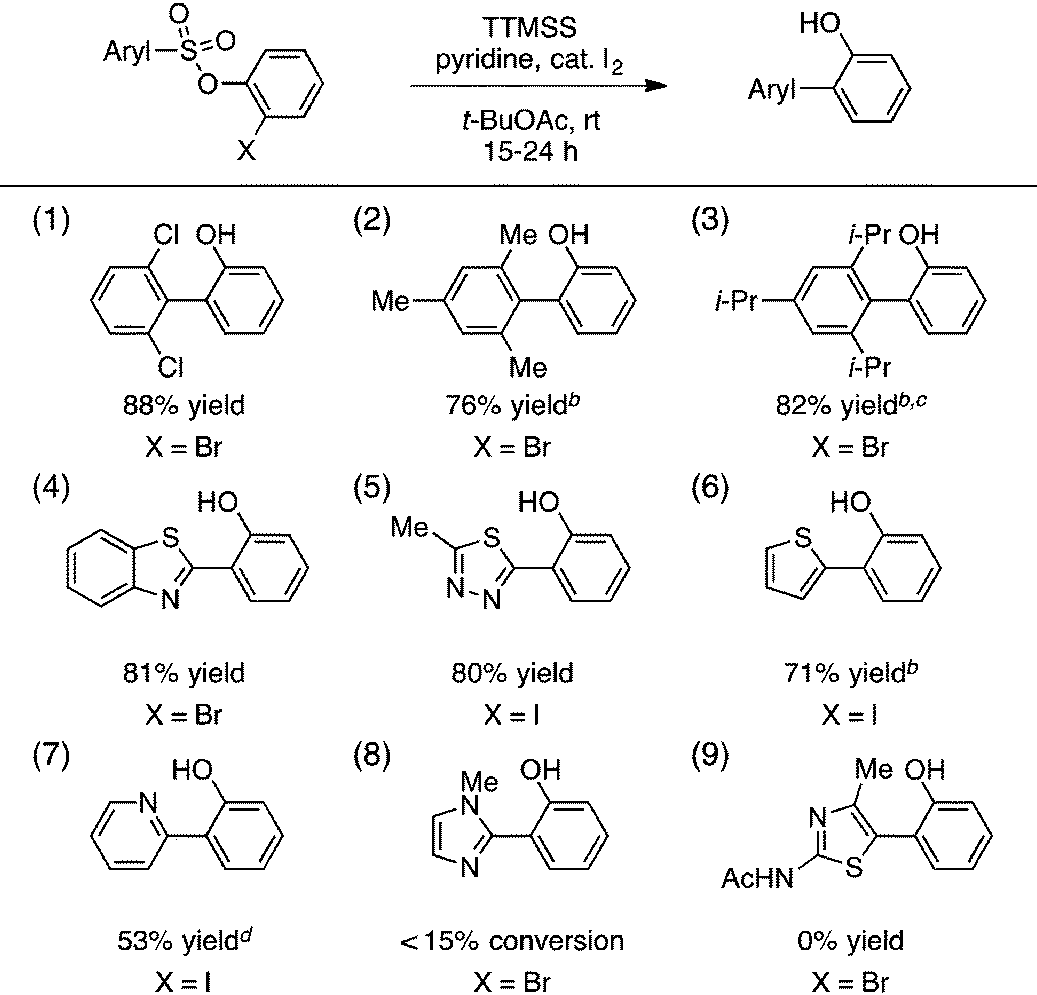
A portion of our study of the aryl sulfonate scope is shown in Table 2. First, we found that a selection of hindered sulfonic acids undergo efficient aryl radical transfer using the optimized conditions, including those with 2,6-diclorophenyl, mesityl, and even 2,4,6-triisopropylphenyl moieties (entries 1–3).14 Various heteroaromatic components were also compatible with this process, giving biaryl products containing benzothiazolyl, 2,4,5-thiadiazolyl, 2-thienyl, and 2-pyridyl fragments in generally good yields (entries 4–7). The production of the latter was lowered by competitive formation of the corresponding sultone (cf.eqn (1)). More electron-rich substrates that would lead to 2-imidazolyl- or 3-acetamido-2,4-thiazolyl-containing products were unreactive, however (entries 8 and 9).
| a Substrates were treated with TTMSS (2 equiv.), pyridine (6 equiv.) and I2 (∼10 mol%) in t-BuOAc (0.01 M) under air. Yields are of purified, isolated products. b Characterized as the 4-bromobenzoate. c TTMSS (4 equiv.) and pyridine (12 equiv.) for 72 h. d TTMSS (3 equiv.) and pyridine (9 equiv.) were used. TTMSS = tris(trimethylsilyl)silane. |
|---|
|
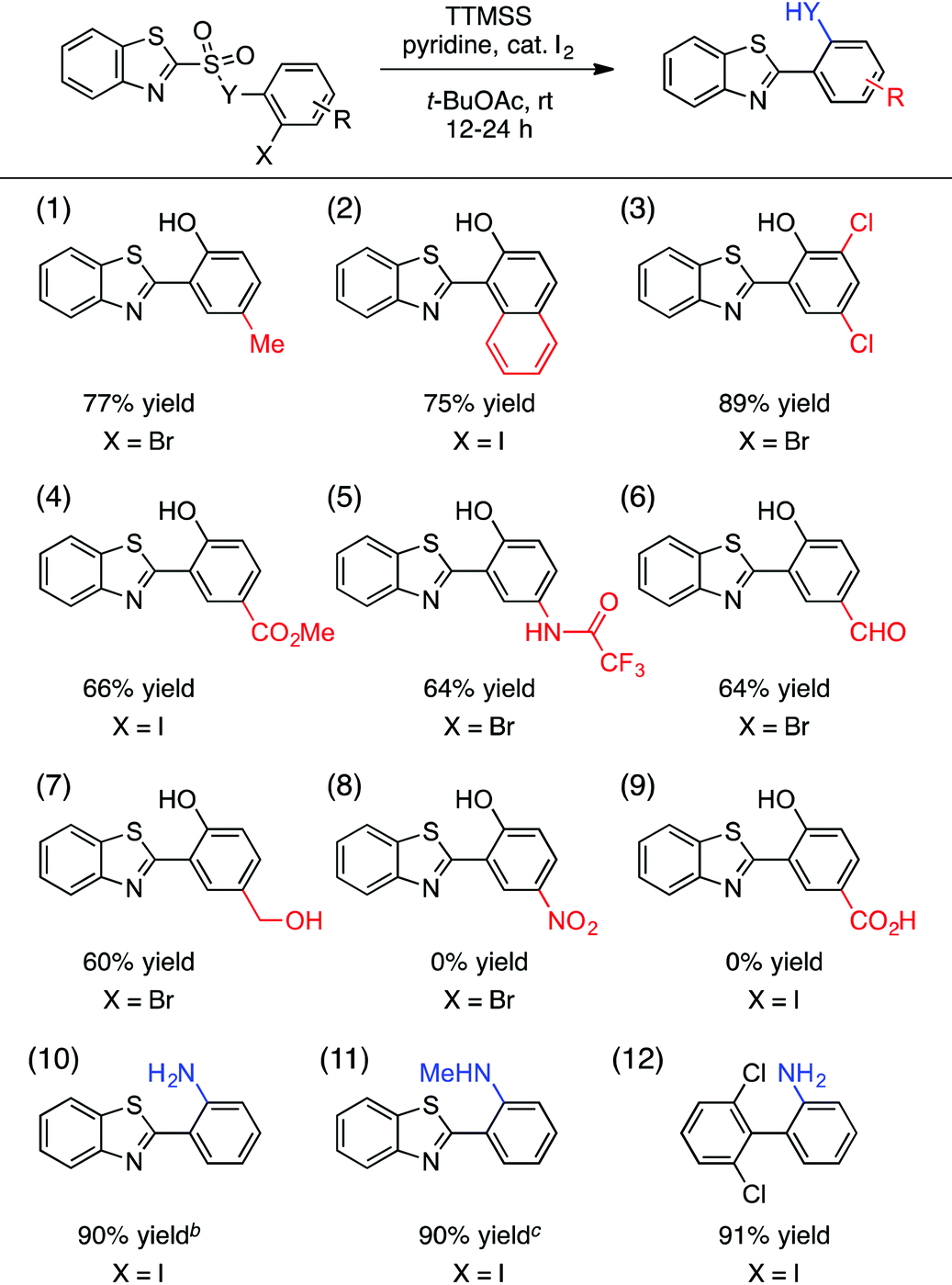
We have also evaluated the scope of the aryl halide component of the substrate (Table 3). A number of substituents on this component were tolerated, giving rise to 4-methyl, 1-naphthyl-fused, 4,6-dichloro, 4-carboxymethyl, and protected 4-amino groups on the 2-(benzothiazolyl)phenol products in good yields (entries 1–5). Useful and potentially reactive handles such as a free aldehyde (entry 6) and a free alcohol (entry 7) were compatible with this process, forming the corresponding products with reasonable efficiency. On the other hand, highly electron-withdrawing substituents such as nitro (entry 8) and carboxylic acid (entry 9) groups impeded the reaction, although the limited solubility of these substrates in t-BuOAc may have played a role in this outcome. In addition, the oxygen linker (Y = O) between the sulfinyl and 2-haloaryl fragments of the substrate in Table 3 was not essential, and both free and N-methylated sulfonamide substrates leading to the corresponding cross-coupled anilines reacted in excellent yields (entries 10 and 11). The 2,6-dichlorophenyl-containing aniline was also obtained in 91% yield (entry 12).
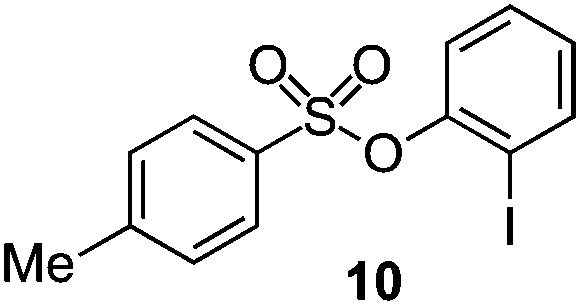
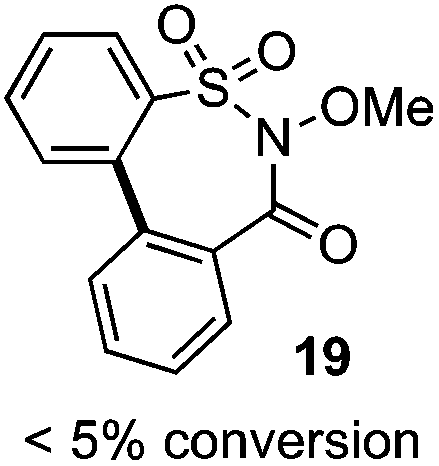
One of the major complications of aryl-radical transfer reactions of this type is the potential to form sultone products, especially with electron-neutral aryl sulfonates bearing ortho C–H groups. We considered whether an alternative carboxylate-derived linker functionality could give rise to selective aryl radical transfer over sultone formation when the aryl sulfinyl component is electronically unbiased. In this case, we reasoned that the desired cross-coupling pathway would be favored over sultone formation, which in this case would require a 7-membered radical cyclization (i.e., 19).
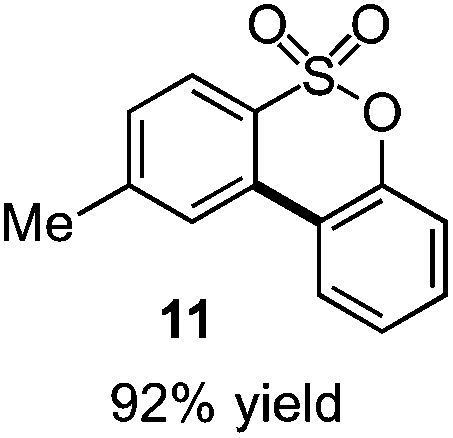
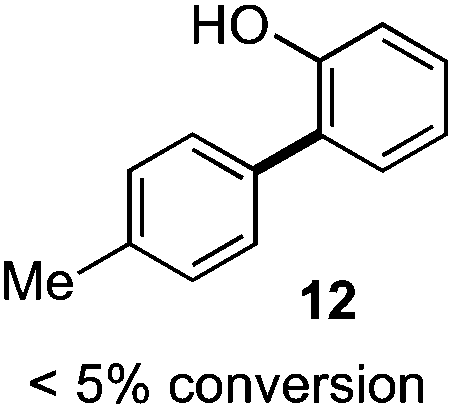
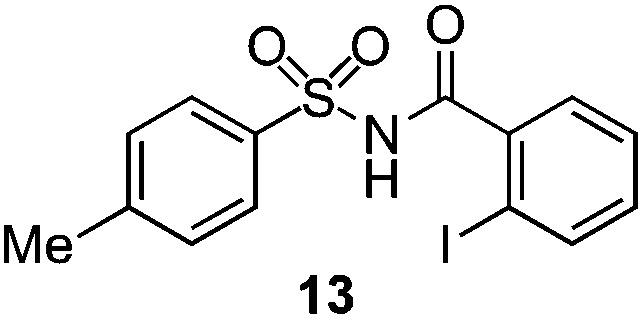
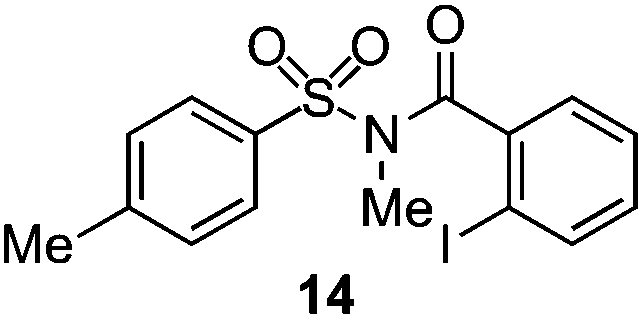
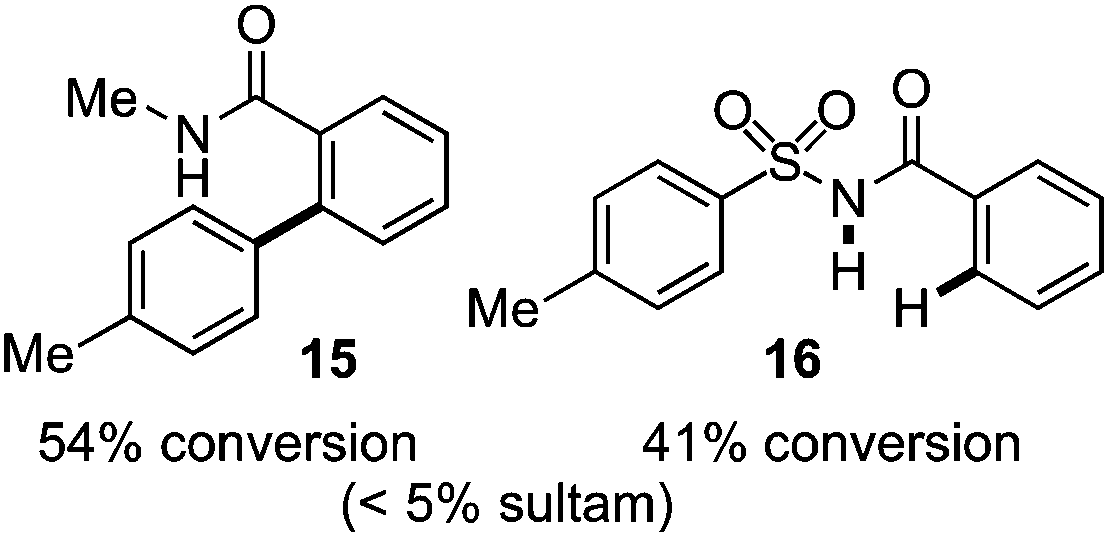
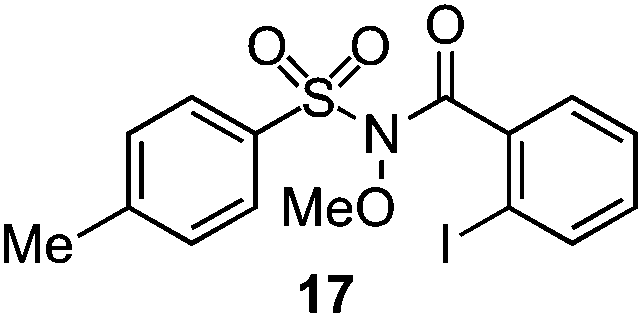
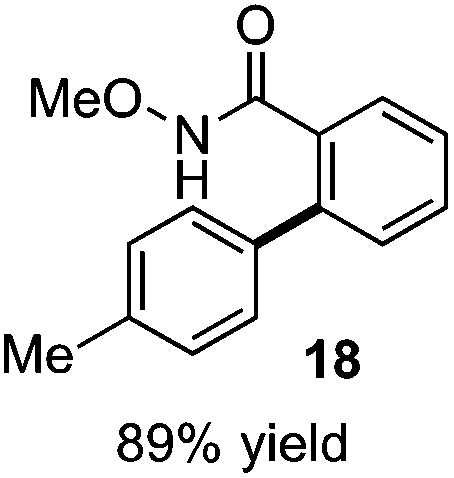
As suggested above, tosylate 10 reacted under optimized ART conditions to form sultone 11 in 92% yield with only trace amounts of biaryl 12 detected (Table 4), as observed with the tin-mediated protocol.10 We thus prepared a series of alternative substrates bearing carboxamide functionality (cf.13, 14, and 17). While mixed imide 13 was unreactive, N-methyl derivative 14 produced the desired biaryl 15 with only minimal amounts of the 7-membered sultam. This result is consistent with that obtained by Motherwell,10 but, as with this tin-mediated precedent, only modest yields were obtained.15 We identified that the material balance in our case was mostly accounted for by dehalogenation/demethylation product 16. On the other hand, N-methoxy amide 17 led to the efficient production of the desired biaryl 18 in 89% yield, with only trace amounts of the cyclized byproduct 19. We thus propose that the SO2–NOMe–CO linker could be a useful solution to this selectivity problem.
|
|
||
|---|---|---|
| Substrate | Major product | Minor product |
| a Substrates were treated with TTMSS (2 equiv.), pyridine (6 equiv.) and I2 (∼ 10 mol%) in t-BuOAc (0.01 M) under air. Yields are of purified, isolated products. Conversions are based on 1H NMR analysis. TTMSS = tris(trimethylsilyl)silane. | ||
|
|
|
|
— | — |
|
|
|
|
|
|
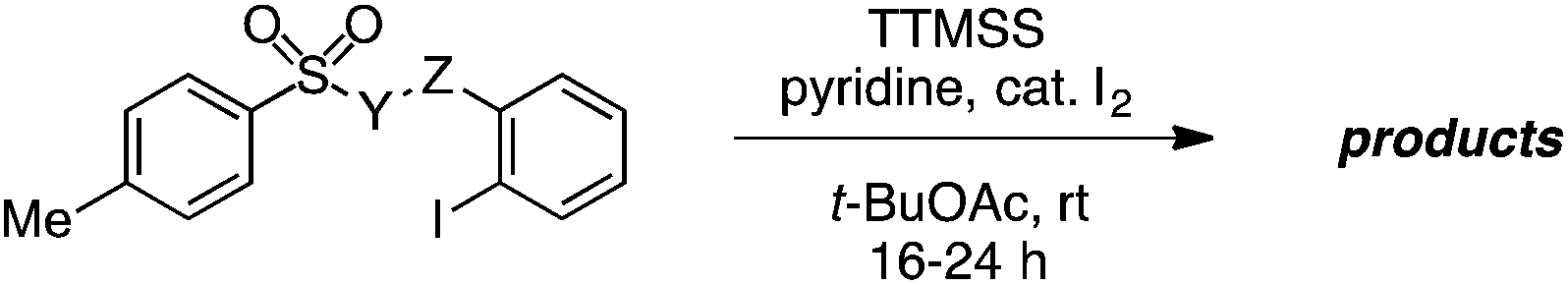
Although the TTMSS-mediated reaction offers a mild and useful approach to ART cross-couplings, we were also interested in the possibility of identifying a catalytic approach to these transformations. Given the demonstrations of Stephenson16 and Lee17 that photoredox catalysis can be used to generate aryl radicals, we evaluated whether such protocols would be applicable to this biaryl formation. Using iodo sulfonamide 10 as a test substrate, we optimized a procedure for the formation of biaryl 11 (Table 5).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Amine | Additive | Solvent | Conc. (M) | Time (h) | Yield (%) |
| a Substrate 20 was treated with Ir catalyst (0.02 equiv.), amine (10 equiv.) and, if indicated, HCO2H (10 equiv.). Yields are based on 1H NMR analysis. b Irradiated using a 20 W CFL. | |||||||
| 1 | 22 | i-Pr2NEt | — | MeCN | 0.01 | 24 | 50 |
| 2 | 22 | i-Pr2NEt | HCO2H | MeCN | 0.01 | 24 | 30 |
| 3 | 23 | i-Pr 2 NEt | — | MeCN | 0.01 | 2 | 85 |
| 4 | 23 | Et3N | — | MeCN | 0.01 | 24 | 74 |
| 5 | 23 | n-Bu3N | — | MeCN | 0.01 | 6 | 67 |
| 6 | 23 | i-Pr2NEt | HCO2H | MeCN | 0.01 | 24 | 77 |
| 7 | 23 | i-Pr2NEt | — | DMSO | 0.01 | 24 | 27 |
| 8 | 23 | i-Pr2NEt | — | DMF | 0.01 | 24 | 25 |
| 9 | 23 | i-Pr2NEt | — | MeCN | 0.03 | 4 | 66 |
| 10 | 23 | i-Pr2NEt | — | MeCN | 0.1 | 4 | 51 |
| 11b | 23 | i-Pr2NEt | — | MeCN | 0.01 | 16 | 90 |
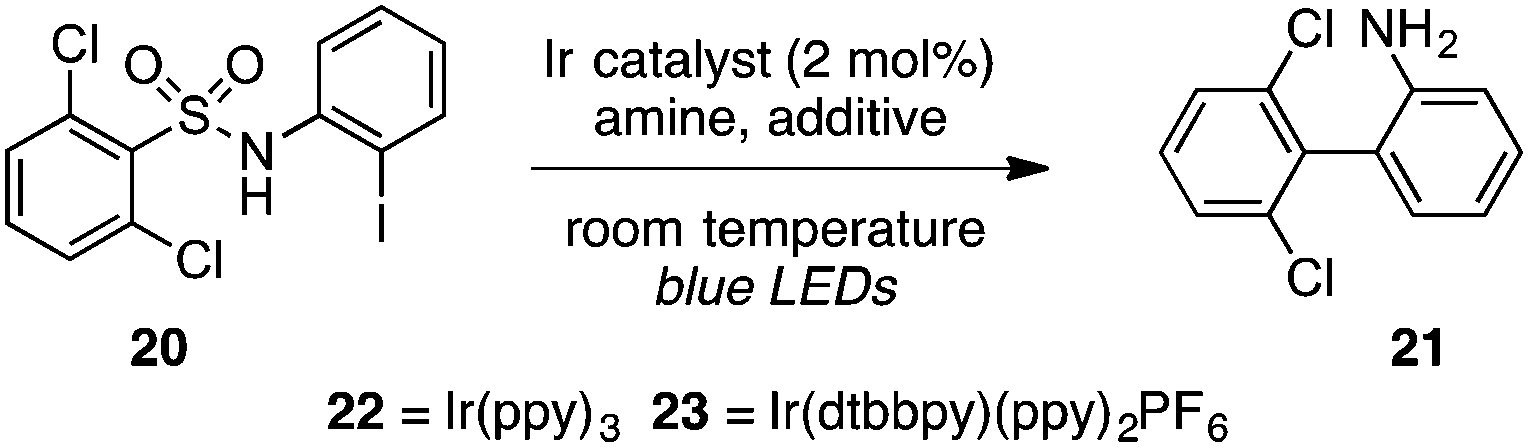
The strongly reducing18 photoredox catalyst Ir(ppy)3 (22) resulted in low to modest yields of the biaryl with or without a formic acid additive, seemingly due to competitive direct cleavage of the C–S bond (entries 1 and 2). Switching to the less reducing19 cationic complex Ir(dtbbpy)(ppy)2PF6 (23, 2 mol%), however, gave the desired biaryl in 85% yield in only 2 h using i-Pr2NEt as the terminal reductant and blue LEDs as the light source in acetonitrile at high dilution (entry 3). Other tertiary amines such as Et3N and n-Bu3N were less efficient (entries 4 and 5), and adding formic acid lowered the yield slightly (entry 6). Other polar solvents such as DMSO or DMF were markedly inferior (entries 7 and 8), as were more concentrated media (entries 9 and 10). A household 20 W CFL could also be used, with the biaryl furnished in 90% yield, although extended reaction times were necessary (entry 11).
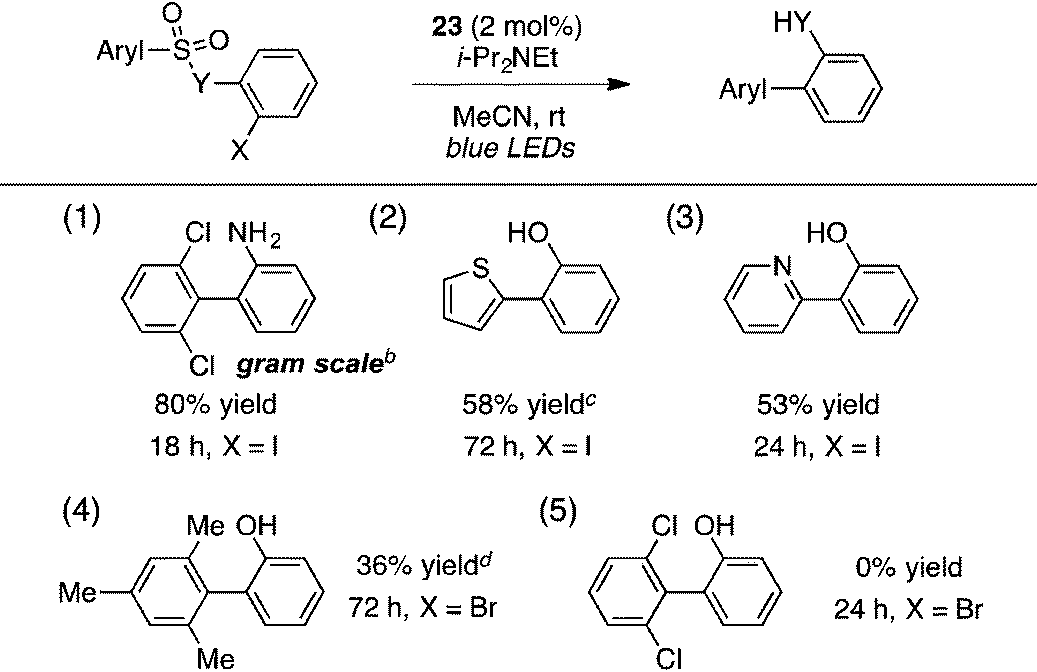
A portion of the scope studies of this photoredox-catalyzed procedure are shown in Table 6. On gram scale, 2-(2,6-dichlorophenyl)aniline (entry 1) was furnished in 80% yield after 18 h at 0.5 mol% catalyst loading. Products incorporating thiophene, pyridine, or mesitylene substituents could also be obtained in modest yields (entries 2–4). However, the 2,6-dichlorophenyl sulfonate substrate did not react at all (entry 5). Furthermore, many of the benzothiazolyl-containing substrates used in the TTMSS-mediated reactions described earlier gave low yields of biaryl compounds, while significant amounts of benzothiazole, resulting from direct cleavage of the sensitive C–S bond, were observed (not shown). This contrast underscores the relative mildness of the TTMSS-mediated conditions.
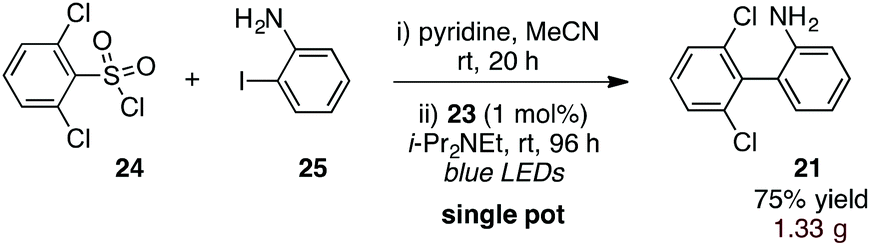
Finally, to further increase the utility of this protocol, we targeted a procedure whereby an activated sulfonic acid derivative and an o-halo substrate could be combined and subjected to the aryl-radical transfer in the same pot. To this end, we found that reaction of sulfonyl chloride 24 and o-iodoaniline (25) in the presence of pyridine, followed by addition of Ir(dtbbpy)(ppy)2PF6 (23, 1 mol%) and i-Pr2NEt to the flask and irradiation furnished 1.33 g of biaryl 21 in 75% yield (eqn (3)).
 | (3) |
Conclusions
In summary, we have developed two new protocols that allow sulfonic acid derivatives to be used as cross-coupling partners under mild conditions. The move away from organotin reagents to TTMSS or photoredox approaches for ART-based transformations significantly increases the attractiveness of these unique bond-forming strategies. Further efforts to further exploit the sulfonate functional group in cross-coupling protocols will surely expand the utility of this venerable functional group.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Funding for this work was provided by the National Science Foundation under CHE-0953259. THL is grateful for an Eli Lilly Grantee Award. EDN is grateful for the Arun Guthikonda Memorial Graduate Fellowship.Notes and references
-
(a) N. Miyaura, K. Yamada and A. Suzuki, Tetrahedron Lett., 1979, 20, 3437 CrossRef
; (b) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS
-
(a) M. Nilsson, Acta Chem. Scand., 1966, 20, 423 CrossRef CAS
-
(a) T. Ikawa, K. Saito and S. Akai, Synlett, 2012, 2241 CAS
- Cross-coupling of ammonium ions:
(a) E. Wenkert, A.-L. Han and C.-J. Jenny, J. Chem. Soc., Chem. Commun., 1988, 975 RSC
- Cross-coupling of sulfinates:
(a) K. Cheng, H.-Z. Yu, B. Zhao, S. Hu, X.-M. Zhang and C. Qi, RSC Adv., 2014, 4, 57923 RSC
- Leading examples of aryl–aryl coupling using partners at the sulfonic acid oxidation state:
(a) E. Wenkert, T. W. Ferreira and E. L. Michelotti, J. Chem. Soc., Chem. Commun., 1979, 637 RSC
- For a review of organosulfur chemistry, see: J. Voss, J. Sulfur Chem., 2009, 30, 167 CrossRef CAS
- A. Studer and M. Bossart, Tetrahedron, 2001, 57, 9649 CrossRef CAS
- R. Loven and W. N. Speckamp, Tetrahedron Lett., 1972, 13, 1567 CrossRef
- For the first use of ART for biaryl formation, see: W. B. Motherwell and A. M. K. Pennell, J. Chem. Soc., Chem. Commun., 1991, 877 RSC
- Related transformations:
(a) L. Eun, L. Chulbom, S. T. Jin, S. W. Ho and S. L. Kap, Tetrahedron Lett., 1993, 34, 2343 CrossRef
- D. P. Curran and A. I. Keller, J. Am. Chem. Soc., 2006, 128, 13706 CrossRef CAS PubMed
-
(a) K. Inoue, A. Sawada, I. Shibata and A. Baba, J. Am. Chem. Soc., 2002, 124, 906 CrossRef CAS PubMed
- ART is especially capable of forming hindered biaryls due to the geometry of the intermediates, see ref. 11c.
- Ref. 11p reports that complex mixtures are obtained, possibly due to the more forcing conditions.
- J. D. Nguyen, E. M. D'Amato, J. M. R. Narayanam and C. R. J. Stephenson, Nat. Chem., 2012, 4, 854 CrossRef CAS PubMed
- H. Kim and C. Lee, Angew. Chem., Int. Ed., 2012, 51, 12303 CrossRef CAS PubMed
- L. Flamigni, A. Barbieri, C. Sabatini, B. Ventura and F. Barigelletti, Top. Curr. Chem., 2007, 281, 143 CrossRef CAS
- M. S. Lowry, J. I. Goldsmith, J. D. Slinker, R. Rohl, R. A. Pascal, G. G. Malliaras and S. Bernhard, Chem. Mater., 2005, 17, 5712 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and product characterization data. See DOI: 10.1039/c7qo00731k |
| This journal is © the Partner Organisations 2018 |