
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, characterization and assessment of hydrophilic oxidized carbon nanodiscs in bio-related applications
Panagiota Zygouri ab,
Theodoros Tsoufisc,
Antonios Kouloumpisab,
Michaela Patilae,
Georgia Potsiab,
Apostolos A. Sevastosd,
Zili Sideratouc,
Fotios Katsarosc,
Georgia Charalambopouloud,
Haralambos Stamatise,
Petra Rudolf*b,
Theodore A. Steriotis*c and
Dimitrios Gournis*a
ab,
Theodoros Tsoufisc,
Antonios Kouloumpisab,
Michaela Patilae,
Georgia Potsiab,
Apostolos A. Sevastosd,
Zili Sideratouc,
Fotios Katsarosc,
Georgia Charalambopouloud,
Haralambos Stamatise,
Petra Rudolf*b,
Theodore A. Steriotis*c and
Dimitrios Gournis*a
aDepartment of Materials Science & Engineering, University of Ioannina, 45110 Ioannina, Greece. E-mail: dgourni@cc.uoi.gr
bZernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: p.rudolf@rug.nl
cInstitute of Nanoscience & Nanotechnology, National Center for Scientific Research “Demokritos”, Athens, GR-15310, Greece. E-mail: t.steriotis@inn.demokritos.gr
dEnvironmental Research Laboratory, INRASTES, National Center for Scientific Research “Demokritos”, Athens, GR-15310, Greece
eBiotechnology Laboratory, Department of Biological Applications and Technologies, University of Ioannina, GR-45110 Ioannina, Greece
First published on 19th December 2017
Abstract
Oxidation of industrially prepared carbon nanodiscs using a simple, versatile, and reproducible approach based on the Staudenmaier method yields a new hydrophilic form of nanocarbon. As a result of the strong acid treatment, which also enables the separation of carbon nanodiscs from the mixed starting material, the graphene planes detach from the discs, while the surface of the carbon nanodiscs is decorated with various oxygen-containing functional polar groups. Thus, the completely insoluble carbon nanodiscs are converted to a hydrophilic derivative dispersable in many polar solvents, including water. The new carbon structure is expected to have a wide range of applications in several fields including bioapplications. To this end, the functionalized carbon nanodiscs exhibit very low cytotoxicity, while they achieve high drug loadings, enabling their application as an effective drug nanocarrier. Furthermore, the carbon disks were evaluated as supports in nanobiocatalytic applications, increasing significantly the stability of the systems, due to carbon disks' nano-sized dimensions.
Introduction
There is no doubt that among the nanomaterials that have been synthesized or isolated in recent decades, carbon nanostructures hold the most prominent position. This is directly related to the intriguing capability of carbon to form many allotropes with bonds based on sp3, sp2 and sp hybridization. Carbon nanomaterials include various low-dimensional forms such as 0D fullerenes, 1D carbon nanotubes and 2D graphene. The importance of their discovery is mirrored by two Nobel prizes awarded in the last two decades (Kroto, Smalley, Curl in 1996 for the discovery of fullerenes and Novoselov and Geim in 2010 for the discovery of graphene). Due to their structure, size, low density, high specific surface area, tunable pore structure, chemical stability together with excellent electronic, thermal and mechanical properties carbon nanostructured materials have been considered as promising candidates for a wide range of applications including organic photovoltaics, biological and medical applications, catalyst supports, field emission devices, nanoprobes, sensors, semiconductor devices, composite materials (polymeric or ceramic), nanoelectronics, gas separations, supercapacitors, and energy storage materials.1–20Carbon nanodiscs (CNDs) represent an interesting alternative to bulk graphite; they are produced through the so-called pyrolytic Kværner Carbon Black & H2 (CB & H) process,21,22 in which a continuous conversion of hydrocarbons (typically heavy oil) is decomposed into carbon and H2 with the aid of an industrial-scale plasma torch generator, operating at a temperature around 2000 °C. CB & H method provides an unusual carbon product, consisting of different turbostratic graphitic microstructures, which may also exhibit disclination defects in their hexagonal network, resulting from the presence of pentagons in the specific seed from which they grew. Flat CNDs (no pentagons) are predominantly present in this mixture together with conical carbon structures (1–5 pentagons) and amorphous carbon (soot), with volume fractions around 82%, 5% and 13%, respectively.23,24 CNDs can be isolated by subjecting the crude material to liquid phase oxidation because soot and conical structures are preferentially oxidized.24
CNDs are ultra-thin, quasi two-dimensional particles with diameter 1–4 μm, while the co-existing carbon cones are of similar size. A combined electron diffraction and electron microscopy study has indicated that both carbon materials exhibit multilayer structures with a graphitic core and outer non-crystalline layers.25 More specifically, it has been proposed that CNDs may start to form during the pyrolytic process through the growth of a thin planar graphite crystal with regular facets, which is then progressively encapsulated by additional non-crystalline carbon layers. Although a different number of carbon layers may be deposited, depending on the local growth conditions, each CND is considered to have homogeneous thickness, typically in the range 10–30 nm.25 The degree of graphitization can be greatly promoted by post heat-treatment at 2700 °C, under argon.26 Indeed, AFM and XRD analysis of thermally treated nanodiscs and cones has shown that the annealing process under such conditions, leads to high structural organization with only few surface defects, not affecting the geometry (and the thickness) of the particles. Annealed CNDs and cones have appeared to be almost single crystalline in the c-direction and comprise a limited number of stacked graphene layers (usually <100). CNDs may thus be considered as the thinnest carbon structure after the well reported few layers graphene nanomaterial.
CNDs have similar features with the other carbon nanostructured materials like fullerenes, CNTs and graphene, due to the presence of fully conjugated π-electrons confined in either zero-, quasi-one- or two-dimensions.27 However, their disc shaped structure differentiates significantly certain of CNDs' properties, enabling their applicability in new fields. Within this context CNDs, which are more close to graphene, differ from fullerenes' curved structure in both shape and wall thickness,28 which in the case of CNDs may range from a few up to several tens of graphene layers. Additionally, the CNDs, due to their stacking properties possess a well-defined structure, which is associated with improved heterogeneous electron transfer rate at the graphene edges,29 and are thus expected to exhibit improved electrochemical properties, compared to other graphitic materials.30 Finally, regarding Carbon Nano Spheres (CNSs), which are widely used in catalysis, electrochemistry, energy storage and biomedical applications,31 1D and 2D carbon materials, like CNDs, exhibit enhanced graphitization degree and in some cases larger surface areas.32
Compared to graphene oxide (the analogous chemical derivative of graphene), oxidized CNDs (oxCNDs) possess a series of advantages including: (i) a significantly narrower size distribution,26 coupled with a well-defined shape,33 (ii) a lower C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O atomic ratio resulting to higher density of functional (oxygen-containing) surface groups, and, (iii) a higher population of highly-reactive structural sites (located mainly at the round edges of the discs) giving rise to enhanced functionalization potential and affording further doping capabilities with hetero-atoms.26,34 Similarly, oxidized CNDs are expected to be superior to 1D oxidized carbon nanotubes and flexible graphene oxides when used as fillers to improve for instance barrier properties since they provide a rigid, flat, functional yet non-permeable surface.35
:O atomic ratio resulting to higher density of functional (oxygen-containing) surface groups, and, (iii) a higher population of highly-reactive structural sites (located mainly at the round edges of the discs) giving rise to enhanced functionalization potential and affording further doping capabilities with hetero-atoms.26,34 Similarly, oxidized CNDs are expected to be superior to 1D oxidized carbon nanotubes and flexible graphene oxides when used as fillers to improve for instance barrier properties since they provide a rigid, flat, functional yet non-permeable surface.35
Up to day, and due to their unique combination of morphological and structural characteristics, CNDs have been proposed as very attractive candidate nanomaterials for applications relative to electrochemistry,26 improvement of friction properties,36 magnetic properties28 and fillers for composite materials.37
The particular size of these disc-like carbon nanoflakes is crucial for maintaining important properties, such as electrical conductivity and on the other hand, small enough or even ideal in some cases, for application in bionanotechnology, nanomedicine and drug delivery, micromanipulation, nanochemistry etc. Like all other carbon structures, CNDs are insoluble in polar solvents, and thus industrial processing is significantly hampered. An additional drawback hindering their applicability is their immiscibility with most media due to their high tendency to establish strong van der Waals and π–π interactions. However, the chemical modification of CNDs' surface could improve their dispersability in both organic solvents and aqueous media, rendering them more compatible with other materials, and facilitating the development of composites. In order to address this technological gap, we report in this study a simple, versatile, and reproducible approach for the chemical oxidation of CNDs, based on the well-known Staudenmaier method38 that has been applied with great success for the chemical oxidation of graphite.39,40 As a result of the strong acid treatment, which also enables purification of CNDs from the mixed starting material, graphene planes detach from the discs, while the surface of the CNDs becomes decorated with various oxygen-containing functional polar groups such as hydroxyl, carboxyl and epoxy groups. Consequently the completely insoluble CNDs can be converted to a hydrophilic derivative, which is dispersible in many polar solvents including water, extending the range of their diverse applications in several fields such as chemical industry, pharmaceutics, electronics, etc. Furthermore, a variety of analytic techniques was used to fully characterize the produced nanocarbons, namely Fourier transform infrared (FTIR), micro-Raman and X-ray photoelectron spectroscopy (XPS), X-ray diffraction (XRD), thermal analysis (differential thermal (DTA) and thermogravimetric analysis (TGA)), as well as atomic force (AFM), transmission electron (TEM) and scanning electron (SEM) microscopy. We further focus on representative case studies addressing cutting edge processes of great importance in biomedicine and biocatalysis, namely the applicability of the developed nanostructures as efficient drug delivery systems and as supports for nanobiocatalytic systems were evaluated.
Experimental section
Pristine material
The nanodiscs sample (also containing a small fraction of conical structures and amorphous carbon) produced from the CB & H process and further annealed at 2500–2700 °C was purchased from Strem Chemicals, Inc. (France). The predominance of nanodiscs in the material is demonstrated by the SEM image shown in Fig. 1. | ||
| Fig. 1 SEM image of the as purchased pristine annealed CNDs sample. | ||
Oxidation of carbon nanodiscs – oxCNDs
Oxidization of carbon nanodiscs was achieved using a modified Staudenmaier method.39,40 In a spherical flask, 500 mg of CNDs were dispersed in a mixture of 20 mL H2SO4 (95–97%) and 10 mL HNO3 (65%), while placed in an ice-water bath (0 °C) and stirred for 20 minutes. 10 g of KClO3 were then added in small portions to the mixture under vigorous stirring and the reaction was completed after 18 hours. The oxidation product (oxCNDs) was separated by centrifugation (3500 rpm, 10 min) and washed several times with distilled water until a pH of 6 was reached. The resulting solid material was air-dried on a glass plate.Characterization techniques
XRD patterns were collected on a D8 Advance Bruker diffractometer by using Cu Kα (40 kV, 40 mA) radiation and a secondary beam graphite monochromator. The patterns were recorded in the 2-theta (2θ) range from 2 to 80°, in steps of 0.02° with a counting time of 2 s per step. Samples were in the form of films supported on glass substrates. For the preparation of the films, aqueous suspensions of oxCNDs were deposited on glass plates and the solvent was allowed to evaporate slowly at ambient temperature. FTIR spectra in the range 400–4000 cm−1 were measured with a Perkin–Elmer Spectrum GX infrared spectrometer, equipped with a deuterated triglycine sulphate (DTGS) detector. A total of 64 scans were averaged for each sample with 2 cm−1, using KBr pellets containing ca. 2 wt% sample. The spectra were obtained against a single-beam spectrum of the pure KBr pellet. Raman spectra were recorded with a micro-Raman system RM 1000 Renishaw, with excitation at 532 nm (Nd-YAG), in the range 1000–2400 cm−1. A power of 1 mW was concentrated in a 1 μm focus spot in order to avoid photodecomposition of the samples. TGA and DTA were performed using a Perkin Elmer Pyris Diamond TG/DTA. Samples of approximately 5 mg were heated in air from 25 °C to 850 °C, at a rate of 5 °C min−1. For the XPS measurements, the samples were dispersed in water and after very short sonication a small droplet of the suspension was drop-casted onto a gold substrate and left to dry at room temperature. The samples were then introduced in an ultrahigh vacuum (UHV) system via a loadlock and kept under UHV for at least 12 h before measurement, allowing for the removal of volatile substances and/or solvents. The spectra were recorded using a SPECS GmbH system, equipped with a twin Al–Mg anode X-ray source and a multi-channel hemispherical sector electron analyzer (HSA-Phoibos 100). The base pressure in the spectrometer was 9 × 10−9 Torr during all measurements. The pass energy was set at 20 eV providing a full-width at half maximum (FWHM) value of 1.1 eV and thus minimizing sample exposure to the X-ray beam (measuring time). The photoelectron take-off angle was 90°. All binding energies were referenced to the C 1s core level line of the C–C bond at 285.0 eV and are given ±0.1 eV.41 Spectral analysis included a Shirley background subtraction and peak fitting using mixed Gaussian–Lorentzian functions in a least-squares curve-fitting program (Winspec) developed in the LISE laboratory of the University of Namur, Belgium. As substrates freshly evaporated gold films supported on mica were used; four points were measured on each sample in order to guarantee the reproducibility of the results. The stability of all analyzed samples against damage induced by the X-ray beam and by secondary electrons produced by photoemission in the underlying substrate was verified by monitoring the lineshape and relative intensity of the C 1s core-level as a function of irradiation time. No evidence of structural degradation was observed even when irradiation was continued twice as long as the acquisition time used for the obtained spectra. Scanning Electron Microscopy (SEM) images were recorded using a Jeol JSM 7401F Field Emission Scanning Electron Microscope equipped with Gentle beam mode. Gentle beam technology can reduce charging and improve resolution, signal-to-noise, and beam brightness, especially at low beam voltages (down to 0.1 kV). TEM images were recorded by a Philips CM12 microscope operating at 120 kV on samples prepared by drop casting a stable aqueous dispersion of oxCNDs over formvar/carbon copper grids (200 mesh). TEM images were recorded with a CCD camera (Gatan 791). Atomic force microscopy images were obtained in tapping mode with a 3D Multimode Nanoscope (Bruker), using Tap-300G silicon cantilevers with a tip radius <10 nm and a force constant of ≈20–75 N m−1. Samples were deposited onto silicon wafers (P/Bor, single side polished, Si-Mat) by drop casting of aqueous dispersions.Study of cytotoxicity in vitro
000 rpm for 45 min, and repeated rinsing. The resulting oxCNDs-DOX was collected after lyophilization and then stored at 4 °C until used. To estimate the DOX loading capacity, the supernatant was sampled and the concentration of unloaded DOX was determined using a Cary 100 Conc UV-visible spectrophotometer (Varian Inc.) at 480 nm with the standard absorbance–concentration calibration curve at the same wavelength.Effect of oxCNDs on the catalytic characteristics of cytochrome c
Results and discussion
The strong oxidation of carbon nanodiscs is expected to generate a derivative similar to graphene oxide, with oxygen-containing groups covalently attached to its layers. In detail, the oxidized carbon nanodiscs are expected to exhibit a lamellar structure with randomly distributed unoxidized aromatic regions (sp2-carbon atoms), six-membered aliphatic regions (sp3-carbon atoms) as a result of oxidation together with a high concentration of oxygen-containing functional groups, such as hydroxyl, epoxy, and carboxyl, grafted to layers and edges. A first indication for the successful oxidation arises from the exceptional solubility of the functionalised nanodiscs in water Fig. 2(a and b). Further proof for the successful oxidation of the carbon nanodiscs is the Tyndall scattering effect for a clear oxCND aqueous colloid (2 mg mL−1) using the beam of a laser pointer as shown in Fig. 2-right. The laser beam becomes visible passing through the liquid because of the light scattering by the highly dispersed nanoparticles. The oxygen-containing groups attached to the surface of the discs and to its edges impart an enhanced dispersability in water and other polar solvents. | ||
| Fig. 2 Left panel: photographs of pristine carbon nanodiscs (a) insoluble in water and of water-dispersible oxidized carbon nanodiscs (b); right panel: photograph of an aqueous colloidal dispersion (2 mg mL−1) of oxCND (right) showing the Tyndall scattering effect. | ||
FTIR and XPS spectroscopies were employed to confirm the presence of oxygen-containing functional groups covalently attached to the oxCNDs. The FTIR spectra of pristine and oxidized carbon nanodiscs are shown in Fig. 3 (Left panel). The untreated CNDs does not show any peaks in all the frequency range. On the contrary, the oxidized carbon nanodiscs present intensive vibrational bands in the region of 1000–1750 cm−1 as well at around 3400 cm−1 due to the oxygen containing functional groups. More specifically, the band at 1060 cm−1 is assigned to stretching vibrations of C–O groups, while the peak at 1412 cm−1 is attributed to bending vibrations (deformation) of hydroxyl groups C–OH groups.44–46 The bands at 1630 cm−1 and 1733 cm−1 are due to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations of the –COOH groups.44–47 The weak band at 1224 cm−1 is assigned to asymmetric stretching of C–O–C bridges in epoxy groups and/or to deformation vibrations of O–H in the carboxylic acid groups. Finally, the appearance of the peak at 3424 cm−1 is ascribed to the hydroxyl stretching vibrations of C–OH groups.44–46 The presence of all these characteristic peaks confirms the successful oxidation of the pristine carbon nanodiscs. XPS results further support the formation of an oxygen-rich derivative. Fig. 3 (Right panel) shows the C 1s core level region of the XPS spectrum of oxCNDs. The spectral analysis entailed mathematically reconstructing the spectrum with a minimum number of peaks consistent with the raw data and the molecular structure. Four major contributions to the carbon 1s core-level region can be identified. The first peak at a binding energy of 285 eV is assigned to the C–C bonds of the graphitic framework48,49 and accounts for 28% of overall carbon intensity. A second peak at 286.1 eV is due to C–O bonds and corresponds to 16% of the overall intensity.50,51 Additional contributions at 287.2 eV and 288.4 eV stem from CO bonds52,53 (43% of the total C 1s intensity) and O–CO bonds54,55 (13% of the overall intensity) respectively. It is important to highlight that similar contributions from hydroxyl, epoxy, carbonyl and carboxylate groups are widely reported in the corresponding XPS spectra of graphene oxide sheets produced via acidic treatment of pristine graphite similar to the one applied here to the CNDs.56–60
O stretching vibrations of the –COOH groups.44–47 The weak band at 1224 cm−1 is assigned to asymmetric stretching of C–O–C bridges in epoxy groups and/or to deformation vibrations of O–H in the carboxylic acid groups. Finally, the appearance of the peak at 3424 cm−1 is ascribed to the hydroxyl stretching vibrations of C–OH groups.44–46 The presence of all these characteristic peaks confirms the successful oxidation of the pristine carbon nanodiscs. XPS results further support the formation of an oxygen-rich derivative. Fig. 3 (Right panel) shows the C 1s core level region of the XPS spectrum of oxCNDs. The spectral analysis entailed mathematically reconstructing the spectrum with a minimum number of peaks consistent with the raw data and the molecular structure. Four major contributions to the carbon 1s core-level region can be identified. The first peak at a binding energy of 285 eV is assigned to the C–C bonds of the graphitic framework48,49 and accounts for 28% of overall carbon intensity. A second peak at 286.1 eV is due to C–O bonds and corresponds to 16% of the overall intensity.50,51 Additional contributions at 287.2 eV and 288.4 eV stem from CO bonds52,53 (43% of the total C 1s intensity) and O–CO bonds54,55 (13% of the overall intensity) respectively. It is important to highlight that similar contributions from hydroxyl, epoxy, carbonyl and carboxylate groups are widely reported in the corresponding XPS spectra of graphene oxide sheets produced via acidic treatment of pristine graphite similar to the one applied here to the CNDs.56–60
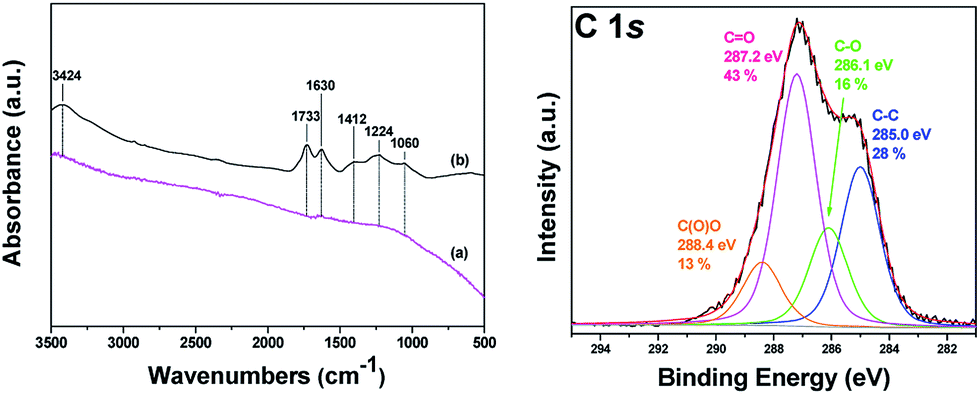 | ||
| Fig. 3 (Left panel) FT-IR spectra of (a) pristine and (b) oxidized carbon nanodiscs (oxCNDs). (Right panel) XPS spectrum and fit of the C 1s core level region of oxidized carbon nanodiscs (oxCNDs). | ||
The successful oxidation of pristine CNDs was further supported by Raman spectroscopy. The Raman spectra of pure and oxidized carbon nanodiscs are shown in Fig. 4. Pristine and oxidized CNDs show the characteristic first-order G and D bands at around 1600 and 1350 cm−1, respectively. The G-band originates from the doubly degenerate zone center E2g mode, associated with sp2-hybridized carbon atoms. On the other hand, the D band, which is correlated with sp3 hybridized carbon, requires a defect for its activation, by double resonance. Thus, it indicates the presence of lattice defects and distortions.61,62 The ratio between the D and G band intensities (ID/IG) is indicative of the quality of the graphitic lattice and was found be equal to 0.57 for pristine CNDs. Upon oxidation, the CND sheets show a noticeable increase in the D/G ratio from 0.57 to 0.93, which is attributed to the change in hybridization of the carbon atoms from sp2 to sp3 when oxygen-containing groups (hydroxyl, carboxyl or epoxide) are created. This is in line with the obtained by FTIR and XPS.
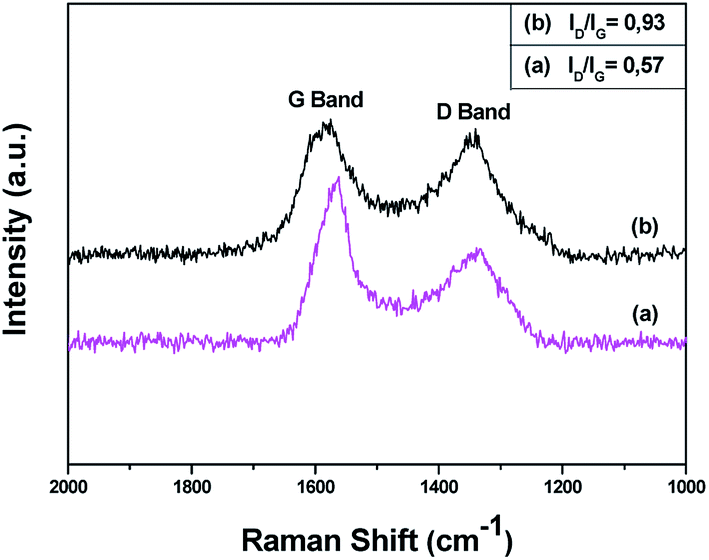 | ||
| Fig. 4 Raman spectra of (a) pristine (CNDs) and (b) oxidized carbon nanodiscs (oxCNDs). | ||
Thermogravimetric analysis (TGA) and thermal analysis (DTA) measurements were performed on pristine and oxidized carbon nanodiscs and the results are shown in Fig. 5. For pristine CNDs, a sharp exothermal peak at 635 °C, followed by the complete decomposition of the material, indicates the thermal destruction of the graphitic network. In the case of oxidized carbon nanodiscs a rather continuous weight loss upon heating is observed in the temperature range between 140 and 320 °C which can be attributed to the removal of the oxygen containing groups (hydroxyl, carboxyl, epoxy) covalently attached to the graphitic layers. The loss in the mass is estimated to be 30 wt%, indicating the high degree of functionalization that has occurred upon oxidation of the pristine CNDs. Moreover, the decomposition of graphitic lattice, identified by the sharp exothermic peak at 440 °C takes place at significantly lower temperatures compared to pristine CNDs. This can be attributed to the presence of oxygenated species and defects that facilitate the combustion of the graphitic network of the oxidized CNDs. From the weight loss up to 120 °C, the percentage of adsorbed water was estimated to be ∼20 wt%, which is a very high value for carbonaceous materials. Finally, the amount of adsorbed water deduced from the TGA data points to the largely increased hydrophilic nature of these novel oxidized CNDs, compared to pristine material.
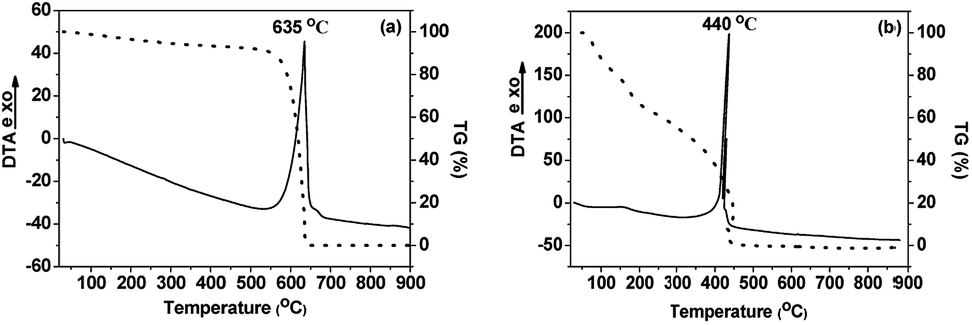 | ||
| Fig. 5 DTA/TG curves of (a) pristine (CNDs) and (b) oxidized carbon nanodiscs (oxCNDs). | ||
The X-ray diffraction patterns of pristine and oxidized carbon nanodiscs are presented in Fig. 6. Pristine CNDs display a well-defined peak, which is attributed to the 002 reflection of the graphite lattice (HOPG) at 26.6° and corresponds to a d002 spacing of 3.4 Å. After oxidation, this diffraction peak disappears and a new sharp one emerges at lower angles (∼11.7°). The latter is due to the principal 001 reflection and corresponds to a basal spacing of d001 = 7.5 Å, consistent with the successful oxidation of graphitic layers and the creation of oxygen-containing groups that are randomly distributed on the basal planes and the edges of the oxidized nanodiscs.
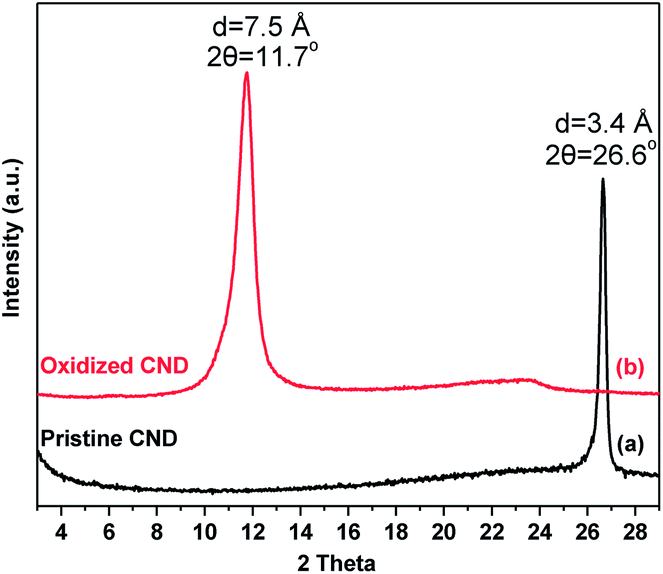 | ||
| Fig. 6 X-ray diffraction patterns of (a) pristine CNDs and (b) oxidized CNDs (oxCNDs). | ||
The morphology of the oxidized nanodiscs was examined thoroughly with a combination of SEM, TEM and AFM microscopies (Fig. 7). Isolated disc-like nanoflakes with a mean diameter in the range of 1 and 2 μm are clearly visible in both TEM and SEM micrographs. The majority of these nanodiscs have a uniform round shape while some of them are decorated by carbon nanoparticles presumably produced during the synthesis of the pristine nanodiscs that endured the oxidation process. Moreover, as a result of the strong acid treatment, single graphene layers have detached from the discs as evidenced by AFM images (see, for instance Fig. 7g together with the topographical height profile). Isolated single layer graphene nanodiscs like this one with thickness 0.8 nm can be seen all over the scanned area. However, nanodiscs with thicknesses between 2 and 5 nm are also present indicating that exfoliation of few layer graphene discs also occurs during the oxidation process.
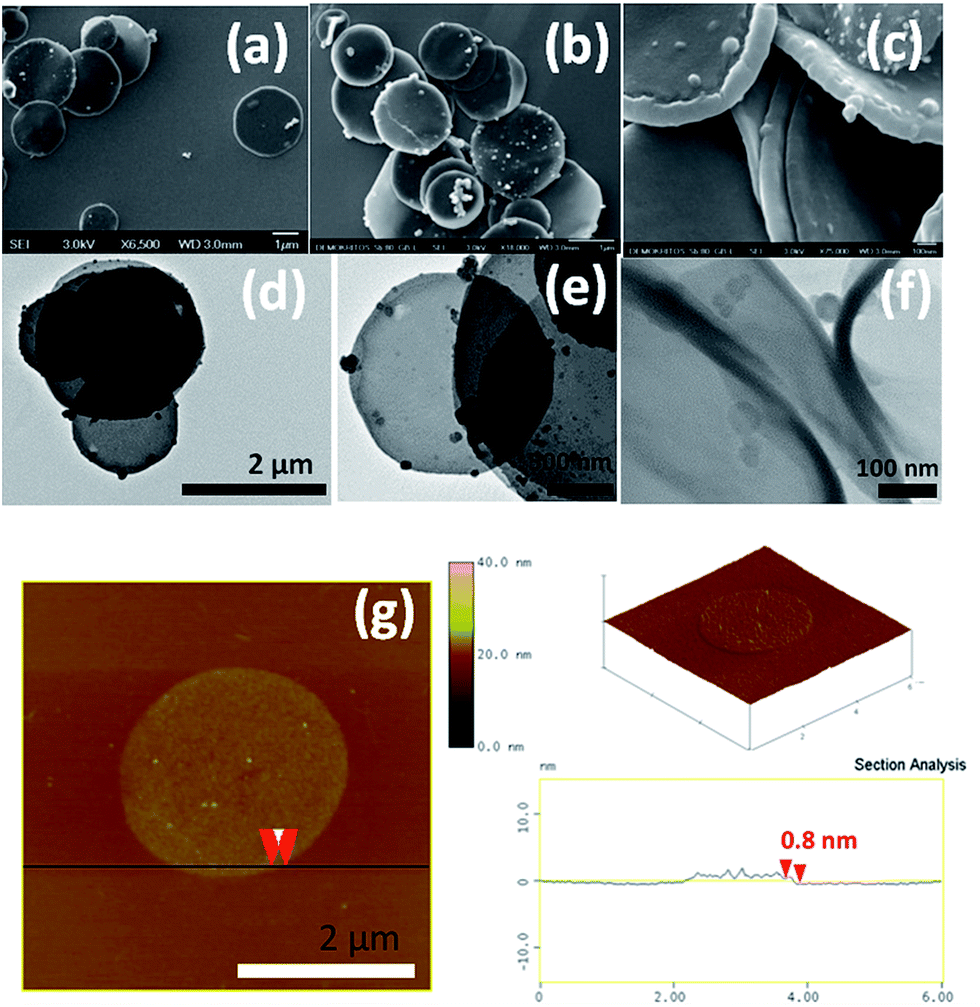 | ||
| Fig. 7 SEM (a–c), TEM (d–f) and AFM (g) micrographs of oxCNDs. | ||
In view of possible applications for these new hydrophilic nanocarbons we tested the ability of oxCNDs to be used as effective drug delivery system. Doxorubicin hydrochloride (DOX), which is a widely used anticancer drugs,63 was employed as a model drug in order to investigate the loading capacity of oxCNDs. Due to the structural similarities of oxCNDs with graphene oxide, it is expected that DOX can be effectively loaded on oxCNDs.64 Specifically, it is known that strong π–π stacking interactions between quinone part of DOX and the graphitic layers65 are formed. Further ionic interactions between the carboxyl groups of GO and the amino groups of DOX, enhances doxorubicin loading capacity of GO.
The loading of DOX onto oxCNDs was determined from the absorbance of the unloaded drug in the supernatant, by employing UV-vis spectroscopy. The DOX-loading capacity was consistently found to be ca. 45% w/w, which is much higher than that of other widely used drug carriers, such as liposomes,66 where the loading capacity is usually below 10%. DOX loading also affected the ζ-potential of oxCNDs. Indeed, while the ζ-potential value of unloaded oxCNDs was found to be −24 ± 1 mV, for the DOX-loaded oxCNDs a value of 20 ± 1 mV was derived, as the binding of doxorubicin induces a positive charge on oxCNDs [isoelectric point of DOX = 8.25].67
Following physicochemical characterization, we proceeded with the administration of oxCNDs-DOX to A549 cells. Our first aim was to prove that the nanocarriers effectively deliver the DOX cargo to the cells. To achieve this, we employed optical fluorescence microscopy and exploited the inherent fluorescence of DOX. The representative images of fluorescence microscopy on A549 cells incubated with DOX and oxCNDs-DOX are shown in Fig. 8. Cells incubated with free DOX (Fig. 8, left) as well as with oxCNDs-DOX (Fig. 8, Right) clearly show DOX-fluorescence in their nuclei. Therefore, oxCNDs did not inhibit DOX to internalize into cells and also DOX was efficiently delivered to the cell nuclei, where it is expected to retain its action.68
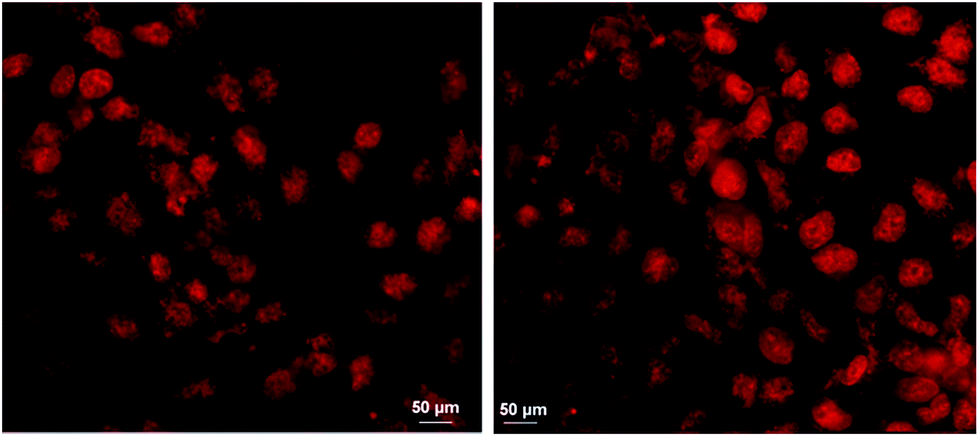 | ||
| Fig. 8 A549 cells incubated with free DOX (left) and oxCNDs-DOX (Right) for 5 h; the DOX concentration was 5 μM in both cases. | ||
We also investigated the anti-tumor efficacy of oxCNDs-DOX on A549 cells. For this purpose, A549 cells were incubated with oxCNDs-DOX and free DOX in various concentrations. Standard MTT assays were employed to assess the viability of the cells at 24, 48 and 72 h post incubation. The cell viability was found to be dose dependent for both oxCNDs-DOX and free DOX as shown in Fig. 9. The MTT cell viability assay confirmed that both oxCNDs-DOX and DOX efficiently kill A549 cancer cells in vitro. The toxicity of both oxCNDs-DOX and free DOX increased gradually with the concentration and the post incubation time. It was found that the maximum cytocidal activity of free DOX is ∼80% (∼20% survival) for a 10 μM concentration and 72 h post incubation, while oxCNDs-DOX at the same concentration (10 μM) and after the same post incubation time (72 h) exhibited significantly higher toxicity (4% survival) compared to free DOX. Thus, it is obvious that the use of oxCNDs for efficient delivery of DOX enhanced its cytocidal activity. Additionally, unloaded oxCNDs was found to be almost non-cytotoxic for A549 cells since in Fig. 9 showed that oxCNDs exhibited high cell viability (∼90%) against A549 cells even at high concentration (7 μg mL−1) and 72 h post incubation. It is known that drug nanocarriers should fulfill some requirements, among others low toxicity, loading efficiency and reduced drug toxicity, in order to be characterized as efficient drug delivery systems. Therefore, based on our results, oxCNDs is a very promising new material able to be used as potential drug delivery system.
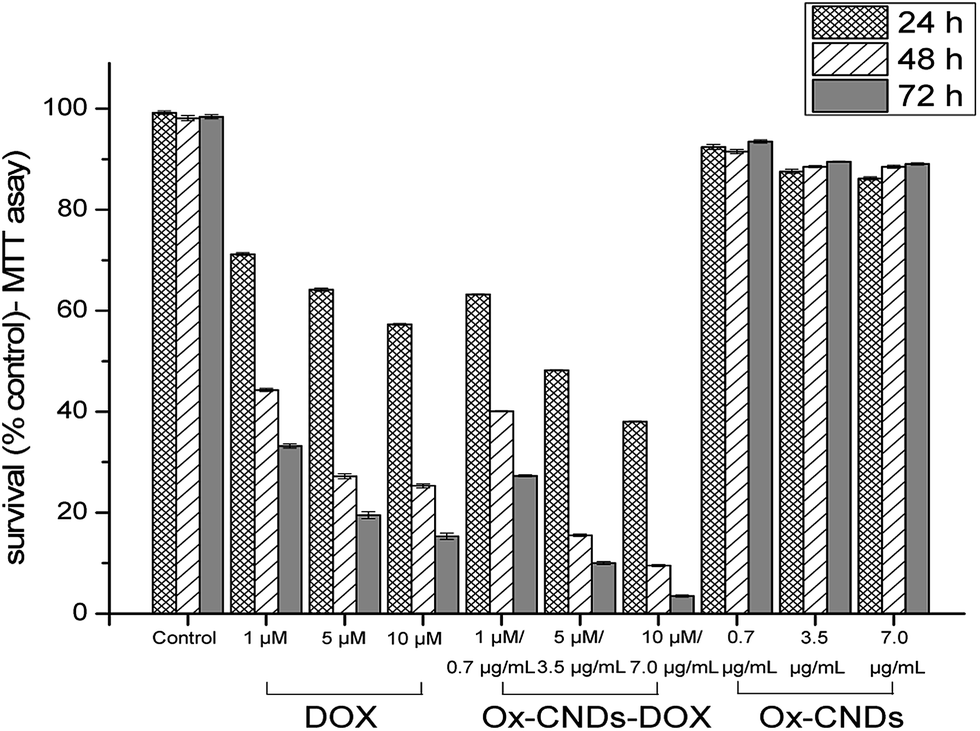 | ||
| Fig. 9 Comparative cytotoxicity of oxCNDs, free DOX and oxCNDs-DOX. The cytotoxicity was assessed at 24, 48 and 72 h post-incubation by standard MTT assay. The survival rates are relative to media only controls. MTT data are shown as means of at least twelve independent values with error bars representing one standard deviation. | ||
Another potential application of oxCNDs involves their use as supports for the development of nanobiocatalytic systems. In this case, cytochrome c (cyt c) was used as a model protein to investigate the effect of oxCNDs on the catalytic behavior (peroxidase activity and stability) of the protein. Cyt c is a small heme protein, present in mitochondria, which is related with the respiratory chain. In the presence of an electron acceptor, such as hydrogen peroxide, it is able to catalyze peroxidase-like reactions, in vitro. Due to its excellent catalytic attributes,69 cyt c is among the best characterized redox proteins and appears to be an excellent model protein to quantitatively assess the effect of carbon nanomaterials on the structure and the catalytic behavior of proteins and enzymes.70
To study the effect of oxCNDs on the stability and peroxidase activity of cyt c, the protein was incubated at 40 °C in a buffer solution containing guaiacol (25 mM) and the remaining peroxidase activity was estimated at predetermined time intervals, by addition of H2O2 and monitoring the oxidation of guaiacol. As seen in Fig. 10a, the presence of oxCNDs significantly increases the thermal stability of cyt c. A denaturation half time of 40 h was calculated for cyt c in the presence of oxCNDs. This is almost 4-fold higher than the denaturation half time observed in the absence of the nanomaterials. The stabilizing effect of oxCNDs was even more pronounced when cyt c was incubated in the presence of H2O2. It is well known that cyt c is rapidly deactivated by an excess of H2O2 in the absence of any reducing substrate.71 As seen in Fig. 10b, in the presence of oxCNDs, cyt c almost totally retained its peroxidase activity after 30 min of incubation with H2O2, while in the absence of oxCNDs, the remaining activity of cyt c was less than 20%. Similar stabilizing effects by various carbon nanomaterials for different enzymes have recently been observed and attributed to specific electrostatic and hydrophobic interactions between protein molecules and nanomaterials.70,72,73 It is interesting to note that the stabilizing effect observed for the small oxCNDs is higher than that observed for significantly larger GO nanomaterials70 indicating that the size of nanomaterials is crucial for this effect. Compared to GO, the small-sized oxCNDs are expected to interact stronger (due to higher number of active sites) with cyt c molecules and these interactions could lead to a more rigid structure of the protein molecule and thus to an increased stability.74,75
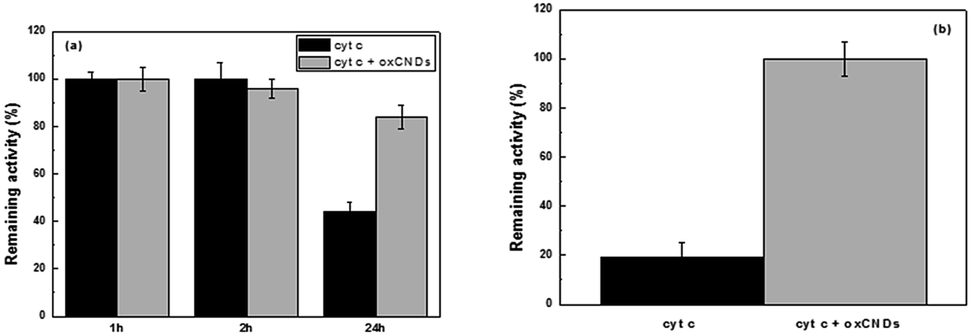 | ||
| Fig. 10 Effect of oxCNDs on the (a) thermal stability of cyt c after incubation up to 24 hours at 40 °C and (b) stability of cyt c against H2O2 after incubation for 30 min at 40 °C. 100% indicates the activity at t = 0 min. | ||
Conclusions
Insoluble carbon nanodiscs produced through the so-called pyrolytic Kværner Carbon Black & H2 process were successfully oxidized using a simple, versatile and reproducible approach based on the well-known Staudenmaier method. The strong acid treatment, which also leads to a partial purification of CNDs (i.e. removal of nanocones and soot found in the starting material), enables the decoration of CNDs surface with various oxygen-containing functional polar groups such as hydroxyl, carboxyl and epoxy groups. Thus, the completely insoluble CNDs can be transformed into a hydrophilic derivative, which is dispersible in many polar solvents, including water. X-ray diffraction in conjunction with FTIR, XPS and Raman spectroscopies were successfully applied to confirm the chemical functionalization and the presence of oxygen-containing functional groups, covalently attached to the oxCNDs. Thermogravimetric and differential thermal analysis illustrated that a high degree of functionalization can be achieved upon oxidation of the pristine CNDs. Light scattering confirmed the increased hydrophilic nature of these novel oxidized nanodiscs compared to pristine CNDs, which show negligible hydrophilicity. The percentage of adsorbed water in oxCNDs was estimated to be high (20 wt%) for carbonaceous materials, revealing the hydrophilic character of oxCNDs. The morphology of the derived oxidized nanodiscs was examined thoroughly with a combination of SEM, TEM and AFM techniques. The abundance of isolated round graphene layers with a mean diameter in the range of 1 and 2 μm and thickness of 0.8 Å indicates that the oxidation occurs together with partial exfoliation. These hydrophilic nanostructures were tested as a potential nanocarrier for the efficient delivery of doxorubicin. High doxorubicin loading efficiency (45%) was achieved due to the strong interactions between the drug and the graphitic structure of oxCNDs. DOX-loaded oxCNDs enhanced cell proliferation inhibition compared to free DOX, while pristine oxCNDs showed almost non-cytotoxic activity on A549 cells, indicating that the oxidized carbon nanodiscs is a very promising non-toxic material suitable to be used as an effective drug delivery system. Finally, oxCNDs were tested as supports for the development of nanobiocatalytic systems. The oxCNDs was found to increase the thermal stability of cytochrome c as well as the stability in the presence of H2O2. The stabilizing effect observed by the small-sized oxCNDs is even higher than that observed for significantly larger oxidized graphite-based nanomaterials (GO) reported in the literature indicating that the size of nanomaterials is crucial for this stabilization effect.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was co-financed by the European Union (European Social Fund – ESF) and Greek national funds through the Operational Program “Education and Lifelong Learning” of the National Strategic Reference Framework (NSRF) – Research Funding Program: THALES. Investing in knowledge society through the European Social Fund. This work was also supported by the “Graphene based electronics” research program of the Stichting voor Fundamenteel Onderzoek der Materie (FOM), part of the Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO). G. P. acknowledges the Ubbo Emmius Program for a PhD fellowship. M. P. is very thankful to the IKY Foundation for the post-doc financial support.References
- J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718–747 CrossRef CAS PubMed.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- P. K. Ang, S. Wang, Q. L. Bao, J. T. L. Thong and K. P. Loh, ACS Nano, 2009, 3, 3587–3594 CrossRef CAS PubMed.
- A. Kumar, A. L. M. Reddy, A. Mukherjee, M. Dubey, X. B. Zhan, N. Singh, L. Ci, W. E. Billups, J. Nagurny, G. Mital and P. M. Ajayan, ACS Nano, 2011, 5, 4345–4349 CrossRef CAS PubMed.
- I. V. Pavlidis, M. Patila, U. T. Bornscheuer, D. Gournis and H. Stamatis, Trends Biotechnol., 2014, 32, 312–320 CrossRef CAS PubMed.
- M. S. Dresselhaus, G. Dresselhaus and P. Eklund, Science of Fullerenes and Carbon Nanotubes, Academic Press, San Diego, 1996 Search PubMed.
- N. F. Goldshleger, Fullerene Sci. Technol., 2001, 9, 255–280 CrossRef CAS.
- M. Inagaki and L. R. Radovic, Carbon, 2002, 40, 2279–2282 CrossRef CAS.
- K. M. Kadish and R. S. Ruoff, Fullerenes: Chemistry, Physics and Technology, Wiley-Interscience, New York, 2000 Search PubMed.
- S. Subramoney, Adv. Mater., 1998, 10, 1157–1171 CrossRef CAS.
- D. Tasis, N. Tagmatarchis, A. Bianco and M. Prato, Chem. Rev., 2006, 106, 1105–1136 CrossRef CAS PubMed.
- T. Tsoufis, C. Tampaxis, I. Spanopoulos, T. Steriotis, F. Katsaros, G. Charalambopoulou and P. N. Trikalitis, Microporous Mesoporous Mater., 2018, 262, 68–76 CrossRef CAS.
- R. Andrews, D. Jacques, D. L. Qian and T. Rantell, Acc. Chem. Res., 2002, 35, 1008–1017 CrossRef CAS PubMed.
- R. H. Baughman, A. A. Zakhidov and W. A. De Heer, Science, 2002, 297, 787–792 CrossRef CAS PubMed.
- E. Frackowiak and F. Beguin, Carbon, 2001, 39, 937–950 CrossRef CAS.
- A. Galano, Nanoscale, 2010, 2, 373–380 RSC.
- X. P. Gao, Y. Zhang, X. Chen, G. L. Pan, J. Yan, F. Wu, H. T. Yuan and D. Y. Song, Carbon, 2004, 42, 47–52 CrossRef CAS.
- V. Georgakilas, D. Gournis, V. Tzitzios, L. Pasquato, D. M. Guldi and M. Prato, J. Mater. Chem., 2007, 17, 2679–2694 RSC.
- M. Moniruzzaman and K. I. Winey, Macromolecules, 2006, 39, 5194–5205 CrossRef CAS.
- P. Serp, M. Corrias and P. Kalck, Appl. Catal., A, 2003, 253, 337–358 CrossRef CAS.
- S. Lynum, J. Hugdahl, K. Hox, R. Hildrum and M. Nordvik, Micro-domain graphitic materials and method for producing the same (Patent), PCT/NO1998/000093, Mar 2008, vol. 18.
- S. Lynum, J. Hugdahl, K. Hox, R. Hildrum and M. Nordvik, Micro-domain graphitic materials and method for producing the same (Patent), US Pat., 20030091495 A1, May 2003, vol. 15.
- A. Krishnan, E. Dujardin, M. M. J. Treacy, J. Hugdahl, S. Lynum and T. W. Ebbesen, Nature, 1997, 388, 451–454 CrossRef CAS.
- Hydrogen Storage in Carbon Cones Project (HYCONES), NMP3-CT-2006–032970, (Unpublished data).
- T. Garberg, S. N. Naess, G. Helgesen, K. D. Knudsen, G. Kopstad and A. Elgsaeter, Carbon, 2008, 46, 1535–1543 CrossRef CAS.
- W. Zhang, M. Dubois, K. Guerin, P. Bonnet, E. Petit, N. Delpuech, D. Albertini, F. Masin and A. Hamwi, Carbon, 2009, 47, 2763–2775 CrossRef CAS.
- A. Hirsch, Nat. Mater., 2010, 9, 868–871 CrossRef CAS PubMed.
- J. Černák, G. Helgesen, A. T. Skjeltorp, J. Kováč, J. Voltr and E. Čižmár, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 0144341–01443418 CrossRef.
- T. J. Davies, M. E. Hyde and R. G. Compton, Angew. Chem., Int. Ed., 2005, 44, 5121–5126 CrossRef CAS PubMed.
- J. G. S. Moo and M. Pumera, RSC Adv., 2012, 2, 1565–1568 RSC.
- Y. W. Jiang, G. Gao, X. Zhang, H. R. Jia and F. G. Wu, Nanoscale, 2017, 9, 15786–15795 RSC.
- A. Nieto-Márquez, R. Romero, A. Romero and J. L. Valverde, J. Mater. Chem., 2011, 21, 1664–1672 RSC.
- C. T. Lin, C. Y. Lee, H. T. Chiu and T. S. Chin, Langmuir, 2007, 23, 12806–12810 CrossRef CAS PubMed.
- Y. Ahmad, E. Disa, M. Dubois, K. Guérin, V. Dubois, W. Zhang, P. Bonnet, F. Masin, L. Vidal, D. A. Ivanov and A. Hamwi, Carbon, 2012, 50, 3897–3908 CrossRef CAS.
- I. Ahmad, B. Yazdani and Y. Zhu, Nanomaterials, 2015, 5, 90 CrossRef PubMed.
- P. Thomas, D. Himmel, J. L. Mansot, W. Zhang, M. Dubois, K. Guérin and A. Hamwi, Tribol. Lett., 2011, 41, 353–362 CrossRef CAS.
- Z. Osváth, Z. Vértesy, J. Lábár, P. Nemes-Incze, Z. E. Horváth and L. P. Biró, Thin Solid Films, 2014, 565, 111–115 CrossRef.
- L. Staudenmaier, Ber. Dtsch. Chem. Ges., 1898, 31, 1481 CrossRef CAS.
- R. Y. N. Gengler, A. Veligura, A. Enotiadis, E. K. Diamanti, D. Gournis, C. Jozsa, B. J. van Wees and P. Rudolf, Small, 2010, 6, 35–39 CrossRef CAS PubMed.
- D. V. Stergiou, E. K. Diamanti, D. Gournis and M. I. Prodromidis, Electrochem. Commun., 2010, 12, 1307–1309 CrossRef CAS.
- T. Tsoufis, G. Tuci, S. Caporali, D. Gournis and G. Giambastiani, Carbon, 2013, 59, 100–108 CrossRef CAS.
- R. Supino, Methods in molecular biology, Clifton, N.J., 1995, vol. 43, pp. 137–149 Search PubMed.
- I. V. Pavlidis, D. Gournis, G. K. Papadopoulos and H. Stamatis, J. Mol. Catal. B: Enzym., 2009, 60, 50–56 CrossRef CAS.
- D. W. Lee, V. L. De Los Santos, J. W. Seo, L. L. Felix, D. A. Bustamante, J. M. Cole and C. H. W. Barnes, J. Phys. Chem. B, 2010, 114, 5723–5728 CrossRef CAS PubMed.
- A. B. Bourlinos, D. Gournis, D. Petridis, T. Szabo, A. Szeri and I. Dekany, Langmuir, 2003, 19, 6050–6055 CrossRef CAS.
- K. Spyrou, G. Potsi, E. K. Diamanti, X. Ke, E. Serestatidou, I. I. Verginadis, A. P. Velalopoulou, A. M. Evangelou, Y. Deligiannakis, G. Van Tendeloo, D. Gournis and P. Rudolf, Adv. Funct. Mater., 2014, 24, 5841–5850 CrossRef CAS.
- W. W. Simons, The Sadtler handbook of infrared spectra, Sadtler Research Laboratories, Philadelphia, 1978 Search PubMed.
- D. Yang, A. Velamakanni, G. Bozoklu, S. Park, M. Stoller, R. D. Piner, S. Stankovich, I. Jung, D. A. Field, C. A. Ventrice Jr and R. S. Ruoff, Carbon, 2009, 47, 145–152 CrossRef CAS.
- Z. Cao, T. Tsoufis, T. Svaldo-Lanero, A. S. Duwez, P. Rudolf and K. Loos, Biomacromolecules, 2013, 14, 3713–3722 CrossRef CAS PubMed.
- M. Quintana, K. Spyrou, M. Grzelczak, W. R. Browne, P. Rudolf and M. Prato, ACS Nano, 2010, 4, 3527–3533 CrossRef CAS PubMed.
- S. Seo, J. Lee, S.-Y. Choi and H. Lee, J. Mater. Chem., 2012, 22, 1868–1875 RSC.
- H. He and C. Gao, Chem. Mater., 2010, 22, 5054–5064 CrossRef CAS.
- X.-Z. Tang, Z. Cao, H.-B. Zhang, J. Liu and Z.-Z. Yu, Chem. Commun., 2011, 47, 3084–3086 RSC.
- T. Tsoufis, F. Katsaros, Z. Sideratou, G. Romanos, O. Ivashenko, P. Rudolf, B. J. Kooi, S. Papageorgiou and M. A. Karakassides, Chem. Commun., 2014, 50, 10967–10970 RSC.
- J. W. Chiou, S. C. Ray, S. I. Peng, C. H. Chuang, B. Y. Wang, H. M. Tsai, C. W. Pao, H. J. Lin, Y. C. Shao, Y. F. Wang, S. C. Chen, W. F. Pong, Y. C. Yeh, C. W. Chen, L. C. Chen, K. H. Chen, M. H. Tsai, A. Kumar, A. Ganguly, P. Papakonstantinou, H. Yamane, N. Kosugi, T. Regier, L. Liu and T. K. Sham, J. Phys. Chem. C, 2012, 116, 16251–16258 CAS.
- H. Y. Jeong, J. Y. Kim, J. W. Kim, J. O. Hwang, J.-E. Kim, J. Y. Lee, T. H. Yoon, B. J. Cho, S. O. Kim, R. S. Ruoff and S.-Y. Choi, Nano Lett., 2010, 10, 4381–4386 CrossRef CAS PubMed.
- M. Acik, C. Mattevi, C. Gong, G. Lee, K. Cho, M. Chhowalla and Y. J. Chabal, ACS Nano, 2010, 4, 5861–5868 CrossRef CAS PubMed.
- D. Long, W. Li, L. Ling, J. Miyawaki, I. Mochida and S.-H. Yoon, Langmuir, 2010, 26, 16096–16102 CrossRef CAS PubMed.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- C. Mattevi, G. Eda, S. Agnoli, S. Miller, K. A. Mkhoyan, O. Celik, D. Mastrogiovanni, G. Granozzi, E. Garfunkel and M. Chhowalla, Adv. Funct. Mater., 2009, 19, 2577–2583 CrossRef CAS.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS PubMed.
- F. Torrisi, T. Hasan, W. Wu, Z. Sun, A. Lombardo, T. S. Kulmala, G.-W. Hsieh, S. Jung, F. Bonaccorso, P. J. Paul, D. Chu and A. C. Ferrari, ACS Nano, 2012, 6, 2992–3006 CrossRef CAS PubMed.
- G. Minotti, P. Menna, E. Salvatorelli, G. Cairo and L. Gianni, Pharmacol. Rev., 2004, 56, 185–229 CrossRef CAS PubMed.
- X. Yang, X. Zhang, Z. Liu, Y. Ma, Y. Huang and Y. Chen, J. Phys. Chem. C, 2008, 112, 17554–17558 CAS.
- Z. Liu, X. Sun, N. Nakayama-Ratchford and H. Dai, ACS Nano, 2007, 1, 50–56 CrossRef CAS PubMed.
- W. Sun, N. Zhang, A. Li, W. Zou and W. Xu, Int. J. Pharm., 2008, 353, 243–250 CrossRef CAS PubMed.
- A. Choucair, P. L. Soo and A. Eisenberg, Langmuir, 2005, 21, 9308–9313 CrossRef CAS PubMed.
- K. M. Laginha, S. Verwoert, G. J. R. Charrois and T. M. Allen, Clin. Cancer Res., 2005, 11, 6944–6949 CrossRef CAS PubMed.
- R. Vazquez-Duhalt, J. Mol. Catal. B: Enzym., 1999, 7, 241–249 CrossRef CAS.
- M. Patila, I. V. Pavlidis, E. K. Diamanti, P. Katapodis, D. Gournis and H. Stamatis, Process Biochem., 2013, 48, 1010–1017 CrossRef CAS.
- J. A. Villegas, A. G. Mauk and R. Vazquez-Duhalt, Chem. Biol., 2000, 7, 237–244 CrossRef CAS PubMed.
- A. A. Vertegel, R. W. Siegel and J. S. Dordick, Langmuir, 2004, 20, 6800–6807 CrossRef CAS PubMed.
- W. Shang, J. H. Nuffer, V. A. Muñiz-Papandrea, W. Colón, R. W. Siegel and J. S. Dordick, Small, 2009, 5, 470–476 CrossRef CAS PubMed.
- I. V. Pavlidis, T. Vorhaben, T. Tsoufis, P. Rudolf, U. T. Bornscheuer, D. Gournis and H. Stamatis, Bioresour. Technol., 2012, 115, 164–171 CrossRef CAS PubMed.
- T. Xue, S. Jiang, Y. Qu, Q. Su, R. Cheng, S. Dubin, C.-Y. Chiu, R. Kaner, Y. Huang and X. Duan, Angew. Chem., Int. Ed., 2012, 51, 3822–3825 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |