
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComparative analysis of oxidative mechanisms of phloroglucinol and dieckol by electrochemical, spectroscopic, cellular and computational methods†
Di Zhang a,
Chengtao Wang*b,
Lingqin Shen*c,
Hyeon-Cheol Shind,
Kyung Bok Leee and
Baoping Jif
a,
Chengtao Wang*b,
Lingqin Shen*c,
Hyeon-Cheol Shind,
Kyung Bok Leee and
Baoping Jif
aSchool of Food and Biological Engineering, Jiangsu University, Zhenjiang, China
bBeijing Engineering and Technology Research Center of Food Additives, Beijing Technology & Business University (BTBU), Beijing, China. E-mail: wangchengtao@th.btbu.edu.cn
cSchool of Chemistry and Chemical Engineering, Jiangsu University, No. 301 Xue Fu Road, Jingkou District, Zhenjiang 212013, China. E-mail: lshen@ujs.edu.cn
dCenter for Molecular Intelligence, SUNY Korea, Incheon, South Korea
eCollege of Medicine, Konyang University, Daejeon 302-718, South Korea
fBeijing Key Laboratory of Functional Food from Plant Resources, College of Food Science and Nutritional Engineering, China Agricultural University, Beijing, China
First published on 9th January 2018
Abstract
Numerous studies have been carried out on the redox activities of phenolic compounds from terrestrial plants, however, the redox pathway of phlorotannins, a type of marine algae-derived polyphenol, is far from clear. In the present study, the redox mechanisms of two phlorotannins, phloroglucinol (PL) and dieckol (DL), were comparatively scrutinized. Differential pulse voltammetry was conducted in the pH range 2.0–12.0 to determine the acid–base dissociation constant (pKa) and the number of electrons and protons involved in the redox reactions of two phlorotannins. Cyclic voltammetry was applied to obtain the heterogeneous electron transfer rate constant (k0). By means of computational calculation, UV-vis spectroscopy, and electrochemical analysis, it is proposed that PL oxidation in the whole pH range undergoes two steps which are dominated by proton-coupled electron transfer (PCET) (pH ≤ 9) and sequential proton-loss electron transfer (SPLET) mechanisms (pH > 9), respectively. In contrast, the multiple steps taking place in the DL oxidation process rely on PCET (pH ≤ 5), mixed SPLET/PCET (5 < pH ≤ 10), and electron transfer (pH > 10) mechanisms, respectively. Also, the lower proton affinity and ionization potential values of DL, which are attributed to its conjugated Cπ–O–Cπ moieties, lead to relatively higher redox activity as compared to PL in various chemical and cellular models. These findings may provide useful insights into the oxidative conversion of phlorotannins in their biological and chemical processes.
1. Introduction
Currently the global market of natural products is driven by health and anti-aging products, which in most cases play an essential role against oxidative stress.1 The antioxidant activity of many natural polyphenols has gained great attention due to their promising biological relevance. Beside the polyphenolic compounds from terrestrial plants, the phlorotannins (seaweed phenols), a class of polyphenolic secondary metabolites abundantly present in brown algae, have recently attracted considerable research attention.2 Except for phloroglucinol (PL, 1,3,5-trihydroxybenzene), other phlorotannins are primarily derived through the polymerization of PL monomer units.3 However, only a very few of the partially isolated polymerized PL have been subsequently characterised due to the difficulty in separating these highly polar compounds during the extraction process.4Dieckol (DL), a hexamer of PL (Fig. 1), is one of the very limited successfully isolated phlorotannins from dietary brown algae. Hence, DL and PL can be used as good model molecules to make a deep insight into a series of biochemical processes of dietary phlorotannins.5,6 To date, DL and PL have been proposed to exhibit a broad range of biological activities including antioxidant, anti-inflammation, anti-hyperlipidemia, anti-radiation, and anti-tumor activities.7–9 Most of these biological activities are closely related to electrochemical feature since abundant lines of evidences demonstrated that redox or electron transfer process plays an important role in physiological performance of polyphenols.10 Nevertheless, the current research on the electron transfer processes of phenol compounds have been intensively focused on terrestrial plants-derived polyphenols such as flavonoids and phenolic acids.11,12 To the best of our knowledge, there is very few literature on electrochemical oxidation processes of DL or other phlorotannins. Consequently, the structural characteristics which determine the varied redox activities of different phlorotannins are still to be investigated.
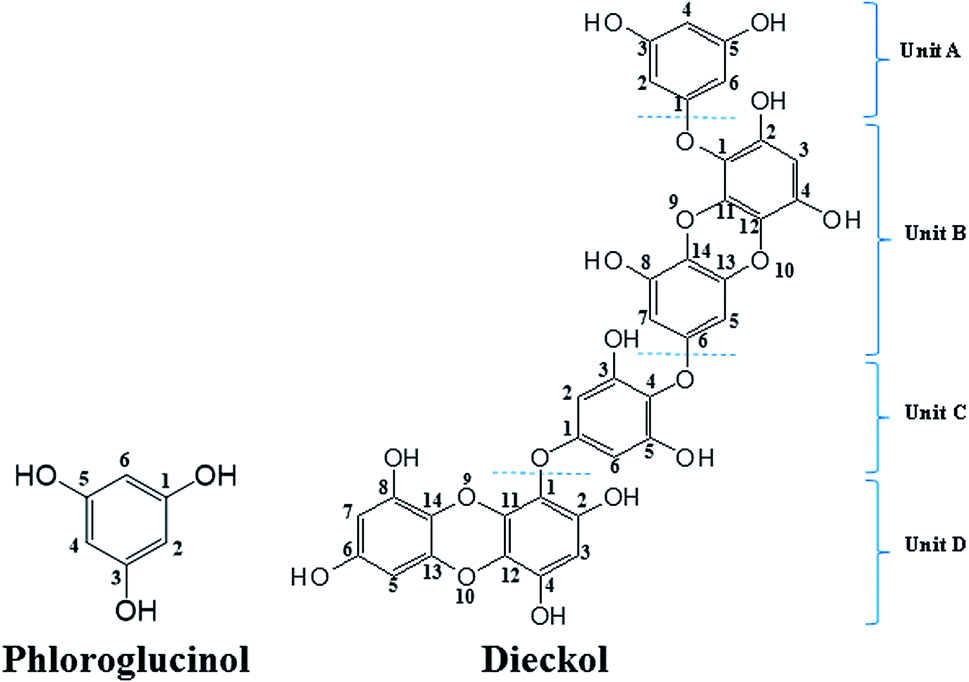 | ||
| Fig. 1 Chemical structures of phloroglucinol and dieckol. | ||
Due to lack of fundamental research on oxidation processes of phlorotannins, we presented a comprehensive investigation on the redox mechanisms of PL and DL, which are two representative phlorotannins. The proton dissociation and electron transfer processes of PL and DL in different pH conditions were comparatively studied in electrochemical assays, accompanied by spectroscopic, computationally theoretical, and oxidants scavenging analyses. The obtained information and resulting conclusions should be helpful in basic understanding the oxidative conversion of phlorotannins in their biological and chemical processes. Additionally, the structure–redox activity relationship of phlorotannins is also analyzed in present study.
2. Experimental
2.1 Chemicals and solutions
DL was prepared and kindly supplied by BotaMedi, Inc. (Jeju, Korea). Briefly, the whole plant of Ecklonia cava was collected from the coast of Jeju Island, Korea. The dried Ecklonia cava powder was extracted with 70% aqueous ethanol (EtOH) and then partitioned to water and ethyl ether fractions. The ether fraction was subjected to octadecylsilyl (ODS) column chromatography to isolate DL (purity ≥ 99%). By means of 1H-NMR spectra and comparison with published data, DL was identified as follows: dieckol: white solid; UV (EtOH) λmax 225 nm; 1H-NMR (DMSO-d6, 500 MHz) δ 5.71 (d, J = 2.40, 2H), 5.79 (t, J = 1.8, 1H), 5.81 (d, J = 1.8, 1H), 5.93 (s, 1H), 5.97 (d, J = 3.0, 1H), 6.01 (d, J = 3.0, 1H), 6.13 (s, 1H), 6.15 (s, 1H), 9.17 (s, OH), 9.24 (s, OH), 9.25 (s, OH), 9.29 (s, OH), 9.37 (s, OH), 9.45 (s, OH), 9.5 (s, OH), 9.62 (s, OH), 9.70 (s, OH); 13C-NMR (DMSO-d6, 125 MHz) δ 93.6, 93.7, 94.1, 94.5, 96.2, 98.1, 98.2, 98.4, 98.5, 146.0, 146.1, 122.2, 122.3, 122.6, 123.2, 123.3,124.0, 124.2, 137.1, 137.2, 141.9, 142.0, 142.6, 142.9, 145.9, 146.1, 151.2, 153.1, 154.2, 155.9, 158.8, 160.3. Fast atom bombardment mass spectrometry (FABMS) m/z 743.1 (M + H)+.Acetic acid, trolox(6-hydroxy-2,5,7,8-tetramethyl-chroman-2-carboxilic acid), PL, 2′,7′-dichlorofluorescin diacetate (DCFH-DA) were purchased from Sigma-Aldrich, Inc. (St. Louis, MO). Tetra-n-butylammonium hexafluorophosphate (Bu4NPF6) was obtained from Tokyo Chemical Industry Co. (Tokyo, Japan). Dimethylsulfoxide (DMSO), sodium hypochlorite solution (NaClO) (available chlorine 10–13%), acetonitrile (ACN), and absolute ethanol were purchased from Sinopharm Chemical Reagent Co. (Beijing, China). All the other chemicals used were of analytical grade.
2.2 Cyclic and differential pulse voltammetry (CV and DPV)
The electrochemical behaviors of DL and PL in CV and DPV were conducted on a Model CHI 660 electrochemical analyzer (CHENHUA, Shanghai, China). CV and DPV measurements were done using a three-electrode system. The working electrode is a glassy carbon electrode (GCE) with an Ag/AgCl electrode as reference. A platinum foil served as the auxiliary electrode. The glassy carbon working electrode was polished successively with 1, 0.3, and 0.05 μm alumina powder before each scanning. The electrolyte solution was 0.2 M phosphate buffers (PB) at different pH values plus 0.3 M KCl. DL and PL stock solutions were diluted to various concentrations using a mixing solvents of PB and ethanol (80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20, v/v). Prior to each run, the dissolved oxygen in electrolyte was removed by bubbling with N2 for about 15 min.
:20, v/v). Prior to each run, the dissolved oxygen in electrolyte was removed by bubbling with N2 for about 15 min.
2.3 Oxidants scavenging activities in electrochemical and cellular systems
The scavenging of hypochlorite was directly measured by the cathodic current decay resulting from its reduction in the presence of increasing concentrations of DL or PL. The main procedure was based on our previous report.13 Briefly, the required volume of phlorotannin solution was added into a mixture solution of supporting electrolyte and hypochlorite (500 μM) in the electrochemical cell. The reaction mixture was left to react for 5 min in darkness at room temperature. The voltammogram was measured immediately to minimize decomposition of hypochlorite. The running parameters were as follows: scan range, from +0.9 to −0.3 V; amplitude, 0.05 V; pulse width, 0.06 s; step size, 4 mV; pulse period, 0.5 s; and sample width, 0.02 s.The method used for assessing scavenging capacity toward superoxide anion has been developed by Blanc et al.14 For the measurements, a solution of 10 mL of DMSO containing the supporting electrolyte 0.1 M Bu4NPF6 was saturated with N2 for about 15 min. Aliquots of each stock solution of DL or PL were successively added to the 10 mL oxygen solution. After each aliquot addition, CV of the oxygen solution was recorded at a scan rate of 0.1 V s−1 with the initial potential at 0.0 V and the reverse one at −1.2 V vs. Ag/AgCl.
The cellular antioxidant activity (CAA) was measured based on the method reported by Wolfe et al.15 Briefly, HepG2 cells were cultured in a 96-well plate at a density of 6 × 104 per well and were incubated at 37 °C for 24 h. After 24 h of incubation, medium was removed and cells were washed with PBS. Then, 25 μM DCFH-DA plus various concentrations of PL or DL were placed in the cell culture plate incubated for 1 h at 37 °C. After DCFH-DA was removed, the cells were washed thoroughly for at least 2 times by PBS. Then, cells were incubated with 1.0 mM AAPH for 30 min. The fluorescence of the cells from each well was recorded by a plate reader (SpectraMax M2e, Molecular Devices, USA). Emission at 540 nm was measured after excitation at 485 nm every 5 min for 1 h. The area under the curve (AUC) of fluorescence (fluorescence unit, FU) versus time (min) was integrated to calculate the CAA value.
2.4 Theoretical calculation
All the theoretical calculations reported were performed using Gaussian 03, Revision D.01.16 The density functional theory (DFT) framework implemented in this computational package was used. The B3LYP hybrid functional (UB3LYP for the corresponding radicals) in conjunction with the 6-31G (d,p) basis set was adopted to optimize the geometrical structures. Harmonic frequency analyses were used to verify the optimized minima. The total enthalpy at 298 K consisted of the thermal correction to the enthalpy and single point energy (SPE) values calculated within the 6-311++ G (d, p) basis set. From the total enthalpies calculated, ionization potential (IP), proton dissociation enthalpy (PDE), proton affinity (PA), and electron transfer enthalpy (ETE) were determined by the following expressions:Proton-coupled electron transfer (PCET)
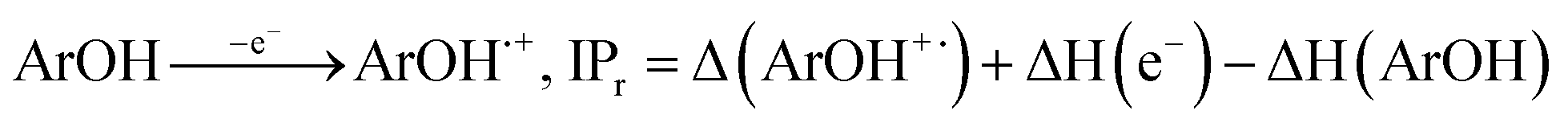
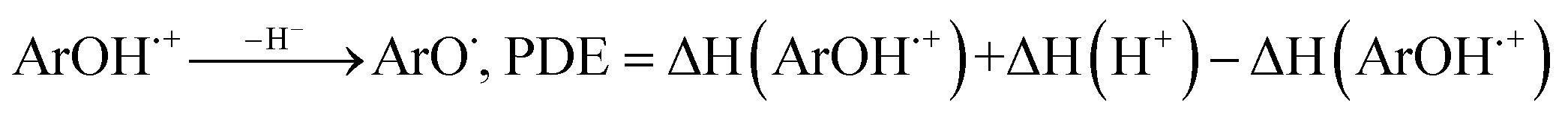
Sequential proton-loss electron transfer (SPLET)
| ArOH ⇌ ArO− + H+, PA = ΔH(ArO−) + ΔH(H+) − ΔH(ArOH) |
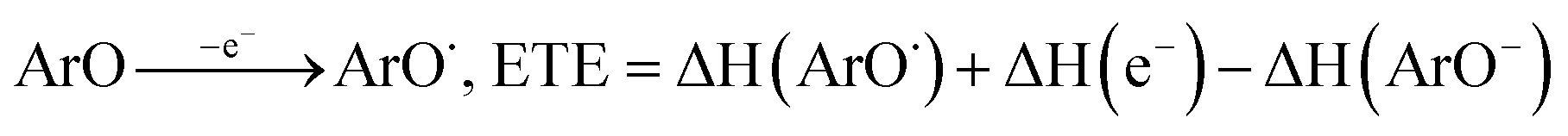
The published enthalpy values of 6.197 kJ mol−1 for H(H+) and 3.145 kJ mol−1 for H(e−)17 were used during the calculation.
3. Results & discussion
3.1 The pH responsive electrochemical behavior
The electron transfer involved in electrochemical oxidation of PL and DL were characterized by CV and DPV. As depicted in Fig. 2A and B, at the pH 2.0, there was a remarkable anodic peak related to the oxidation of PL in both cyclic and DP voltammograms. In contrast to PL, two sequential anodic signals of DL could be observed in the two voltammograms in acidic medium, suggesting causative moieties to get oxidized at potentials closed to each other (Fig. 3A and B). In addition, all of the anodic peaks from PL and DL were irreversible in cyclic voltammograms, indicating that their electrochemical oxidations are followed by chemical reactions.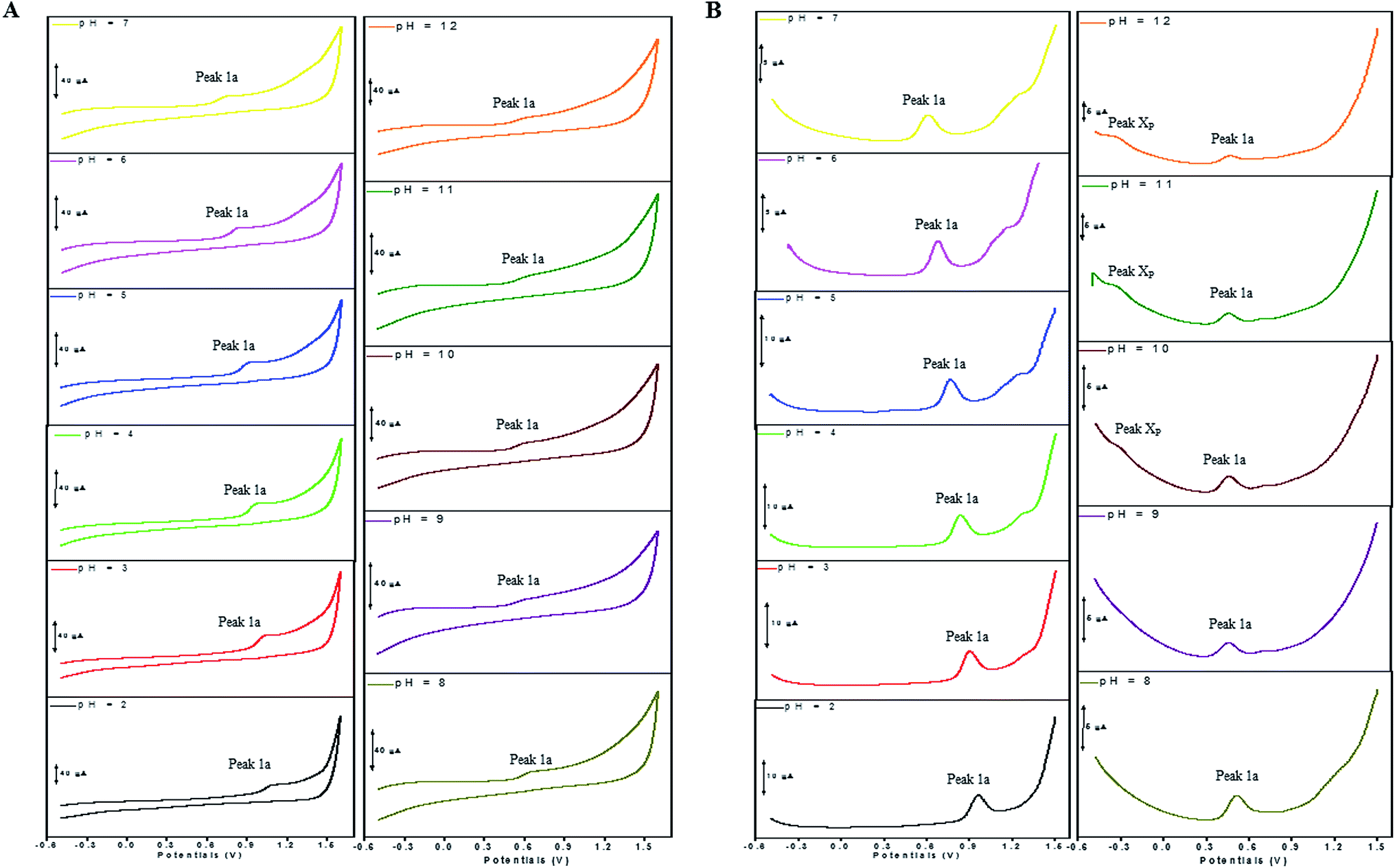 | ||
| Fig. 2 CVs (A) and DPVs (B) obtained for PL (100 μM) in media of various pH ranging from pH 2.0 to 12.0. The scan rate in CVs was 300 mV s−1. The running parameter in DPVs were amplitude, 0.05 V; pulse width, 0.06 s; step size, 5 mV; pulse period, 0.1 s; sample width, 0.02 s. | ||
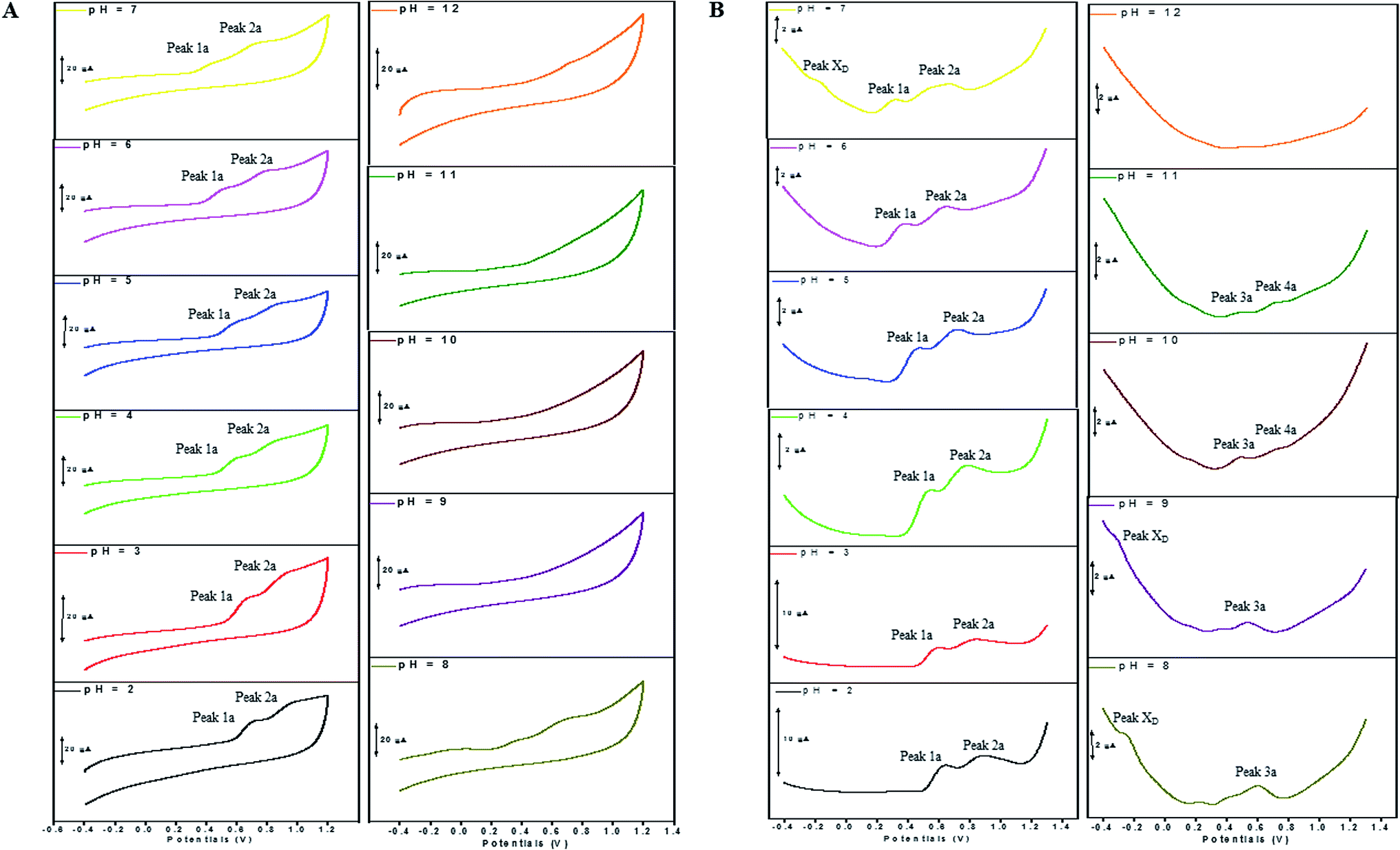 | ||
| Fig. 3 CVs (A) and DPVs (B) obtained for DL (100 μM) in media of various pH ranging from pH 2.0 to 12.0. The running parameters of CVs and DPVs were same with those in Fig. 2. | ||
The anodic peak potentials of PL and DL in cyclic and DP voltammograms shifted cathodically with the increasing pH of the media (Fig. 2 and 3), indicating facile electron abstraction in weak acidic or neutral condition. To quantitatively reflect the rules of pH responsive electrochemical behaviors, DPV was chosen to depict the variation of anodic peak potential in the whole pH interval since this technique has the ability of minimizing the charging current and extracting faradaic current which leads to improved sensitivity and greater accuracy.
The DP voltammograms of PL solution showed a linear region in the Ep vs. pH plot for Ep1a. The linear fitting for the range of 2.0 ≤ pH ≤ 9.0 was Ep (V) = 1.118 − 0.073 pH (Fig. 4A). The number of electrons (n) and protons (m) involved in PL oxidation can be obtained from half peak width (W1/2) of DPVs and slope of the plots of Ep as a function of pH using equations18
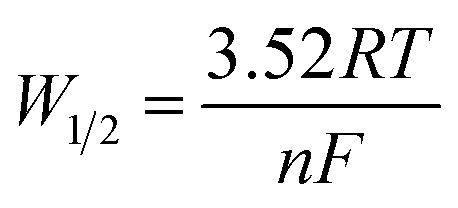 | (1) |
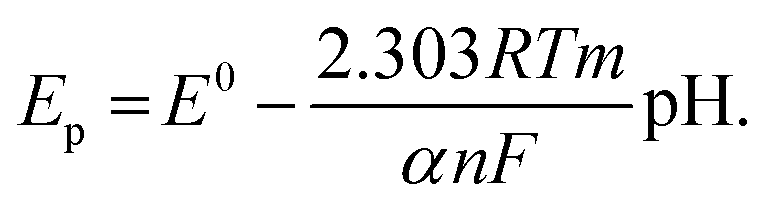 | (2) |
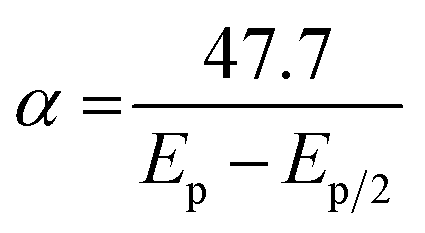 | (3) |
:n) involved in the oxidation process of PL was calculated as 0.9:1 ≈ 1:1. Hence, the oxidation of one molecule of PL occurred by the abstraction of 1H+ and 1e− under the conditions of pH ≤9.0.
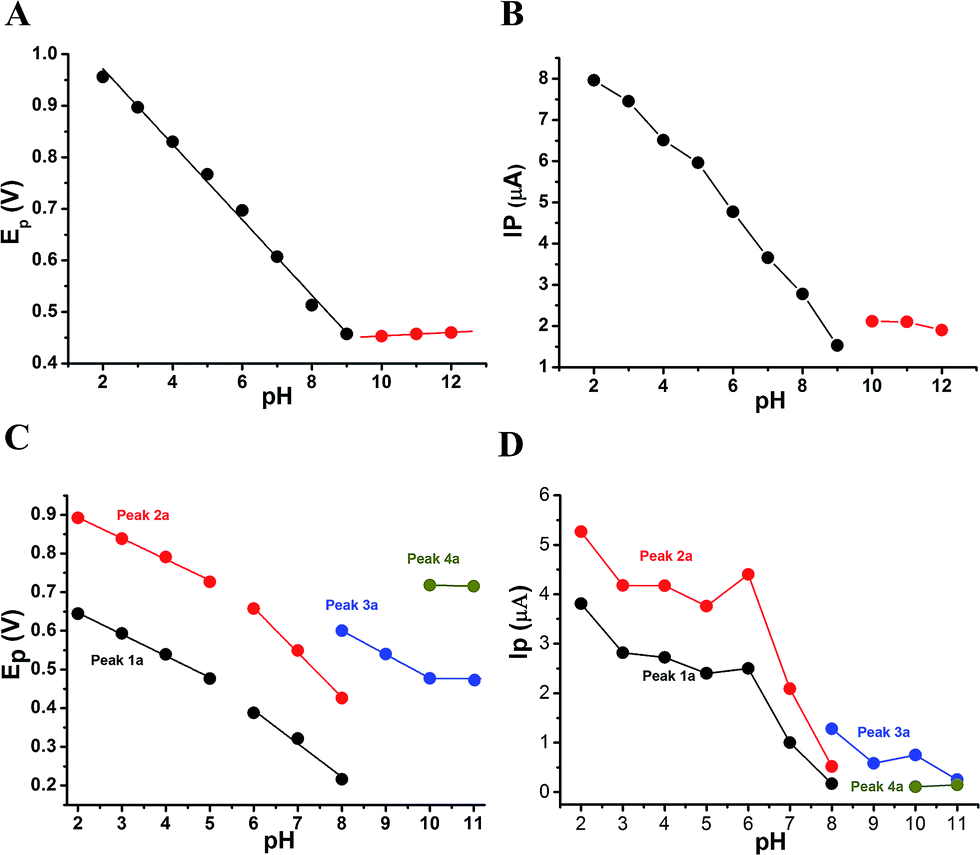 | ||
| Fig. 4 Influence of pH on the anodic peak potential (Ep) and intensity (Ip) of PL and DL. Plots of Ep vs. pH (A) and Ip vs. pH (B) were obtained for oxidation of 0.4 mM PL. Plots of Ep vs. pH (C) and Ip vs. pH (D) were derived from oxidation of 100 μM DL. | ||
In solutions above pH 9.0, the Epa versus pH plot of PL (Fig. 4A) demonstrated the switching of mechanism. The oxidation potential of anodic peak1a exhibited no influence with further increase in alkalinity of the medium thus, providing evidence of electron transfer without proton accompaniment in this condition. Therefore, chemical deprotonation of PL took place around pH 9.0 thus, the acid–base dissociation constant (pKa1) value of PL was obtained as 9.0. This is in good agreement with pKa value measured by previous report.21 Interestingly, a new anodic peak (Peak XP, Epa = −0.30 V) in DP voltammogram of PL was observed in the condition of pH ≥10.0 (Fig. 2B). This may be ascribed to oxidative response of hydroxylated PL, a very reactive product possibly generated from the auto-oxidation of ionized PL in aqueous solution.21,22
Further observation of Fig. 4B revealed that the current intensity of peak 1a was remarkably decreased within the increasing pH ranging from 2.0 to 9.0, yet it was not significantly changed in the range of 10.0 ≤ pH ≤ 12.0. The variation of anodic peak currents can be associated with the changes of the adsorption of PL onto GCE. In acid media the OH groups increase PL hydrophobicity and adsorption on the hydrophobic GCE surface. Under alkaline conditions, the OH groups were almost or fully deprotonated, increasing PL hydrophilicity and consequently decreasing PL adsorption on the GCE surface. At pH above 9.0, the protons were not involved in oxidation reaction of PL on GCE, therefore, the peak current intensity was not obviously changed.
The Epa–pH plots for anodic peaks 1a and 2a of DL were shown in Fig. 4C. The plots clearly consisted of two linear segments in the 2.0–8.0 pH interval, suggesting the existence of two successive overlapping equilibria. In addition, the intersection point of the two lineal segments is at pH 5.0. This determined that the chemical deprotonation possibly occurs at the pKa value of 5.0. In solutions of pH 2.0–5.0 (i.e. first lineal segment), the slopes of Epa–pH plot corresponding to peaks 1a and 2a were very close, 56 mV and 54 mV per pH unit, respectively (Table S1†). Considering the W1/2 and α values for peaks 1a and 2a as listed in Table S1,† showing the involvement of 0.5 protons and one electron transfer in both the two anodic peaks of DL. For second lineal segment of Epa versus pH plot, the slopes were calculated as 86 mV and 116 mV per pH unit for peaks 1a and 2a, respectively. The ratios of proton-transfer number to electron-transfer number were calculated as 0.8:1 and 0.9:1, respectively. These ratios were almost equal to unity, so that it offered the evidence of one electron transfer with one proton accompaniment in the two successive oxidation of DL at pH 6.0–8.0.
Furthermore, the anodic peaks 1a and 2a decreased intensely when the pH of electrolyte solution increased from 6.0 to 8.0, disappearing in pH >8.0 (Fig. 4D). In pH ≥8, new anodic peaks (peaks 3a and 4a) could be observed in DPV of DL. This may have been due to the varied orientation of the oxidizable moieties of DL in solutions of different pH. In pH <9, the most oxidizable hydroxyl groups are expected to have closer accessibility to the electrode surface while in pH ≥9 other oxidizable electrophore may penetrate deeper into the electrical double layer. In addition, the peak 3a shifted its position within pH ranging from 8.0 to 10.0 and deprotonation reaction possibly took place at pH 10.0 as evidenced by the intersection of two linear segments of Epa3–pH plot (Fig. 4C, Table S1†). However, peak 4a only appeared in basic conditions at pH ≥10.0 and its electron transfer occurred without accompaniment of proton.
In accordance with PL, the hydroxylation of DL also took place in aqueous solution, evidenced by the new defined peak (peak XD) appearing at −0.16 V, −0.25 V and −0.33 V, respectively in the range of 7.0 ≤ pH ≤ 9.0. It was demonstrated that the auto-oxidation of DL occurred at neutral or moderate alkaline medium, suggesting that it may exhibit as a mild pro-oxidant at physiological conditions (around pH 7.4). This property can be contributed to the activation of Nrf2–ARE signaling pathway in HepG2 cells23 since exogenous oxidants stimulation is an essential factor triggering induction of intracellular Nrf2-related antioxidant enzymes.
3.2 Kinetic process of electro-oxidation
The kinetic nature of redox process can be assessed from the value of heterogeneous electron transfer rate constant (k0) in aqueous solution. Firstly, the cyclic voltammograms of PL and DL at different scan rates from 20 to 400 mV s−1 (Fig. 5) were examined in pH 6.0. A linear relationship was observed between the anodic peak intensity ip (Ipa1) and the scan rates ν (Fig. 5A, inset), suggesting that the electrode process of PL is controlled by adsorption rather than diffusion. Similarly, within the variable scan rates, the currents of anodic peaks 1a and 2a in DL were linearly depended on the scan rate (ν) (Fig. 5B inset). This indicates that the electrode process of DL is also adsorption-controlled.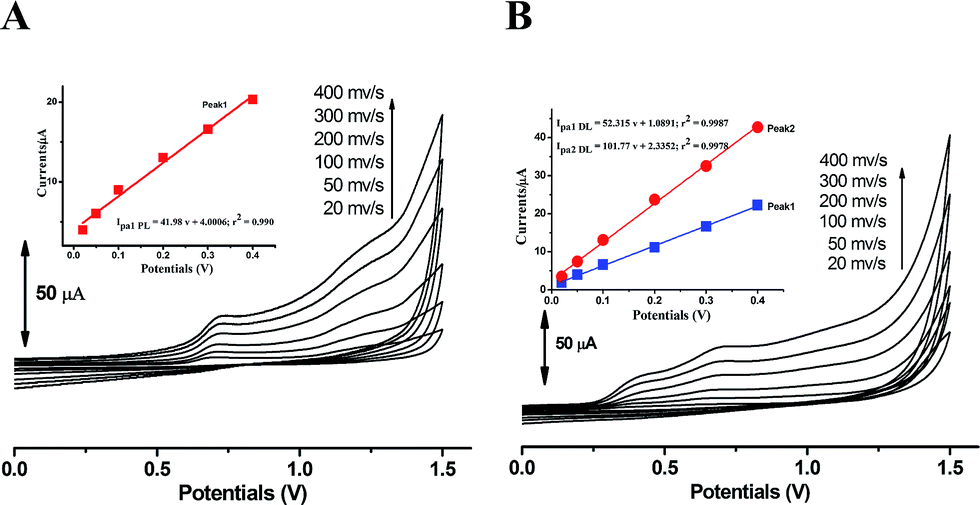 | ||
| Fig. 5 Overlapped cyclic voltammograms of PL and DL obtained at scan rates of 20, 50, 100, 200, 300, and 400 mV s−1. All lines are the representative of three independent experiments. Inset shows the plot of anodic peak current intensity versus scan rate. | ||
In addition, the peak potentials of PL and DL drifted towards more positive values with the rise in scan rates, which points to a degree of irreversibility in their electrochemical processes. As for an adsorption-controlled irreversible electrode process, k0 can be calculated with the help of the following equation:24
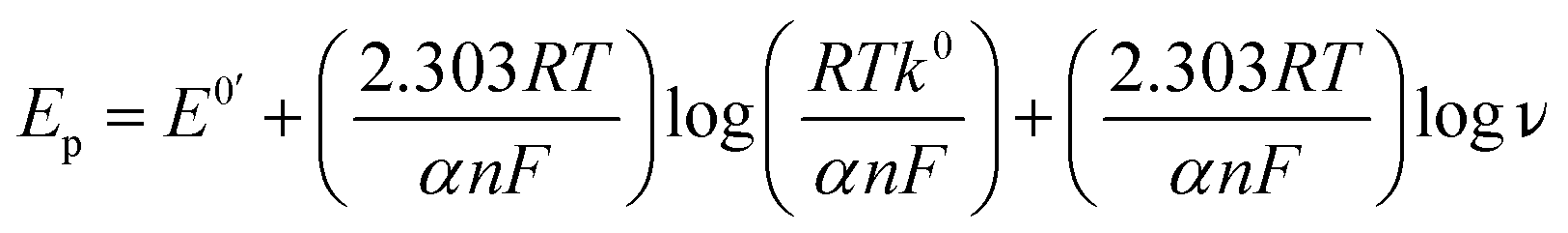 | (4) |
480. According to eqn (4), an intercept from which k0 can be calculated if the value of E0′ was derived. The value of E0′ in eqn (4) can be obtained from the intercept of the Ep vs. ν curve by extrapolation to the vertical axis at ν = 0.25
The linear relationships between peak potential and logarithm of scan rate for PL and DL can be expressed as Epa1 PL (V) = 0.741 + 0.0215 logν (r2 = 0.9961), Epa1 DL (V) = 0.419 + 0.024 logν (r2 = 0.9926), and Epa2 DL (V) = 0.715 + 0.049 logν (r2 = 0.9913). The E0′ values for peak 1a of PL and peaks 1a and 2a of DL were obtained as 0.695 V, 0.340 V, and 0.622 V, respectively. Therefore, the k0 value was calculated to be 0.91 × 103 s−1 for PL. The k0 value for peak 1a of DL was obtained to be 2.58 × 103 s−1. Similarly for peak 2a, the k0 value was calculated to be 0.32 × 103 s−1. Hence, the comparatively higher k0 value for peak 1a of DL represents the faster kinetic process in electron donation from DL, which may be meaningful for the competitive protection for biological macromolecules against free radicals.
3.3 Electronic absorption spectroscopic analysis
By correlating the absorption spectra recorded in solutions of various pH, the UV-vis spectroscopic measurement was conducted for reflecting electron donating properties of moieties and exploring the structural differences between DL and PL.The spectra of PL were displayed in Fig. 6A for the pH ranging from 2.0 to 12.0. In acidic and neutral conditions, the absorption peak at 268 nm (band I), which is attributed to the π–π* transitions of the phenolic ring, did not suffer obvious shifts. By increasing the pH, evident changes were observed in the spectra of PL at pH 9.0, concurring with the pKa value obtained by DPV experiments. The intense absorption bands (II–V) emerging around 224, 252, 276 and 348 nm in pH 9.0–12.0 can be assigned to deprotonation of hydroxyl group in PL as well as its corresponding auto-oxidation products.26 In addition, the spectrum of PL obtained at pH 11.0 was practically identical to that of pH 12.
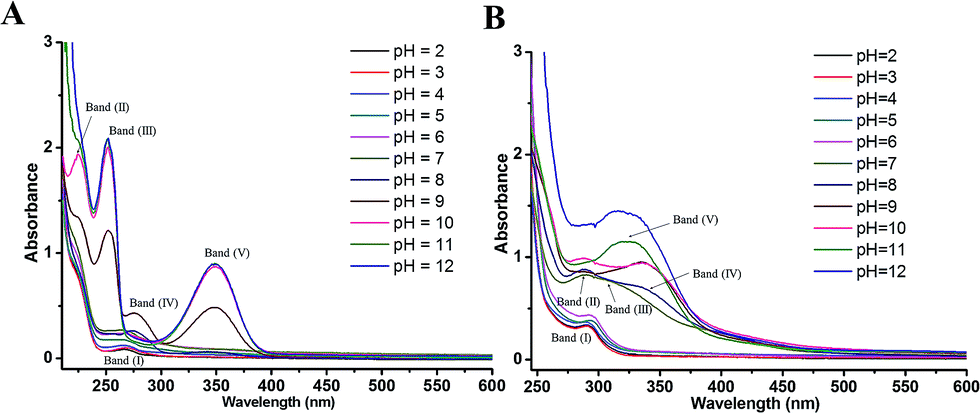 | ||
| Fig. 6 UV-visible spectra of 50 μM PL (A) and 80 μM DL (B) in PB solutions with different pH ranging from pH 2.0 to 12.0. | ||
Comparing with PL, the changes in electronic spectra of DL were more complicated in different pH conditions (Fig. 6B). In range 2.0 ≤ pH ≤ 4.0, there was a relatively higher absorption peak at 290 nm (band I) as compared to the absorption signal of PL at 268 nm. This can be attributed to higher extent of conjugation moieties in DL molecule. However, no obvious shifts for band (I) were observed in this pH range. By the pH increasing and inducing the deprotonation of OH groups, absorption band (I) became a slight red shift toward 295 nm at pH 5.0 and 6.0. The deprotonation at the pH 5.0 was also evidenced from color change of dieckol (Fig. S1†). Following this, a much broader absorption band (II) at 290 nm and its shoulder band (III) at 310 nm were emerged at pH 7.0, suggesting the coexisting mixture of ionic and auto-oxidation products of DL in the solution. This is in good agreement with the observation of new anodic peak (peak XD) in DP voltammograms under neutral condition.
As the pH of solution was adjusted to 8.0, a new adsorption band (IV) at 336 nm coupled with the band (II) at 290 nm can be observed. Also, the intensity of adsorption band (IV) was further highly increased at pH 9.0 and 10.0. With the pH of solution was over 10.0, band (II) and (IV) seemed to be merged into a new adsorption band (V) appearing at 324 nm. Overall, the UV-vis spectra of DL was governed by four equilibria within the tested pH interval. This is coordinated with DL's electrochemical oxidation process where anodic peak signals will be changed every time a new production of deprotonated DL becomes dominant in the system (vide supra). Therefore, the variable pH of media solution, triggering the successive deprotonation of DL, had caused the changes of conjugated system as well as the electron-donating process.
3.4 Computational studies
Computational studies were conducted to further investigate the redox-related electronic descriptors in PL and DL molecules. Considering the three hydroxyl groups in PL molecule are structurally equivalent thus, C1–OH of PL was chosen as the representative of these hydroxyls to be investigated. In addition, as a hexamer of PL, there are four independent planar structures (i.e. rings A, B, C, and D) in DL molecule. Rings A and C are composed of dimeric PL moiety while rings B and D are formed only by one PL ring. Since there are eleven hydroxyls distributed in the four structural units of DL, it is significant to compare the electron transfer activities of these hydroxyls.Based on the theory by Litwinienko and Ingold,27 the electron transfer process of phenols in ionizing-supporting solutions was dominated by two pathways: (i) the sequential proton-loss electron transfer (SPLET) where deprotonation of a phenol in solvent is followed by electron transfer from its phenoxyl anion to free radical. (ii) The proton-coupled electron transfer (PCET) where phenol transfers an electron followed by proton abstraction. Therefore, electronic parameters based on PCET and SPLET mechanisms were discussed as follows.
When the pH value of solution was lower than pKa1, the phlorotannin compounds should be in the presence of its protonated or neutral form in aqueous solution, evidenced by corresponding UV-vis absorption spectra. In this condition, the oxidation of the two phlorotannin species predominately followed PCET pathway in which they provides for electrons to be donated, coupled with protons abstraction from cation radical. Thus, the IP and PDE values of PL and DL were calculated to reflect the ease of electron and proton transfer in PCET pathway. It was first noted that the IP value of DL was obviously lower than that of PL, representing that the direct electron donating by DL is much easier than that by PL. In favor to this also goes more negative Epa1 value of DL in comparison to the value for PL in DPV.
The PDE values of the two phlorotannins were listed in Table 1. In case of DL molecule, 2-OH group in B unit exhibited the lowest PDE value, indicating this moiety in ring B is the most probable site of deprotonation. However, in comparison with PL, the higher PDE value of DL demonstrated its harder proton donating thus, supporting the less proton (0.5 protons transfer per one electron transfer) involved in electrochemical oxidation of DL in acidic condition.
| Compounds | PCET | SPLET | ||
|---|---|---|---|---|
| IP (kcal mol−1) | PDE (kcal mol−1) | PA1 (kcal mol−1) | ETE1 (kcal mol−1) | |
| Dieckol | 150.58 | |||
| Unit A | ||||
| 3-OH | 241.97 | 326.71 | 65.84 | |
| 5-OH | 242.91 | 327.78 | 65.71 | |
| Unit B | ||||
| 2-OH | 236.68 | 326.36 | 60.91 | |
| 4-OH | 237.41 | 325.60 | 62.39 | |
| 8-OH | 241.34 | 322.68 | 69.25 | |
| Unit C | ||||
| 3-OH | 244.08 | 325.48 | 69.18 | |
| 5-OH | 243.49 | 325.56 | 68.51 | |
| Unit D | ||||
| 2-OH | 240.82 | 330.60 | 60.80 | |
| 4-OH | 238.73 | 329.80 | 59.51 | |
| 6-OH | 240.21 | 337.15 | 53.64 | |
| 8-OH | 243.02 | 335.57 | 58.02 | |
|
||||
| Phloroglucinol | ||||
| 1-OH | 185.79 | 209.40 | 340.77 | 54.42 |
On the other hand, the chemical deprotonation of the most acidic hydroxyl group in phlorotannins occurred favorably when the pH was greater than pKa1. Therefore, the oxidation pathway of PL and DL should be shifted from PCET to SPLET. In SPLET pathway, the PA value reflects the acidity of hydroxyl groups and determines the kinetic aspect of SPLET. As listed in Table 1, the C8–OH in unit B of DL presented the lowest PA value, suggesting that it is the most acidic site in DL molecule. Hence, in comparison to PL, the less PA1 value which points its easier proton donating is due to better charge distribution on molecular structures of DL (mulliken charge in Fig. 7). This is in line with its lower pKa1 and higher k0 values presented in electrochemical assays. Moreover, the ETE value from SPLET can represent the extent of electron donating from two deprotonated phlorotannins namely, C1–O− anion of PL and C8–O− anion of DL. A significant difference in ETE values of the tested two monoanionic phlorotannins was presented in Table 1, explaining the greater magnitude of oxidation peak current of PL than that of DL.
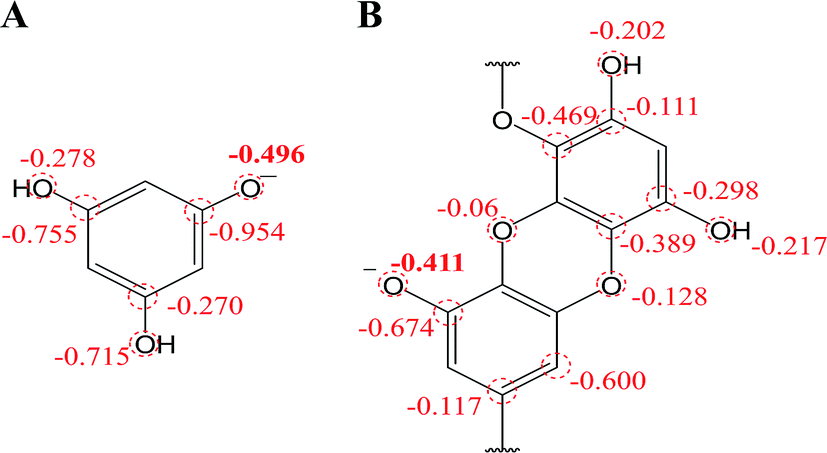 | ||
| Fig. 7 Mulliken charge distribution on monoanionic form of (A) phloroglucinol and (B) dieckol (B unit in its molecule). | ||
Besides, in previous electrochemical assay it has been evidenced that protons are not involved in electro-oxidation process of PL in the solutions of pH > 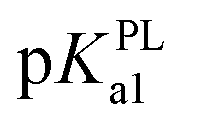 (i.e. pH ≥ 9). In contrast with PL, it is interesting that protons are involved in the electron transfer process of DL in the pH over its pKa1 value (i.e. pH > 5). To explain the above phenomena, the PA2 values from the deprotonation of different hydroxyls in C8–O radical of DL were calculated as well (Table S2†). Specifically, the acidity of all hydroxyl groups in C8–O radical of DL is dramatically enhanced thereby suggesting the occurrence of deprotonation after the first SPLET process. Furthermore, C2–OH in B unit exhibited the lowest PA value among these hydroxyls, implying that it is the secondarily possible site for proton abstraction. In previous experimental and theoretical studies, this property has also been presented in flavonoid molecules due to the strongly electron-withdrawing ability of the O atom with unpaired electron. Therefore, DL possibly undergoes the combined SPLET/PCET pathway in pH range of 6.0–8.0. This means the oxidation of DL in these pH conditions can be described as sequential proton-loss and proton-coupled electron transfer (SPL-PCET). Nevertheless, it may be very difficult for PL to make the secondary proton transfer since its PA2 value is much higher than those derived from most hydroxyls in DL (Table S2†). This is the reason why there is no proton involvement in its electron-oxidation when the pH ranges from 10.0 to 12.0. The comparison of proposal oxidation pathways of the two phlorotannins will be discussed in the following section.
(i.e. pH ≥ 9). In contrast with PL, it is interesting that protons are involved in the electron transfer process of DL in the pH over its pKa1 value (i.e. pH > 5). To explain the above phenomena, the PA2 values from the deprotonation of different hydroxyls in C8–O radical of DL were calculated as well (Table S2†). Specifically, the acidity of all hydroxyl groups in C8–O radical of DL is dramatically enhanced thereby suggesting the occurrence of deprotonation after the first SPLET process. Furthermore, C2–OH in B unit exhibited the lowest PA value among these hydroxyls, implying that it is the secondarily possible site for proton abstraction. In previous experimental and theoretical studies, this property has also been presented in flavonoid molecules due to the strongly electron-withdrawing ability of the O atom with unpaired electron. Therefore, DL possibly undergoes the combined SPLET/PCET pathway in pH range of 6.0–8.0. This means the oxidation of DL in these pH conditions can be described as sequential proton-loss and proton-coupled electron transfer (SPL-PCET). Nevertheless, it may be very difficult for PL to make the secondary proton transfer since its PA2 value is much higher than those derived from most hydroxyls in DL (Table S2†). This is the reason why there is no proton involvement in its electron-oxidation when the pH ranges from 10.0 to 12.0. The comparison of proposal oxidation pathways of the two phlorotannins will be discussed in the following section.
3.5 Redox mechanisms and SAR analysis
In the light of computational findings and experimental results obtained using electrochemical techniques, redox pathways of the tested phlorotannins were suggested. The oxidation pathway for PL and DL can be divided into two and four segments, respectively. It is proposed that both two phlorotannin compounds proceeded PCET pathway when the pH was lower than each pKa1 values. However, the significant variation in the ratio of proton-transfer number to electron transfer number was observed as detailed in Fig. 8.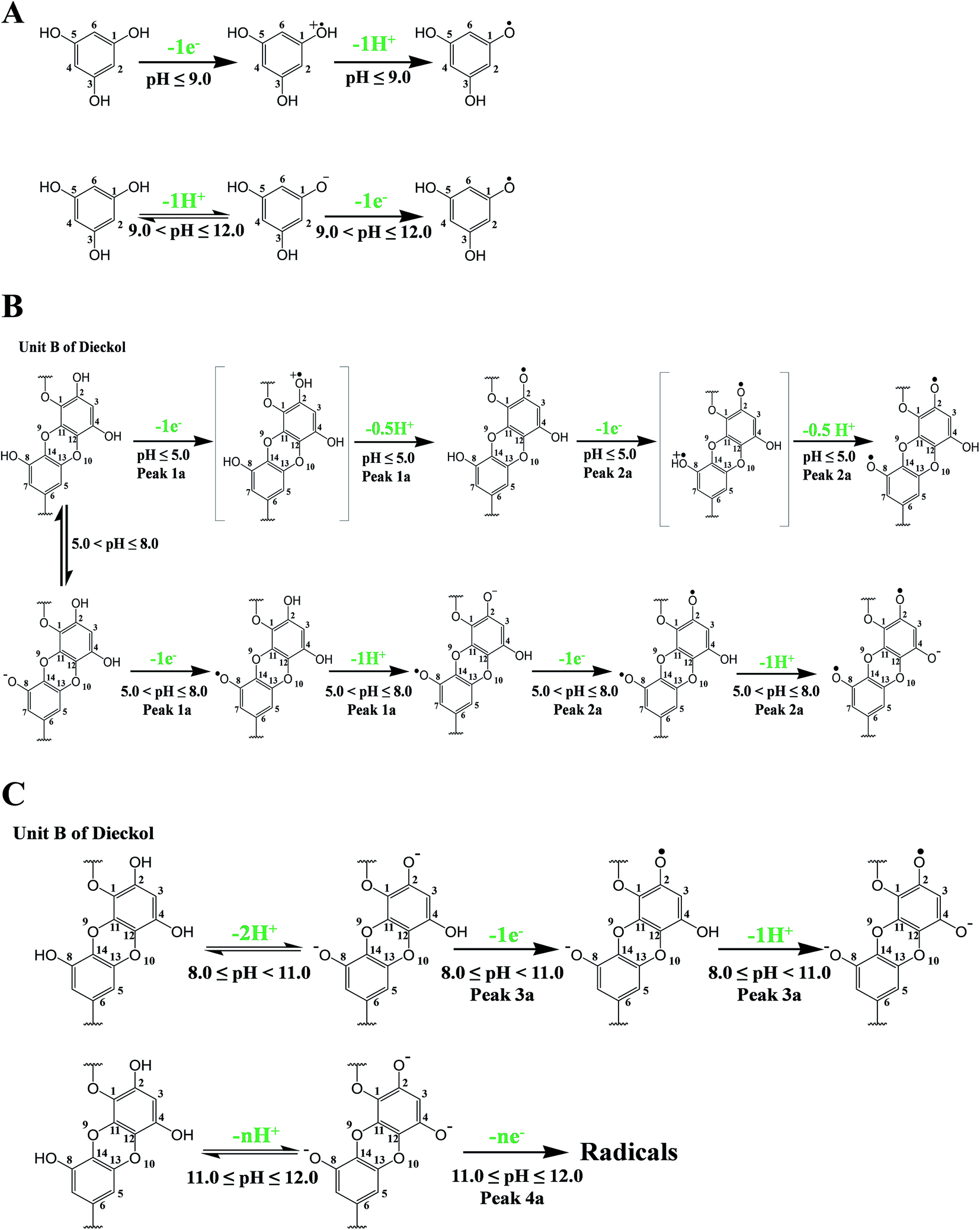 | ||
| Fig. 8 Proposed oxidation mechanism of PL (A) and DL corresponding to peaks 1a and 2a (B) and to peaks 3a and 4a (C). | ||
As depicted in Fig. 8A and B, the oxidation of PL and DL are dominated by SPLET and SPL-PCET pathway if the pH broke through pKa1 points. The difference between two pathways is dependent on whether proton transfer occurred in phlorotannin-derived radical. Finally, the third and fourth oxidation segments of DL are much more complicated than previous two segments. In pH >8, the predominant species in bulk solution may be di- or trianionic forms of DL, which was evidenced by UV-vis spectra of DL in corresponding conditions. These ionic molecules are very unstable and will severely contaminate the oxidation peaks in DPV due to their oxidation products. Consequently, it is an arduous task to show a complete pathway regarding the third and fourth oxidation segments of DL (i.e. peak 3a and peak 4a). However, a hypothetically ‘pure’ pathway presented by us has been shown in Fig. 8C. The dianionic DL undergoes electron transfer followed by a further proton abstraction, while the oxidation of trianionic DL only exhibits electron transfer (ET).
As compared with PL molecule, the conjugation systems in DL molecule have been greatly enhanced since monomeric PL unit are linked by both ether bonds and dibenzodioxin moieties. These structural characteristics of DL leads to the following redox activities: (i) much lower IP value of DL was observed in theoretical calculation, which is coordinated with its easier electron donating activity of neutral form. (ii) The hydroxyls in DL presented more acidic (lower pKa1) due to better deformation of the electron cloud in anionic molecule with the help of Cπ–O–Cπ moiety in ortho-position to deprotonated hydroxyl group. Hence, the aforementioned structural properties make DL much higher electron transfer ability. However, not all the natural polymerized products from phenols could cause the enhancement in redox activity in comparison to parent molecules. For example, computerized IP and PA values from procyanidins (PCs) were similar with those from subunit molecules namely, (−)-epicatechin and (+)-catechin.28 It should stress that the catechin subunits in PCs are linked by Cπ–Cσ bond with the intermolecular hyperconjugative interactions whereas the PL rings are linked by Cπ–O–Cπ moiety with p–π conjugation. Thus, the electronic interaction between the different subunit rings is limited, which is in contrast to the conjugation of interphloroglucinol in DL molecule. Therefore, the unique interphloroglucinol linkage in DL is significant for redox activity improvement in comparison with PL molecule.
3.6 Oxidants scavenging activity in ex vivo models
The comparative analysis in redox activity of two phlorotannins was further clarified in the following three oxidants scavenging activity assays. The scavenging capacity of DL and PL toward two physiological oxidants (i.e. hypochlorite and superoxide anion) were measured by electrochemical system.In the superoxide anion scavenging assay, DL behaved as an excellent superoxide anion scavengers (Fig. 9). In the anodic potential range, the peak current of superoxide anion (Ip) was decreased linearly with the increase of DL concentrations (100 μM to 500 μM). On the basis of standard calibration curve of PL (ΔIp (μA) = 0.0007XPL conc. (μM) + 3.1108) thus, the PL equivalent (PE) value of DL was calculated to be 1421 ± 63 μmol PE/100 μmol DL, suggesting that the superoxide anion scavenging activity of DL was around thirteen times higher than that of PL. Similar ordering was also exhibited in the hypochlorite assay (Fig. 10) where the DL presented higher scavenging capacity toward hypochlorite than PL did (PE value was calculated to be 233 ± 61 μmol PE/100 μmol DL). Hence, the reducing capacity of DL against the physiological oxidants was much stronger than that of PL molecule.
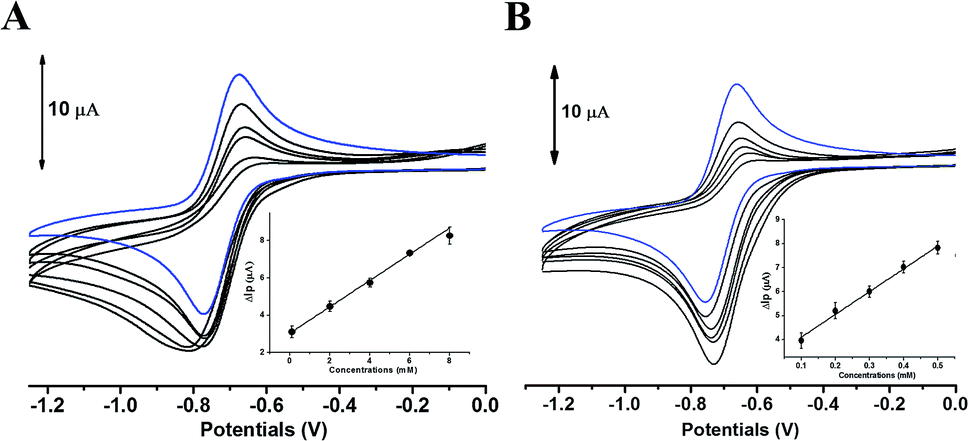 | ||
| Fig. 9 Cyclic voltammograms obtained without (blue line) and further addition (black line) of PL (A) or DL (B) in DMSO containing 0.1 mM Bu4NPF6. All lines are the representative of three independent experiments. Inset: variation of ΔIp versus phlorotannin sample concentration. | ||
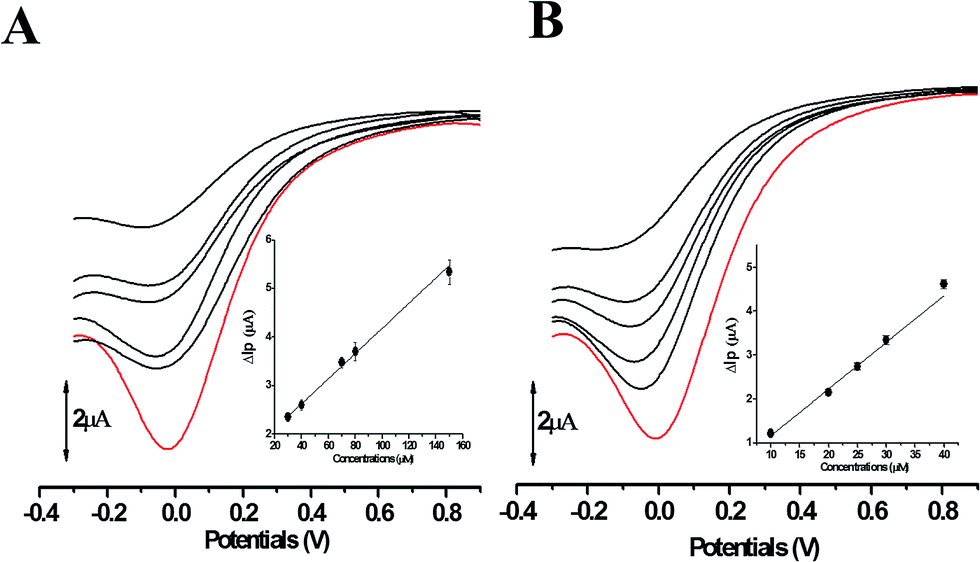 | ||
| Fig. 10 The differential pulse voltammograms of hypochlorite in the presence of PL (A) and DL (B). Red line represents the differential pulse voltammograms of hypochlorite obtained without addition of phlorotannins. Black line is the cathodic wave of hypochlorite after reduction by phlorotannins. All lines are the representative of three independent experiments. Inset: variation of ΔIp versus phlorotannin sample concentration. | ||
Furthermore, in the CAA assay (Fig. 11), the inhibitory effects of PL and DL against AAPH-induced oxidants were measured in intracellular circumstance. It was clearly observed that the AUCDL value (2.44 × 103 FU min) was significantly lower than AUCPL (3.62 × 103 FU min), evidencing that the scavenging of intracellular oxidants by DL was more favorable in competitive scavenging reaction toward oxidants. This is also in good agreement with the observation in the heterogeneous electron transfer reactions of DL and PL on the surface of GCE.
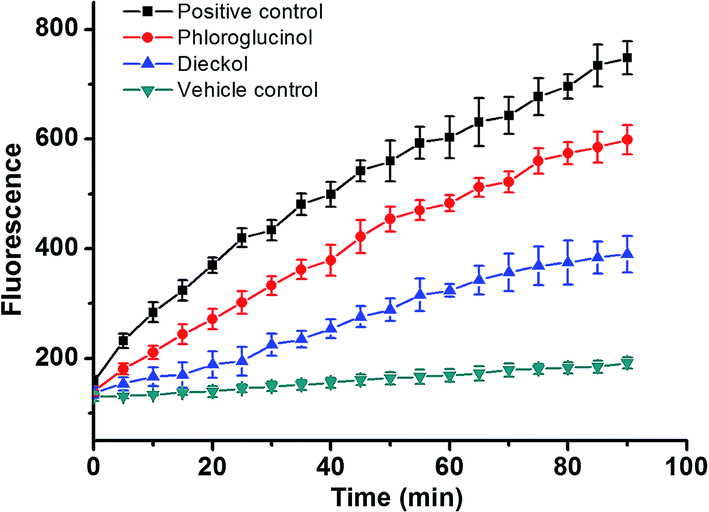 | ||
| Fig. 11 AAPH-induced oxidation of DCFH in HepG2 cells and the inhibition of oxidation by 50 μM PL and 50 μM DL compounds over time. The positive control group: the cells were treated by 1.0 mM AAPH in the absence of phlorotannin compounds. | ||
4. Conclusions
It is the first report on the oxidative mechanisms of phlorotannin compounds. Both the phloroglucinol (PL) and dieckol (DL) presented pH-dependent and adsorption-controlled irreversible mode of oxidation at GCE in aqueous solution. However, the unique interphloroglucinol linkage in DL molecular structure is significant for its specific oxidation pathway and redox activity improvement in comparison with PL molecule. Based on the findings from UV-vis spectrum, electrochemical analysis, and computational calculation, it can be proposed that the multiple steps occurred in DL oxidation are relied on PCET, mixed SPLET/PCET, and ET mechanisms, respectively, which is more complicated than PL oxidation pathway. These findings may give useful insights about oxidative conversion of phlorotannins in biochemical processes and their structure–redox activity relationship. However, beside PL and DL, more detailed information on the oxidation mechanisms of other phlorotannin compounds should be further investigated.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for Profs. Zhixiang Wang and Suci Meng's assistances in theoretical calculation. Authors acknowledge a kind donation of dieckol from BotaMedi, Inc. (Jeju, Korea). This work was supported by National Natural Science Foundation of China (21506082) and Natural Science Foundation of Jiangsu Province (BK20150494); DZ and LS are grateful for China Postdoctoral Science Foundation (2016M591786; 2015M571699), Jiangsu Planned Projects for Postdoctoral Research Funds (1601084B), and Research Foundation of Jiangsu University (15JDG026; 15JDG058). The fund of the Beijing Engineering and Technology Research Center of Food Additives, Beijing Technology & Business University (BTBU) is also acknowledged.Bibliographic references & notes
- A. Argyropoulou, N. Aligiannis, I. P. Trougakos and A. L. Skaltsounis, Nat. Prod. Rep., 2013, 30, 1412–1437 RSC
.
- D. Pádua, E. Rocha, D. Gargiulo and A. A. Ramos, Phytochem. Lett., 2015, 14, 91 CrossRef
- L. Montero, A. D. P. Sánchez-Camargo, E. Ibáñez and B. Gilbert-López, Curr. Med. Chem., 2017, 24 DOI:10.2174/0929867324666170523120101
- D. A. Kirke, T. J. Smyth, D. K. Rai, O. Kenny and D. B. Stengel, Food Chem., 2017, 221, 1104–1112 CrossRef CAS PubMed
- J. H. Kwak, Z. Yang, B. Yoon, Y. He, S. Uhm, H. C. Shin, B. H. Lee, Y. C. Yoo, K. B. Lee, S. Y. Han and J. S. Kim, Blood-brain barrier-permeable fluorone-labeled dieckols acting as neuronal ER stress signaling inhibitors, Biomaterials, 2015, 61, 52–60 CrossRef CAS PubMed
- J. Ryu, R. Zhang, B. H. Hong, E. J. Yang, K. A. Kang, M. Choi, K. C. Kim, S. J. Noh, H. S. Kim, N. H. Lee, J. W. Hyun and H. S. Kim, PLoS One, 2013, 8, e71178 CAS
- H. J. Choi, J. H. Park, B. H. Lee, H. Y. Chee, K. B. Lee and S. M. Oh, Suppression of NF-κB by dieckol Extracted from Ecklonia cava Negatively Regulates LPS Induction of Inducible Nitric Oxide Synthase Gene, Appl. Biochem. Biotechnol., 2014, 173, 957–967 CrossRef CAS PubMed
- H. S. Choi, H. J. Jeon, O. H. Lee and B. Y. Lee, Mol. Nutr. Food Res., 2015, 59, 1458–1471 CAS
- M. J. Piao, M. J. Ahn, K. A. Kang, K. C. Kim, J. W. Cha, N. H. Lee and J. W. Hyun, DNA Repair, 2015, 28, 131–138 CrossRef CAS PubMed
- R. Stocker, Arch. Biochem. Biophys., 2016, 595, 136–139 CrossRef CAS PubMed
- D. Zhang, L. Chu, Y. Liu, A. Wang, B. Ji, W. Wu, F. Zhou, Y. Wei, Q. Cheng, S. Cai, L. Xie and G. Jia, J. Agric. Food Chem., 2011, 59, 10277–10285 CrossRef CAS PubMed
- A. Masek, M. Zaborski and E. Chrzescijanska, Food Chem., 2011, 127, 699–704 CrossRef CAS PubMed
- D. Zhang, R. Y. Yin, Y. X. Liu, X. Li, F. Zhou and B. P. Ji, Food Analytical Methods, 2015, 8, 2218–2227 CrossRef
- N. Blanc, D. Hauchard, L. Audibert and E. A. Gall, Talanta, 2011, 84, 513–518 CrossRef CAS PubMed
- K. L. Wolfe and R. H. Liu, J. Agric. Food Chem., 2007, 55, 8896–8907 CrossRef CAS PubMed
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez, and J. A. Pople, Gaussian 03, Revision D.02, Gaussian, Inc., Wallingford CT, 2004 Search PubMed
- E. Klein and V. Lukes, J. Phys. Chem. A, 2006, 110, 12312–12320 CrossRef CAS PubMed
- C. M. A. Brett and A. M. Oliveira Brett, Electrochemistry: Principles, Methods and Aplications, Oxford Science University Publications, Oxford, 1993 Search PubMed
- R. Nimal, S. Aftab, U. A. Rana, A. Lashin, S. U. D. Khan, S. Ali, H. B. Kraatz and A. Shah, J. Electrochem. Soc., 2016, H871–H880 CrossRef CAS
- A. J. Bard, and L. R. Faulkner, Electrochemical Methods, Fundamentals and Applications, John Wiley & Sons, New York, 2nd edn, 2001 Search PubMed
- D. Wang, K. Hildenbrand, J. Leitich, H. P. Schuchmann and C. V. Sonntag, Z. Naturforsch., B: J. Chem. Sci., 1993, 48, 478–482 CAS
- H. Hotta, H. Sakamoto, S. Nagano, T. Osakai and Y. Tsujino, Biochim. Biophys. Acta, 2001, 1526, 159–167 CrossRef CAS
- M. S. Lee, B. Lee, K. E. Park, T. Utsuki, T. Shin, C. W. Oh and H. R. Kim, Food Chem., 2015, 174, 538–546 CrossRef CAS PubMed
- E. Laviron, J. Electroanal. Chem., 1979, 101, 19–28 CrossRef CAS
- Y. Wu, X. Ji and S. Hu, Bioelectrochemistry, 2004, 64, 91–97 CrossRef CAS PubMed
- L. Fang, L. Ji and Q. Chen, Chin. J. Biological. Pharms., 1994, 15, 12–15 Search PubMed
- G. Litwinienko and K. U. Ingold, Accounts Chem. Res., 2007, 40, 222–230 CrossRef CAS PubMed
- A. M. Mendoza-Wilson, S. I. Castro-Arredondo and R. R. Balandrán-Quintana, Food Chem., 2014, 161, 155–161 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra10875c |
| This journal is © The Royal Society of Chemistry 2018 |