
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSilylated oligomeric poly(ether-azomethine)s from monomers containing biphenyl moieties: synthesis and characterization
Alain Tundidor-Cambaae,
Carmen M. González-Henríquezb,
Mauricio A. Sarabia-Vallejosc,
Luis H. Taglea,
René A. Hauyóna,
Patricio A. Sobarzoa,
Alexis Gonzáleza,
Pablo A. Ortiza,
Eva M. Maya d and
Claudio A. Terraza*ae
d and
Claudio A. Terraza*ae
aResearch Laboratory for Organic Polymers (RLOP), Department of Organic Chemistry, Pontificia Universidad Católica de Chile, P.O. Box. 306, Post 22, Santiago, Chile. E-mail: cterraza@uc.cl
bLaboratory of Nanotechnology and Advanced Materials (LNnMA), Chemistry Department, Universidad Tecnológica Metropolitana, P.O. Box 9845, Post 21, Santiago, Chile
cDepartamento de Ingeniería Estructural y Geotecnia, Escuela de Ingeniería, Pontificia Universidad Católica de Chile, Santiago, Chile
dInstituto de Ciencia de Materiales de Madrid, Consejo Superior de Investigaciones Científicas, CSIC, Spain
eUC Energy Research Center, Pontifica Universidad Católica de Chile, Chile
First published on 2nd January 2018
Abstract
In this study, four new silicon-containing poly(ether-azomethine)s with linear structures were prepared using original silicon and biphenyl moiety-containing monomers: two diamines and two dialdehydes. The oligomeric natures of the samples were established by GPC analysis, which showed chains containing 3 to 5 repetitive units. The monomers and the oligomeric samples were structurally characterized by NMR and FT-IR spectroscopy. The solubilities of the samples in common organic solvents and their thermal behavior enable improvement of their industrial and technological processability. The optical band gaps of the oligomeric samples were estimated from optical measurements (UV-vis), and their electrical behavior in films was determined using the four-point method. The surface arrangements and morphological characteristics of the films were determined via atomic force microscopy measurements. The roughness, area increase percentage and layer stiffness of the films were also measured using this technique.
Introduction
Conjugated polymers have recently been used as photovoltaic compounds in organic bulk heterojunctions due to their intrinsic properties, such as their small bandgaps and good hole transportation. Their ease of processing and low cost in comparison to inorganic semiconductors, namely silicon, make them excellent alternatives for this purpose.1 On this basis, the synthesis of organometallic polymers with silicon atoms in their structures may unify the features of a suitable band gap with electron delocalization, high voltage, and low sub-bandgap absorption, among other properties.Conjugated poly(azomethine)s (PAMs) are a family of polymers which possess an imine repeating unit, R–HC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–R′, where R and R′ are moieties that allow conjugation (e.g. aromatic moieties).2 These materials have been the subject of extensive investigation, especially in the last three decades; due to their optoelectronic, non-linear optic and electroluminescent behaviors, they are excellent candidates as materials to be used in the fabrication of organic light-emitting diodes (OLEDs), photovoltaic cells, and pH sensors, among other applications.3
N–R′, where R and R′ are moieties that allow conjugation (e.g. aromatic moieties).2 These materials have been the subject of extensive investigation, especially in the last three decades; due to their optoelectronic, non-linear optic and electroluminescent behaviors, they are excellent candidates as materials to be used in the fabrication of organic light-emitting diodes (OLEDs), photovoltaic cells, and pH sensors, among other applications.3
Early studies on the synthesis of PAMs led to low molecular weight polymers and intractable materials (both infusible and insoluble); these characteristics hampered their potential applications due to their low processability.4 This, in turn, excluded the possibility of fabricating films, thus limiting the characterization of these materials apart from high-temperature processability leading to the fabrication of fibers.5 The ability to form films is desirable today for the fabrication of devices such as OLEDs.6
In recent years, many approaches have been used to solve this problem: namely, the introduction of side groups in the polymer chain7 and their complexation with Brønsted acids such as diphenyl-phosphate and di-m-cresyl phosphate and Lewis acids such as gallium chloride to interact with the nitrogen atom of the imine functional group.5 These strategies enhanced the solubility of PAMs in common organic solvents and affected the thermal behavior of the polymers, achieving meltability without compromising the high thermal stability that characterizes this family of materials.
Another widely used strategy to achieve better processability of polymers is the introduction of flexible –Ar–Si(R1R2)–Ar– units, in which the R1 and R2 groups are alkyl or phenyl units. In this sense, this strategy has already proved useful in the preparation of highly thermally stable poly(ester)s and poly(amide)s with glass transition temperatures (Tg) below their thermal decomposition temperatures (TDT) and with high solubility in common organic solvents.8
The –Ar–Si(R1R2)–Ar– moiety has not only been investigated in the improvement of the processability of polymers. More recently, this unit has been used in the creation of materials for phosphorescent organic light-emitting diodes (PHOLEDs),9–13 separation and capture of CO2 gas,14,15 membrane fabrication for gas separation applications,16 fabrication of low-cost polymer solar cells17,18 and synthesis of porous aromatic frameworks.19
In the case of PAMs, Iwan and Sek3 have provided an extensive review that considers the applications of these polymers, ranging from the aerospace industry to catalyst carriers, ion complexing agents, and anticorrosion materials as well as in the fabrication of photovoltaic cells and OLEDs.
Considering the interesting properties of both the –Ar–Si(R1R2)–Ar– unit and the PAMs family, it is quite worthwhile to investigate their use in the same macromolecular material. The scientific literature provides very few studies devoted to this subject. In fact, in studies in which the silicon atom is incorporated into the poly(azomethine) backbone, it is not part of a –Ar–Si(R1R2)–Ar– unit. In general, the silicon atom is part of a siloxane moiety, which implies a Si–O bond.20–25 In 2014, Zaltariov et al.26 reported a PAM with a central silicon atom bonded to four carbon atoms complexed with different metal ions. However, they used a methylene unit instead of an aromatic moiety bonded to a silicon atom which, along with other structural features, rendered the polymer too flexible; thus, the Tg measured for the polymer was 48 °C. For a material to be used for charge transport in a PHOLED device, its Tg value must be over 100 °C to avoid deterioration of its mechanical properties under high temperature conditions.9
The aim of this work is the synthesis and chemical and structural characterization of four oligomeric poly(ether-azomethine)s, designated PAM-I, PAM-II, PAM-III and PAM-IV, which contain silicon atoms and imino functions in their main chains in addition to methyl or phenyl groups in their lateral segments. Thus, the structural properties of the materials were studied using nuclear magnetic resonance (NMR) spectroscopy, Fourier transform infrared (FT-IR) spectroscopy and elemental analyses. The optical properties of the samples were measured via UV-vis spectroscopy (optical absorption). Their solubilities were tested in several organic solvents, and their thermal behavior was established by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC). Finally, two different films were prepared using the spin-coating technique for deposition, using silicon wafers previously treated with piranha solution (H2SO4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O2) as the substrate. The samples selected were PAM-I and PAM-II; both dissolved in DMAc. The thicknesses of the films were determined by ellipsometry using the refractive index of each layer, determined through an Abbe refractometer. Morphological representations of the film surfaces were obtained by atomic force microscopy (AFM). Additionally, conductivity values for the solid films were also determined through the four-point method, using a silicon wafer as the calibration material.
:H2O2) as the substrate. The samples selected were PAM-I and PAM-II; both dissolved in DMAc. The thicknesses of the films were determined by ellipsometry using the refractive index of each layer, determined through an Abbe refractometer. Morphological representations of the film surfaces were obtained by atomic force microscopy (AFM). Additionally, conductivity values for the solid films were also determined through the four-point method, using a silicon wafer as the calibration material.
Experimental section
Materials
n-Butyllithium solution (2 M in cyclohexane), dimethyldichlorosilane, diphenyldichlorosilane, bis(triphenylphosphine) palladium(II) dichloride, anhydrous N,N-dimethylacetamide (DMAc), anhydrous calcium chloride, p-toluenesulfonic acid, 1-fluoro-4-nitrobenzene, hydrazine monohydrate (80%) and Pd/C (10% w/w) were obtained from Aldrich Chemical (Milwaukee, WI, USA). 4-Hydroxyphenylboronic acid and 4-formylphenylboronic acid were purchased from AK Scientific, Inc. (San Francisco, USA). All other reagents and solvents were purchased commercially as analytical grade (Aldrich Chemical, WI, USA or Merck, Darmstadt, Germany). Immediately before use, diethyl ether was refluxed with sodium under nitrogen atmosphere and then distilled.Measurements
NMR spectra were acquired on a 400 MHz instrument (Bruker AC-200, Germany) using CDCl3 or DMSO-d6 as solvent for the precursors and monomers, while DMAc-acetone-d6 was employed for the oligomeric samples. In all cases, TMS was added as an internal standard. FT-IR spectra (KBr pellets) were recorded on a Perkin-Elmer (Fremont CA, USA) 1310 spectrophotometer over the range of 4000 to 450 cm−1. The melting points of the precursors and monomers were obtained using a Stuart Scientific SMP3 instrument. Elemental analyses were conducted on Fisons EA 1108-CHNS-O equipment (Thermo Scientific, Waltham/MA, USA). Viscosimetric measurements were performed in a Desreux–Bischof-type dilution viscosimeter at 25 °C (c = 0.5 g dL−1). The weight-average (Mw) and number-average (Mn) molecular weights, degrees of polymerization (DP) and polydispersity indices (PDI) of the oligomeric samples relative to polyethylene glycol oxide standards were determined at 40 °C using a GPC 150cv system (Waters, USA) equipped with a refractive index detector. For this, solutions of oligomeric samples in DMF-0.05 M LiBr (c = 0.5 mg mL−1) were filtered through micro-filters with pores of 2 μm; then, 100 μL of sample was injected at 1 mL min−1 in a MesoPore column (300 × 75 mm), which is suitable for samples with molecular weights up to 25000. Glass transition temperatures were obtained with a Mettler-Toledo (Greifensee, Switzerland) DSC 821 calorimetric system (20 °C min−1 under N2 flow) after the second heating scan. Thermogravimetric analyses were carried out on a Mettler (Switzerland) TA-3000 calorimetric system equipped with a TC-10A processor and a TG-50 thermobalance with a Mettler MT5 microbalance. Samples of 6 to 10 mg were placed in an alumina sample holder, and the thermogravimetric measurements were carried out between 25 °C and 800 °C with a heating rate of 20 °C min−1 under N2 flow. The absorption spectra of the polymers were recorded at room temperature between 270 and 440 nm using a Lambda 35 model UV-vis spectrophotometer (Perkin Elmer, USA) using solutions of DMSO (c = 0.5 g L−1).
A spin coater, model KW-4A from Chemat Scientific, coupled with an oil-free vacuum pump (Rocker Chemker 410) was used for deposition of the polymer dissolved in DMAc over the pretreated silicon wafer. This instrument possesses two spinning stages: (1) 500 to 2500 rpm (2 to 18 seconds) and (2) 800 to 8000 rpm (3 to 60 seconds) with a tunable acceleration rate. A Bresser Trino Researcher II (40-1000X) trinocular microscope coupled with a CCD color camera (5 Mp, Bresser) and a cold light source (Optika CL-41) was used as the first approach for visualizing the topographies of the polymeric films. The surface topographies of the polymers were obtained at room temperature using an NTEGRA Prima AFM (NT-MDT Co.) in intermittent contact mode at different scan ranges (25 × 25 μm2 and 50 × 50 μm2). Force spectroscopy measurements were performed with a NaioAFM atomic force microscope (Nanosurf Inc.) using a conical contact mode tip. These results were analyzed through the model proposed by Hertz for tip indentation over soft surfaces according to Roa et al.27 Images were treated and analyzed using the off-line software Gwyddion.28 A multi-angle laser ellipsometer, model SE 400adv, from SENTECH Instruments GmbH was used to perform optical measurements with variable incidence angles from 30° to 90° in steps of 0.5°; the equipment possessed an attached motorized goniometer (Hüber Diffraktionstechnik GmbH & Co. KG) for control incidence angle variation. A stabilized He–Ne laser (λ = 633 nm) permitted a precision of 0.1 Å in the thin film thickness measurements. This equipment was used to measure the thicknesses of the polymeric films (PAM-I and PAM-II). Refractive indices of these polymers were obtained using an Abbe-Refractometer model AR4 from A. KRÜSS Optronic GmbH in DMAc solution at room temperature. The four-point method was carried out using a digital multimeter from GW Instek, model GDM-8255, which includes a 4 W test lead for conduction measurements as an optional accessory. A DC power supply from GW Instek, model PLR 20-18, was used to administer a constant current to the system (6 A).
Polymeric film preparation
:H2O2; 7:3 vol/vol) for 30 minutes at 80 °C.29,30 Afterward, the wafers were washed with HPLC water and slightly sonicated in order to eliminate traces of sulfuric acid. Then, the wafers were dried with an ultra-pure nitrogen gas jet. This substrate treatment is important to improve the adherence of the polymer when the film is formed.Precursors and monomer synthesis
 C); 1373 (Si–Ph); 1253 (Si–CH3); 832 (arom. p-subst.); 500 (C–Br). 1H NMR (CDCl3, δ, ppm): 0.44 (s, 6H, H5); 7.27 (d, J = 7.91 Hz, 4H, H2); 7.39 (d, J = 7.86 Hz, 4H, H3). 13C NMR (CDCl3, δ, ppm): −2.42 (C5); 124.38 (C4); 131.23 (C3), 135.82 (C2), 136.55 (C1). 29Si NMR (CDCl3, δ, ppm): −7.00. Elem. anal. calcd for C14H14Br2Si; (370.17): C, 45.42%; H, 3.82%. Found: C, 45.39%; H, 3.79%.C); 1427, 729 (Si–Ph); 809 (arom. p-subst); 512 (C–Br). 1H NMR (CDCl3, δ, ppm): 7.37 (m, 4H, H7); 7.39 (m, 4H, H2); 7.44 (m, 2H, H8); 7.51 (m, 8H, H3, H6). 13C NMR (CDCl3, δ, ppm): 123.94 (C4); 127.07 (C7); 128.97 (C8); 130.18 (C3), 131.62 (C1), 131.97 (C5), 135.18 (C6), 136.78 (C2). 29Si NMR (CDCl3, δ, ppm): −14.01. Elem. anal. calcd for C24H18Br2Si; (494.31): C, 58.31%; H, 3.68%. Found: C, 58.26%; H, 3.61%.
C); 1373 (Si–Ph); 1253 (Si–CH3); 832 (arom. p-subst.); 500 (C–Br). 1H NMR (CDCl3, δ, ppm): 0.44 (s, 6H, H5); 7.27 (d, J = 7.91 Hz, 4H, H2); 7.39 (d, J = 7.86 Hz, 4H, H3). 13C NMR (CDCl3, δ, ppm): −2.42 (C5); 124.38 (C4); 131.23 (C3), 135.82 (C2), 136.55 (C1). 29Si NMR (CDCl3, δ, ppm): −7.00. Elem. anal. calcd for C14H14Br2Si; (370.17): C, 45.42%; H, 3.82%. Found: C, 45.39%; H, 3.79%.C); 1427, 729 (Si–Ph); 809 (arom. p-subst); 512 (C–Br). 1H NMR (CDCl3, δ, ppm): 7.37 (m, 4H, H7); 7.39 (m, 4H, H2); 7.44 (m, 2H, H8); 7.51 (m, 8H, H3, H6). 13C NMR (CDCl3, δ, ppm): 123.94 (C4); 127.07 (C7); 128.97 (C8); 130.18 (C3), 131.62 (C1), 131.97 (C5), 135.18 (C6), 136.78 (C2). 29Si NMR (CDCl3, δ, ppm): −14.01. Elem. anal. calcd for C24H18Br2Si; (494.31): C, 58.31%; H, 3.68%. Found: C, 58.26%; H, 3.61%.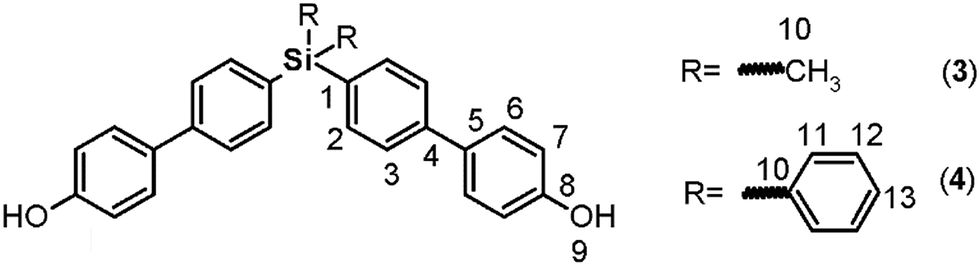 C); 1434 (Si–Ph); 1247 (Si–CH3); 819 (arom. p-subst); 1H NMR (DMSO-d6, δ, ppm): 0.56 (s, 6H, H10); 6.88 (d, J = 8.54 Hz, 4H, H7); 7.50 (d, J = 8.53 Hz, 4H, H6); 7.55 (d, J = 8.15 Hz, 4H, H2), 7.58 (d, J = 8.19 Hz, 4H, H3), 9.59 (s, 2H, H9). 13C NMR (DMSO-d6, δ, ppm): −2.57 (C10); 115.78 (C7); 125.45 (C3); 127.79 (C6); 130.73 (C5); 134.43 (C2); 135.44 (C1); 140.96 (C4); 157.34 (C8). 29Si NMR (DMSO-d6, δ, ppm): −8.64. Elem. anal. calcd for C26H24O2Si; (396.59): C, 78.74%; H, 6.11%. Found: C, 78.64%; H, 6.08%.C); 1427 (Si–Ph); 815 (arom. p-subst.). 1H NMR: (CDCl3, δ, ppm): 6.89 (d, J = 8.06 Hz, 4H, H7); 7.38 (d, J = 6.68 Hz, 4H, H12); 7.42 (d, J = 6.97 Hz, 2H, H13); 7.47 (d, J = 8.22 Hz, 4H, H6); 7.54 (d, J = 7.65 Hz, 4H, H3); 7.62 (d, J = 7.03 Hz, 8H, H2 y H11). 5.11 (s, 2H, H9). 13C NMR (CDCl3, δ, ppm): 115.77 (C7); 126.10 (C3); 127.92 (C12); 128.38 (C6); 129.62 (C13); 132.21 (C1); 133.48 (C5); 134.35 (C10); 136.42 and 136.87 (C2 and C11); 141.78 (C4), 155.41 (C8). 29Si NMR (CDCl3, δ, ppm): −14.46. Elem. anal. calcd for C36H28O2Si; (520.73): C, 83.03%; H, 5.43%. Found: C, 82.98%; H, 5.37%.:1 vol/vol), and an ochre solid was formed. The product was filtered and washed with water and then purified through silica column chromatography. For isolation of 3, an n-hexane–chloroform mixture (1:1 vol/vol)) was employed as the mobile phase. The residue obtained after solvent evaporation was recrystallized from ethyl acetate to afford a light yellow solid (88%). For isolation of 4, a petroleum ether–chloroform mixture (1:1 vol/vol) was used, and the obtained residue was treated with diethyl ether to produce a light yellow solid (74%).
C); 1434 (Si–Ph); 1247 (Si–CH3); 819 (arom. p-subst); 1H NMR (DMSO-d6, δ, ppm): 0.56 (s, 6H, H10); 6.88 (d, J = 8.54 Hz, 4H, H7); 7.50 (d, J = 8.53 Hz, 4H, H6); 7.55 (d, J = 8.15 Hz, 4H, H2), 7.58 (d, J = 8.19 Hz, 4H, H3), 9.59 (s, 2H, H9). 13C NMR (DMSO-d6, δ, ppm): −2.57 (C10); 115.78 (C7); 125.45 (C3); 127.79 (C6); 130.73 (C5); 134.43 (C2); 135.44 (C1); 140.96 (C4); 157.34 (C8). 29Si NMR (DMSO-d6, δ, ppm): −8.64. Elem. anal. calcd for C26H24O2Si; (396.59): C, 78.74%; H, 6.11%. Found: C, 78.64%; H, 6.08%.C); 1427 (Si–Ph); 815 (arom. p-subst.). 1H NMR: (CDCl3, δ, ppm): 6.89 (d, J = 8.06 Hz, 4H, H7); 7.38 (d, J = 6.68 Hz, 4H, H12); 7.42 (d, J = 6.97 Hz, 2H, H13); 7.47 (d, J = 8.22 Hz, 4H, H6); 7.54 (d, J = 7.65 Hz, 4H, H3); 7.62 (d, J = 7.03 Hz, 8H, H2 y H11). 5.11 (s, 2H, H9). 13C NMR (CDCl3, δ, ppm): 115.77 (C7); 126.10 (C3); 127.92 (C12); 128.38 (C6); 129.62 (C13); 132.21 (C1); 133.48 (C5); 134.35 (C10); 136.42 and 136.87 (C2 and C11); 141.78 (C4), 155.41 (C8). 29Si NMR (CDCl3, δ, ppm): −14.46. Elem. anal. calcd for C36H28O2Si; (520.73): C, 83.03%; H, 5.43%. Found: C, 82.98%; H, 5.37%.:1 vol/vol), and an ochre solid was formed. The product was filtered and washed with water and then purified through silica column chromatography. For isolation of 3, an n-hexane–chloroform mixture (1:1 vol/vol)) was employed as the mobile phase. The residue obtained after solvent evaporation was recrystallized from ethyl acetate to afford a light yellow solid (88%). For isolation of 4, a petroleum ether–chloroform mixture (1:1 vol/vol) was used, and the obtained residue was treated with diethyl ether to produce a light yellow solid (74%).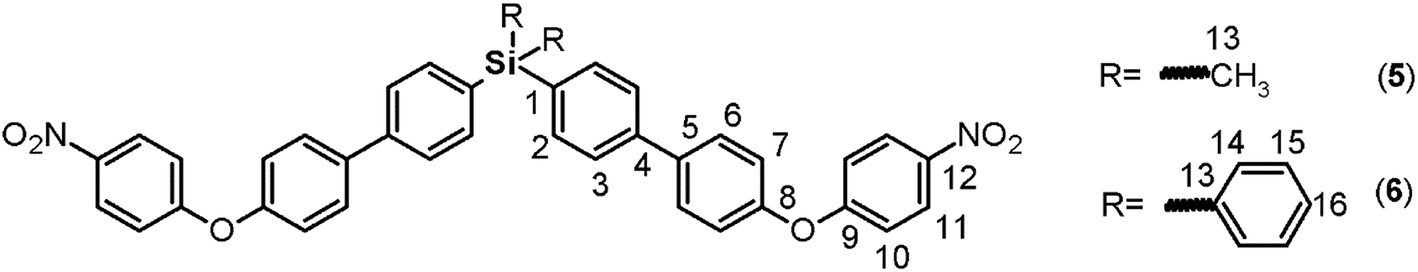 C); 1518, 1345 (NO2); 1246 (C–O–C); 1110 (Si–Ph); 847 (Si–CH3); 819 (arom p-subst.). 1H NMR (CDCl3, δ, ppm): 0.64 (s, 6H, H13); 7.07 (d, J = 9.06, 4H, H10); 7.16 (d, J = 8.51, 4H, H7); 7.60 (d, J = 7.76, 4H, H3); 7.65 (m, 8H, H2, H6); 8.21 (d, J = 9.13, 4H, H11). 13C NMR (CDCl3, δ, ppm): −2.33 (C13); 117.27 (C10); 120.77 (C7); 125.98 (C11); 126.46 (C3); 128.96 (C6); 134.81 (C2); 137.22 (C1); 138.28 (C5); 140.77 (C4); 142.78 (C12); 154.35 (C8); 163.21 (C9). 29Si NMR (CDCl3, δ, ppm): −7.83. Elem. anal. calcd for C38H30N2O6Si; (638.79): C, 71.44%; H, 4.74%; N, 4.39%. Found: C, 71.40%; H, 4.68%, N, 4.32%.C); 1517, 1343 (NO2); 1246 (C–O–C), 1111 (Si–Ph), 818 (arom. p-subst.). 1H NMR (CDCl3, δ, ppm): 7.06 (d, J = 9.10 Hz, 4H, H10); 7.16 (d, J = 8.43 Hz, 4H, H7); 7.41 (t, J = 7.40 Hz, 4H, H15); 7.47 (m, 2H, H16); 7.62 (d, J = 8.10 Hz, 4H, H3); 7.64 (d, J = 9.44 Hz, 4H, H14), 7.67 (d, J = 9.04 Hz, 4H, H6); 7.70 (d, J = 7.31 Hz, 4H, H2), 8.21 (d, J = 9.12 Hz, 4H, H11). 13C NMR (CDCl3, δ, ppm): 117.29 (C10); 120.79 (C7); 125.98 (C11); 126.47 (C3); 128.03 (C15); 128.98 (C6); 129.81 (C16); 133.27 (C1); 133.94 (C13); 136.39 (C14); 137.00 (C2); 138.10 (C5); 141.13 (C4); 142.79 (C12); 154.45 (C8); 163.17 (C9). 29Si NMR (CDCl3, δ, ppm): −14.37. Elem. anal. calcd for C48H34N2O6Si; (762.93): C, 75.56%; H, 4.50%; N, 3.67%. Found: C, 75.51%; H, 4.44%, N, 3.60%.
C); 1518, 1345 (NO2); 1246 (C–O–C); 1110 (Si–Ph); 847 (Si–CH3); 819 (arom p-subst.). 1H NMR (CDCl3, δ, ppm): 0.64 (s, 6H, H13); 7.07 (d, J = 9.06, 4H, H10); 7.16 (d, J = 8.51, 4H, H7); 7.60 (d, J = 7.76, 4H, H3); 7.65 (m, 8H, H2, H6); 8.21 (d, J = 9.13, 4H, H11). 13C NMR (CDCl3, δ, ppm): −2.33 (C13); 117.27 (C10); 120.77 (C7); 125.98 (C11); 126.46 (C3); 128.96 (C6); 134.81 (C2); 137.22 (C1); 138.28 (C5); 140.77 (C4); 142.78 (C12); 154.35 (C8); 163.21 (C9). 29Si NMR (CDCl3, δ, ppm): −7.83. Elem. anal. calcd for C38H30N2O6Si; (638.79): C, 71.44%; H, 4.74%; N, 4.39%. Found: C, 71.40%; H, 4.68%, N, 4.32%.C); 1517, 1343 (NO2); 1246 (C–O–C), 1111 (Si–Ph), 818 (arom. p-subst.). 1H NMR (CDCl3, δ, ppm): 7.06 (d, J = 9.10 Hz, 4H, H10); 7.16 (d, J = 8.43 Hz, 4H, H7); 7.41 (t, J = 7.40 Hz, 4H, H15); 7.47 (m, 2H, H16); 7.62 (d, J = 8.10 Hz, 4H, H3); 7.64 (d, J = 9.44 Hz, 4H, H14), 7.67 (d, J = 9.04 Hz, 4H, H6); 7.70 (d, J = 7.31 Hz, 4H, H2), 8.21 (d, J = 9.12 Hz, 4H, H11). 13C NMR (CDCl3, δ, ppm): 117.29 (C10); 120.79 (C7); 125.98 (C11); 126.47 (C3); 128.03 (C15); 128.98 (C6); 129.81 (C16); 133.27 (C1); 133.94 (C13); 136.39 (C14); 137.00 (C2); 138.10 (C5); 141.13 (C4); 142.79 (C12); 154.45 (C8); 163.17 (C9). 29Si NMR (CDCl3, δ, ppm): −14.37. Elem. anal. calcd for C48H34N2O6Si; (762.93): C, 75.56%; H, 4.50%; N, 3.67%. Found: C, 75.51%; H, 4.44%, N, 3.60%.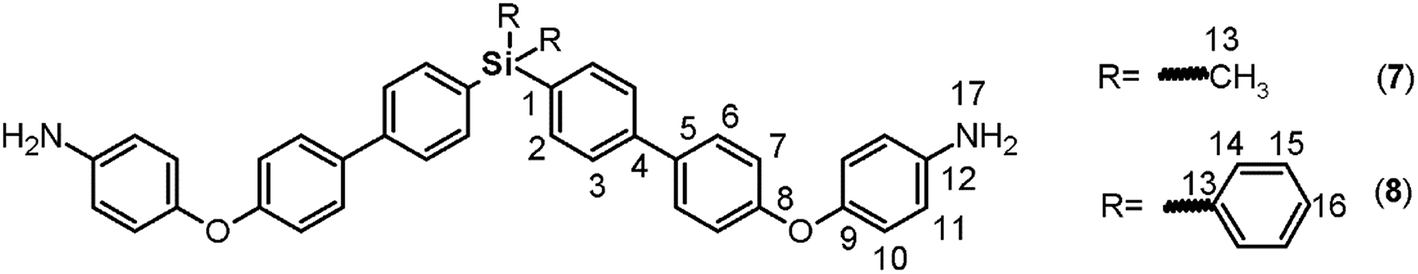 C); 1239 (C–O–C); 1111 (Si–Ph); 879 (Si–CH3); 811 (arom. p-subst.). 1H NMR (CDCl3, δ, ppm): 0.59 (s, 6H, H13); 3.57 (s, 4H, H17); 6.68 (d, J = 8.33 Hz, 4H, H11); 6.90 (d, J = 7.14 Hz, 4H, H10); 6.98 (d, J = 7.36 Hz, 4H, H7); 7.50 (d, J = 7.39 Hz, 4H, H6); 7.54 (d, J = 7.41 Hz, 4H, H3); 7.59 (d, J = 6.91 Hz, 4H, H2). 13C NMR (CDCl3, δ, ppm): −2.30 (C13); 116.24 (C11); 117.36 (C7); 121.24 (C10); 126.24 (C3); 128.22 (C6); 134.67 (C2); 134.89 (C5); 136.41 (C1); 141.34 (C4); 142.84 (C12), 148.38 (C9); 158.67 (C8). 29Si NMR (CDCl3, δ, ppm): −8.09. Elem. anal. calcd for C38H34N2O2Si; (578.83): C, 78.84%; H, 5.93%; N, 4.84%. Found: C, 78.81%; H, 5.89%, N, 4.81%.C); 1235 (C–O–C); 1111 (Si–Ph); 816 (arom. p-subst.). 1H NMR (CDCl3, δ, ppm): 3.49 (s, 4H, H17); 6.68 (d, J = 8.61 Hz, 4H, H11); 6.90 (d, J = 8.62 Hz, 4H, H10); 6.99 (d, J = 8.56 Hz, 4H, H7), 7.39 (m, 4H, H15); 7.44 (m, 2H, H16); 7.52 (d, J = 8.58 Hz, 4H, H6); 7.56 (d, J = 7.88 Hz, 4H, H3), 7.62 (d, J = 6.26 Hz, 4H, H14); 7.64 (d, J = 7.62 Hz, 4H, H2). 13C NMR (CDCl3, δ, ppm): 116.26 (C11); 117.40 (C7); 121.27 (C10); 126.24 (C3); 127.93 (C15); 128.25 (C6); 129.64 (C16); 132.42 (C1); 134.28 (C13); 134.74 (C5); 136.41 (C14); 136.87 (C2); 141.71 (C4); 142.83 (C12); 148.41 (C9); 158.78 (C8). 29Si NMR (CDCl3, δ, ppm): −14.52. Elem. anal. calcd for C48H38N2O2Si; (698.93): C, 82.48%; H, 4.91%; N, 4.01%. Found: C, 82.42%; H, 4.85%, N, 3.96%.:1 vol/vol) was added; then, the mixture was heated at 85 °C for 12 hours. Then, water was added, and a dark brown solid was obtained which was filtered under vaccum. This product was treated with THF, and the remaining solids were removed by filtration through celite. The obtained solution was dried and concentrated to obtain a black solid, which was purified by column chromatography (silica/petroleum ether–chloroform, 1:1 vol/vol) and subsequent recrystallization from ethyl acetate. Following this procedure, 9 and 10 were obtained as white solids in yields of 56% and 51%, respectively.
C); 1239 (C–O–C); 1111 (Si–Ph); 879 (Si–CH3); 811 (arom. p-subst.). 1H NMR (CDCl3, δ, ppm): 0.59 (s, 6H, H13); 3.57 (s, 4H, H17); 6.68 (d, J = 8.33 Hz, 4H, H11); 6.90 (d, J = 7.14 Hz, 4H, H10); 6.98 (d, J = 7.36 Hz, 4H, H7); 7.50 (d, J = 7.39 Hz, 4H, H6); 7.54 (d, J = 7.41 Hz, 4H, H3); 7.59 (d, J = 6.91 Hz, 4H, H2). 13C NMR (CDCl3, δ, ppm): −2.30 (C13); 116.24 (C11); 117.36 (C7); 121.24 (C10); 126.24 (C3); 128.22 (C6); 134.67 (C2); 134.89 (C5); 136.41 (C1); 141.34 (C4); 142.84 (C12), 148.38 (C9); 158.67 (C8). 29Si NMR (CDCl3, δ, ppm): −8.09. Elem. anal. calcd for C38H34N2O2Si; (578.83): C, 78.84%; H, 5.93%; N, 4.84%. Found: C, 78.81%; H, 5.89%, N, 4.81%.C); 1235 (C–O–C); 1111 (Si–Ph); 816 (arom. p-subst.). 1H NMR (CDCl3, δ, ppm): 3.49 (s, 4H, H17); 6.68 (d, J = 8.61 Hz, 4H, H11); 6.90 (d, J = 8.62 Hz, 4H, H10); 6.99 (d, J = 8.56 Hz, 4H, H7), 7.39 (m, 4H, H15); 7.44 (m, 2H, H16); 7.52 (d, J = 8.58 Hz, 4H, H6); 7.56 (d, J = 7.88 Hz, 4H, H3), 7.62 (d, J = 6.26 Hz, 4H, H14); 7.64 (d, J = 7.62 Hz, 4H, H2). 13C NMR (CDCl3, δ, ppm): 116.26 (C11); 117.40 (C7); 121.27 (C10); 126.24 (C3); 127.93 (C15); 128.25 (C6); 129.64 (C16); 132.42 (C1); 134.28 (C13); 134.74 (C5); 136.41 (C14); 136.87 (C2); 141.71 (C4); 142.83 (C12); 148.41 (C9); 158.78 (C8). 29Si NMR (CDCl3, δ, ppm): −14.52. Elem. anal. calcd for C48H38N2O2Si; (698.93): C, 82.48%; H, 4.91%; N, 4.01%. Found: C, 82.42%; H, 4.85%, N, 3.96%.:1 vol/vol) was added; then, the mixture was heated at 85 °C for 12 hours. Then, water was added, and a dark brown solid was obtained which was filtered under vaccum. This product was treated with THF, and the remaining solids were removed by filtration through celite. The obtained solution was dried and concentrated to obtain a black solid, which was purified by column chromatography (silica/petroleum ether–chloroform, 1:1 vol/vol) and subsequent recrystallization from ethyl acetate. Following this procedure, 9 and 10 were obtained as white solids in yields of 56% and 51%, respectively.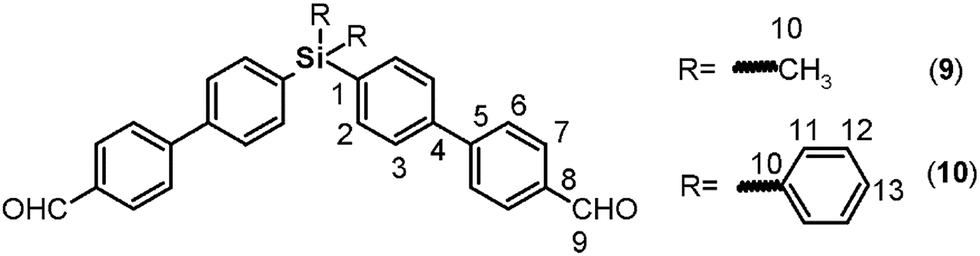 O); 1604, 1572 (CC); 1387 (Si–CH3); 1166, 1111 (Si–Ph); 813 (arom. p-subst.). 1H NMR (DMSO-d6, δ, ppm): 0.64 (s, 6H, H10); 7.70 (d, J = 8.07 Hz, 4H, H2); 7.74 (d, J = 8.04 Hz, 4H, H3); 7.86 (d, J = 8.16 Hz, 4H, H6); 7.97 (d, J = 8.23 Hz, 4H, H7); 10.08 (s, 2H, H9). 13C NMR (DMSO-d6, δ, ppm): −3.48 (C10); 125.72 (C3); 126.55 (C6); 129.08 (C7); 133.83 (C2); 134.98 (C8); 137.55 (C1), 139.12 (C4); 145.14 (C5); 191.12 (C9). 29Si NMR (DMSO-d6, δ, ppm): −7.85. Elem. anal. calcd for C28H24O2Si; (420.61): C, 79.95%; H, 5.76%. Found: C, 79.91%; H, 5.70%.O); 1572, 1604 (CC); 1111, 1429 (Si–Ph); 810 (arom. p-subst.). 1H NMR (CDCl3, δ, ppm): 7.42 (d, J = 7.17 Hz, 4H, H12); 7.48 (m, 2H, H13); 7.63 (d, J = 7.28 Hz, 4H, H11); 7.67 (d, J = 7.79 Hz, 4H, H3); 7.72 (d, J = 7.64 Hz, 4H, H2); 7.78 (d, J = 7.88 Hz, 4H, H6); 7.96 (d, J = 7.85 Hz, 4H, H7); 10.06 (s, 2H, H9). 13C NMR (CDCl3, δ, ppm): 126.84 (C3); 127.72 (C6); 128.09 (C12); 129.92 (C13); 130.32 (C7); 133.60 (C10); 134.50 (C1); 135.44 (C8); 136.38 (C11); 137.05 (C2); 140.86 (C4); 146.81 (C5); 191.83 (C9). 29Si NMR (CDCl3, δ, ppm): −14.36. Elem. anal. calcd for C38H28O2Si; (544.75): C, 83.78%; H, 5.19%. Found: C, 83.72%; H, 5.13%.
O); 1604, 1572 (CC); 1387 (Si–CH3); 1166, 1111 (Si–Ph); 813 (arom. p-subst.). 1H NMR (DMSO-d6, δ, ppm): 0.64 (s, 6H, H10); 7.70 (d, J = 8.07 Hz, 4H, H2); 7.74 (d, J = 8.04 Hz, 4H, H3); 7.86 (d, J = 8.16 Hz, 4H, H6); 7.97 (d, J = 8.23 Hz, 4H, H7); 10.08 (s, 2H, H9). 13C NMR (DMSO-d6, δ, ppm): −3.48 (C10); 125.72 (C3); 126.55 (C6); 129.08 (C7); 133.83 (C2); 134.98 (C8); 137.55 (C1), 139.12 (C4); 145.14 (C5); 191.12 (C9). 29Si NMR (DMSO-d6, δ, ppm): −7.85. Elem. anal. calcd for C28H24O2Si; (420.61): C, 79.95%; H, 5.76%. Found: C, 79.91%; H, 5.70%.O); 1572, 1604 (CC); 1111, 1429 (Si–Ph); 810 (arom. p-subst.). 1H NMR (CDCl3, δ, ppm): 7.42 (d, J = 7.17 Hz, 4H, H12); 7.48 (m, 2H, H13); 7.63 (d, J = 7.28 Hz, 4H, H11); 7.67 (d, J = 7.79 Hz, 4H, H3); 7.72 (d, J = 7.64 Hz, 4H, H2); 7.78 (d, J = 7.88 Hz, 4H, H6); 7.96 (d, J = 7.85 Hz, 4H, H7); 10.06 (s, 2H, H9). 13C NMR (CDCl3, δ, ppm): 126.84 (C3); 127.72 (C6); 128.09 (C12); 129.92 (C13); 130.32 (C7); 133.60 (C10); 134.50 (C1); 135.44 (C8); 136.38 (C11); 137.05 (C2); 140.86 (C4); 146.81 (C5); 191.83 (C9). 29Si NMR (CDCl3, δ, ppm): −14.36. Elem. anal. calcd for C38H28O2Si; (544.75): C, 83.78%; H, 5.19%. Found: C, 83.72%; H, 5.13%.Silylated oligomeric poly(ether-azomethine)s synthesis
A mixture of dialdehyde (9 or 10), anhydrous calcium chloride, p-toluenesulfonic acid (p-TsOH) and anhydrous DMAc was placed under a nitrogen atmosphere with vigorous stirring and heated until reflux. When the dialdehyde was dissolved, a solution containing the diamine (7 or 8) in anhydrous DMAc prepared under nitrogen flow was slowly added; the reaction was then maintained under reflux for 24 hours. Then, the mixture was added to methanol under stirring and the obtained solid was filtered, extensively washed with hot methanol, and dried at 100 °C under vacuum for 24 hours.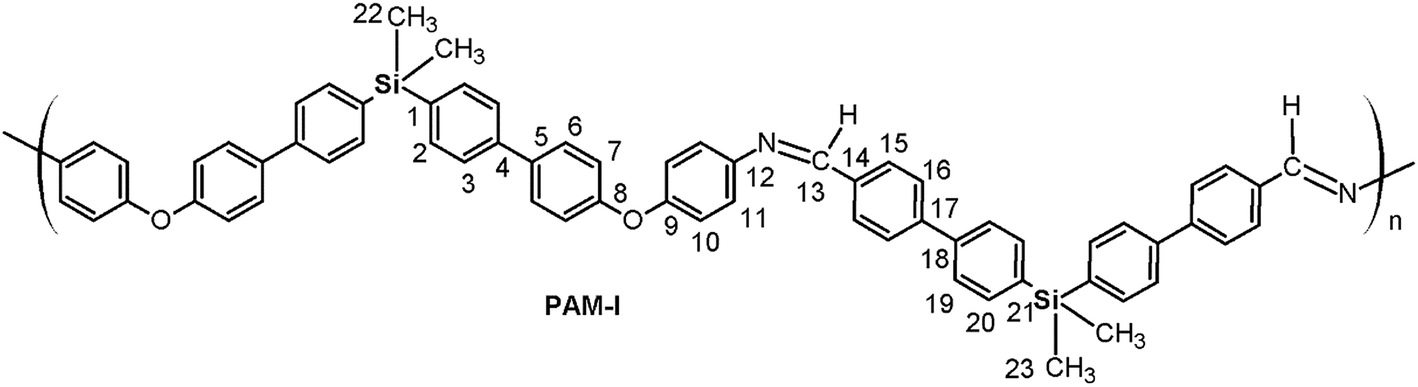 O); 1624 (CN); 1603, 1496, 1487 (CC); 1244 (C–O–C); 1114 (Si–Ph); 872 (Si–CH3); 810 (arom. p-subst.). 1H NMR (DMAc, acetone-d6 internal reference (int. ref.), δ, ppm): 0.47 (m, 12H, H22, H23); 7.14 (d, J = 7.25 Hz, 4H, H7), 7.29 (m, 4H, H10); 7.55 (m, 8H, H11, H20); 7.61–7.84 (m, 16H, H2, H3, H6, H19); 7.97 (m, 8H, H15, H16); 8.80 (s, 2H, H13). 13C NMR (DMAc, acetone-d6 int. ref., δ, ppm): −3.333; −3.329 (C22, C23); 121.55 (C7); 122.82 (C10); 126.36 (C3); 126.46 (C19); 127.46, 127.56, 127.79 (C6, C11, C16); 129.04 (C15); 132.13 (C5); 133.21; 133.22 (C2, C20); 133.61, 133.70, 134.70, 135.63 (C1, C14, C18, C21); 139.31 (C4); 144.34 (C17); 148.94 (C12); 154.28 (C9); 156.95 (C8); 164.05 (C13). 29Si NMR (DMAc, acetone-d6 int. ref., δ, ppm): −9.82 (diamino moiety); −9.79 (dialdehyde moiety).
O); 1624 (CN); 1603, 1496, 1487 (CC); 1244 (C–O–C); 1114 (Si–Ph); 872 (Si–CH3); 810 (arom. p-subst.). 1H NMR (DMAc, acetone-d6 internal reference (int. ref.), δ, ppm): 0.47 (m, 12H, H22, H23); 7.14 (d, J = 7.25 Hz, 4H, H7), 7.29 (m, 4H, H10); 7.55 (m, 8H, H11, H20); 7.61–7.84 (m, 16H, H2, H3, H6, H19); 7.97 (m, 8H, H15, H16); 8.80 (s, 2H, H13). 13C NMR (DMAc, acetone-d6 int. ref., δ, ppm): −3.333; −3.329 (C22, C23); 121.55 (C7); 122.82 (C10); 126.36 (C3); 126.46 (C19); 127.46, 127.56, 127.79 (C6, C11, C16); 129.04 (C15); 132.13 (C5); 133.21; 133.22 (C2, C20); 133.61, 133.70, 134.70, 135.63 (C1, C14, C18, C21); 139.31 (C4); 144.34 (C17); 148.94 (C12); 154.28 (C9); 156.95 (C8); 164.05 (C13). 29Si NMR (DMAc, acetone-d6 int. ref., δ, ppm): −9.82 (diamino moiety); −9.79 (dialdehyde moiety).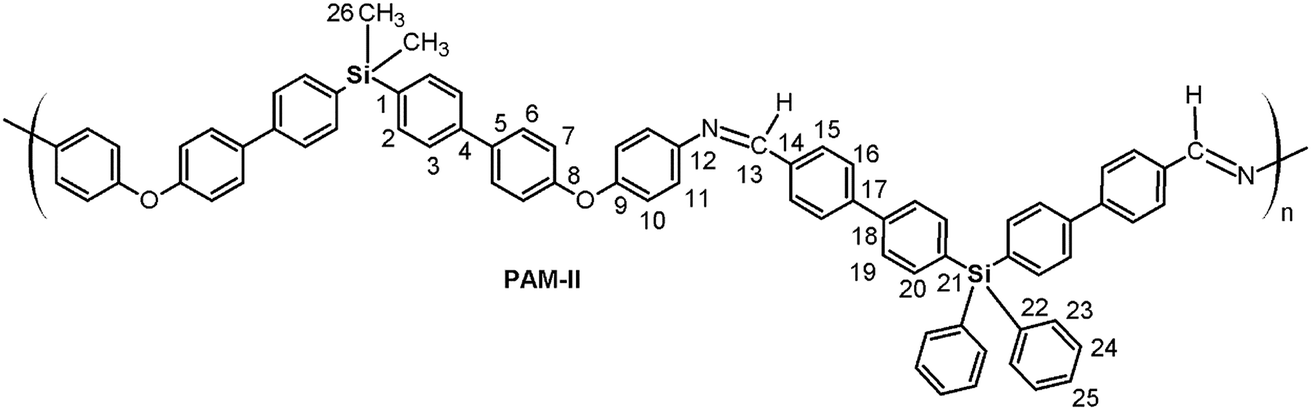 O); 1624 (CN); 1603, 1495, 1486 (CC); 1240 (C–O–C); 1112 (Si–Ph); 871 (Si–CH3); 811 (arom. p-subst). 1H NMR (DMAc, acetone-d6 int. ref., δ, ppm): 0.63 (s, 6H, H26); 7.15 (m, 8H, H7, H10); 7.47 (d. J = 8.11 Hz, 4H, H11); 7.53 (m, 10H, H24, H25); 7.58–7.81 (m, 20H, H2, H3, H6, H20, H23); 7.94 (m, 8H, H16, H19); 8.13 (m, 4H, H15); 8.82 (s, 2H, H13). 13C NMR (DMAc, acetone-d6 int. ref., δ, ppm): −2.56 (C26); 119.15 (C7); 120.15 (C10); 123.28 (C11); 126.39 (C3); 127.02 (C19); 127.59 (C16); 128.65 (C24); 128.76 (C6); 129.72 (C15); 130.53 (C25); 133.95, 134.02, 134.13 (C14, C21, C22); 135.09 (C2); 135.83 (C5); 136.50 (C23); 136.99 (C1); 137.22 (C20); 141.00 (C4); 141.47 (C18); 143.09 (C17); 147.84 (C12); 155.59 (C9); 157.63 (C8); 159.62 (C13). 29Si NMR (DMAc, acetone-d6 int. ref., δ, ppm): −15.34 (dialdehyde moiety); −9.11 (diamine moiety).
O); 1624 (CN); 1603, 1495, 1486 (CC); 1240 (C–O–C); 1112 (Si–Ph); 871 (Si–CH3); 811 (arom. p-subst). 1H NMR (DMAc, acetone-d6 int. ref., δ, ppm): 0.63 (s, 6H, H26); 7.15 (m, 8H, H7, H10); 7.47 (d. J = 8.11 Hz, 4H, H11); 7.53 (m, 10H, H24, H25); 7.58–7.81 (m, 20H, H2, H3, H6, H20, H23); 7.94 (m, 8H, H16, H19); 8.13 (m, 4H, H15); 8.82 (s, 2H, H13). 13C NMR (DMAc, acetone-d6 int. ref., δ, ppm): −2.56 (C26); 119.15 (C7); 120.15 (C10); 123.28 (C11); 126.39 (C3); 127.02 (C19); 127.59 (C16); 128.65 (C24); 128.76 (C6); 129.72 (C15); 130.53 (C25); 133.95, 134.02, 134.13 (C14, C21, C22); 135.09 (C2); 135.83 (C5); 136.50 (C23); 136.99 (C1); 137.22 (C20); 141.00 (C4); 141.47 (C18); 143.09 (C17); 147.84 (C12); 155.59 (C9); 157.63 (C8); 159.62 (C13). 29Si NMR (DMAc, acetone-d6 int. ref., δ, ppm): −15.34 (dialdehyde moiety); −9.11 (diamine moiety).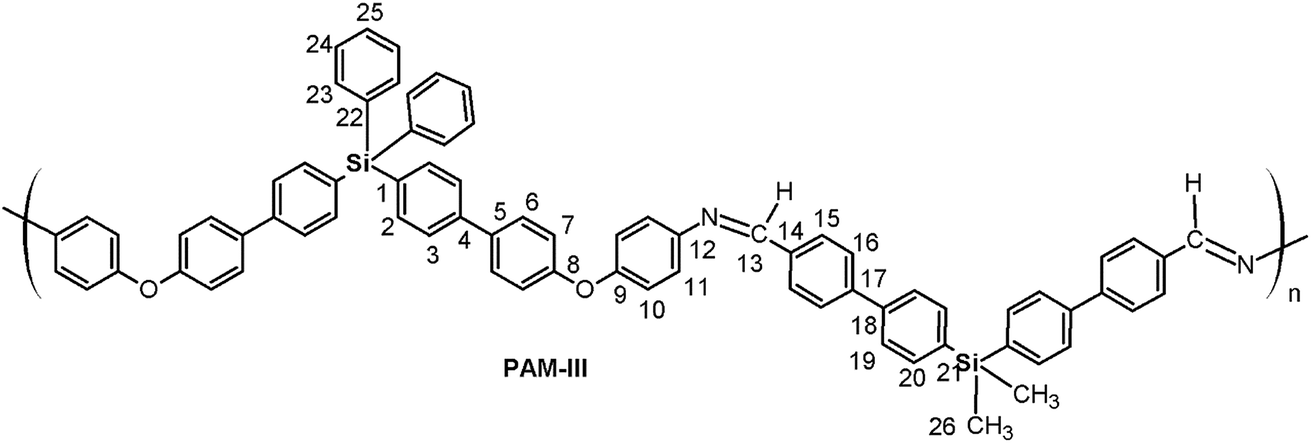 O); 1623 (CN); 1603, 1496, 1486 (CC); 1239 (C–O–C); 1112 (Si–Ph); 872 (Si–CH3); 808 (arom. p-subst). 1H NMR (DMAc, acetone-d6 int. ref., δ, ppm): 0.66 (s, 6H, H26); 7.17 (m, 8H, H7, H10); 7.40–5.56 (m, 10H, H11, H24, H25); 7.63 (m, 4H, H23); 7.68 (m, 4H, H2); 7.74 (m, 4H, H20), 7.75–7.87 (m, 12H, H3, H6, H19); 7.90 (m, 4H, H16), 8.10 (m, 4H, H15); 8.80 (s, 2H, H13). 13C NMR (DMAc, acetone-d6 int. ref., δ, ppm): −2.67 (C26), 119.19 (C7), 120.20 (C10); 123.29 (C11); 126.62 (C3); 126.80 (C19); 127.54 (C16); 128.61 (C24); 128.87 (C6); 129.71 (C15); 130.52 (C25); 132.88 (C1); 134.38 (C22); 135.20 (C20); 135.51 (C5); 136.49 (C23); 137.16 (C2); 138.10 (C21); 140.35 (C18); 140.88 (C14); 141.63 (C4); 143.42 (C17); 147.94 (C12); 155.54 (C9); 157.84 (C8); 159.71 (C13). 29Si NMR (DMAc, acetone-d6 int. ref., δ, ppm): −15.35 (diamine moiety); −8.33 (dialdehyde moiety).
O); 1623 (CN); 1603, 1496, 1486 (CC); 1239 (C–O–C); 1112 (Si–Ph); 872 (Si–CH3); 808 (arom. p-subst). 1H NMR (DMAc, acetone-d6 int. ref., δ, ppm): 0.66 (s, 6H, H26); 7.17 (m, 8H, H7, H10); 7.40–5.56 (m, 10H, H11, H24, H25); 7.63 (m, 4H, H23); 7.68 (m, 4H, H2); 7.74 (m, 4H, H20), 7.75–7.87 (m, 12H, H3, H6, H19); 7.90 (m, 4H, H16), 8.10 (m, 4H, H15); 8.80 (s, 2H, H13). 13C NMR (DMAc, acetone-d6 int. ref., δ, ppm): −2.67 (C26), 119.19 (C7), 120.20 (C10); 123.29 (C11); 126.62 (C3); 126.80 (C19); 127.54 (C16); 128.61 (C24); 128.87 (C6); 129.71 (C15); 130.52 (C25); 132.88 (C1); 134.38 (C22); 135.20 (C20); 135.51 (C5); 136.49 (C23); 137.16 (C2); 138.10 (C21); 140.35 (C18); 140.88 (C14); 141.63 (C4); 143.42 (C17); 147.94 (C12); 155.54 (C9); 157.84 (C8); 159.71 (C13). 29Si NMR (DMAc, acetone-d6 int. ref., δ, ppm): −15.35 (diamine moiety); −8.33 (dialdehyde moiety).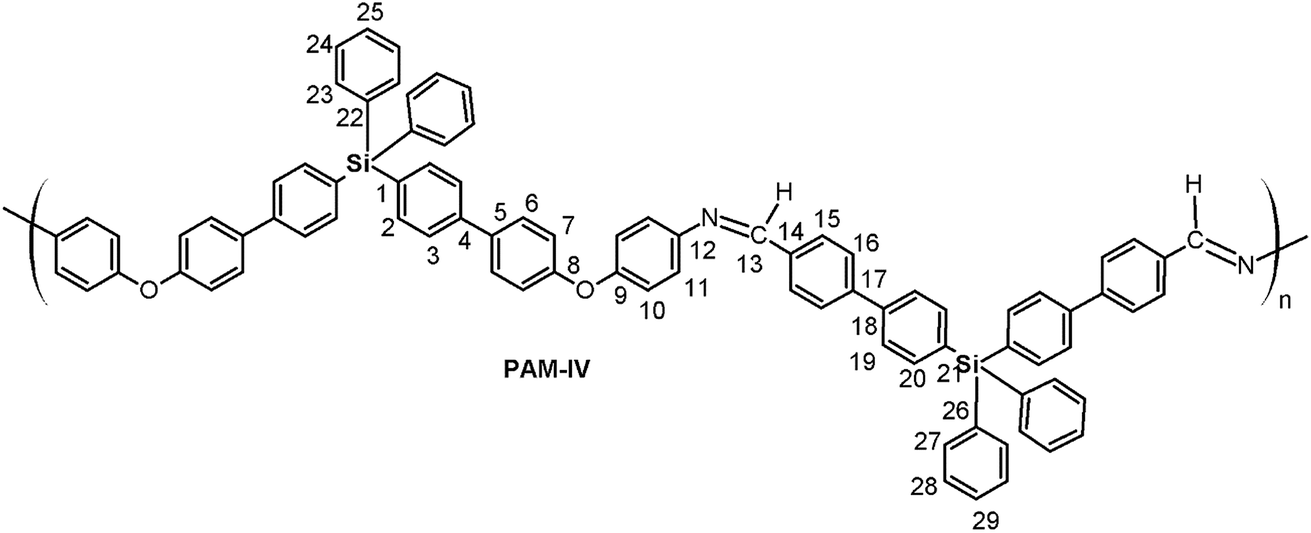 O); 1623 (CN); 1603, 1496, 1486 (CC); 1238 (C–O–C); 1110 (Si–Ph); 813 (arom. p-subst). 1H NMR (DMAc, acetona-d6 int. ref., δ, ppm): 7.18 (m, 8H, H7, H10); 7.47 (m, 16H, H11, H24, H25, H28, H29); 7.65 (m, 12H, H2, H23, H27); 7.75 (m, 4H, H20); 7.82 (m, 8H, H3, H6); 7.95 (m, 8H, H16, H19); 8.13 (d, J = 6.13 Hz, 4H, H15), 8.82 (s, 2H, H13). 13C NMR (DMAc, acetone-d6 int. ref., δ, ppm): 119.19 (C7); 120.19 (C10); 123.29 (C11); 126.62 (C3); 127.04 (C19); 127.60 (C16); 128.60 (C24); 128.67 (C28); 128.86 (C6); 129.74 (C15); 130.34, 130.52 (C25, C29); 132.87 (C1); 134.03 (C21); 134.35, 134.38 (C22, C26); 135.52 (C5), 136.49 (C23, C27); 137.16 (C2); 137.23 (C20); 140.96 (C18); 141.49 (C14); 141.62 (C4); 143.11 (C17); 147.90 (C12); 155.56 (C9); 157.82 (C8); 159.66 (C13). 29Si NMR (DMAc, acetone-d6 int. ref., δ, ppm): −15.33; −15.31.
O); 1623 (CN); 1603, 1496, 1486 (CC); 1238 (C–O–C); 1110 (Si–Ph); 813 (arom. p-subst). 1H NMR (DMAc, acetona-d6 int. ref., δ, ppm): 7.18 (m, 8H, H7, H10); 7.47 (m, 16H, H11, H24, H25, H28, H29); 7.65 (m, 12H, H2, H23, H27); 7.75 (m, 4H, H20); 7.82 (m, 8H, H3, H6); 7.95 (m, 8H, H16, H19); 8.13 (d, J = 6.13 Hz, 4H, H15), 8.82 (s, 2H, H13). 13C NMR (DMAc, acetone-d6 int. ref., δ, ppm): 119.19 (C7); 120.19 (C10); 123.29 (C11); 126.62 (C3); 127.04 (C19); 127.60 (C16); 128.60 (C24); 128.67 (C28); 128.86 (C6); 129.74 (C15); 130.34, 130.52 (C25, C29); 132.87 (C1); 134.03 (C21); 134.35, 134.38 (C22, C26); 135.52 (C5), 136.49 (C23, C27); 137.16 (C2); 137.23 (C20); 140.96 (C18); 141.49 (C14); 141.62 (C4); 143.11 (C17); 147.90 (C12); 155.56 (C9); 157.82 (C8); 159.66 (C13). 29Si NMR (DMAc, acetone-d6 int. ref., δ, ppm): −15.33; −15.31.Results and discussion
Monomer synthesis
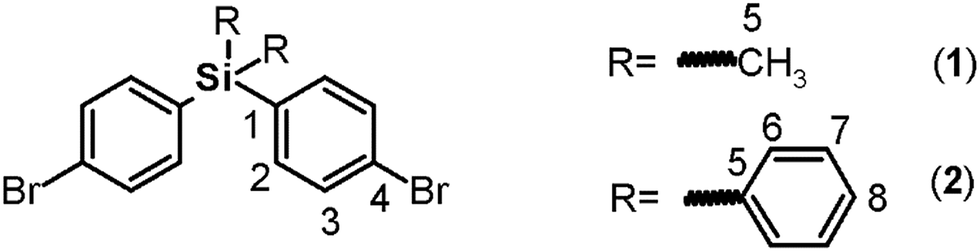
Both monomers use dibromo derivatives (1 or 2) as common precursors (bis(4-bromophenyl)di-R-silanes); these were directly obtained by means of controlled mono-lithiation of 1,4-dibromobenzene and then reacted with the respective dichloro-silanes (Fig. 1, step a). In this last step, the nucleophilic substitution on the central silicon atom is favored because the Si–X distances (X = Cl, C) in the organo-silane compounds are longer than the C–X bond distances. Thus, the steric hindrance for SN2 attack was negligible, and yields of over 70% were obtained.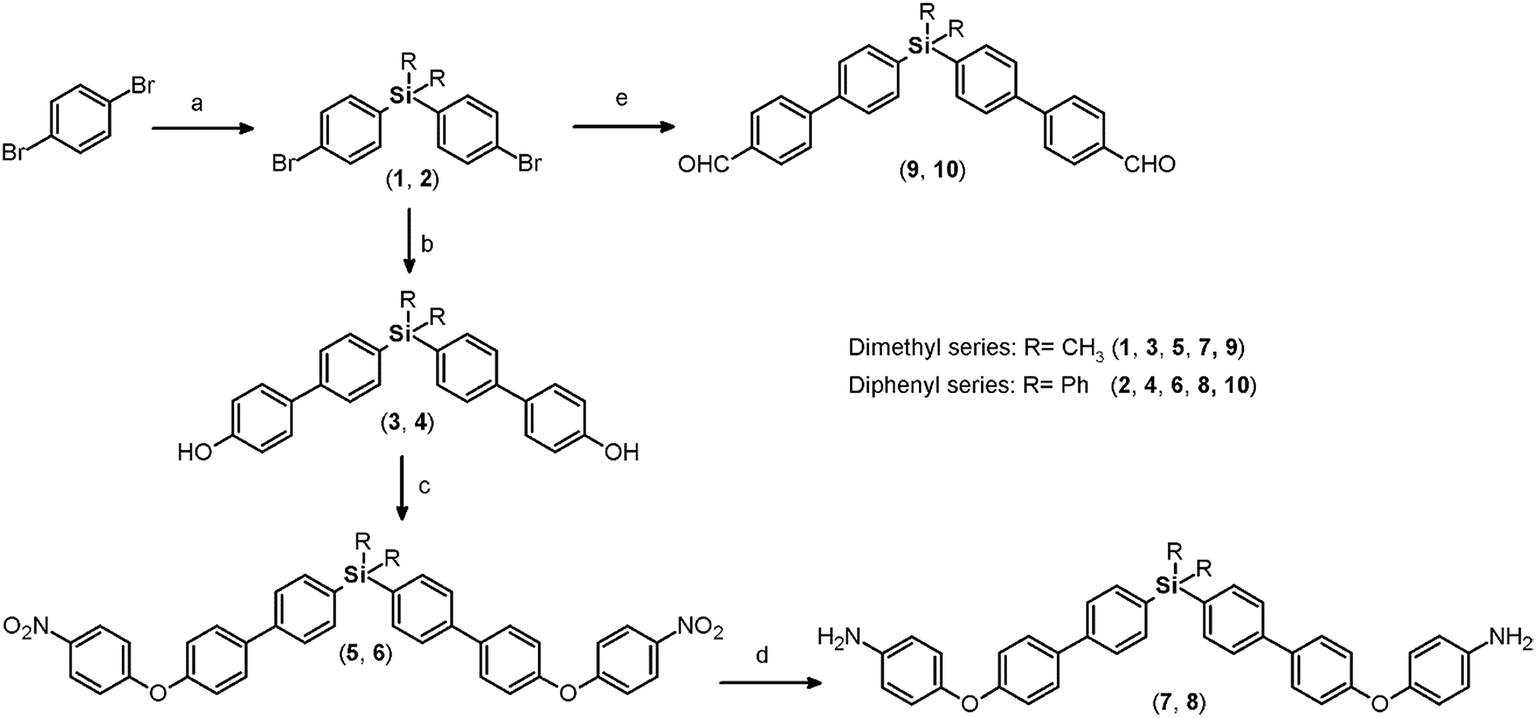 | ||
| Fig. 1 Synthesis of monomers containing silicon and biphenyl moieties. (a) (1) n-BuLi, Et2O anh., 0 °C. (2) R2SiCl2 (R = CH3, Ph), Et2O anh.; (b) 4-hydroxyphenylboronic acid, Pd(PPh3)2Cl2, K2CO3 (aq.) 2 M, 1,4-dioxane, 85 °C; (c) 1-fluoro-4-nitrobenzene, K2CO3, DMF, 60 °C; (d) H2N–NH2 (80%), Pd/C (10%), THF, reflux; (e) 4-formylphenylboronic acid, Pd(PPh3)2Cl2, K2CO3 (aq.) 2 M, 1,4-dioxane, 85 °C. | ||
Elemental analysis, FT-IR spectroscopy and NMR spectroscopy techniques, including COSY, HMQC, and HMBC, were employed to establish the structures of the dibromo compounds and of all the precursors and monomers. In this case, the main IR bands are associated with the C–Si bonds at 1373 to 1427 cm−1 and 1253 cm−1 according to the nature of the carbon atom, aromatic or aliphatic, respectively, and the C–Br stretching band observed at approximately 510 cm−1. The 1H and 13C NMR spectra clearly showed displacement due to the high magnetic fields exerted by the silicon atoms over their environment. Thus, the H and C nuclei of the methyl group of 1 appear at 0.44 ppm and −2.42 ppm, respectively. Also, the aromatic carbon atoms bonded directly to the silicon atom were observed at high field. This effect can be observed in the spectra of all the silylated precursors and monomers.
The biphenyl moieties were incorporated into the monomers and, therefore, the final polymers by Suzuki coupling reactions. Thus, dibromo compounds 1 or 2 were reacted with the corresponding boronic acid derivatives to produce the dialdehyde monomers 9 and 10 (Fig. 1, step e) and the diphenols 3 and 4 (Fig. 1, step b). The FT-IR spectra of the latter compounds showed the characteristic phenolic O–H band between 3500 and 3400 cm−1. Likewise, the 1H NMR spectra evidenced this function, with a narrow singlet at 9.59 ppm for 3 and a broad singlet centered at 5.11 ppm for 4. In the latter case, the electronic contribution of the lateral phenyl groups bonded to the silicon atom probably promotes electronic shielding of this nucleus.
By means of arylation of the oxygen atoms using a potassium carbonate aqueous solution as a basic medium and 1-fluoro-4-nitrobenzene, the dinitro derivatives 5 and 6 were prepared with yields of 88% and 74%, respectively (Fig. 1, step c). For these compounds, IR bands originating from the NO2 groups were observed at 1518 and 1344 cm−1 and from the stretching ether linkage (C–O–C) at about 1236 cm−1. The aromatic zones of the NMR spectra of both dinitro compounds showed new signals for the Ph-NO2 moieties, like an AB system, at 6.68 and 6.90 ppm, without evidence of the signals at about 9.6 and 5.1 ppm attributed to the hydroxylic groups of the respective precursors.
The reduction of the NO2 groups to produce diamine monomers 7 and 8 was carried out following a classic procedure which involves the use of hydrazine and Pd/C (Fig. 1, step d). In both cases, chromatographic techniques were employed to purify the products. In the case of diamine 7, additional recrystallization from acetonitrile was required. The amino group was easily detected in the FT-IR spectra through two stretching bands between 3340 and 3450 cm−1 (Fig. 2). Also, the 1H NMR spectra reported evidence of the reduction. A narrow singlet with an integration of 4H associated with NH2 groups was observed at 3.57 ppm for 7; meanwhile, for diamine 8, a broad and flat singlet centered at 3.49 ppm also showed an integration of 4H (Fig. 3). Likewise, in these spectra and the 13C spectra, the aromatic zone was altered when NH2 replaced NO2 due to the contrasting electronic effects mainly exerted on the adjacent phenyl groups.
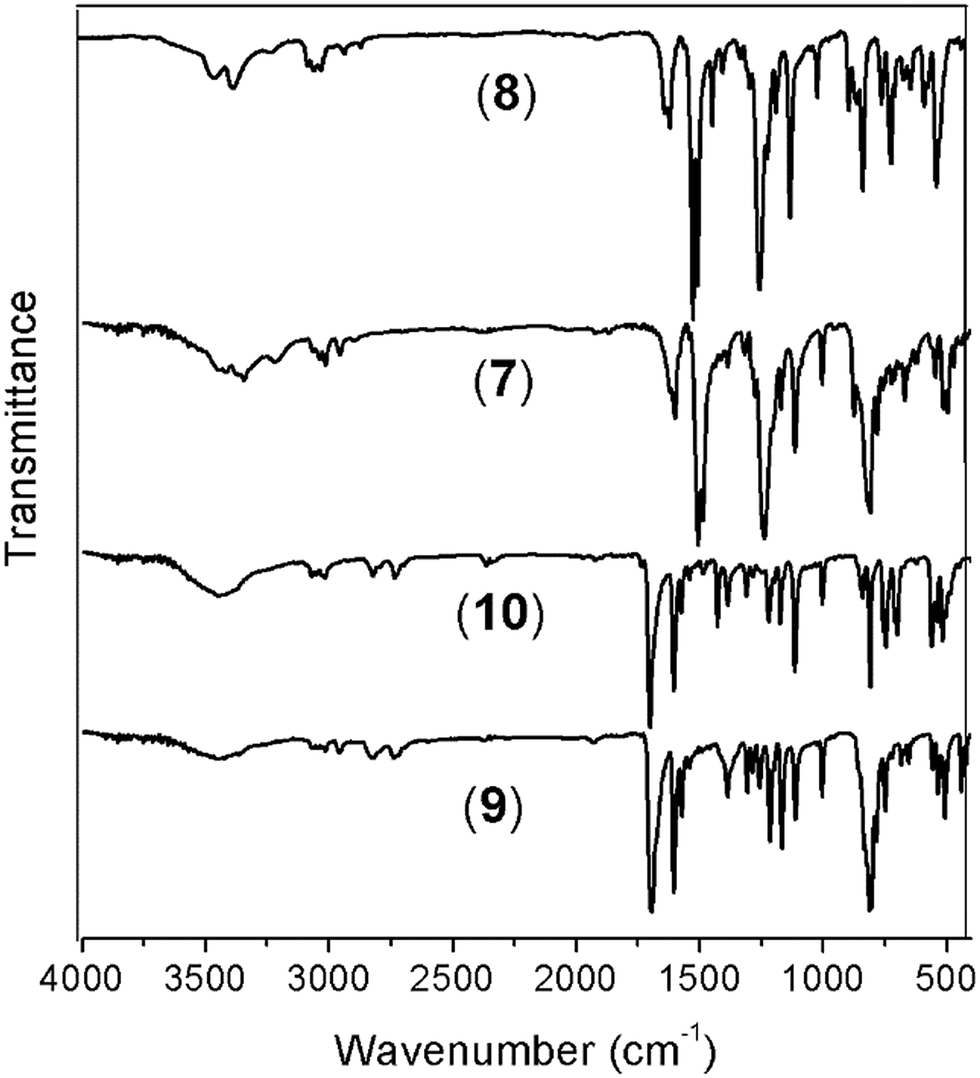 | ||
| Fig. 2 FT-IR spectra (vertically shifted) of the silylated monomers. Diamines 7 and 8; dialdehydes 9 and 10. | ||
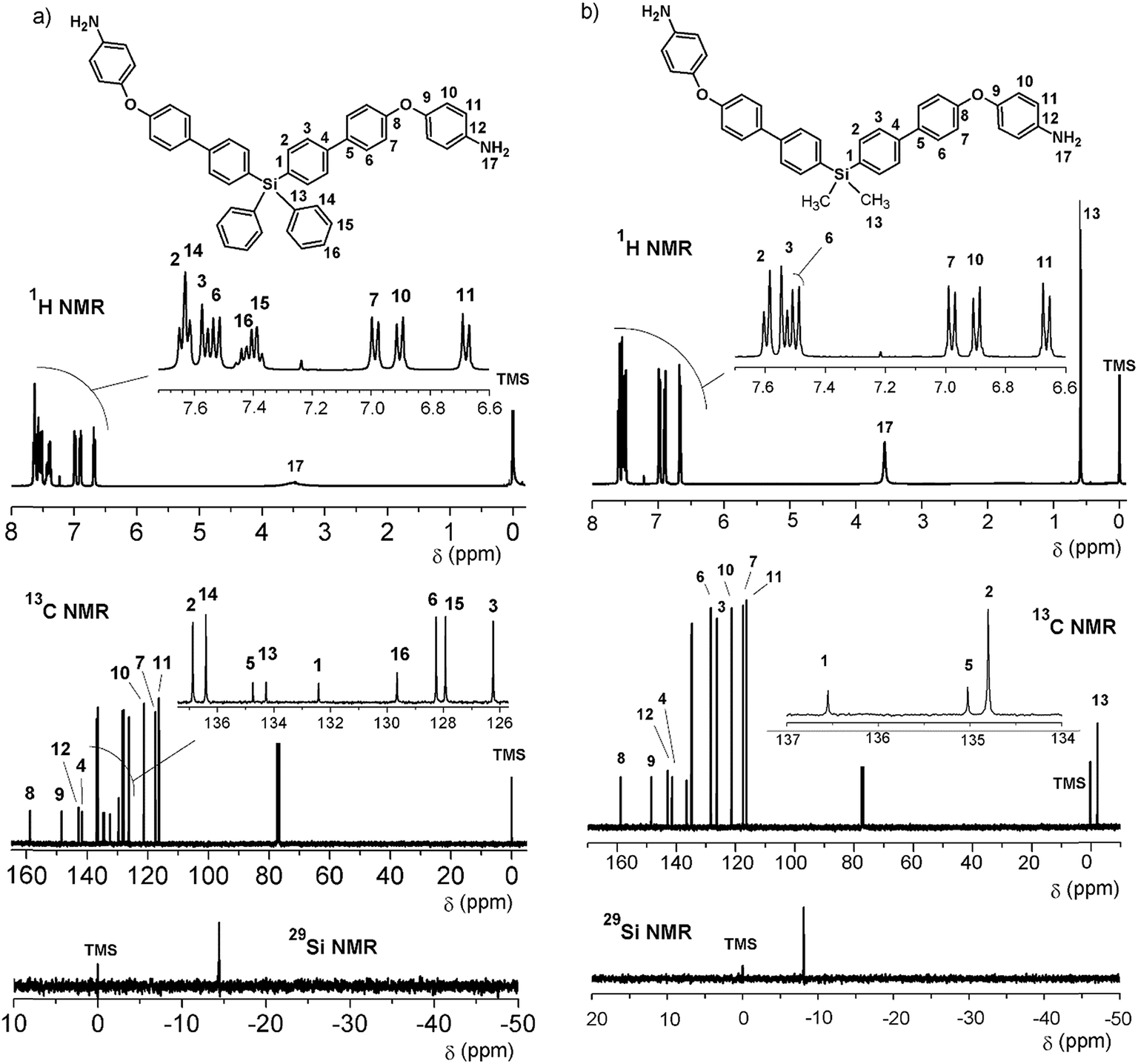 | ||
| Fig. 3 NMR spectra (CDCl3) of diamines 7 (right) and 8 (left). | ||
On the other hand, for the silicon-containing dialdehydes, the FT-IR spectra showed the characteristic stretching band for carbonyl groups close to 1700 cm−1, independently of the R group bonded to the silicon atom (Fig. 2). The 1H NMR spectra demonstrated the proton of the aldehyde group at 10 ppm, while the 13C NMR spectra showed the carbonylic carbon at 191 ppm (Fig. 4).
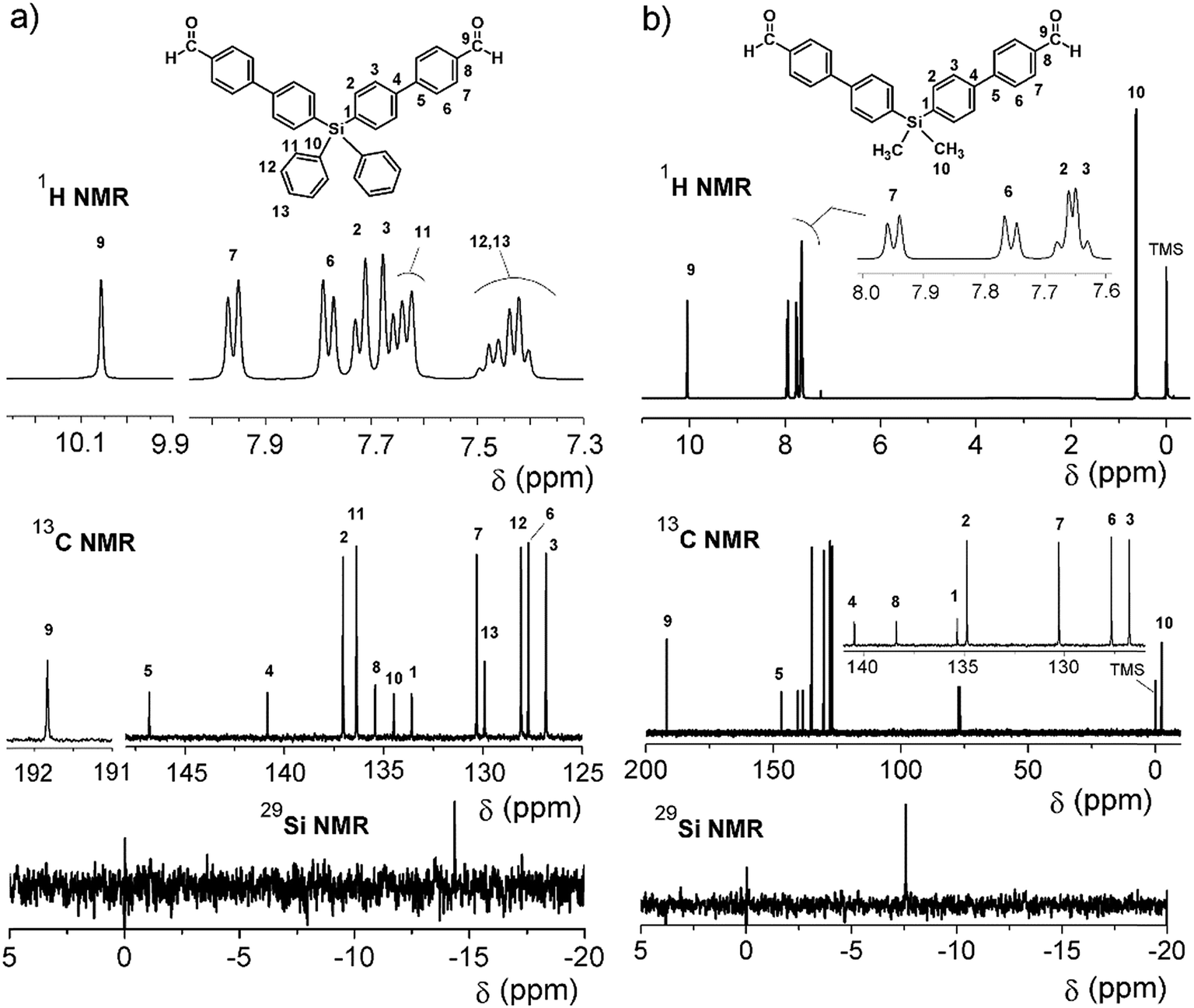 | ||
| Fig. 4 NMR spectra of the dialdehydes 9 (DMSO-d6) (right) and 10 (CDCl3) (left). | ||
In all cases (precursors and monomers), the 29Si NMR spectra showed two different signals. For the dimethyl series (1, 3, 5, 7 and 9), a band could be observed between −7.00 ppm and −8.64 ppm; meanwhile, for the diphenyl series (2, 4, 6, 8 and 10), this signal was observed close to −14 ppm. These values are associated with the –Ph–Si(CH3)2–Ph– and –Ph–Si(Ph)2–Ph– moieties, respectively, and can be explained according to the particular electronic effects of the methyl and phenyl groups. This phenomenon has been previously observed in several monomer and polymer families synthesized by our research group.31–33
Oligomeric poly(ether-azomethine)s synthesis and spectroscopic characterization
Four new silylated oligomeric poly(ether-azomethine)s were prepared through a polycondensation procedure in DMAc solution and under an inert atmosphere. The pH of the reaction was adjusted to 5.0 by adding p-TsOH, and anhydrous CaCl2 was employed to capture water, which is the byproduct of the dehydration of the carbonylamine intermediate (Fig. 5). The solutions obtained after heating at 180 °C over 24 h were added to methanol, precipitating light beige solids in all cases. The yields were between 65% and 86%.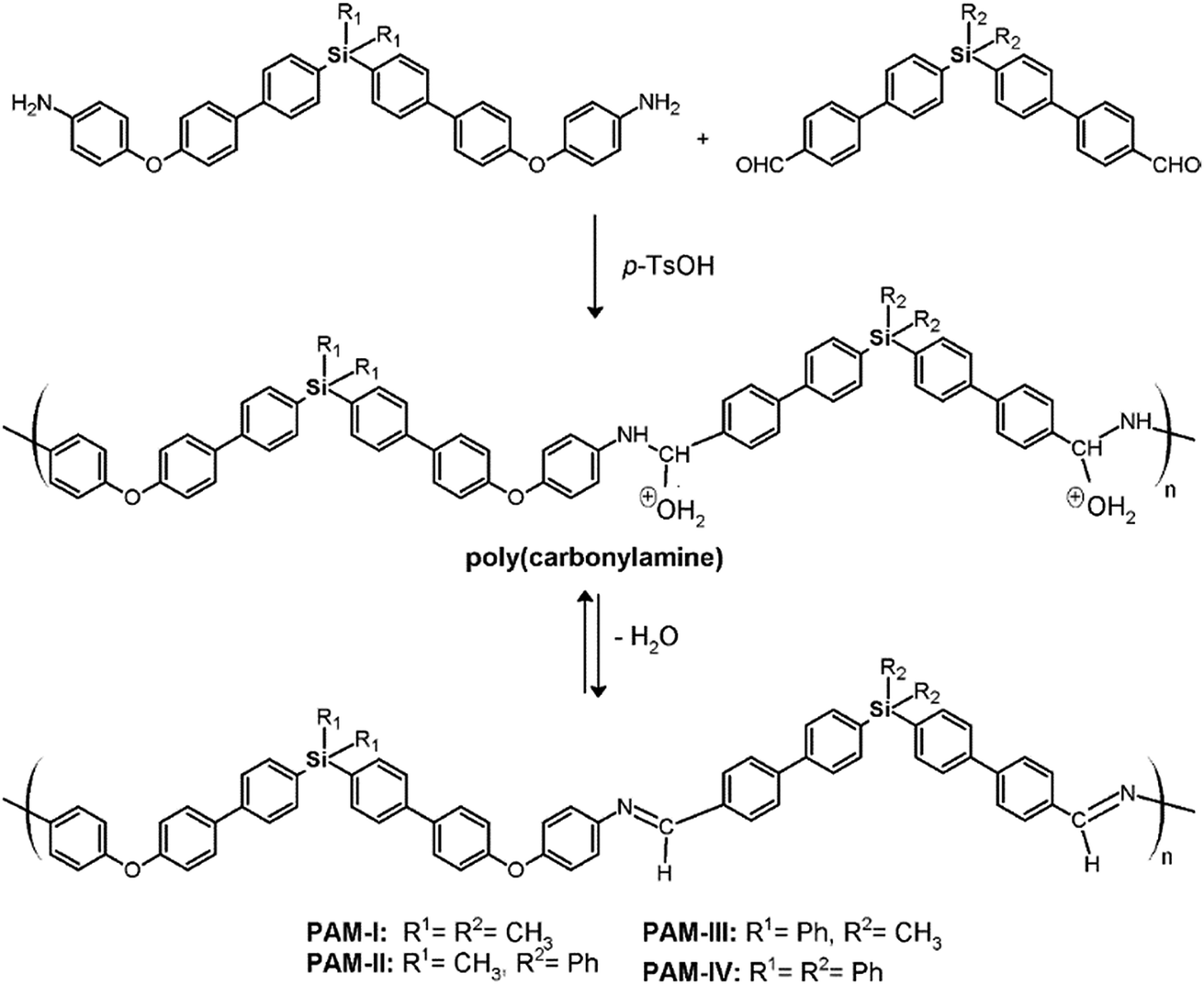 | ||
| Fig. 5 Synthesis of silylated oligomeric poly(ether-azomethines). | ||
The structural characterization of the samples was performed using spectroscopic techniques. KBr pellets were prepared, and the IR main bands were assigned (see Experimental section). Thus, the stretching CN of the imine group was identified at 1623 cm−1. In all cases, a CO band of variable intensity was observed close to 1700 cm−1; this is not attributed to residual monomer due to the exhaustive washing procedure of the samples after the isolation step. Additionally, TLC analysis using CHCl3 as the mobile phase did not show a monomer signal, indicating that the IR band is associated with terminal groups of short chains (oligomers). In this sense, the presence of NH2 chain terminal groups is not clearly defined in the spectra, probably due to their lower intensity in relation to the carbonyl band.
Fig. 6 shows a representative FT-IR spectrum of the obtained materials and the definition of the dCO and dCN distances, which are associated with the intensity of the carbonyl and imine bands, respectively. Using these values, it is possible to establish the aldehyde average contribution parameters inside the chains (AC):
| AC = dCO/(dCO + dCN) |
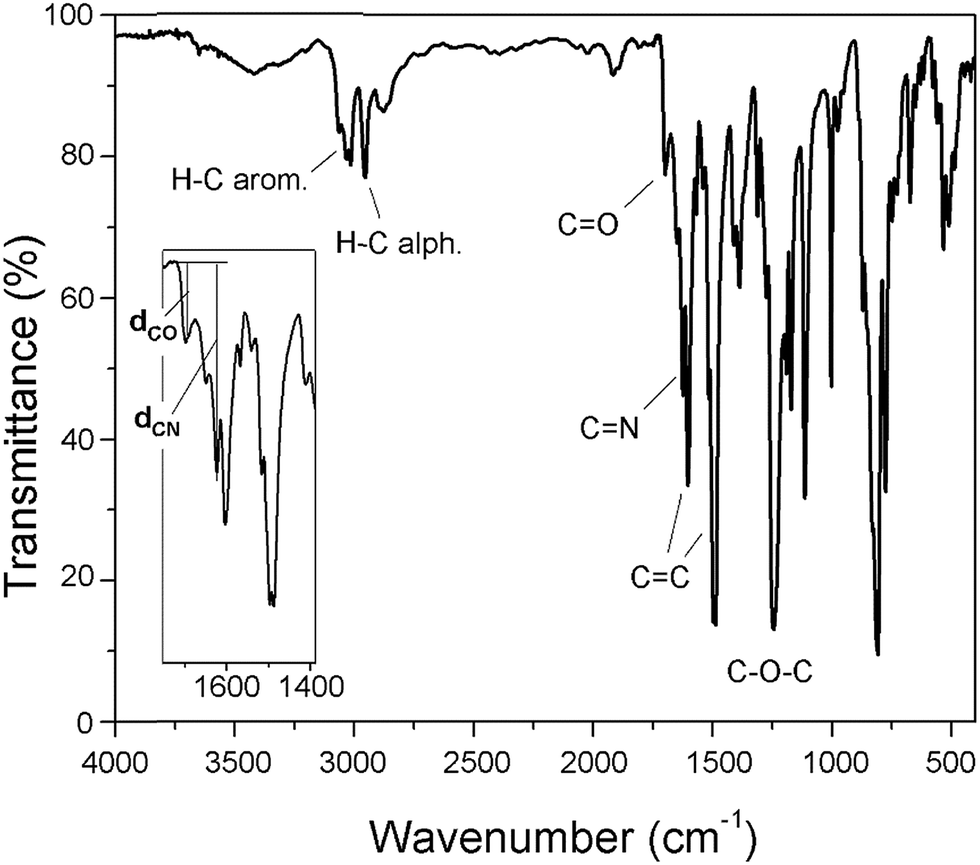 | ||
| Fig. 6 The FT-IR spectrum of PAM-I with details regarding the stretching of the CO and CN bands. | ||
Thus, the samples PAM-I to PAM-IV showed values of 0.28, 0.36, 0.52 and 0.51, respectively. According to these data, PAM-I and PAM-II have lower proportions of CHO in their chains and are the longest oligomers. On the other hand, PAM-III and PAM-IV show similar aldehyde contributions and have comparable lengths.
Initially, DMSO-d6 was used as the solvent to perform structural characterization of the samples. The resolution of the obtained NMR spectra was inadequate; for this reason, the samples were analyzed in DMAc solution using a capillary with acetone-d6 as an internal reference. In this sense, the complexity of the aromatic zone in the NMR spectra was resolved with the aid of DEPT-135° and 2D NMR (COSY, HMQC, and HMBC) analysis, enabling correct assignment of the signals. As an example, representative 2D NMR and 29Si NMR spectra of PAM-II are shown in Fig. 7.
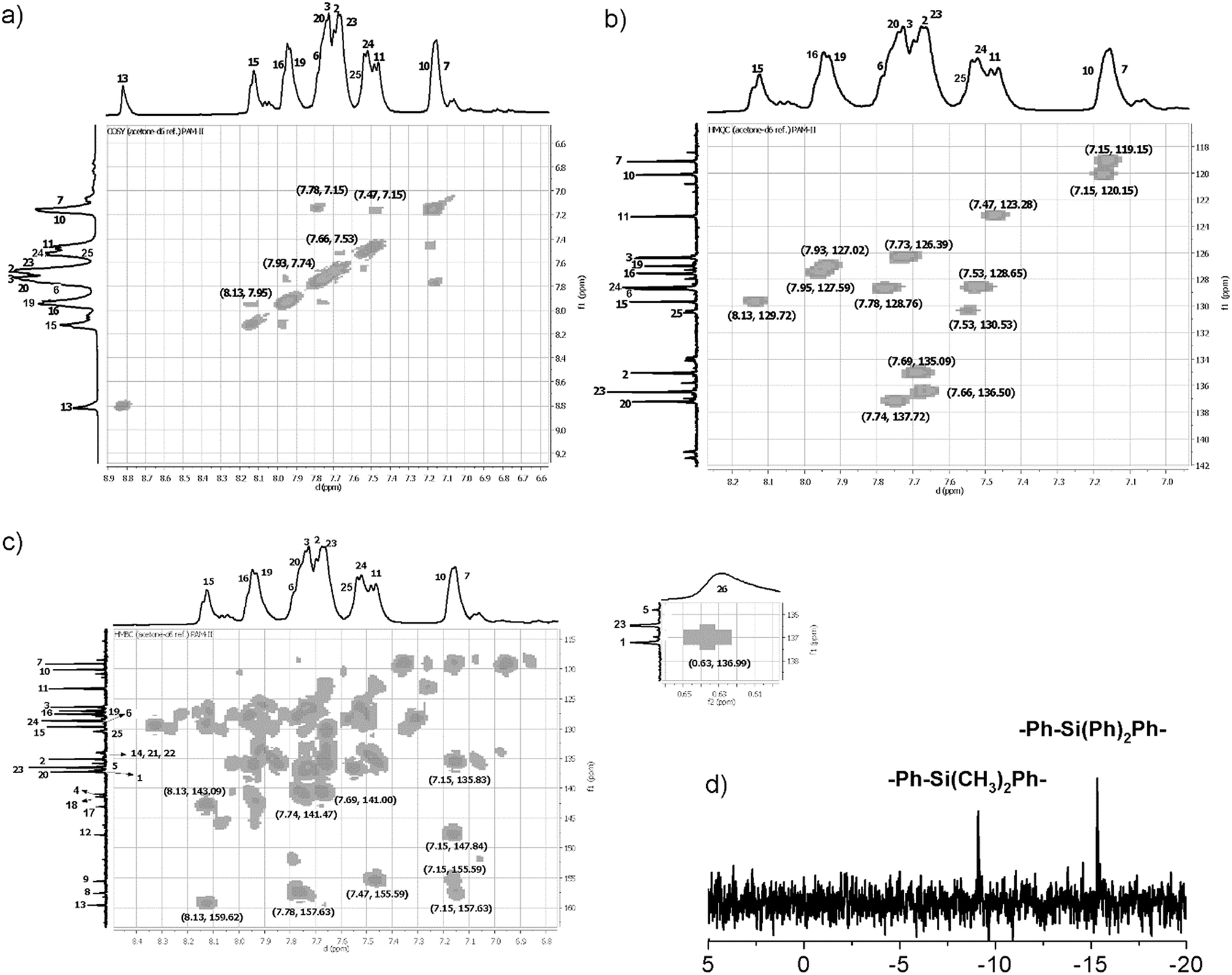 | ||
| Fig. 7 NMR spectra (DMAc-acetone-d6) of PAM-II. (a) COSY, (b) HMQC, (c) HMBC and (d) 29Si NMR spectra. | ||
In all the 1H NMR spectra, a singlet at ∼8.8 ppm was observed, while the 13C NMR analyses evidenced a signal between 160 and 164 ppm, corroborating the formation of imine groups (–CHN–).
Solubility, inherent viscosity and molecular weight
The samples were tested for their ability to form solutions with several solvents; a concentration of 5 mg mL−1 at 19 °C was used, and the time to dissolve each sample with sporadic stirring of similar intensity was recorded. The behavior was classified as soluble/time, partially soluble and insoluble. For these last two classifications, the samples were heated at 40 °C for 24 hours. The results are shown in Table 1.| Solubility | ηinh (dL g−1) | |||||
|---|---|---|---|---|---|---|
| DMSO | DMAc | DMF | m-cresol | H2SO4 conc. | DMAc | |
| a Ins: insoluble; Part. sol.: partially soluble; Sol.: soluble; ηinh: inherent viscosity measured at 25 °C (c = 0.5 g dL−1). | ||||||
| PAM-I | Part. sol. | Sol. in >180 min. | Ins. | Ins. | Sol. in 40 min. | 0.32 |
| PAM-II | Part. sol. | Sol. in 15 min. | Part. sol. | Sol. in 40 min. | Sol. in 40 min. | 0.20 |
| PAM-III | Part. sol. | Sol. in >180 min. | Ins. | Sol. in 40 min. | Sol. in 180 min. | 0.14 |
| PAM-IV | Part. sol. | Sol. in 15 min. | Sol. in 15 min. | Sol. in 40 min. | Sol. in >180 min. | 0.10 |
PAM-I was soluble in DMAc in >180 minutes, while in conc. H2SO4, it dissolved in less time (40 minutes). The other samples showed higher solubility in common polar organic solvents. Probably, the lower volumes of the four methyl groups bonded to the silicon atoms in PAM-I promote large packing forces between the chains; this results in π-stacking interactions, decreasing the solubility. In this sense, the ether linkages promoted by the diamine moiety and the corresponding flexibility of the chains did not increase the solubility of the sample. Among the samples, PAM-I shows the highest inherent viscosity value (Table 1); this is also associated with higher average size of the chains (see the GPC results) and, therefore, with lower solubility. At the tested concentration, all the samples were partially soluble in DMSO and insoluble in THF and CHCl3.
In general terms, PAM-II and PAM-III, whose repetitive units are structural isomers, showed solubility in DMAc and m-cresol; this is probably due to the introduction of bulky phenyl groups in one of the silicon atoms and therefore to diminished packing forces relative to those operating in PAM-I. On the other hand, the average molecular weights of these samples are lower than that of PAM-I. The structural effect is more evident in PAM-IV, in which all the lateral groups are phenyl rings. This sample, with an average length-chain similar to that of PAM-III, was the only one that showed solubility in DMF under the tested conditions. It is clear that the molecular weight and the structural composition of the repetitive units of the samples strongly affected the solubility behavior, where the possibility to establish interactions between the chains was limited or favored according to the volume of the groups bonded to the silicon atoms.
Additionally, a modification to the solubility test was developed by adding LiBr (0.05 M) to dilute polymer-DMF systems (0.05 mg mL−1) at 40 °C in order to obtain solutions. In all cases, clear solutions were obtained and then used in the GPC analysis.
Because no reports exist regarding the synthesis and characterization of poly(ether-azomethine)s containing –PhSiR2Ph– moieties in their chains (R = methyl, phenyl), the solubility behavior of the new materials was compared with results obtained for non-silylated poly(ether-azomethine)s.34,35 These polymers have similar structural elements to the repetitive units of the prepared PAMs, including oxyether linkages and high aromatic content. Likewise, the –PhSiR2Ph– moieties are replaced by bulky and/or flexible central units promoted by the symmetric monomer moieties (Fig. 8).
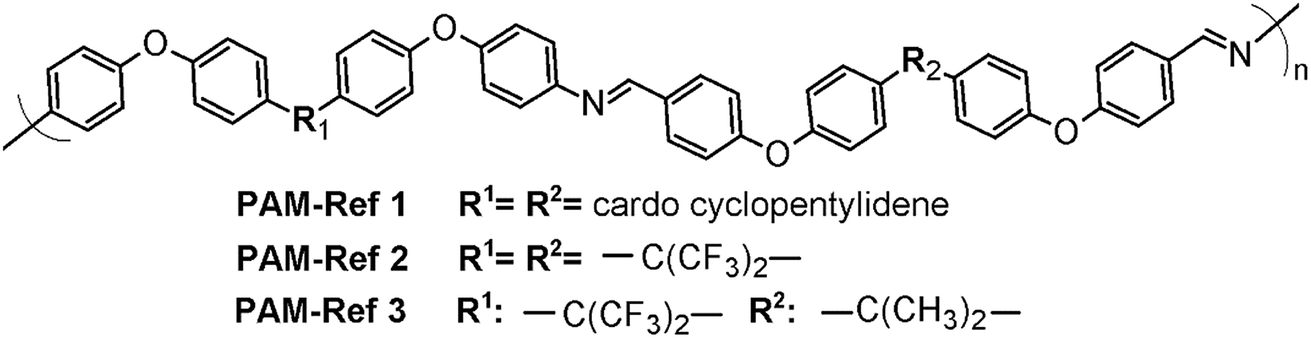 | ||
| Fig. 8 General structure of the poly(ether-azomethine) used for comparison, emphasizing the natures of the central elements (R1 and R2).34,35 | ||
In all cases, the authors report higher solubility in common organic solvents than those observed for the silylated PAMs. The PAMs-ref showed solubility even in THF and CHCl3 at room temperature. These differences can be mainly associated with the presence of oxyether bonds in the dialdehyde moiety, which favors the solubility process. Also, the biphenyl moieties of the silylated PAMs hinder the solubility of the chains.36 In this sense, the respective central elements of the silylated samples and of the PAMs-ref may have similar effects on the studied properties because they are bulky elements that affect the packing of the polymeric chains.
Inherent viscosity measurements were used to establish an order of the size chain in the series. Although the samples showed solubility in concentrated sulfuric acid, this parameter was not determined in this medium because hydrolysis of the imine function was promoted by this reagent. According to a report by Morgan et al.,7 although these measurements are made quickly, it is not possible to avoid hydrolysis of the chains, a process that is enhanced by the action of light.
Thus, the inherent viscosity was recorded at 25 °C in DMAc (c = 0.5 g dL−1); values between 0.10 and 0.32 dL g−1 were obtained, which are characteristic of oligomers (Table 1). According to these data, PAM-I and PAM-II correspond to samples with larger sizes, while PAM-III and PAM-IV are smaller in size but are similar to each other. This observation is in accordance with the results of the FT-IR analysis because the AC parameter is related to the relative size of the chains; the smaller the AC value, the larger the chain. On the other hand, the GPC results obtained from DMF-LiBr solutions generally ratify these observations (Table 2). The obtained oligomers have 3 to 5 repetitive units, where PAM-I shows the highest molecular weight and PDI.
Thermal analysis
The thermal properties of the oligomers were evaluated by TGA and DSC techniques at 20 °C min−1 in nitrogen atmosphere.All the samples showed similar TG behaviors, with an initial decomposition temperature of around 450 °C (Tonset) (Fig. 9a). The 10% weight loss (TDT10%) occured between 490 °C and 510 °C. The char yields at 720 °C were similar in all samples, between 55% and 60%; this is common for silylated samples, principally due to the formation of non-volatile silicon oxides and non-oxidized organic material.30,37,38
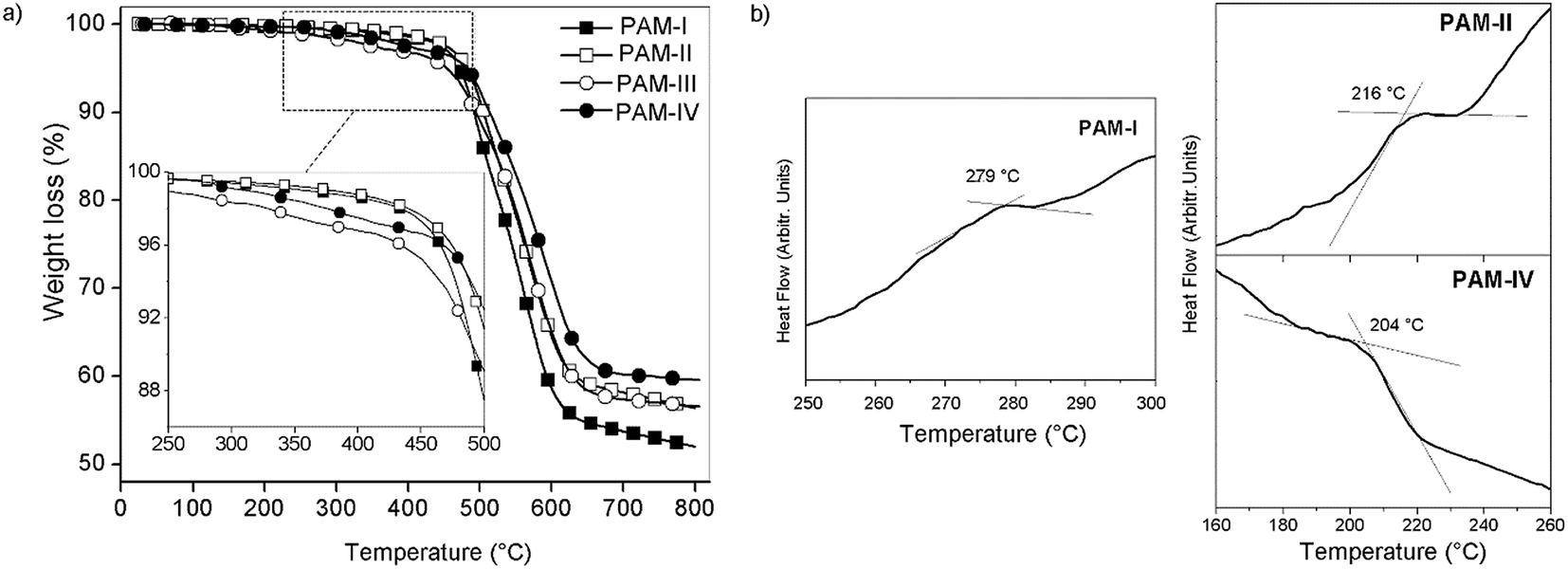 | ||
| Fig. 9 Thermal studies: (a) TGA and (b) DSC of the samples. | ||
Thermal stability studies performed on the poly(ether-azomethines)s based on the decomposition point indicate that they are thermally stable.39 The high aromatic content due to the biphenyl moieties of both monomers allows inter-chain interactions that increase the thermal stability in a similar way to the effect discussed in the solubility section. When the lateral groups bonded to the silicon atoms were the same (R1 = R2, CH3 or phenyl group; PAM-I and PAM-IV), the samples were more stable than those with different groups (R1 ≠ R2, CH3 or phenyl group; PAM-II and PAM-III). This behavior can be attributed to the structural symmetry of the repetitive units and the consequently higher packing forces between the chains. These results are summarized in Table 3.
| DSC | Thermogravimetric analysis (TGA) | |||
|---|---|---|---|---|
| Tgonset (°C) | TDT10% (°C) | Tonset (°C) | Charge yield at 720 °C (%) | |
| a Tonset: temperature at which polymer decomposition starts; n.o.: not observed.; n.r.: not reported. | ||||
| PAM-I (R1: –CH3, R2: –CH3) | 279 | 505 | 465 | 53 |
| PAM-II (R1: –CH3, R2: –Ph) | 216 | 490 | 475 | 58 |
| PAM-III (R1: –Ph, R2: –CH3) | n.o. | 495 | 455 | 57 |
| PAM-IV (R1: –Ph, R2: –Ph) | 204 | 515 | 475 | 60 |
| PAM-Ref 1 | 192 | 462 | n.r. | n.r. |
| PAM-Ref 2 | 211 | 481 | 420 | 0 (at 630 °C) |
| PAM-Ref 3 | 215 | 458 | 250 | 0 (at 630 °C) |
The DSC curves (Fig. 9b) show transitions between 279 °C and 204 °C, which are attributed to the glass transition temperature (Tg); however, for PAM-III, this thermal transition was not observed. No melting temperatures were observed in the analyses. In accordance with the results, the introduction of the flexible oxyether bond as a common structural element lowers the rigidity of the chains, resulting in low Tg values despite the high aromatic content of the repetitive units.
Inside the series, the order of the Tg values correlates to the nature of the groups bonded to the silicon atoms and the sizes of the chains, which were established by GPC analysis. Thus, PAM-I, which contains only lateral methyl groups and has the highest average chain size, showed the highest Tg and therefore the greatest chain rigidity; meanwhile, PAM-IV, which contains four lateral phenyl groups and has the lowest average size, showed the lowest Tg. In this sense, the smaller volume of a methyl group compared with a phenyl group promotes the interaction between chains, diminishing the internal rotation.
It is clear that the decomposition temperatures and the Tg values showed by the samples demonstrate an adequate thermal window in view of processing of this type of aromatic material.
If these thermal results are compared with those of non-silylated-PAMs, similar tendencies in the Tg values can be seen in selected references34,35 (Fig. 8); the bulkiest central element results in the lowest Tg. On the other hand, if PAM-I is compared with PAM-Ref 1 or PAM-Ref 2, where the main difference is the atom of the central unit (Si vs. C, respectively), the silylated sample shows a higher Tg. This result is contrary to the relationship of the volume of the atoms and can also be explained by the higher number of oxyether linkages in the reference samples, which promote higher flexibility of the chains. Regarding the TDT10%, the silylated samples show higher values, in accordance with the biphenyl moieties present in their repetitive units.
UV/vis spectroscopy
UV/vis spectroscopy was carried out in dilute DMSO solutions (c = 0.5 mg mL−1) in the wavelength range between 270 and 440 nm (Fig. 10). The PAMs showed bands within 280 to 290 nm that can be assigned to the π → π* transition of the aromatic rings of the main chain. On the other hand, a broad absorption can be found between 351 nm and 356 nm, which was assigned to the n → π transition of the imine groups.40,41 The slight difference between these bands can be attributed to the modification of the chain planarity. That is, the conjugation is disrupted due to the presence of sterically hindered structures such as the phenyl rings as lateral groups in PAM-IV, showing bands centered at a lower wavelength compared with the other samples (structural variation of the molecule rigid backbone). These results are shown in Table 4.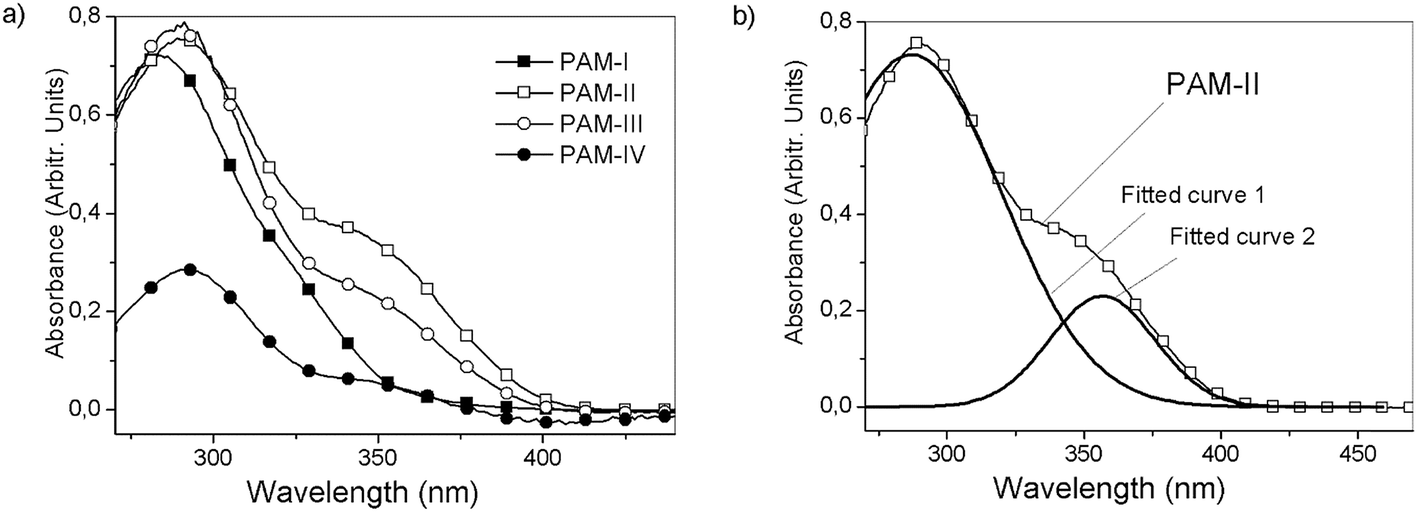 | ||
| Fig. 10 (a) UV/vis spectra for PAM-I to PAM-IV in DMSO and (b) fitted curves for PAM-II. | ||
| 1° band | 2° band | λonset (nm) | Eg (eV) | |
|---|---|---|---|---|
| a Cutoff: Lo DMSO: 330 nm and L1: 285 nm. | ||||
| PAM-I | 279.9 | — | 357.0 | 3.5 |
| PAM-II | 287.0 | 356.6 | 398.5 | 3.1 |
| PAM-III | 287.0 | 354.0 | 392.2 | 3.2 |
| PAM-IV | 290.6 | 351.7 | 377.5 | 3.3 |
From the UV-vis spectra, optical band gap values were obtained using eqn (1).42,43
| Eg = 1242/λonset | (1) |
As can be observed, the optical band gaps are between 3.1 and 3.5 eV; these values are related to internal effects due to morphology and carrier transport, among other factors.44 PAM-I and PAM-IV showed the highest optical band gaps because the two groups bonded to their silicon atoms are identical (R1 = R2 = methyl and phenyl, respectively). This structural symmetry around the silicon atom can strongly modulate the morphological effects on the measured parameter in a similar way to that proposed in the thermal section. On the other hand, small Eg values were obtained in the analysis of PAM-II and PAM-III. This behavior can be attributed, among other factors, to the reduction of steric hindrance and the greater delocalized π-electron density in the aromatic segments of the chains (conjugated polymers), which may involve electronic delocalization.
According to these results, it can be seen that the incorporation of phenyl groups can dramatically decrease the planarity of the system and therefore diminish the π-electron delocalization along the backbone.
Atomic force microscopy and force spectroscopy
PAM-I and PAM-II were chosen for the preparation of films using a spin-coating technique due to their high Mn values of 4800 and 4200, respectively. DMAc solutions of the samples were deposited over an activated silicon wafer substrate by spin-coating with several cycles at a constant speed. Afterward, the films were subjected to vacuum to remove the remaining solvent.Four experiments were performed by varing the rotational speed and time (speed in rpm/time in seconds): PAM-I (1) 2000/18 and 4000/60; PAM-II (2) 1000/18, 1500/18, 2000/18 and 4000/60, (3) 1500/18, 2000/18 and 4000/60, (4) 2000/18 and 4000/60.
In this preparation, the homogeneity of the layers is controlled by the physical structure of the sample. Additionally, the viscosity of the solvent and the rotational speed used for its deposition are factors that must be considered.
Fig. 11 shows AFM images of the PAM-I (1) and PAM-II (4) films. For PAM-I (1), the morphological structure presents a relatively homogeneous film formed by large clusters with shared boundaries, probably due to the high degree of solubility at the moment of material deposition. Also, it can be observed that the clusters are formed of small particles with similar sizes. On the other hand, PAM-II (4) shows a homogeneous surface, but with smaller clusters. Probably, the smaller size of the chains and the replacement of the methyl group by a phenyl group with greater volume do not promote the formation of higher structures.
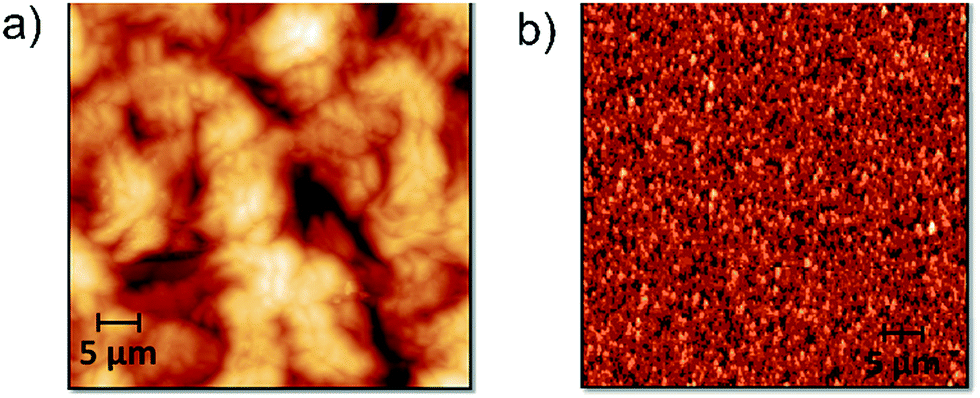 | ||
| Fig. 11 AFM topographic images (50 × 50 μm2) of spin-coated films obtained from DMAc solutions: (a) PAM-I (1) and (b) PAM-II (4). | ||
During the spin coating process, solvent evaporation reduces the spin radius and increases the viscosity of the material; thus, it is impossible for centrifugal force to remove the remaining material from the wafer. Fig. 12a shows three different AFM images of PAM-II and their respective magnifications. The variation in the topography of the sample films is due to changes in the spinning speed during the deposition process. Fig. 12b shows the trends followed by the Young's modulus, the roughness, and the area increase percentage according to the initial spin speed used for the formation of each film.
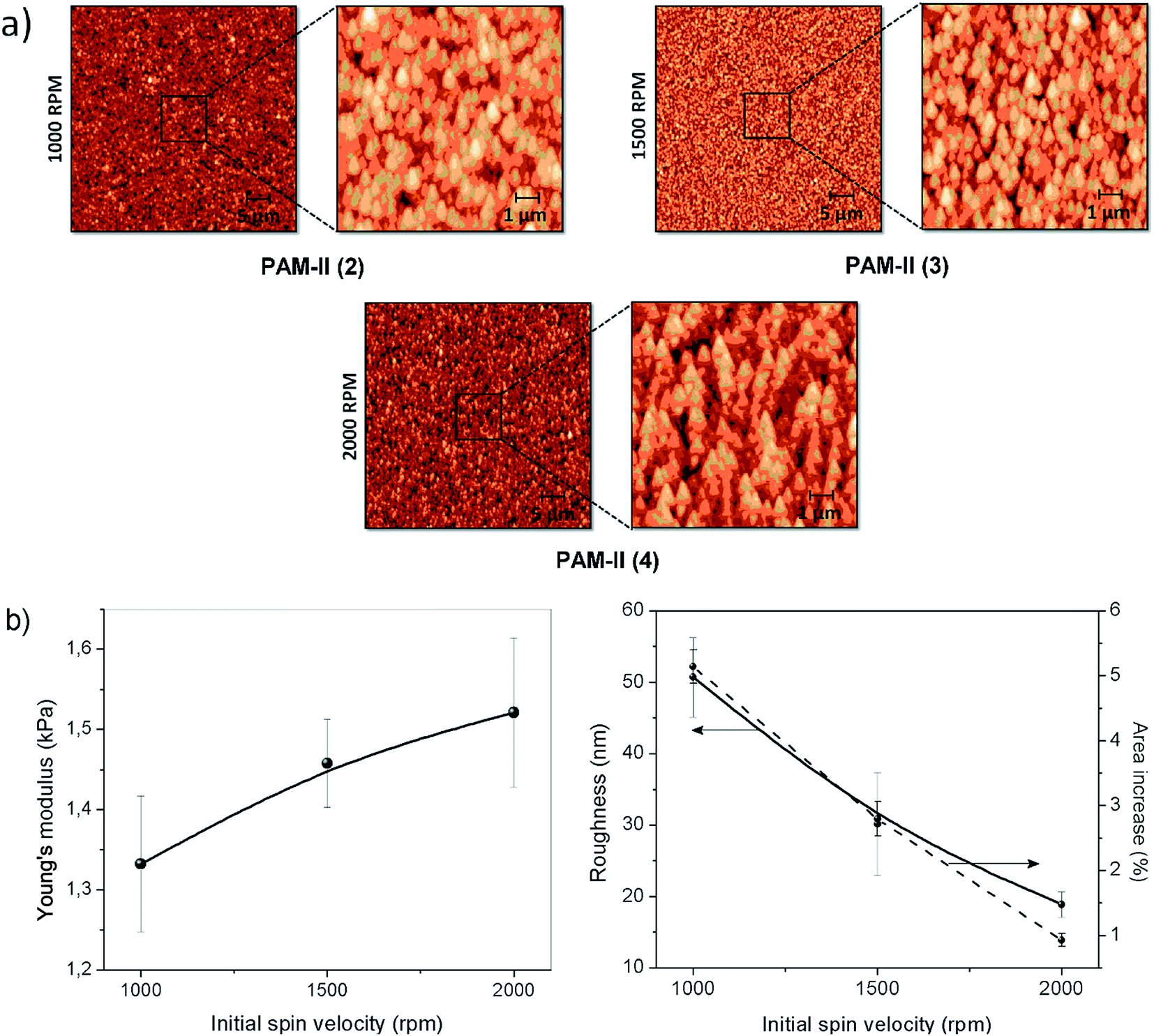 | ||
| Fig. 12 (a) AFM topographic images (50 × 50 μm2 and 10 × 10 μm2) of spin-coated PAM-II films obtained from DMAc solutions and (b) Young's modulus, roughness, and area increase percentage obtained from the analysis of the images. | ||
At higher initial spinning speeds, the stiffness of the surface increases. This may be indirectly related to the roughness of the film. Higher spinning speeds produce homogeneous surfaces, decreasing the roughness but maintaining the particle size. The homogeneity of the films is also indicative of the presence of interconnected microstructures inside the material, producing network or boundary interactions that increase the Young's modulus of the surface. In the same way, by definition, the roughness increase produces an increase in the percentage of exposed surface area; both plots follow a similar tendency.
The four-point probe technique was used to measure the resistivities of the thin films. The setup consists of four point collinear probes, where a constant current is applied between the two outer probes and the voltage variation is measured in the inner probes. The surface resistivity was determined by eqn (2), where CF corresponds to a corrector factor based on the ratio of the probe and the substrate/film dimensions.45,46
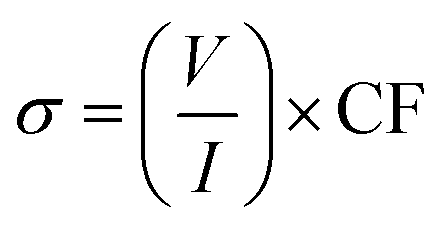 | (2) |
The experiments were carried out using the following parameters: I = 6 A; w (substrate thickness) = 0.0525 cm, and s (separation between the probes) = 0.1 cm. Silicon wafer was used as a calibration standard and also as a substrate. The resistivity results obtained for the two samples are summarized in Table 5.
| Voltage (V) | Resistivity (Ω cm) | Average (Ω cm) | |
|---|---|---|---|
| PAM-I (1) | 3.56 | 2.19 | 2.19 |
| PAM-II (2) | 32.24 | 19.85 | 20.45 |
| PAM-II (3) | 35.84 | 22.06 | |
| PAM-II (4) | 31.59 | 19.45 | |
| Silicon wafer_1 (bare) | 15.32 | 9.43 | 10.02 |
| Silicon wafer_2 (bare) | 17.24 | 10.61 |
The refractive indices of PAM-I and PAM-II were 1.433 and 1.440, respectively; these were measured using an Abbe refractometer. These values are important for measuring the thicknesses of the films; using an ellipsometer, thicknesses near 500 nm were measured. These values were used to determine the resistivities of the films. The surface resistivities of the calibration standards (silicon wafers) are certified by the company (Siegert Wafer GmbH).
Scientific reports have shown that the resistivity of a specimen increases with decreasing thickness.47 However, in this case, the thickness of the sample was not strongly affected by the rate of deposition, probably due to the high viscosity of the polymer-low volatile solvent (DMAc) solution. Table 5 shows the results obtained for PAM-I and PAM-II. The first sample has low resistivity (2.19 Ω cm), while PAM-II showed a resistivity close to 20 Ω cm. As can be seen, these results are not related to the rotation speed used for their deposition. Also, these results are in disagreement with those obtained from the UV-vis analysis (Table 4), mainly due to intrinsic differences of the samples under study. That is, a sample deposited over a silicon wafer as a thin film presents structural differences that are reflected in its morphology at the surface level; the sample in solution, observed by UV/vis spectroscopy, does not possess this property (Fig. 10).
Conclusions
From four bifunctional monomers, two diamines and two dialdehydes containing diphenylsilane and biphenyl moieties, four oligomeric poly(ether-azomethine)s were synthesized and structurally characterized using NMR and FT-IR spectroscopy. The FT-IR spectroscopy results suggested that the qualitative relationship between the sizes of the chains of these samples agreed with their inherent viscosity values and GPC results, which established that PAM-I is the sample with the highest molecular weight. The solubility results showed an effect of the nature of the organic groups bonded to the silicon atoms, which modify the packing forces between the chains. In this sense, the replacement of the lateral methyl groups by phenyl groups with greater volume enhanced the solubilities of the samples. Optical properties measured by UV-vis (optical absorption) showed band gap values between 3.1 and 3.5 eV, which are insufficient for potential applications in the optoelectronics field. Additionally, the thermal analysis showed that all the samples were highly stable, with TDT10% > 490 °C and an adequate thermal window of processability between TDT and Tg.Two samples were used to generate films from DMAc solutions using a spin-coating technique. Thus, morphological and resistivity studies were performed using AFM and the four-probe method, respectively. The speed of rotation did not affect the thickness of the films; therefore, no significant changes in their conductivities were observed. However, the surface morphology of the films was affected by the structure of the oligomeric poly(ether-azomethine)s and also by the spin speed used for deposition; an increase in the stiffness of the surface was observed with increasing initial spin speed.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial assistance from Fondo Nacional de Desarrollo Científico y Tecnológico, FONDECYT, through Project 1150157, and FONDEQUIP, through Project EQM120021. P. A. Sobarzo thanks CONICYT for fellowship No. 21151317.References
- M. L. Petrus, R. K. Bouwer, U. Lafont, D. H. K. Murthy, R. J. Kist, M. L. Böhm, Y. Oliver, T. J. Savenije, L. D. A. Siebbeles, N. C. Greenham and T. J. Dingemans, Polym. Chem., 2013, 4, 4182–4191 RSC.
- M. Grigoras and C. O. Catanescu, J. Macromol. Sci., Polym. Rev., 2004, 44, 131–173 CrossRef.
- A. Iwan and D. Sek, Prog. Polym. Sci., 2008, 33, 289–345 CrossRef CAS.
- G. F. Dálelio, J. V. Crivello, R. K. Schoenig and T. F. Huemmer, J. Macromol. Sci., Part A, 1967, 1, 1161–1249 CrossRef.
- C.-J. Yang and S. A. Jenekhe, Macromolecules, 1995, 28, 1180–1196 CrossRef CAS.
- R. H. Friend, R. W. Gymer, A. B. Holmes, J. H. Burroughes, R. N. Marks, C. Taliani, D. D. C. Bradley, D. A. Dos Santos, J. L. Bredas, M. Logdlund and W. R. Salaneck, Nature, 1999, 397, 121–128 CrossRef CAS.
- P. W. Morgan, S. L. Kwolek and T. C. Pletcher, Macromolecules, 1987, 20, 729–739 CrossRef CAS.
- M. Bruma and B. Schulz, J. Macromol. Sci., Part C: Polym. Rev., 2006, 41, 1–40 Search PubMed.
- K. S. Yook and J. Y. Lee, Adv. Mater., 2012, 24, 3169–3190 CrossRef CAS PubMed.
- Q. Zhao, W. Zhang, Z. Fan, J. Li, X. Chen, G. Luo and X. Zhang, Synth. Met., 2015, 204, 70–75 CrossRef CAS.
- F. Xu, J. H. Kim, H. U. Kim, J. H. Jang, K. S. Yook, J. Y. Lee and D. H. Hwang, Macromolecules, 2014, 47, 7397–7406 CrossRef CAS.
- D. Sun, X. Zhou, H. Li, X. Sun, Y. Zheng, Z. Ren, D. Ma, M. R. Bryce and S. Yan, J. Mater. Chem. C, 2014, 2, 8277–8284 RSC.
- H. C. Yeh, C. H. Chien, P. I. Shih, M. C. Yuan and C. F. Shu, Macromolecules, 2008, 41, 3801–3807 CrossRef CAS.
- P. Cui, X. F. Jing, Y. Yuan and G. S. Zhu, Chin. Chem. Lett., 2016, 27, 1479–1484 CrossRef CAS.
- P. Puthiaraj and W. S. Ahn, Ind. Eng. Chem. Res., 2016, 55, 7917–7923 CrossRef CAS.
- J. Moncada, C. A. Terraza, L. H. Tagle, D. Coll, P. Ortiz, G. Pérez, J. G. de la Campa, C. Alvarez and A. Tundidor-Camba, Eur. Polym. J., 2017, 91, 354–367 CrossRef CAS.
- Y. Liu, J. Y. L. Lai, S. Chen, Y. Li, K. Jiang, J. Zhao, Z. Li, H. Hu, T. Ma, H. Lin, J. Liu, J. Zhang, F. Huang, D. Yu and H. Yan, J. Mater. Chem. A, 2015, 3, 13632–13636 CAS.
- J. Yang, W. He, K. Denman, Y.-B. Jiang and Y. Qin, J. Mater. Chem. A, 2015, 3, 2108–2119 CAS.
- L. Li, H. Ren, Y. Yuan, G. Yu and G. Zhu, J. Mater. Chem. A, 2014, 2, 11091–11098 CAS.
- B. Zhang, P. Zhang, H. Zhang, C. Yan, Z. Zheng, B. Wu and Y. Yu, Macromol. Rapid Commun., 2017, 38, 1700110 CrossRef PubMed.
- I. F. MacSim, E. N. Dragoi, M. Cazacu and S. Curteanu, J. Appl. Polym. Sci., 2015, 132, 42552 CrossRef.
- M. F. Zaltariov, M. Cazacu, C. Racles, V. Musteata, A. Vlad and A. Airinei, J. Appl. Polym. Sci., 2015, 132, 41631 Search PubMed.
- A. Airinei, M. Homocianu, A. Vlad and M. Cazacu, J. Mol. Liq., 2013, 186, 171–178 CrossRef CAS.
- C. Racles, M. Silion, A. Arvinte, M. Iacob and M. Cazacu, Des. Monomers Polym., 2013, 16, 425–435 CrossRef CAS.
- M. Palewicz, A. Iwan, M. Sibinski, A. Sikora and B. Mazurek, Energy Procedia, 2011, 3, 84–91 CrossRef CAS.
- M.-F. Zaltariov, M. Cazacu, S. Shova, C.-D. Varganici, L. Vacareanu, V. Musteata and A. Airinei, Des. Monomers Polym., 2014, 17, 668–683 CrossRef CAS.
- J. J. Roa, G. Oncins, J. Díaz, F. Sanz and M. Segarra, Recent Pat. Nanotechnol., 2011, 5, 27–36 CrossRef CAS PubMed.
- P. Klapetek, M. Valtr, D. Neḉas, O. Salyk and P. Dzik, Nanoscale Res. Lett., 2011, 6, 514 CrossRef PubMed.
- I. M. Tidswell, B. M. Ocko, P. S. Pershan, S. R. Wasserman, G. M. Whitesides and J. D. Axe, Phys. Rev. B, 1990, 41, 1111–1128 CrossRef CAS.
- G. W. Gale, R. J. Small and K. A. Reinhardt, Aqueous Cleaning Surface Conditioning Processes, in Handbook on Silicon Wafer Cleaning Technology, William Andrew Inc., USA, 2nd edn, 2008 Search PubMed.
- C. A. Terraza, L. H. Tagle, F. Concha and L. Poblete, Des. Monomers Polym., 2007, 10, 253–261 CrossRef CAS.
- C. A. Terraza, L. H. Tagle, A. Tundidor-Camba, C. M. González-Henríquez, D. Coll and M. A. Sarabia, RSC Adv., 2015, 5, 28515–28526 RSC.
- C. A. Terraza, L. H. Tagle, C. Contador, A. Tundidor-Camba and C. M. González-Henríquez, Polym. Bull., 2014, 71, 1101–1115 CrossRef CAS.
- S. S. Ankushrao, V. N. Kadam, V. P. Ubale and A. A. Ghanwat, International Journal of Engineering and Science Invention, 2016, 5, 67–73 CrossRef.
- P. K. Gutch, S. Banerjee, D. C. Gupta and D. K. Jaiswall, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 383–388 CrossRef CAS.
- S. Banerjee, P. K. Gutch and C. Saxena, Des. Monomers Polym., 1999, 2, 135–142 CrossRef CAS.
- C. A. Terraza, L. H. Tagle, A. Tundidor-Camba, C. M. González-Henríquez, M. A. Sarabia-Vallejos and D. Coll, RSC Adv., 2016, 6, 49335–49347 RSC.
- C. A. Terraza, L. H. Tagle, D. Muñoz, A. Tundidor-Camba, P. A. Ortiz, D. Coll, C. M. González-Henríquez and I. A. Jessop, Polym. Bull., 2016, 73, 1103–1117 CrossRef CAS.
- I. Kaya, A. Avci and K. Temizkan, Korean J. Chem. Eng., 2015, 31, 777–786 CrossRef.
- D. Şenol and K. İsmet, J. Saudi Chem. Soc., 2017, 21, 505–516 CrossRef.
- D. Dilek, F. Doğan, A. Bilici and İ. Kaya, Thermochim. Acta, 2011, 518, 72–81 CrossRef CAS.
- M. M. Ahmida and S. H. Eichhorn, ECS Trans., 2010, 25, 1–10 CAS.
- T. Daimon and E. Nihei, Materials, 2013, 6, 1704–1717 CrossRef CAS PubMed.
- H. Cha, C. E. Park, S. K. Kwon and T. K. An, Org. Electron., 2017, 45, 263–272 CrossRef CAS.
- F. M. Smits, Measurements of Sheet Resistivity with the Four-Point Probe, Bell Syst. Tech. J., 1958, 37, 711–718 CrossRef.
- L. B. Valdes, Proc. IRE, 1954, 420–427 CrossRef.
- D. B. Tanner and D. C. Larson, Phys. Rev., 1968, 3, 652–655 CrossRef.
| This journal is © The Royal Society of Chemistry 2018 |