
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEthyl pyruvate attenuates delayed experimental cerebral vasospasm following subarachnoid haemorrhage in rats: possible role of JNK pathway
Ke Jin†
 ,
Hui Wu†,
Tao Lv,
Jiong Dai,
Xiaohua Zhang* and
Yichao Jin*
,
Hui Wu†,
Tao Lv,
Jiong Dai,
Xiaohua Zhang* and
Yichao Jin*
Department of Neurosurgery, Renji Hospital, Shanghai Jiaotong University, School of Medicine, 160, Pujian Road, Shanghai 200127, People's Republic of China. E-mail: zhangxh1969@hotmail.com; honam612@163.com; Fax: +86-21-58394262; Tel: +86-21-68383707 Tel: +86-21-34506561
First published on 16th February 2018
Abstract
The pathophysiology of delayed cerebral vasospasm (CVS) after subarachnoid haemorrhage (SAH) is multifaceted and involves endothelial apoptosis and inflammation. Ethyl pyruvate (EP) could attenuate early brain injury following SAH via anti-inflammation and inhibition of the c-Jun N-terminal kinase (JNK) signalling pathway. However, the role of EP in the delayed CVS has yet to be determined. In this study, we examined the effect of EP on endothelial apoptosis and inflammation and explore possible signalling pathways. We found that EP could significantly attenuate the delayed CVS. Possible mechanisms include a decrease in the endothelial cell apoptosis of the basilar artery and alleviation of endothelial inflammation. The JNK signalling pathway may play an important role in the neuroprotective effects of EP on delayed CVS. The results suggest that EP may be a possible therapy for delayed CVS, and the JNK signalling pathway should be targeted for therapeutic purposes in the future.
1. Introduction
Subarachnoid haemorrhage (SAH) is a neurological emergency. Delayed ischaemic neurological deficit is one of the main causes of poor outcomes after SAH and is probably caused, at least in part, by cerebral vasospasm (CVS).1 The pathophysiology of CVS is multifaceted and involves both endothelial apoptosis and inflammation.2–7 However, the development of therapeutic interventions has been largely hindered by the fact that the underlying pathological mechanisms of CVS remain poorly understood.Pyruvate is a key intermediate metabolite of metabolism and has antioxidant and anti-inflammatory properties that can affect numerous pathological processes.8 However, the pyruvate is too unstable to be a therapeutic agent. Ethyl pyruvate (EP), which is a lipophilic ester derivative of pyruvate, is safer and more stable. It has been demonstrated that EP has protective effects in several animal models, including severe sepsis, acute pancreatitis and acute coronary artery ischaemia.9 Some researchers found that EP can decrease the incidence of apoptosis by inhibiting the expression of P53, Bax and caspase-3.10,11 Additionally, numerous studies have confirmed the anti-inflammatory effects of EP via inhibition of pro-inflammatory signalling pathways, including NF-κB, P38 mitogen-activated protein kinase (MAPK), Janus kinase (JAK)/signal transducers and activator of transcription (STAT) as well as protein kinase B (AKT).9,12 EP also demonstrates a neuroprotective effect on SAH, traumatic brain injury, cerebral ischaemia, neurodegenerative diseases, etc.12–14 EP could attenuate early brain injury following SAH via anti-inflammation and inhibition of the c-Jun N-terminal kinase (JNK) signalling pathway.15,16 However, the effect of EP on the delay of CVS and its possible mechanisms are poorly understood.
To explore the effects of EP on delayed CVS, a rat cisterna magna double-injection model of SAH was introduced. Meanwhile, the possible mechanisms were also investigated.
2. Experiment
2.1 Ethics statement
All animal procedures were approved by the Animal Care and Experimental Committee of the School of Medicine, Shanghai Jiaotong University (Shanghai, China). All animal experiments were carried out at Shanghai Jiaotong University School of Medicine following National Institutes of Health guidelines and local Institutional Animal Care and Use Committee regulations. Adult male Sprague-Dawley (SD) rats were used in this study. Rats were housed in individual cages in a temperature- and humidity-controlled animal facility with a 12 h light/dark cycle. Rats were housed in the animal facility for at least 7 days before surgery and were given free access to food and water during this period.2.2 Experimental design and EP treatment
A total of 54 adult male SD rats weighing 280–320 g were divided into three groups randomly: sham + vehicle (n = 18), SAH + vehicle (n = 18) and SAH + EP (n = 18). The 24 h of the first SAH induction was defined as day 1. All rats were sacrificed on day 7. For haematoxylin and eosin (H&E) staining, terminal deoxynucleotidyl transferase (TdT)-mediated biotin16-dUTP nick-end labelling (TUNEL) staining and immunohistochemical analysis, 18 (6 for each group) rats were perfused transcardially with 4% paraformaldehyde in phosphate-buffered saline (PBS) on day 7. Specimens were removed and fixed further at 4 °C overnight; for western blotting, 18 (6 for each group) rats were perfused transcardially with saline and then specimens were removed and preserved in a −80 °C freezer. For quantification of the mRNA expression of IL-1β and TNF-α, 18 (6 for each group) rats were used for collecting cerebrospinal fluid (CSF).EP was purchased from Sigma-Aldrich (St Louis, MO, USA) and dissolved in 0.9% saline. Rats were given EP (100 mg kg−1) at 6 h, 24 h, 48 h, 72 h, and 96 h after second SAH induction via intraperitoneal injection. The sham + vehicle group and SAH + vehicle group received the same intraperitoneal (i.p.) volume at the corresponding time points after injury.
2.3 Animal models for delayed CVS
Experimental CVS was induced using a modified rat double haemorrhage model. Rats were anaesthetized by i.p. injection of 10% chloral hydrate (3.3 ml kg−1). 0.3 ml fresh arterial blood were withdrawn and injected into the cisterna magna via a stereotactic apparatus. 0.1 ml CSF was drawn by no. 30 Terumo needle to make sure the tip of the needle had been in subarachnoid space. The animals were placed in a ventral recumbent position for 20 min to facilitate clot formation. After monitoring for respiratory distress and giving mechanical ventilation if needed, the animals were returned to the vivarium when they were fully awake. A habitat with a 12 h light–dark cycle was offered with access to food and water ad libitum. Forty-eight hours after 1st SAH, the animals received a secondary injection of SAH to maintain the tendency of vasoconstriction. The sham + vehicle group was injected with 0.3 ml saline at 0 h and 48 h in the same manner.2.4 Neurological score
The neurological scores were evaluated on day 7. Briefly, the evaluation consists of six tests that can be scored from 0–3 or 1–3 and includes the following variables: spontaneous activity, symmetry in the movement of four limbs, forepaw outstretching, climbing, body proprioception, and the response to vibrissae touch. The potential scores ranged from 3 to 18. All tests were evaluated by an observer who was blind to the treatment conditions.2.5 H&E staining
On day 7, the rats were perfused with PBS (150 ml), followed by 4% paraformaldehyde (100 ml) in 0.1 M PBS. The brainstem containing the basilar artery was immediately removed and post-fixed in the same fixative solution for 24 h.The entire length of the basilar artery (BA) was divided into proximal, middle, and distal thirds. After dissection, each section was deparaffinized, hydrated, washed, and stained with H&E staining. The cross-sectional area and thickness of each BA were measured and calculated using ImageJ software (NIH) by an investigator who was blinded to the animal groups. The inner perimeter (p) of the vessel was measured by tracing the luminal surface of the intima, and the area was calculated (area = p2/4π), to correct for vessel deformation and off-transverse sections. Three sections (midpoint of proximal, middle and distal) from each vessel were measured and averaged for one value. The BA wall thickness was measured as the distance from the luminal surface of the intima to the outer border of the media at four different points of one section from each vessel (3, 6, 9, and 12 o'clock), and the adventitia was excluded from the measurement. The four measurements were averaged for one score.
2.6 TUNEL
The rats were killed and intracardially perfused with PBS (pH 7.4) and 4% paraformaldehyde (pH 7.4) on day 7. The BAs were collected and immersed in 4% paraformaldehyde at 4 °C for 12 h and then immersed in a 30% sucrose solution until the tissue sank (3 days). Then, the BAs were frozen in tissue-freezing media and cut in 20 μm sections. TUNEL staining was performed according to the manufacturer's protocol (Roche Inc, Basel, Switzerland), negative controls replaced a mixture of enzyme and label solution with label solution, and the samples were examined under a laser scanning confocal microscope (LSM-710; Zeiss). The apoptotic index was calculated as the number of immunoreactive nuclei per total number of endothelial cells and expressed as a percentage.2.7 Immunohistochemical analysis
The 20 μm-thick formalin-fixed OCT-embedded sections were subjected to immunofluorescence analysis to determine the immunoreactivity of cysteinyl aspartate specific proteinase-3 (caspase-3) and phosphorylated c-Jun N-terminal kinase (p-JNK). Endogenous peroxidase was blocked with 3% H2O2 for 5 min followed by a brief rinse in distilled water and a 15 min wash in PBS. The sections were cooled at room temperature for 20 min and rinsed in PBS. Nonspecific protein binding was blocked with an incubation in 5% horse serum for 40 min. The sections were incubated with primary antibodies anti-caspase-3 [CST9662] and anti-phospho-JNK [CST4668] and anti-IgG, which were all diluted 1:200 (from Suzhou Ard Biological Co., Ltd.) for 1 h at room temperature, followed by a 15 min wash in PBS. The sections were incubated with FITC/Cy3-conjugated IgG (1:500 dilution; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) for 60 min at room temperature. For negative controls, the sections were incubated in the absence of a primary antibody. From each BA, ten microscopic fields per section from five to eight BA sections separated by at least 150 μm were photographed using a random number generator for counting the caspase-3 and p-JNK-positive cells (400× magnifications, Nikon TE300, Japan).2.8 Western blotting analysis
The frozen tissue was mechanically lysed in 20 mM Tris (pH 7.6) containing 0.2% sodium dodecyl sulfate (SDS), 1% Triton X-100, 1% deoxycholate, 1 mM phenylmethylsulfonyl fluoride, and 0.11 IU ml−1 aprotinin (all purchased from Sigma-Aldrich, Inc.). The lysates were centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 20 min at 4 °C. The protein concentration was estimated using the Bradford method. The samples (60 μg per lane) were separated by 12% SDS polyacrylamide gel electrophoresis and electro-transferred onto a polyvinylidene-difluoride membrane (Bio-Rad Lab, Hercules, CA). The membrane was blocked with 5% skimmed milk for 1 h at room temperature and incubated with primary antibodies against phospho-JNK (1:1000, CST), caspase-3 (1:500, Abcam), Bcl-2 (1:1000, Santa Cruz Biotechnology) and Bax (1:1000, Santa Cruz Biotechnology) at 4 °C overnight. β-Actin (diluted in 1:50000, Sigma-Aldrich) was used as the loading control. After the membrane was washed three times in TBS + Tween-20 (TBST) for 10 min each, it was incubated in the appropriate horseradish peroxidase-conjugated secondary antibody (diluted 1:10000 in TBST) for 2 h. Then, the membrane was washed three times in TBS + Tween-20 (TBST) for 10 min each. The blotted protein bands were visualized by enhanced chemiluminescence western blot detection reagents (Millipore, IL) and exposed to X-ray film. The developed films were digitized using an Epson Perfection 2480 scanner (Seiko Corp, Nagano, Japan). The results were quantified by Quantity One Software (BioRad) and normalized to a loading control, β-actin. Values from the sham + vehicle group were used as 100%.
000g for 20 min at 4 °C. The protein concentration was estimated using the Bradford method. The samples (60 μg per lane) were separated by 12% SDS polyacrylamide gel electrophoresis and electro-transferred onto a polyvinylidene-difluoride membrane (Bio-Rad Lab, Hercules, CA). The membrane was blocked with 5% skimmed milk for 1 h at room temperature and incubated with primary antibodies against phospho-JNK (1:1000, CST), caspase-3 (1:500, Abcam), Bcl-2 (1:1000, Santa Cruz Biotechnology) and Bax (1:1000, Santa Cruz Biotechnology) at 4 °C overnight. β-Actin (diluted in 1:50000, Sigma-Aldrich) was used as the loading control. After the membrane was washed three times in TBS + Tween-20 (TBST) for 10 min each, it was incubated in the appropriate horseradish peroxidase-conjugated secondary antibody (diluted 1:10000 in TBST) for 2 h. Then, the membrane was washed three times in TBS + Tween-20 (TBST) for 10 min each. The blotted protein bands were visualized by enhanced chemiluminescence western blot detection reagents (Millipore, IL) and exposed to X-ray film. The developed films were digitized using an Epson Perfection 2480 scanner (Seiko Corp, Nagano, Japan). The results were quantified by Quantity One Software (BioRad) and normalized to a loading control, β-actin. Values from the sham + vehicle group were used as 100%.
2.9 Quantification of mRNA expression of IL-1β and TNF-α
The ABI PRISM®7900 System (Applied Biosystems, Foster City, Calif., USA) was used to examine the expressed mRNA (RT-PCR). According to the manufacturer's instructions, the mRNAs for IL-1β and TNF-α expression were analysed, while 18S was assigned as a housekeeping gene due to its stability. Briefly, the sample was launched into a TaqMan® Human Cytokine Card that enclosed probes and primers for specific targets such as the 18S ribosomal RNA as a control. This procedure was operated through an affixed filling reservoir and a vacuum loading process via the ABI PRISM Card Filling Station.Target mRNAs were standardized nearly to the reference gene (18S) as a relative multiple from baseline values. Comparative mRNA expression was set by the ΔCT method of Livak and Schmittgen. The results were analysed at least with a 5-fold increase in the mRNA levels compared to baseline to allow for data consistency.
2.10 Statistical analysis
Data are expressed as the means ± standard error (SEM). SPSS 17.0 was used for statistical analysis of the data. For group comparisons, an analysis of variance with the Bonferroni post hoc test was used. A difference at a probability value less than 0.05 was considered significant.3. Results
3.1 EP could improve neurological deficits in rats with delayed CVS
Compared to rats in the sham + vehicle group, neurological scores were significantly lower in the SAH + vehicle group (17 ± 0.5 vs. 10.7 ± 0.9, p < 0.001). EP treatment significantly increased the neurological scores compared to rats in the SAH + vehicle group (13.3 ± 0.5 vs. 10.7 ± 0.9, p < 0.05) (Fig. 1).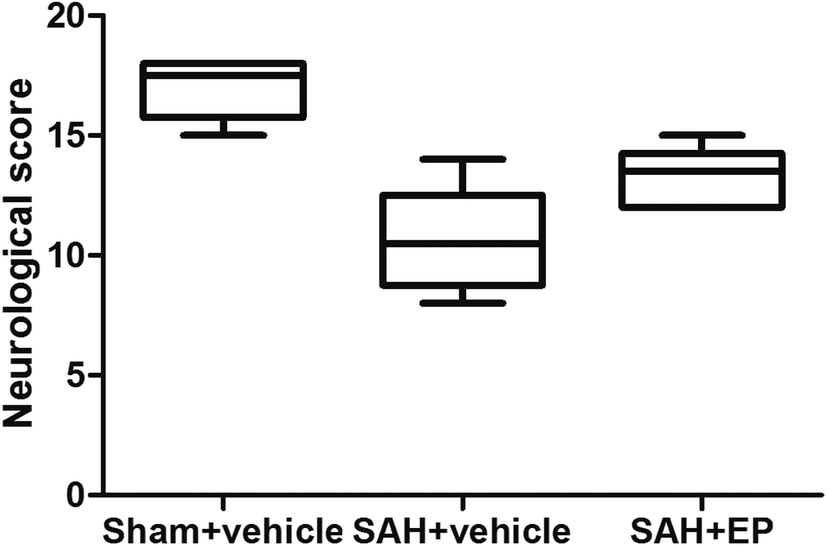 | ||
| Fig. 1 Neurological scores of rats in different groups, including sham + vehicle, SAH + vehicle and SAH + EP, at day 7 after first SAH. SAH impaired the neurological function of rats and EP treatment significantly improved the neurological functions. | ||
3.2 EP could ameliorate CVS
As shown in Fig. 2, there was a significant difference in the cross-sectional area of BA among all groups. A significant difference was detected between the sham + vehicle group (56258 ± 3332 μm2) and SAH + vehicle group (31617 ± 2179 μm2) (p < 0.001). There was also a significant difference in the basilar arterial cross-sectional area between the SAH + vehicle and SAH + EP group (31617 ± 2179 μm2 vs. 44000 ± 2436 μm2, p < 0.05).
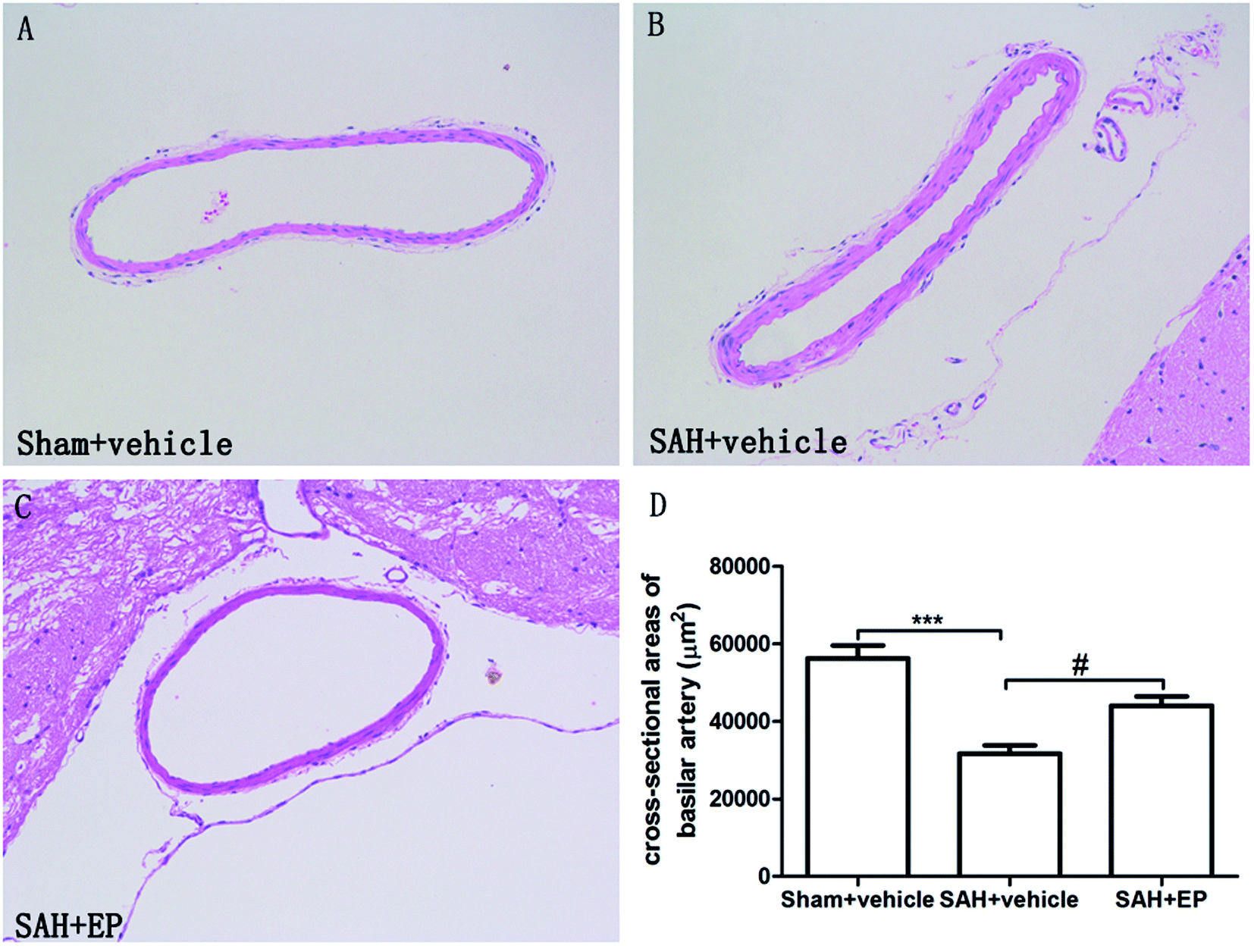 | ||
| Fig. 2 Cross-sectional area of BA in different groups at day 7 after first SAH. SAH reduced the cross-sectional area of BA and EP treatment could improve the area. | ||
As shown in Fig. 3, there was a significant difference in the arterial wall thickness of BA among all groups. A significant difference was detected between the sham + vehicle group (11 ± 1 μm) and the SAH + vehicle group (26 ± 1.9 μm) (p < 0.001). There was also a significant difference in the basilar arterial cross-sectional area between the SAH + vehicle and SAH + EP group (26 ± 1.9 μm vs. 17.4 ± 1.5 μm, p < 0.01).
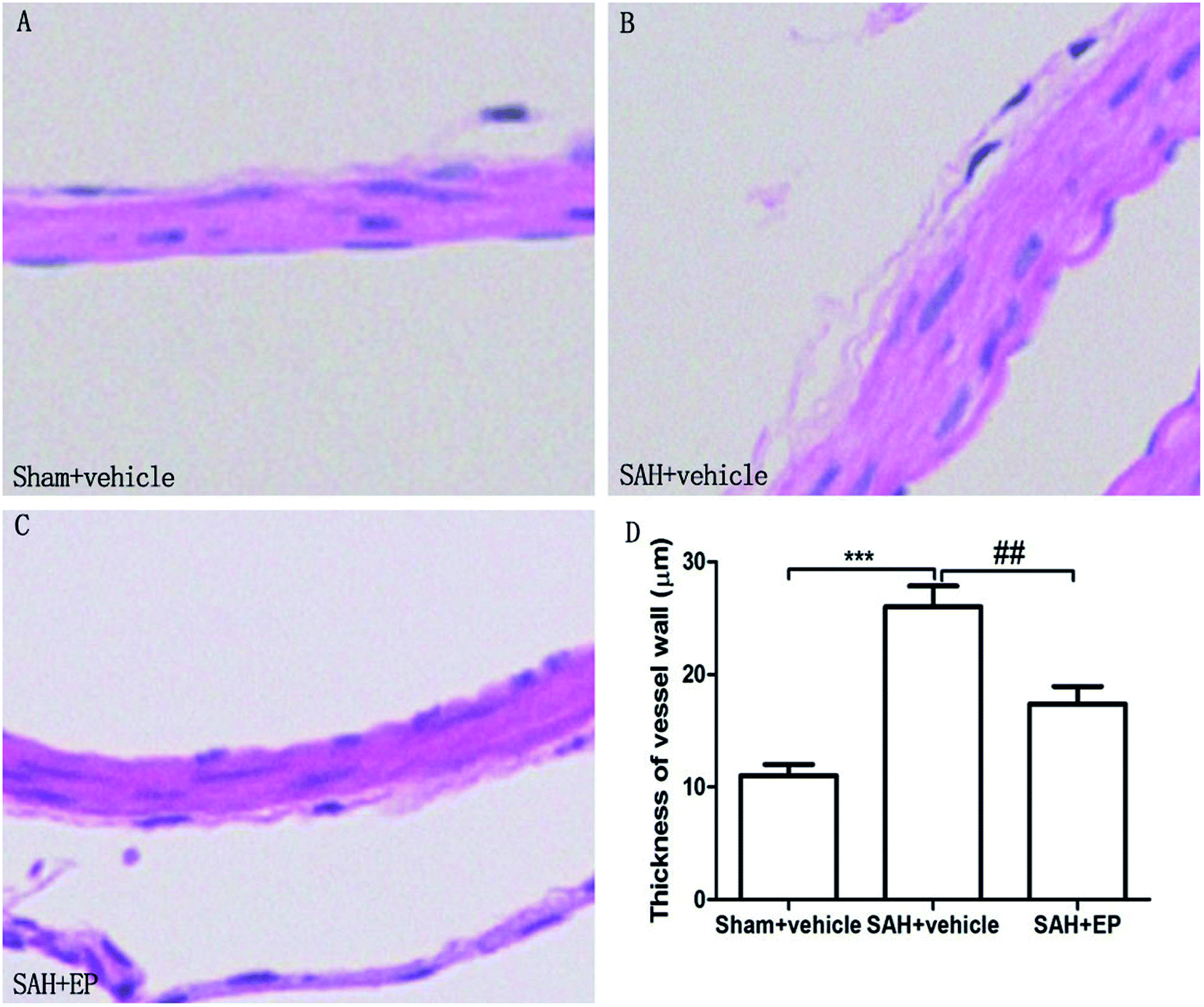 | ||
| Fig. 3 Arterial wall thickness of BA in different groups at day 7 after first SAH. SAH increased the arterial wall thickness of BA and EP treatment decreased the wall thickness. | ||
3.3 EP reduced endothelial apoptosis in BA
Counting of TUNEL-positive in BA sections confirmed a significant elevation in the number of apoptotic cells in the SAH + vehicle group compared to the sham + vehicle (62.4 ± 2.2 vs. 31.9 ± 2.5, p < 0.001). Ethyl pyruvate could significantly reduce apoptotic cells in the endothelial cells of the basilar artery (49.1 ± 2.4 vs. 62.4 ± 2.2, p < 0.01) (Fig. 4).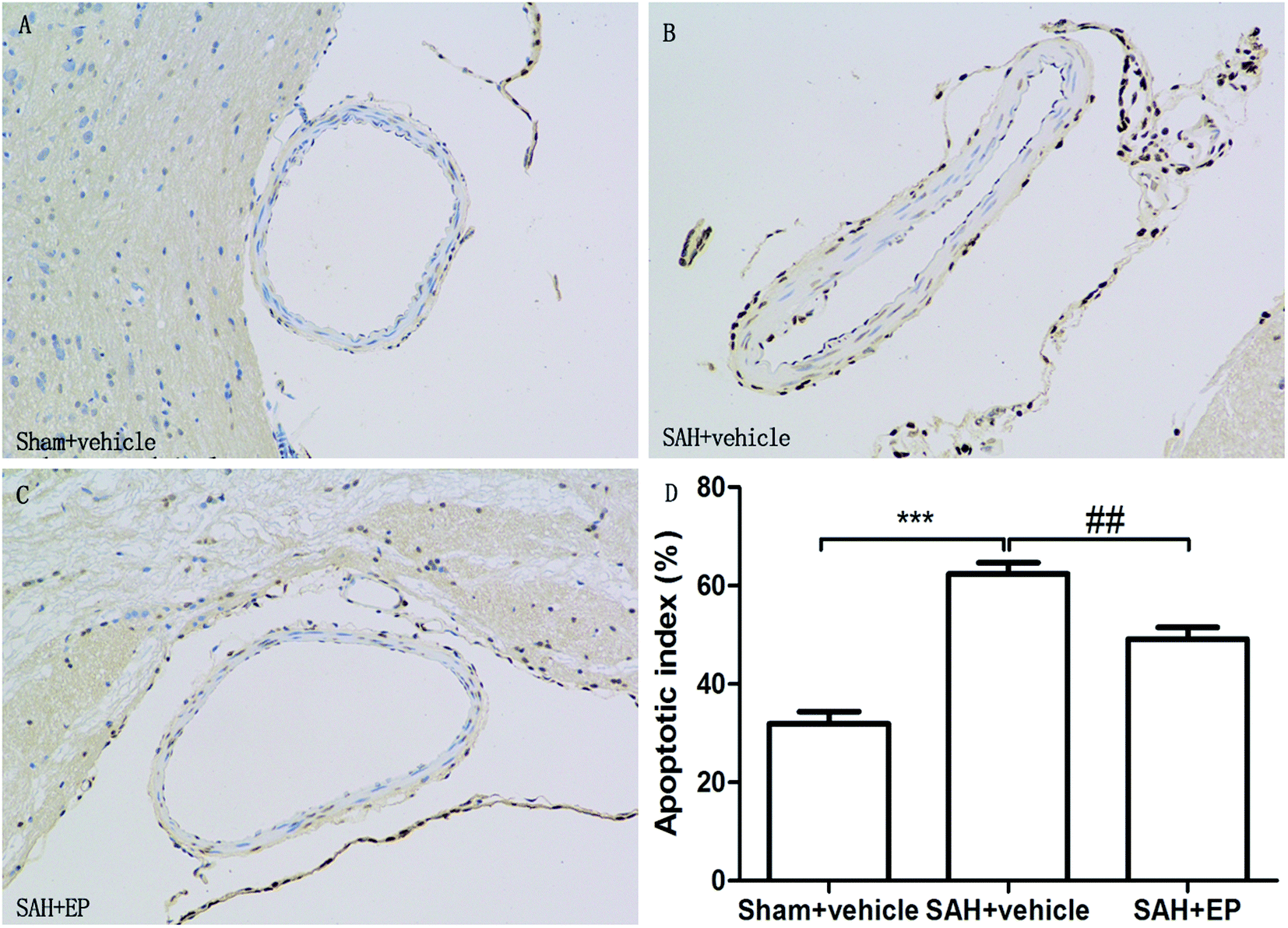 | ||
| Fig. 4 TUNEL staining of endothelial cell of BA. EP markedly reduced the apoptotic index and protected the endothelial integrity. | ||
3.4 Caspase-3 protein expression in BA
Caspase-3 protein expression in BA endothelial cells was detected using immunostaining and western blotting. Caspase-3 staining was stronger in the SAH + vehicle group compared to the sham + vehicle and SAH + EP groups (14.8 ± 1.3 vs. 1.2 ± 0.5 and 10.8 ± 1, p < 0.001 and p < 0.05, respectively) (Fig. 5). A western blot showed that the caspase-3 expression was higher in BA sections from the SAH + vehicle group compared to the sham + vehicle group (p < 0.001), and EP significantly decreased the expression (p < 0.001) (Fig. 6A).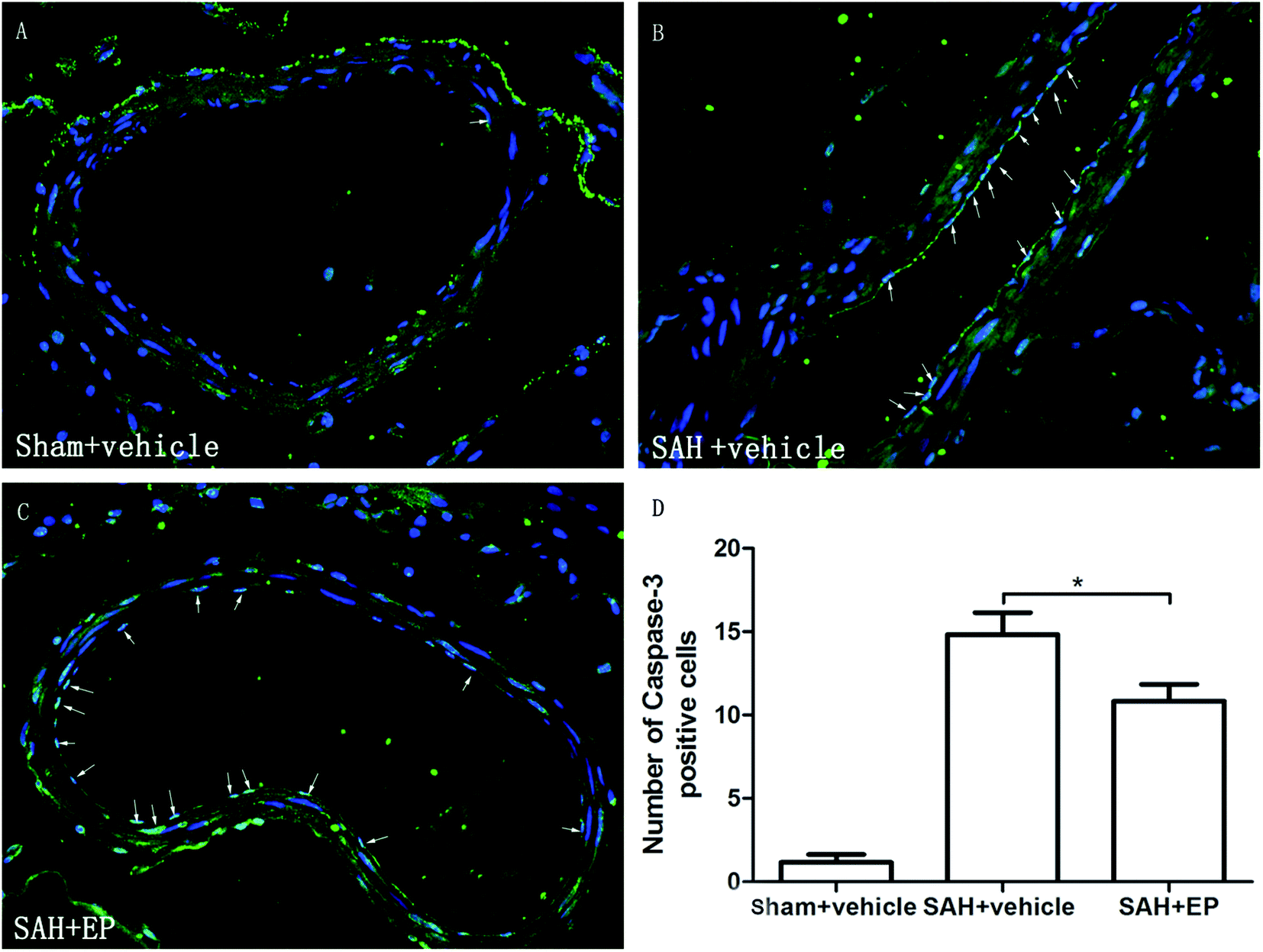 | ||
| Fig. 5 Effect of EP on caspase-3 expression in BA. SAH increased the expression of caspase-3 in endothelium and EP treatment significantly reduced the expression of caspase-3 in endothelium of BA. | ||
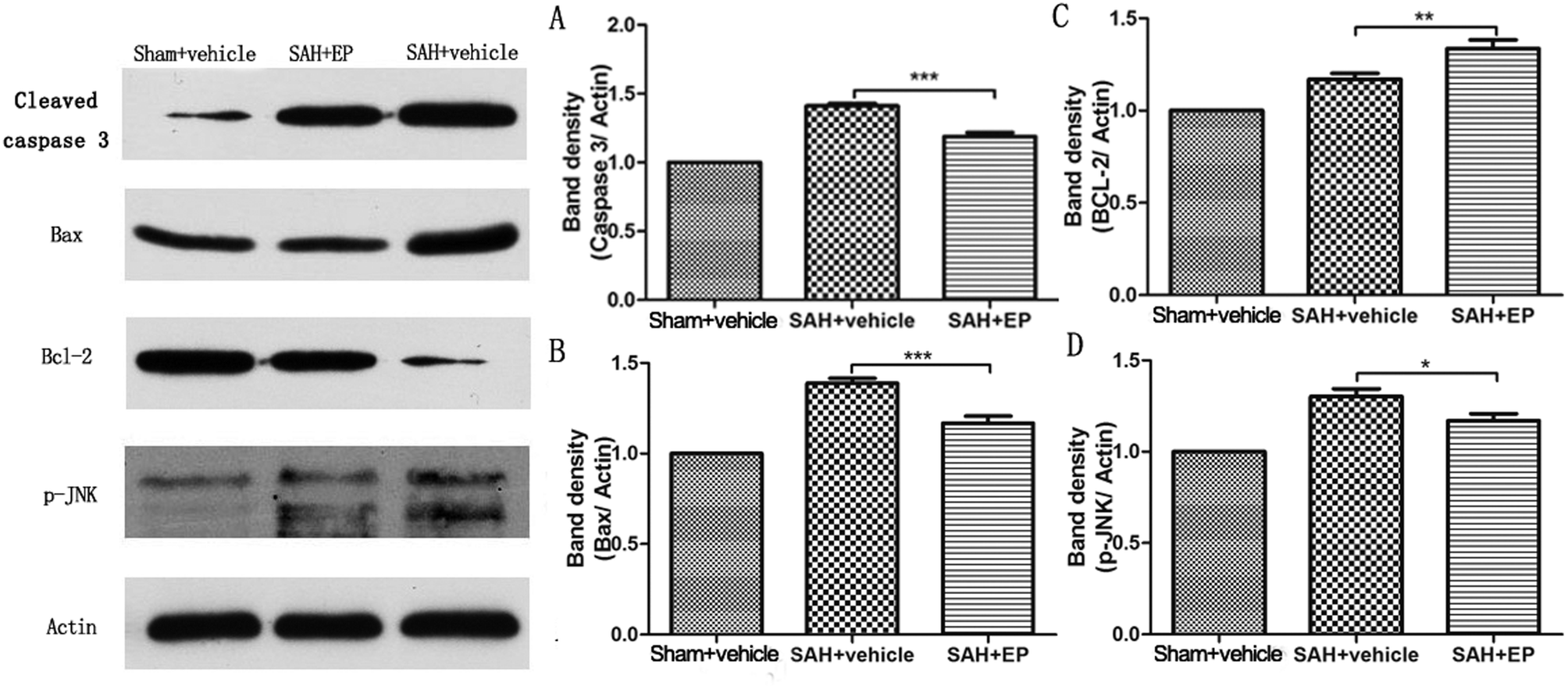 | ||
| Fig. 6 Effect of EP on expression of different proteins in BA. EP markedly decreased expression of caspase-3 (A), Bax (B) and p-JNK (D) while the level of Bcl-2 showed opposite trend (C). | ||
3.5 Influence of EP on expression of Bax and Bcl-2
Western blot analysis of Bax and Bcl-2 in the basilar artery showed that the level of Bax was increased at 7 d after SAH and reduced by EP treatment (p < 0.001), whereas the level of Bcl-2 showed an opposite trend (p < 0.01) (Fig. 6B and C).3.6 p-JNK protein expression in BA
p-JNK protein expression in BA endothelial cells was detected using immunostaining and western blotting. p-JNK staining was stronger in the SAH + vehicle group than in the sham + vehicle and SAH + EP groups (18.3 ± 1.6 vs. 1.2 ± 0.3 and 8 ± 1.2, p < 0.001 and p < 0.001, respectively) (Fig. 7). A western blot showing that the p-JNK expression was higher in BA sections from the SAH + vehicle group compared to the sham + vehicle group (p < 0.001), and EP significantly decreased the expression (p < 0.05) (Fig. 6D).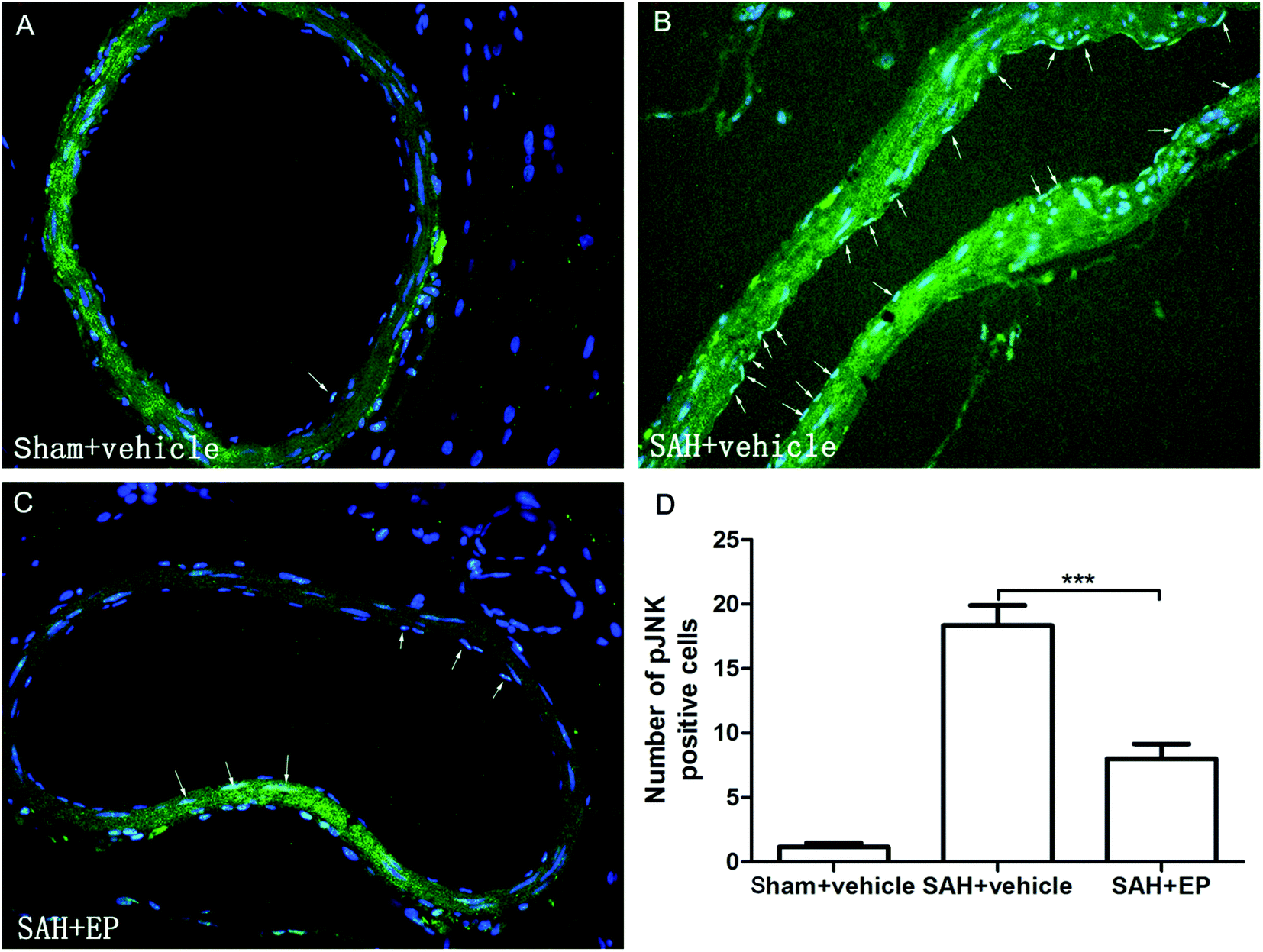 | ||
| Fig. 7 Immunofluorescent imaging of p-JNK expression in BA. EP inhibited the expression of p-JNK. | ||
3.7 Effects of EP on TNF-α and IL-β in CSF
SAH was found to significantly increase the CSF concentrations of TNF-α and IL-β, while EP treatment could decrease the levels of TNF-α and IL-β (p < 0.001) (Fig. 8).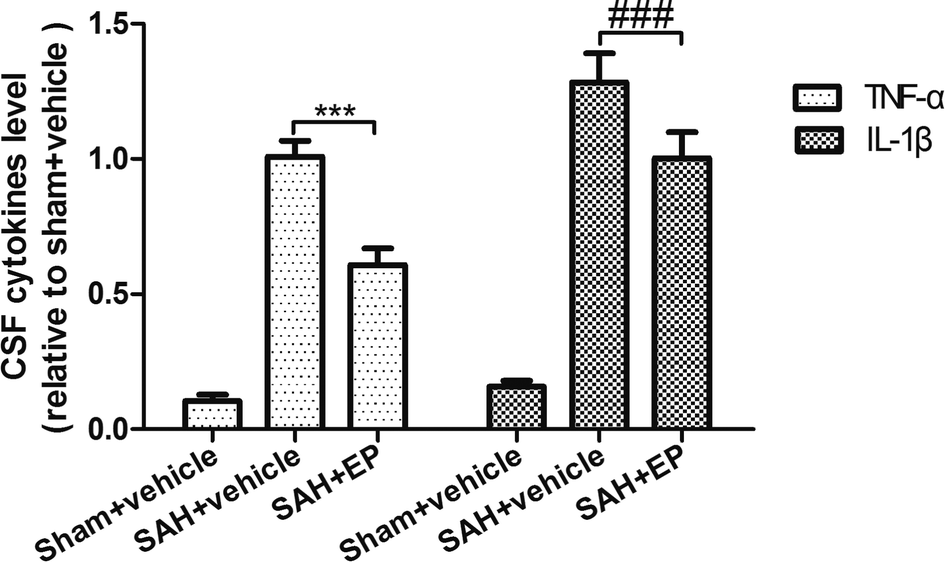 | ||
| Fig. 8 Effect of EP on TNF-α and IL-β. SAH was found to significantly increase the CSF concentrations of TNF-α and IL-β, while EP treatment could decrease the levels of TNF-α and IL-β. | ||
3.8 Discussion
In this study, we found that the rat cisterna magna double-injection model could significantly reduce the BA diameter and increase arterial wall thickness 7 day after the first injection of SAH, which is consistent with the results of previous studies.17–20 EP could significantly attenuate the delayed CVS. Possible mechanisms include a decrease in endothelial cell apoptosis of basilar artery and alleviation of endothelial inflammation. The JNK signalling pathway may play an important role in the neuroprotective effect of EP on delayed CVS.Delayed CVS after SAH is characterized by extended constriction of the large extraparenchymal arteries throughout the cerebral circulation. It has been suggested that delayed CVS is the result of events occurring immediately after the initial haemorrhage that initiate a cascade of pathological mechanisms, including contraction of vascular smooth muscle, formation of microthrombi and inflammation of the vessel wall.1,2 Endothelial damage and activation have been implicated in the pathogenesis of CVS after SAH.3,4,21 Inflammatory reactions in the arterial wall may contribute the pathological damage initiated by SAH.2 Upregulation of many different inflammatory molecules has been observed after SAH, including endothelial adhesion molecules and cytokines, including intercellular adhesion molecule-1, major histocompatibility complex II, tumour necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6 and inducible NO synthase.22 The upregulation of these molecules has been attributed to the local effects of blood and blood clots in the subarachnoid space, the reduction of cerebral blood flow initiating ischaemic inflammation and oxidative-reperfusion stress.22
Inflammation causing delayed CVS mainly arises from the interaction between leukocytes and cell adhesion molecules in the vascular endothelium.23,24 In this process, JNK signalling pathway was activated and inhibition of the JNK signalling pathway reduced CVS through suppression of the inflammatory response.25
Apoptosis could also play an important role in the development and maintenance of CVS.7,26,27 The activation of JNK also causes the phosphorylation of c-Jun, other transcription factors, and cellular proteins, particularly those associated with apoptosis (for example, Bcl-2, P53).28
JNK could activate apoptotic signalling through upregulation of pro-apoptotic genes via transactivation of specific transcription factors or by directly modulating the activities of mitochondrial pro- and anti-apoptotic proteins through distinct phosphorylation events. Upon activation by upstream MAPK2Ks, the phosphorylated JNK translocates to the nucleus (where it phosphorylates and transactivates c-Jun).29 Phosphorylation of c-Jun leads to the formation of AP-1, which is involved in the transcription of a wide variety of proteins with some of them that are known as pro-apoptotic proteins.30–32 It has been noted that the JNK-AP-1 pathway is involved in the increased expression of pro-apoptotic genes, such as TNF-α, Fas-L, and Bak.33 In addition, it has been observed that JNK is required for the apoptosis of central nervous system neurons.34 An alternate pathway contributing to JNK-mediated apoptosis involves the phosphorylation of p53 family of proteins by JNK.35
Previous researchers have focused on methods for attenuating the delayed cerebral ischaemia targeting endothelial apoptosis and inflammation. Some therapies have attenuated vasospasm via anti-inflammatory mechanisms, such as 4′-O-β-D-glucosyl-5-O-methylvisamminol, L-citrulline, glycyrrhizin, curcumin, 6-mercaptopurine, atorvastatin, and so on.36–41 These treatments could decrease the release of pro-inflammatory cytokines that participate in the pathophysiological mechanisms of CVS. There were also drugs with anti-apoptotic effects, including proanthocyanidin, N-acetylcysteine, mexiletine, immunoglobulin, telmisartan, 2,2′-dipyridyl, recombinant human erythropoietin, human tissue kallikrein, alpha lipoic acid and so on.6,42–50 Signalling pathways participating in the anti-apoptotic effects include the Akt pathway, JAK2/STAT3 pathway, and others. Based on the above results, we could conclude that anti-apoptotic and anti-inflammatory mechanisms play an important role in the attenuation of CVS after SAH.
In this study, we found that EP ameliorates CVS after SAH through anti-apoptotic and anti-inflammatory mechanisms. The JNK pathway may at the crossroads of apoptotic and inflammatory effects of SAH, which causes the delayed CVS. The JNK signalling pathway should be targeted for therapeutic purposes.
4. Conclusion
The mechanisms of delayed CVS after SAH include apoptosis and inflammation of endothelial cells, whereas EP could achieve neuroprotective effects through anti-apoptotic and anti-inflammatory mechanisms. JNK signalling pathway may be in the crossroad of the neuroprotective effect of EP.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This project was supported by the National Nature Science Foundation of China (No. 81471333, 81601061), Seed Fund of Renji Hospital (No. RJZZ13-007) and the project to construct clinical ability of the third class hospital in suburban of Shanghai Shen-Kang Hospital Development Center (No. SHDC12013906).References
- M. Gybel-Brask, R. Rasmussen, J. Stensballe, P. I. Johansson and S. R. Ostrowski, Acta Neurochir., 2017, 159, 1073–1078 CrossRef PubMed.
- D. L. Penn, S. R. Witte, R. J. Komotar and E. Sander Connolly Jr, J. Clin. Neurosci., 2015, 22, 1–5 CrossRef PubMed.
- C. J. Frijns, K. M. Kasius, A. Algra, R. Fijnheer and G. J. Rinkel, J. Neurol., Neurosurg. Psychiatry, 2006, 77, 863–867 CrossRef CAS PubMed.
- S. H. van Ierssel, V. M. Conraads, E. M. Van Craenenbroeck, Y. Liu, A. I. Maas, P. M. Parizel, V. Y. Hoymans, C. J. Vrints and P. G. Jorens, J. Neurosci. Res., 2015, 93, 866–872 CrossRef CAS PubMed.
- Z. Guo, L. Xu, X. Wang and X. Sun, Neurol Sci., 2015, 36, 1241–1245 CrossRef PubMed.
- J. He, M. Liu, Z. Liu and L. Luo, Brain Res., 2015, 1611, 74–83 CrossRef CAS PubMed.
- C. Zhou, M. Yamaguchi, G. Kusaka, C. Schonholz, A. Nanda and J. H. Zhang, J. Cereb. Blood Flow Metab., 2004, 24, 419–431 CrossRef CAS PubMed.
- S. D. Varma, K. R. Hegde and S. Kovtun, Ophthalmologica, 2006, 220, 52–57 CrossRef CAS PubMed.
- R. Yang, S. Zhu and T. I. Tonnessen, J. Inflammation, 2016, 13, 37 CrossRef PubMed.
- H. S. Shim, J. H. Park, Y. A. Kim, J. Y. Han, M. Park, Y. G. Song, J. S. Kim and I. W. Shin, Singapore Med. J., 2017, 58, 557–561 CrossRef PubMed.
- J. Zeng, J. Liu, G. Y. Yang, M. J. Kelly, T. L. James and L. Litt, Anesthesiology, 2007, 107, 630–640 CrossRef CAS PubMed.
- M. Jang, M. J. Lee and I. H. Cho, Brain, Behav., Immun., 2014, 38, 151–165 CrossRef CAS PubMed.
- S. W. Kim, J. Y. Jeong, H. J. Kim, J. S. Seo, P. L. Han, S. H. Yoon and J. K. Lee, Neurotoxic. Res., 2010, 17, 39–49 CrossRef CAS PubMed.
- H. Shi, H. L. Wang, H. J. Pu, Y. J. Shi, J. Zhang, W. T. Zhang, G. H. Wang, X. M. Hu, R. K. Leak, J. Chen and Y. Q. Gao, CNS Neurosci. Ther., 2015, 21, 374–384 CrossRef CAS PubMed.
- T. Lv, Y. F. Miao, Y. C. Jin, S. F. Yang, H. Wu, J. Dai and X. H. Zhang, Neurochem. Res., 2017, 42, 1044–1056 CrossRef CAS PubMed.
- R. Fang, X. Zheng and M. Zhang, Acta Neurochir., 2016, 158, 1069–1076 CrossRef PubMed.
- Y. Miyagi, R. C. Carpenter, T. Meguro, A. D. Parent and J. H. Zhang, J. Neurosurg., 2000, 93, 471–476 CrossRef CAS PubMed.
- H. Vatter, S. Weidauer, J. Konczalla, E. Dettmann, M. Zimmermann, A. Raabe, C. Preibisch, F. E. Zanella and V. Seifert, Neurosurgery, 2006, 58, 1190–1197 CrossRef PubMed.
- E. Guresir, N. Vasiliadis, S. Dias, P. Raab, V. Seifert and H. Vatter, Acta Neurochir., 2012, 154, 11–19 CrossRef PubMed.
- E. Guresir, N. Vasiliadis, J. Konczalla, P. Raab, E. Hattingen, V. Seifert and H. Vatter, J. Neurol. Sci., 2013, 332, 128–135 CrossRef CAS PubMed.
- K. W. Park, C. Metais, H. B. Dai, M. E. Comunale and F. W. Sellke, Anesth. Analg., 2001, 92, 990–996 CAS.
- G. F. Prunell, N. A. Svendgaard, K. Alkass and T. Mathiesen, Neurosurgery, 2005, 56, 1082–1092 Search PubMed.
- J. Li, J. Chen, H. Mo, C. Qian, F. Yan, C. Gu, Q. Hu, L. Wang and G. Chen, Mol. Neurobiol., 2016, 53, 2668–2678 CrossRef CAS PubMed.
- A. Shao, H. Wu, Y. Hong, S. Tu, X. Sun, Q. Wu, Q. Zhao, J. Zhang and J. Sheng, Mol. Neurobiol., 2016, 53, 3462–3476 CrossRef CAS PubMed.
- H. Yatsushige, M. Yamaguchi, C. Zhou, J. W. Calvert and J. H. Zhang, Stroke, 2005, 36, 1538–1543 CrossRef CAS PubMed.
- J. Cahill, J. W. Calvert, I. Solaroglu and J. H. Zhang, Stroke, 2006, 37, 1868–1874 CrossRef PubMed.
- C. Zhou, M. Yamaguchi, A. R. Colohan and J. H. Zhang, J. Cereb. Blood Flow Metab., 2005, 25, 572–582 CrossRef CAS PubMed.
- A. M. Manning and R. J. Davis, Nat. Rev. Drug Discovery, 2003, 2, 554–565 CrossRef CAS PubMed.
- L. Chang and M. Karin, Nature, 2001, 410, 37–40 CrossRef CAS PubMed.
- D. N. Dhanasekaran and G. L. Johnson, Oncogene, 2007, 26, 3097–3099 CrossRef CAS PubMed.
- M. Raman, W. Chen and M. H. Cobb, Oncogene, 2007, 26, 3100–3112 CrossRef CAS PubMed.
- A. G. Turjanski, J. P. Vaque and J. S. Gutkind, Oncogene, 2007, 26, 3240–3253 CrossRef CAS PubMed.
- M. Fan and T. C. Chambers, Drug Resist. Updates, 2001, 4, 253–267 CrossRef CAS PubMed.
- B. Bjorkblom, J. C. Vainio, V. Hongisto, T. Herdegen, M. J. Courtney and E. T. Coffey, J. Biol. Chem., 2008, 283, 19704–19713 CrossRef CAS PubMed.
- S. Y. Fuchs, V. Adler, M. R. Pincus and Z. Ronai, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 10541–10546 CrossRef CAS.
- C. Z. Chang, S. C. Wu, A. L. Kwan and C. L. Lin, Behav. Brain Funct., 2015, 11, 28 CrossRef PubMed.
- G. Pradilla, T. Garzon-Muvdi, J. J. Ruzevick, M. Bender, L. Edwards, E. N. Momin, R. C. Thompson and R. J. Tamargo, Neurosurgery, 2012, 70, 747–756 CrossRef PubMed.
- C. Z. Chang, S. C. Wu and A. L. Kwan, J. Vasc. Res., 2015, 52, 12–21 CrossRef CAS PubMed.
- C. Wakade, M. D. King, M. D. Laird, C. H. Alleyne Jr. and K. M. Dhandapani, Antioxid. Redox Signaling, 2009, 11, 35–45 CrossRef CAS PubMed.
- C. Z. Chang, C. L. Lin, N. F. Kassel, A. L. Kwan and S. L. Howng, Acta Neurochir., 2010, 152, 861–867 CrossRef PubMed.
- C. Z. Chang, S. C. Wu, C. L. Lin, S. L. Hwang, S. L. Howng and A. L. Kwan, Acta Neurochir., 2010, 152, 1399–1406 CrossRef PubMed.
- C. Yilmaz, T. Cansever, A. Kircelli, O. Isiksacan Ozen, F. Aydemir, A. Akar and H. Caner, Turk. Neurosurg., 2015 DOI:10.5137/1019-5149.
- S. L. Cengiz, M. F. Erdi, M. C. Avunduk, M. Tosun, M. E. Ustun, R. Gokce, A. Yosunkaya and A. Baysefer, Brain Inj., 2011, 25, 965–971 CrossRef PubMed.
- O. Sen, H. Caner, M. V. Aydin, O. Ozen, B. Atalay, N. Altinors and M. Bavbek, Neurol. Res., 2006, 28, 859–863 CrossRef CAS PubMed.
- F. Erdi, F. Keskin, H. Esen, B. Kaya, B. Feyzioglu, I. Kilinc, Y. Karatas, G. Cuce and E. Kalkan, Neurol. Res., 2016, 38, 224–231 CrossRef CAS PubMed.
- M. F. Erdi, O. Guney, A. Kiyici and H. Esen, Turk. Neurosurg., 2011, 21, 527–533 Search PubMed.
- G. Chen, S. Zhang, J. Shi, J. Ai and C. Hang, Cytokine, 2009, 45, 162–168 CrossRef CAS PubMed.
- Y. Yu, Z. Lin, Y. Yin and J. Zhao, Neurol Sci., 2014, 35, 1413–1419 CrossRef PubMed.
- M. Yunchang, D. Qinxue, J. Binbin, H. Xin, Y. Lili, C. Linbi, G. Wujun, Z. Pengbo and W. Junlu, Neurol. Res., 2015, 37, 1082–1089 CrossRef PubMed.
- O. Guney, F. Erdi, H. Esen, A. Kiyici and Y. Kocaogullar, World Neurosurg., 2010, 73, 42–49 CrossRef PubMed.
Footnote |
| † These two authors contribute equally to the study. |
| This journal is © The Royal Society of Chemistry 2018 |