DOI:
10.1039/C7RA11055C
(Paper)
RSC Adv., 2018,
8, 1398-1406
Mesoporous TiO2 coated ZnFe2O4 nanocomposite loading on activated fly ash cenosphere for visible light photocatalysis†
Received
8th October 2017
, Accepted 18th December 2017
First published on 3rd January 2018
Abstract
Several activated fly ash cenosphere (AFAC) supporting TiO2 coated ZnFe2O4 (TiO2/ZnFe2O4/AFAC) photocatalysts were prepared by sol–gel and hydrothermal methods. These photocatalysts were characterized by X-ray diffraction (XRD), scanning electron microscopy (SEM), transmission electron microscopy (TEM), Fourier transform infrared spectroscopy (FT-IR), UV-vis diffuse reflectance spectroscopy (UV-DRS) and nitrogen adsorption analyses for Brunauer–Emmett–Teller (BET) specific surface area measurements. We found that the main components of spherical AFAC were mullite (Al6Si2O13) and SiO2; the crystallite size of the TiO2/ZnFe2O4 nanocomposite was less than 10 nm and its specific surface area was 162.18 m2 g−1. The TiO2/ZnFe2O4 nanocomposite had a band-gap of 2.56 eV, which would photodegrade 95% of rhodamine B (RhB) under visible light within 75 min. When hybridized with 0.02 g AFAC, the TiO2/ZnFe2O4/0.02 g AFAC photocatalyst with a band-gap of 2.50 eV could remove 97.1% of RhB and be reused three consecutive times with minor decrease in photocatalytic performance. However, the photocatalytic performance decreased to 91.0% on increasing the dosage of AFAC to 0.30 g. The mesoporous structure of all the photocatalysts and the strong adsorption ability of AFAC accounted for the notable performance.
1. Introduction
With superior photocatalytic performance, high chemical stability, low cost and low-toxicity, TiO2 has the potential for applications in environmental purification and solar energy conversion.1–3 However, the wide band gap of TiO2 (3.18 eV for anatase), making it absorb no more than 5.0% of sunlight, together with the low quantum efficiency (high recombination rate of photogenerated electron–hole pair) hindered its application.4–6 To extend the visible light response of TiO2, increasing efforts have been directed towards the improvement of the photocatalytic activity of TiO2. Some of these methods are: doping ions, surface modification by noble metals and coupling with photosensitive dye and other semiconductors.7–11 Among these methods, coupling with other semiconductors could efficiently improve the photocatalytic activity of TiO2 under visible light irradiation.12–15 In particular, ZnFe2O4 has been considered as an interesting coupling semiconductor to TiO2, which could prevent the transformation of TiO2 from anatase to rutile.16,17 In addition, ZnFe2O4 can absorb sunlight since its band gap was about 1.8 eV.18,19 Hence, under visible light, the utilization of a TiO2/ZnFe2O4 nanocomposite in aqueous suspension has potential applications in wastewater treatment.18–21 Immobilizing TiO2 on adsorptive or hydrophobic materials would facilitate the oxygenation process throughout the photocatalytic process.22–25 Fly ash cenosphere (FAC) is the by-product generated in coal-fired power plants and could be used as a hydrophobic material or floating substrate,6,26,27 which shows bifunctional properties with adsorption capability in addition to photocatalytic activity in loading semiconductor nanoparticles.28,29
In this study, the TiO2 coated ZnFe2O4 (TiO2/ZnFe2O4) nanocomposite was synthesized by a sol–gel method, and then hybridized with different amounts of AFAC for the first time. Compared with the TiO2/ZnFe2O4 nanocomposite, the TiO2/ZnFe2O4 loaded on AFAC (TiO2/ZnFe2O4/AFAC) photocatalyst exhibited better adsorption activity and photocatalytic activity of removal of RhB under visible light.
2. Experimental
2.1 Materials
Fly ash was obtained from thermal power plants of Lingshou city (Hebei province, China). All reagents including zinc acetate dihydrate (Zn(CH3COO)2·2H2O), sodium hydroxide (NaOH), ferric chloride hexahydrate (FeCl3·6H2O), hydrazine hydrate (N2H4·H2O), ethylene glycol ((CH2OH)2), ethanol (C2H5OH), cetyl trimethyl ammonium bromide (CTAB), tetrabutyl titanate (TBOT), isopropanol ((CH3)2CHOH), nitric acid (HNO3) and rhodamine B (RhB) used in the experiments were analytical grade (purchased from Sinopharm Chemical Regent Co. Ltd) and used without further purification. Deionized water was used throughout the experiment.
2.2 Synthesis
2.2.1 Activating fly ash. The acquired fly ash (10 g) was activated by 200.0 mL HNO3 under stirring at 80 °C for 2 h, and then washed with deionized water until the pH of the solution was 7. The particles suspended in the solution were collected and dried in a vacuum oven at 80 °C for 24 h.
2.2.2 Preparation of ZnFe2O4. The mole ratio of n(Zn)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :n(Fe) = 1:2 of the soluble salts was dissolved in a mixture of ethylene glycol and deionized water solution. This mixture was magnetically stirred for 30 min and named as solution A. A certain amount of NaOH was dissolved in deionized water and mixed with hydrazine hydrate, magnetically stirred for 30 min, and the mixture was labeled as solution B. Solutions A and B were mixed and vigorously stirred for 10 min, with the injection of CTAB; magnetically stirring was continued for 20 min. The reaction mixture was transferred into a 100 mL Teflon-lined stainless steel autoclave and kept at 150 °C for 15 h. The obtained dark green products were washed three times with ethanol and deionized water before being dried at 60 °C for 12 h and then crushed in an agate mortar to obtain ZnFe2O4 powders.
:n(Fe) = 1:2 of the soluble salts was dissolved in a mixture of ethylene glycol and deionized water solution. This mixture was magnetically stirred for 30 min and named as solution A. A certain amount of NaOH was dissolved in deionized water and mixed with hydrazine hydrate, magnetically stirred for 30 min, and the mixture was labeled as solution B. Solutions A and B were mixed and vigorously stirred for 10 min, with the injection of CTAB; magnetically stirring was continued for 20 min. The reaction mixture was transferred into a 100 mL Teflon-lined stainless steel autoclave and kept at 150 °C for 15 h. The obtained dark green products were washed three times with ethanol and deionized water before being dried at 60 °C for 12 h and then crushed in an agate mortar to obtain ZnFe2O4 powders.
2.2.3 Photocatalysts preparation. The TiO2/ZnFe2O4 nanocomposite was prepared using the sol–gel method. Initially, 34.0 mL TBOT was diluted in 8.5 mL isopropoxide solution under stirring. The mixture was added dropwise into 340 mL water with vigorous stirring and the pH was adjusted to 3 with HNO3. The mixture was placed in a 500 mL flask and heated in a water bath at 75 °C for 12 h. Finally, pure TiO2 sol was obtained.30 Further, 0.35 g ZnFe2O4 powder was dispersed in 300.0 mL TiO2 sol in an ultrasonic bath for 30 min. Then, it was dried into a powder in a rotatory evaporator under vacuum at 75 °C. The obtained powder was named as TiO2 coated ZnFe2O4 nanocomposite (TiO2/ZnFe2O4). The final TiO2/ZnFe2O4 loadings on AFAC (TiO2/ZnFe2O4/AFAC) photocatalysts were obtained by triturating TiO2/ZnFe2O4 powder and AFAC with mass ratio of 1:0.02 and 1:0.30, respectively.
2.3 Characterization
The crystalline structure of the samples was determined using X-ray diffraction (XRD MAC Science, MXP18, Japan). The morphology of the as-synthesized samples was characterized by field emission scanning electron microscopy (FESEM, JEOL JEM-2010HR) and transmission electron microscopy (TEM, JEOL JEM-2010HR). N2 adsorption isotherms were measured using an AUTOSORB-IQ (Quantachrome Instruments, USA). UV-vis absorption spectra were acquired with an UV-vis spectrophotometer (UV-5800PC, Shanghai Metash Instruments Co., Ltd).
2.4 Photocatalytic activity test
The photocatalytic performance of the as-prepared photocatalysts was evaluated by the photocatalytic decomposition of rhodamine B (RhB) aqueous solution. In the experiment, 50 mg of the as-prepared photocatalyst was dispersed in 50.0 mL of RhB solution (10 mg L−1) in a 100 mL beaker. A 350 W Xe lamp equipped with a 420 nm cut-off filter was used as a light source along with a cooling device to eliminate the thermal effect. The average light energy density was estimated to be 68 mW cm−2. The distance between the beaker containing the reaction mixture and the light source was fixed at 16 cm. First, the reaction mixture was mechanically stirred for 30 min in the dark to ensure the adsorption/desorption equilibrium of the RhB on the surface of the photocatalysts. The reaction solutions were sampled at 15 min illumination intervals, and the corresponding UV-vis spectra (measured over the range of 300–800 nm) were recorded to monitor the progress of the degradation of RhB using an UV-vis spectrophotometer. All the photocatalytic performances were tested under UV light, visible light and sunlight. To investigate the stability of the TiO2/ZnFe2O4/0.02 g AFAC photocatalyst, the composite was reused three times for the photocatalytic degradation of RhB (10 mg L−1).
3. Results and discussion
3.1 Structure and morphology of photocatalysts
Fig. 1 shows the X-ray diffraction patterns of AFAC, ZnFe2O4, TiO2/ZnFe2O4 nanocomposite and the TiO2/ZnFe2O4/0.02 g AFAC sample. For the AFAC sample, most diffraction peaks belonged to mullite (Al6Si2O13, JCPDF no. 15-0776); the amorphous phase located at 22° primarily composed of SiO2 (JCPDF no. 88-1535). For the as-prepared ZnFe2O4 sample, all the diffraction peaks belonged to ZnFe2O4 (JCPDF no. 82-1049). For the TiO2/ZnFe2O4 nanocomposite, the TiO2 on the surface of ZnFe2O4 exhibited a dominant anatase phase (JCPDF no. 84-1286), while five diffraction peaks of ZnFe2O4 could also be observed at lower intensity, which demonstrated that the amount of ZnFe2O4 accounted for a small proportion. For the TiO2/ZnFe2O4/0.02 g AFAC sample, the diffraction peaks of Al6Si2O13, ZnFe2O4 and TiO2 could all be observed and marked by different symbols. However, the peak intensities of mullite were relatively small, which suggested that the amount of AFAC in the TiO2/ZnFe2O4/0.02 g AFAC sample was small. The crystallite sizes of the above samples were determined from the broadening of the corresponding X-ray diffraction peaks by using Scherer's formula D = (Kλ)/(βcosθ), where λ is the wavelength of the X-ray radiation (λ = 0.15406 nm), K is the Scherer constant (K = 0.9), θ is the X-ray diffraction peak and β is the full-width at half maximum (FWHM) of the peak (in radians), which was corrected for the instrumental broadening (β0 = 0.00122 rad) prior to the calculation of its crystallite size broadening. The crystallite sizes were calculated and listed in Table 1. The crystallite sizes of TiO2/ZnFe2O4, TiO2/ZnFe2O4/0.02 g AFAC and TiO2/ZnFe2O4/0.30 g AFAC samples were all less than 10 nm, which implied that the above samples would have a large specific surface area.
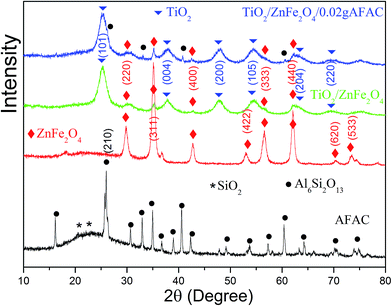 |
| | Fig. 1 The XRD spectra of AFAC, ZnFe2O4, TiO2/ZnFe2O4 and TiO2/ZnFe2O4/0.02 g AFAC samples. | |
Table 1 The crystallite size and band gap of different samples
| Sample |
ZnFe2O4 |
TiO2/ZnFe2O4 |
TiO2/ZnFe2O4/0.02 g AFAC |
TiO2/ZnFe2O4/0.30 g AFAC |
| Crystallite size (nm) |
25.0 ± 2.3 |
8.7 ± 0.6 |
8.4 ± 0.5 |
9.1 ± 0.9 |
| Band gap (eV) |
1.33 |
2.56 |
2.50 |
2.44 |
Fig. 2A–C show the SEM and TEM images of AFAC. In Fig. 2A and B we can observe that the particle shape of AFAC was spherical and their diameters ranged from about 1 μm to nearly 10 μm, which was similar to previous studies.6,26–28 In Fig. 2C, the perfect AFAC sphere is presented in the TEM image with some tiny particles on its surface, which implied that the AFAC had adsorption ability. Fig. 2D shows the SEM image of ZnFe2O4, whose crystallite size is about 20–30 nm, coinciding with the results summarized in Table 1. Fig. 2E and F represent the SEM and TEM images of the TiO2/ZnFe2O4 nanocomposite. It could be observed that the dark ZnFe2O4 particles with a crystallite size of about 25 nm are coated by the outer grey homogeneous layer of TiO2 particles with a crystallite size of less than 10 nm, which coincided with the results summarized in Table 1. Fig. 2G and H show the SEM image and EDS results of the ZnFe2O4/TiO2/0.30 g AFAC sample. It can be observed that the AFAC particle is semi-spherical and the ZnFe2O4/TiO2 particles are loaded on the AFAC surface. The EDS elemental analysis shows that most of the elements are oxygen (70%), silicon (14%), aluminum (11%) and titanium (2.3%), which corresponds to the compounds SiO2, Al6Si2O13 and TiO2, respectively. AFAC primarily consisted of SiO2, Al6Si2O13, and other oxides, such as Fe2O3. Only 0.05% of zinc and 0.29% of iron were found, which corresponded to the compounds Fe2O3 and ZnFe2O4, which might be because the added amount of ZnFe2O4 was small (3 wt%, wt% is an abbreviation of weight percentage) and the ZnFe2O4 particles were coated by TiO2 particles.
 |
| | Fig. 2 SEM images (A, B) and TEM image (C) of AFAC, SEM image (D) of ZnFe2O4, SEM images (E) and TEM image (F) of TiO2/ZnFe2O4 nanocomposite, SEM image (G) and EDS (H) of ZnFe2O4/TiO2/0.30 g AFAC sample. | |
3.2 FT-IR spectrum
FT-IR analysis was performed on TiO2/ZnFe2O4, TiO2/ZnFe2O4/0.02 g AFAC and TiO2/ZnFe2O4/0.30 g AFAC composites to further prove the coating of TiO2 on ZnFe2O4 and the existence of AFAC. Fig. 3 shows their FT-IR spectra recorded over the range of 4000–500 cm−1. In all the spectra, the band centered at 551 cm−1 can be assigned to the Zn–O stretching of ZnFe2O4 (ref. 31) and the band centered at 1384 cm−1 can be attributed to the Ti–O–Ti vibration observed in TiO2.32 The band centered at 1630 cm−1 can be assigned to the bending vibration of the O–H bond of chemisorbed water,33 while the band centered at 1053 cm−1 in the spectra of the TiO2/ZnFe2O4/0.02 g AFAC and TiO2/ZnFe2O4/0.30 g AFAC composites could be assigned to the asymmetric stretching of Si–O–Si groups of AFAC,33 which could not be observed in the spectrum of the TiO2/ZnFe2O4 nanocomposite.
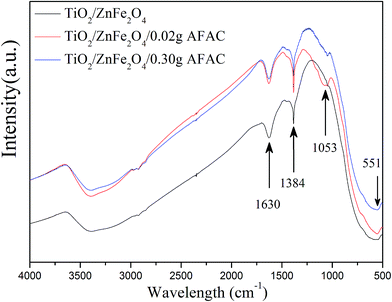 |
| | Fig. 3 The FT-IR spectra of TiO2/ZnFe2O4, TiO2/ZnFe2O4/0.02 g AFAC and TiO2/ZnFe2O4/0.30 g AFAC samples. | |
3.3 Pore structure and surface area determination
N2 adsorption measurement was carried out to characterize the porous structures and specific surface area of the TiO2/ZnFe2O4, TiO2/ZnFe2O4/0.02 g AFAC and TiO2/ZnFe2O4/0.30 g AFAC photocatalysts. Fig. 4A–C display the N2 adsorption–desorption isotherms and the corresponding pore size distribution histogram of the above samples. According to the International Union of Pure and Applied Chemistry (IUPAC) classification, all the samples exhibited reversible type IV isotherms with H3-type hysteresis loop,34,35 which indicated the formation of mesoporous materials.36 The hysteresis loop in the relative pressure range between 0.4 and 0.9 was probably related to the finer intra-aggregated pores formed between intra-agglomerated primary particles in the above composites. The pore diameter distribution histogram was calculated using the adsorption branch of the isotherm, which indicated the presence of mesopores (from 30 to 50 nm) and macropores (from 50 to 120 nm). The mesoporous structure could be related to the pores formed between stacked TiO2 particles coating on the ZnFe2O4 particles. The data summarized in Table 2 indicated that the BET specific surface area of the above photocatalysts were all more than 150.00 m2 g−1 and larger than that reported in some previous studies (less than 100.00 m2 g−1), which inferred that all the composites would have a strong adsorption ability.
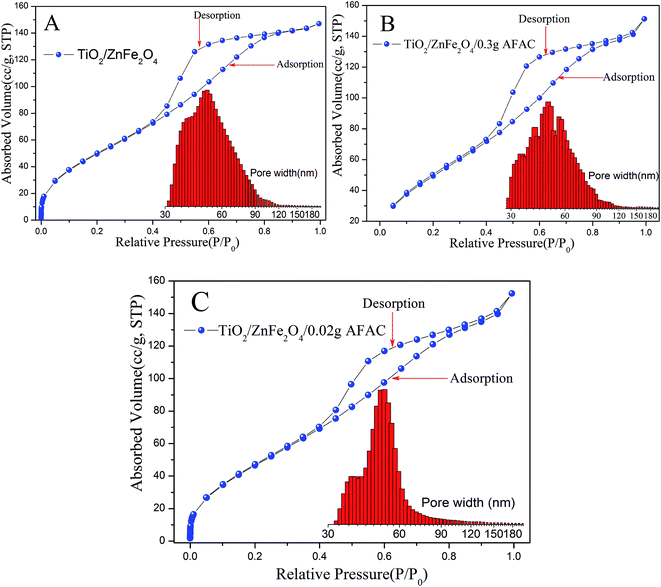 |
| | Fig. 4 BET isotherm and pore diameter distribution of different samples: (A) TiO2/ZnFe2O4; (B) TiO2/ZnFe2O4/0.30 g AFAC; (C) TiO2/ZnFe2O4/0.02 g AFAC (inset illustrates the graph of the corresponding pore diameter distributions). | |
Table 2 BET specific surface area of different samples
| Sample |
Pore volume (cm3 g−1) |
BET specific surface (m2 g−1) |
Half pore width (nm) |
Fitting error |
| TiO2/ZnFe2O4 |
0.213 |
162.183 |
25.935 |
2.495% |
| TiO2/ZnFe2O4/0.02 g AFAC |
0.209 |
151.122 |
24.210 |
2.618% |
| TiO2/ZnFe2O4/0.30 g AFAC |
0.219 |
167.447 |
26.500 |
0.474% |
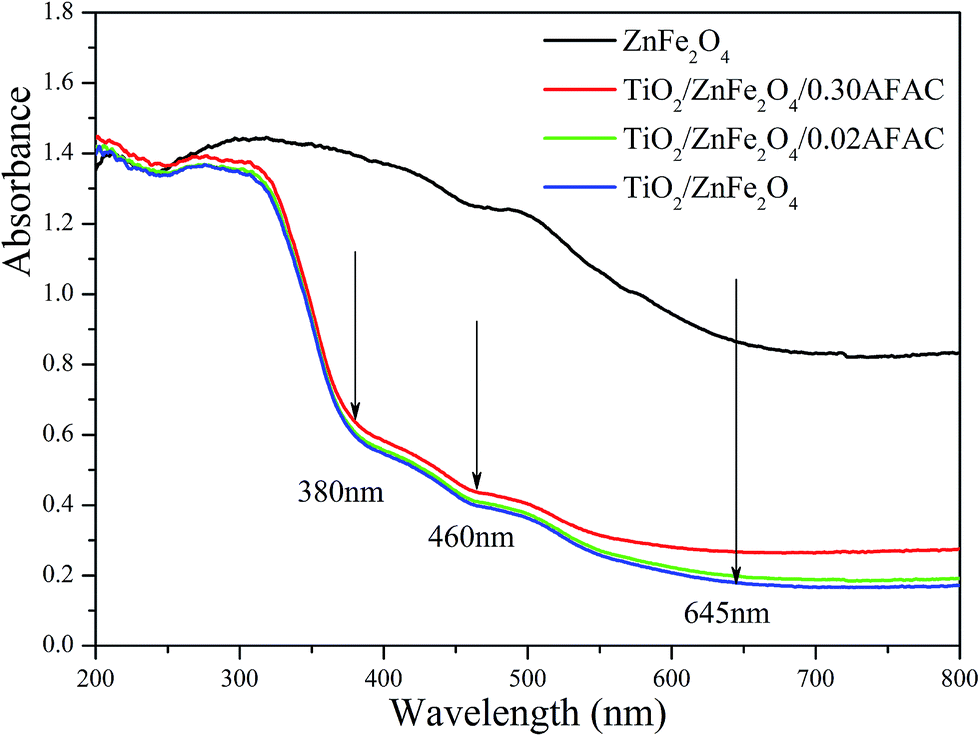
3.4 UV-vis photoresponse and band gap determination
The UV-vis diffuse reflectance spectra (UV-DRS) of the samples are depicted in Fig. 5. It is clearly shown that the as-prepared TiO2/ZnFe2O4/AFAC samples primarily absorbed UV light and part of the visible light. There are three intrinsic adsorption edges located at 380 nm, 460 nm and 645 nm, which correspond to anatase TiO2, TiO2/ZnFe2O4/AFAC sample and the ZnFe2O4 material, respectively. On further addition of AFAC, the light absorption of TiO2/ZnFe2O4/AFAC sample slightly enhanced. Based on the optical absorption edge obtained from UV-DRS, the energy band gaps for direct band-gap semiconductor of different samples were calculated by plotting (Ahν)1/2 to hν,6,27,37 where A is the absorption coefficient, hν is the photon energy and Eg is the energy band gap; the measured energy band gap values of the samples are listed in Table 1. The estimated Eg values of the as-prepared ZnFe2O4, TiO2/ZnFe2O4, TiO2/ZnFe2O4/0.02 g AFAC and TiO2/ZnFe2O4/0.30 g AFAC samples were 1.33 eV, 2.56 eV, 2.50 eV and 2.44 eV, respectively, which indicated that all the above samples would possess photocatalytic ability under visible light irradiation. The energy band-gap of anatase TiO2 is 3.18 eV; when hybridized with 0.35 g narrow band-gap semiconductor ZnFe2O4, it decreases greatly to 2.56 eV in TiO2/ZnFe2O4 nanocomposite, which is similar to previous reports.17–21 When hybridized with 0.30 g AFAC, the band-gap decreases to 2.44 eV, which had a less significant effect on the band-gap than that reported by Huo et al.6 Hence, ZnFe2O4 has more influence on the band gap of TiO2 than that of AFAC in our as-prepared TiO2/ZnFe2O4/AFAC photocatalysts. The visible red shift of the adsorption of TiO2/ZnFe2O4/AFAC samples resulted from two factors. One factor was the mixing effect of band gaps of ZnFe2O4 and TiO2 semiconductors discussed above. Since the added amount of ZnFe2O4 was small (3 wt%), the mixing effect alone cannot account for the entire large red shift. The other factor considered was the interface effect. Due to the interfacial coupling effect between ZnFe2O4 and TiO2 grains, ZnFe2O4 can induce lattice defects on the surface TiO6 octahedra, which may serve as the centers of bound excitons.38 These two factors together resulted in the red shift of the band gap of TiO2/ZnFe2O4/AFAC photocatalysts.
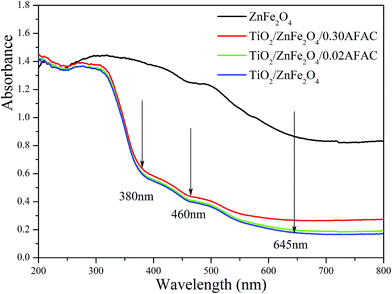 |
| | Fig. 5 UV-vis DRS of ZnFe2O4, TiO2/ZnFe2O4, TiO2/ZnFe2O4/0.02 g AFAC and TiO2/ZnFe2O4/0.30 g AFAC samples. | |
3.5 Adsorption of RhB
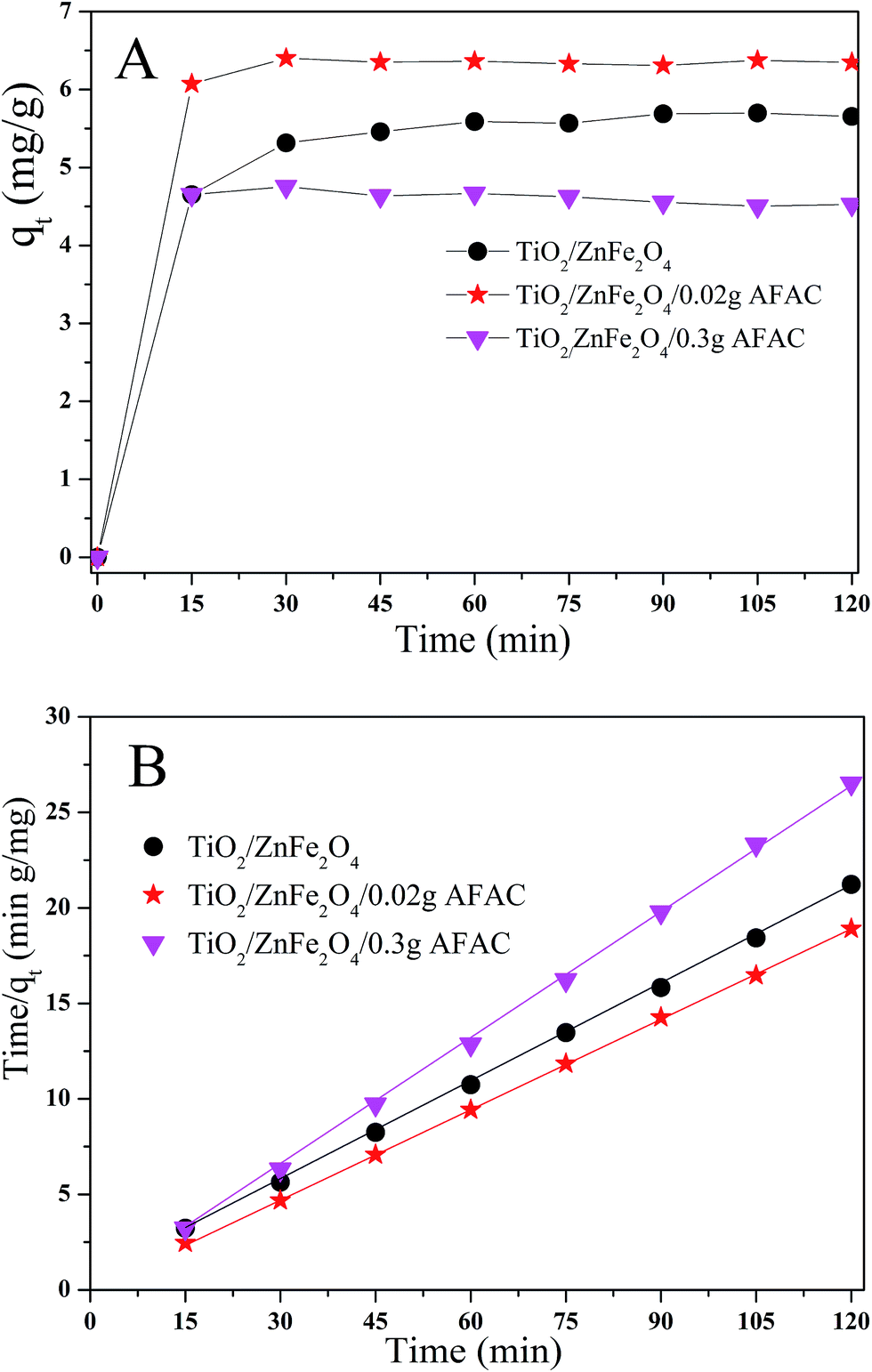
The adsorption ability would affect the photocatalysis performance greatly. Hence, rhodamine B (RhB) was chosen as the simulated pollutant to evaluate the adsorption activity of the as-prepared photocatalysts. Fig. 6A shows the adsorption process of RhB removal on different TiO2/ZnFe2O4/AFAC photocatalysts in the dark for 120 min; all the samples adsorbed the RhB quickly in the first 15 min and achieved maximum adsorption after 30 min. Their adsorption kinetics of RhB shown in Fig. 6B could be fitted well with a pseudo-second-order adsorption model.27 The adsorption pseudo second-order kinetic constants (k2) and their standard error, and the regression coefficient R2 are listed in Table 3. Therefore, the photocatalysts were first placed in dark for 30 min to reach the adsorption equilibrium and then were irradiated by the Xe lamp for 75 min for photocatalytic degradation. It was noteworthy that the adsorption activity of TiO2/ZnFe2O4/0.30 g AFAC photocatalyst was not even as good as that of the TiO2/ZnFe2O4 nanocomposite, which would result in disadvantageous influence on the final removal rate of RhB.
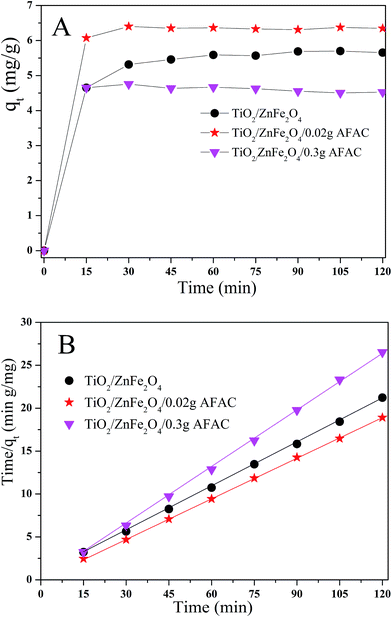 |
| | Fig. 6 (A) Adsorption kinetics of RhB onto TiO2/ZnFe2O4 nanocomposite, TiO2/ZnFe2O4/0.02 g AFAC and TiO2/ZnFe2O4/0.30 g AFAC photocatalysts; (B) pseudo second-order kinetic plots for RhB removal. | |
Table 3 Adsorption pseudo second-order kinetic constant and related parameters of different samples
| Samples |
k2 (g mg−1) |
Standard error |
R2 |
| TiO2/ZnFe2O4 |
0.14284 |
0.00611 |
0.98732 |
| TiO2/ZnFe2O4/0.02 g AFAC |
0.15692 |
0.00152 |
0.99935 |
| TiO2/ZnFe2O4/0.30 g AFAC |
0.20229 |
0.00333 |
0.99810 |
3.6 Photocatalytic degradation of RhB
The photocatalytic activity of the pristine TiO2, TiO2/ZnFe2O4, TiO2/ZnFe2O4/0.02 g AFAC and TiO2/ZnFe2O4/0.30 g AFAC were evaluated by degrading RhB under visible light, simulated sunlight (without the 420 nm cutoff filter) and UV light irradiation. The results are shown in Fig. S1.† Under UV light irradiation, RhB was almost thoroughly degraded in 15 min by pure TiO2; however, it was evident that TiO2 was impotent for the degradation of RhB under visible light irradiation. Hence, we focused on the photocatalytic performance of the as-prepared composites under visible light, which would be enlightening for future application. Fig. 7 shows the processes of RhB removal on different TiO2/ZnFe2O4/AFAC photocatalysts under visible light irradiation. The TiO2/ZnFe2O4 nanocomposite removed 95.1% of the RhB. When hybridized with 0.02 g AFAC, the TiO2/ZnFe2O4/0.02 g AFAC photocatalyst removed 97.1% of the RhB with the highest photocatalytic rate except for the abnormal upturn point located at 30 min, which is demonstrated in Fig. 8. When the amount of AFAC was increased to 0.30 g, the ratio of the removed RhB decreased to 91%, which resulted from its relatively weak adsorption activity of RhB as mentioned before. With the appropriate band gap, large specific surface area and good adsorption ability, the TiO2/ZnFe2O4/0.02 g AFAC photocatalyst had the best photocatalytic performance on the degradation of RhB among all our samples.
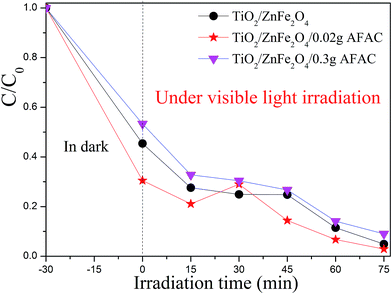 |
| | Fig. 7 The photodegradation rate of RhB with different photocatalysts under visible light. | |
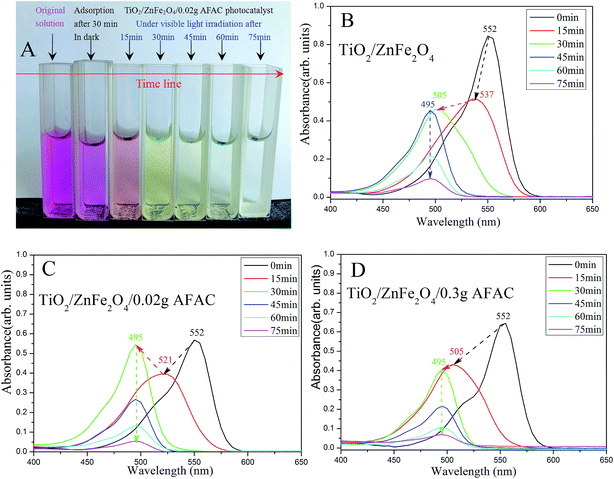 |
| | Fig. 8 (A) The color of the simulated wastewater after different times under visible light by TiO2/ZnFe2O4/0.02 g AFAC photocatalyst; the spectra of photocatalytic degradation of RhB by (B) TiO2/ZnFe2O4; (C) TiO2/ZnFe2O4/0.02 g AFAC and (D) TiO2/ZnFe2O4/0.30 g AFAC photocatalysts. | |
RhB could hardly be photodegraded under visible light irradiation;39 therefore the removal of RhB was primarily attributed to the photocatalytic degradation by the photocatalyst. Fig. 8 shows the spectra of photocatalytic degradation of RhB by the three photocatalysts. When the simulated RhB wastewater was irradiated under visible light, RhB was clearly photodegraded in 75 min. The color of the wastewater changed gradually from magenta to colorless (Fig. 8A). The maximum absorption peak shifted gradually from 552 nm to 495 nm, which implied that there were different intermediate products. Under visible light irradiation, the TiO2/ZnFe2O4 nanocomposite first photodegraded RhB (550 nm) to N,N,N′-triethyl rhodamine (537 nm) after 15 min, then to rhodamine (495 nm) after 30 min and the peak of rhodamine sharply decreased after 75 min, which indicated that RhB was photodegraded. Moreover, the TiO2/ZnFe2O4/0.02 g AFAC photocatalyst photodegraded RhB (550 nm) first to N,N′-diethyl rhodamine (521 nm) after 15 min and then to rhodamine (495 nm) after 30 min. The TiO2/ZnFe2O4/0.30 g AFAC photocatalyst photodegraded RhB (550 nm) first to N-ethyl rhodamine (505 nm) after 15 min and then to rhodamine (495 nm) after 30 min. The above results were similar to those reported by Watanabe et al.,40 in which the absorption of the intermediate products changed from RhB (555 nm), to N,N,N′-triethyl rhodamine (539 nm), N,N′-diethyl rhodamine (522 nm), N-ethyl rhodamine (510 nm) and rhodamine (498 nm). Fig. 8C shows an abnormal upturn from N,N′-diethyl rhodamine (521 nm) to rhodamine (495 nm) after 30 min, corresponding to the abnormal upturn of the TiO2/ZnFe2O4/0.02 g AFAC photocatalyst (Fig. 7), which implied that the formation of rhodamine (495 nm) was critical to the photocatalysis process.
The process of RhB removal on the ZnFe2O4/TiO2/AFAC photocatalyst surface could be summarized as follows: (1) when the ZnFe2O4/TiO2 nanocomposite was loaded on AFAC, the surface adsorption of RhB on the photocatalyst could be enhanced, which causes a concentration effect in photodegrading RhB; (2) the photodegradation of RhB would empty the adsorption sites on the surface of the photocatalyst, which would continue adsorbing molecular RhB. The synergistic effects of the adsorption ability and photocatalytic ability of the ZnFe2O4/TiO2/AFAC photocatalyst improve the removal of RhB in the solution.
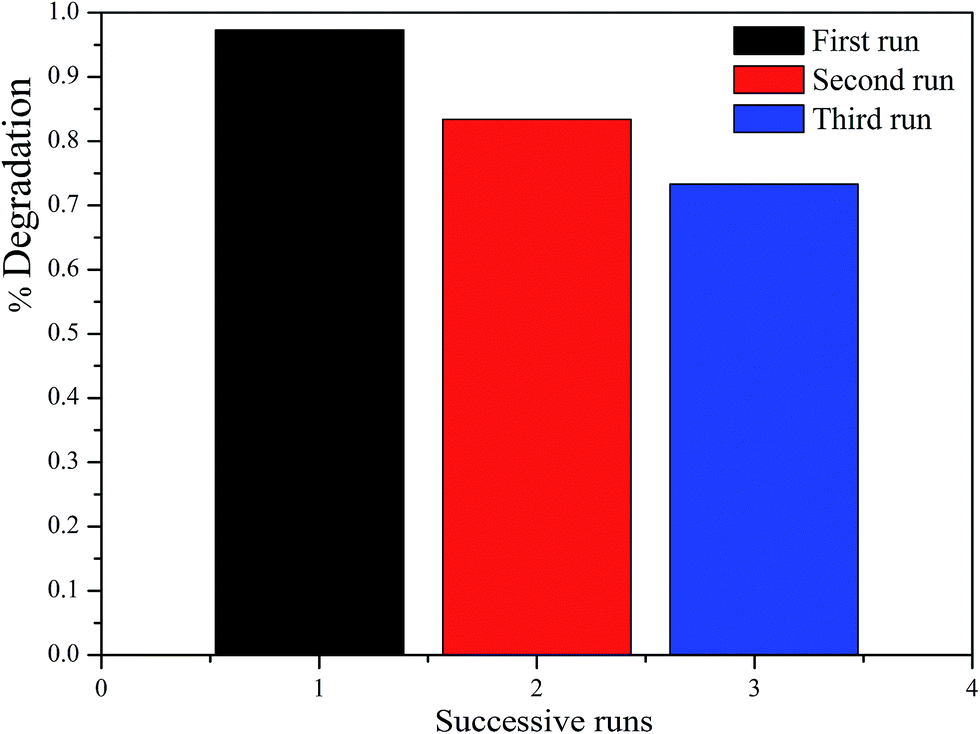
3.7 Recyclability
To evaluate the recyclability of the as-prepared catalysts, the ZnFe2O4/TiO2/0.02 g AFAC photocatalyst was selected to test the recycle experiments. As shown in Fig. 9, after two and three runs, about 83.4% and 73.7% of RhB was photodegraded, respectively. The photocatalytic performance of the as-prepared catalyst decreased slightly during the photodegradation process. The decrease in the RhB removal efficiency might be attributed to the accumulation of RhB on the photocatalyst surface, which blocked the mesopores gradually, and then the adsorption of RhB from the solution was restrained. Therefore, to improve the photocatalytic performance of the reused catalyst, appropriate prolonging of the oxidation time is beneficial.
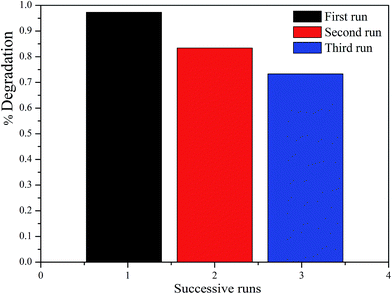 |
| | Fig. 9 The photocatalytic degradation of RhB for three cycles by TiO2/ZnFe2O4/0.02 g AFAC photocatalyst. | |
3.8 Photocatalytic mechanism
Under visible light, the impressive photocatalytic performance of the ZnFe2O4/TiO2/0.02 g AFAC photocatalyst primarily resulted from the following three factors: (1) the crystallite size of the as-prepared anatase TiO2 was less than 10 nm, which indicated that the specific surface area of anatase TiO2 would be large; (2) when TiO2 particles were coated on ZnFe2O4 particles, the band-gap of the TiO2/ZnFe2O4 nanocomposite decreased from 3.18 to 2.56 eV, so the nanocomposite could photodegrade RhB under visible light irradiation; (3) when hybridized with 0.02 g AFAC, the TiO2/ZnFe2O4/0.02 g AFAC exhibited a band-gap of 2.50 eV, the number of the macropores in the photocatalyst decreased and the majority of the pores were mesopores, which resulted in a competitive phenomenon: a dominant positive effect of the enhancement of the adsorption ability and a negligible negative effect of a decrease in the specific surface area from 162.18 m2 g−1 to 151.12 m2 g−1. The combination of the above three positive factors resulted in a synergistic effect, which led to the slight enhancement of the photocatalytic performance from 95% to 97.1%. When hybridized with 0.30 g AFAC, although the TiO2/ZnFe2O4/0.30 g AFAC photocatalyst had a bigger specific surface area of 167.44 m2 g−1 and a smaller band-gap of 2.44 eV, which were both positive effects for photocatalytic performance, the removal efficiency of RhB decreased to 91%. The reason might be as follows: the more AFAC was added, the more chances that AFAC may absorb and reflect the visible light energy, so less energy would be absorbed by the TiO2/ZnFe2O4 photocatalyst and eventually the photocatalytic performance decreased.
A probable photocatalytic mechanism is schematically illustrated in Fig. 10. Under visible light irradiation, the electrons (e−) would be promoted to the conduction band (CB), while some corresponding positive holes (h+) would emerge in the valence band (VB) as described in eqn (1).16,41,42 The photogenerated electrons could migrate to the surface and adsorb molecular oxygen to generate the superoxide radical (˙O2−) (eqn (2)) and the photogenerated holes could react with the adsorbed H2O molecules to generate hydroxyl radical (˙OH) (eqn (3)).43 The RhB adsorbed on the photocatalyst surface could be promoted to its excited state (RhB*);44 then, RhB* would promote an electron to the photocatalyst and ˙RhB+ would be formed (eqn (4)). The generated superoxide radicals, hydroxyl radicals and the photogenerated holes of the valence band could degrade RhB (and ˙RhB+) into carbon dioxide and water (eqn (5)). The above process can be summarized as follows:
| | |
ZnFe2O4/TiO2/AFAC + hν → e− + h+
| (1) |
| | |
RhB + hν → RhB* → ˙RhB+ + e−
| (4) |
| | |
h+, ˙O−2, ˙OH + RhB, ˙RhB+ → N,N,N′-triethyl rhodamine → N,N′-diethyl rhodamine → N-ethyl rhodamine → rhodamine → CO2, H2O, etc.
| (5) |
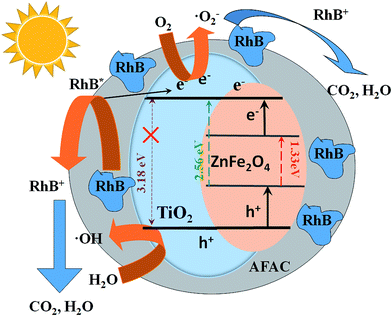 |
| | Fig. 10 Schematic diagram of photocatalytic degradation of RhB on TiO2/ZnFe2O4/AFAC photocatalyst under visible light irradiation. | |
4. Conclusions
A series of TiO2/ZnFe2O4/AFAC photocatalysts were prepared on the basis of the sol–gel and hydrothermal method. By coating ZnFe2O4 nanoparticles with TiO2 nanoparticles, the band gap of TiO2/ZnFe2O4 nanocomposite was narrowed to 2.56 eV, which would possess photocatalytic ability under visible light. When the AFAC was hybridized with the TiO2/ZnFe2O4 nanocomposite, their adsorption capacity and absorption ability were enhanced. Under visible light irradiation for 75 min, the removal rate of RhB on the TiO2/ZnFe2O4 nanocomposite and TiO2/ZnFe2O4/0.02 g AFAC photocatalyst were 95.0% and 97.1%. However, on hybridization with more AFAC, the TiO2/ZnFe2O4/0.30 g AFAC photocatalyst had a lower removal rate of 91%. The impressive photocatalytic performance originated from the synergistic effect of small crystallite size, narrow band-gap and mesoporous structure of the photocatalysts. Recycling experiments indicated that the photocatalyst retained higher activity after three cycles. Therefore, the TiO2/ZnFe2O4/AFAC photocatalysts would have potential application in environmental remediation.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by National Natural Science Foundation of China (Grant No. 61775081, 61475063, 21576111) and the Graduate Innovation Research Fund at the Jilin Normal University (No. 201628).
References
- A. Fujishima, T. N. Rao and D. A. Tryk, J. Photochem. Photobiol., C, 2000, 1, 1–21 CrossRef CAS.
- A. Fujishima, X. T. Zhang and D. A. Tryk, Surf. Sci. Rep., 2008, 63, 515–582 CrossRef CAS.
- Y. Su, P. Chen and F. Wang, et al., RSC Adv., 2017, 7(54), 34096–34103 RSC.
- Y. L. Pang and A. Z. Abdullah, J. Hazard. Mater., 2012, 235, 326–335 CrossRef PubMed.
- J. Lim, A. D. Bokare and W. Choi, RSC Adv., 2017, 7(52), 32488–32495 RSC.
- P. Huo, Y. Yan, S. Li, H. Li and W. Huang, Desalination, 2010, 263, 258–263 CrossRef CAS.
- S. G. Kumar and L. G. Devi, J. Phys. Chem. A, 2011, 115, 13211–13241 CrossRef CAS PubMed.
- L. G. Gao, J. W. Du and T. L. Ma, Ceram. Int., 2017, 43(12), 9559–9563 CrossRef CAS.
- J. Li, S. Jia and G. Sui, et al., RSC Adv., 2017, 7(55), 34857–34865 RSC.
- M. Xing, Y. Wu, J. Zhang and F. Chen, Nanoscale, 2010, 2, 1233–1239 RSC.
- M. Kang, J. Mol. Catal. A: Chem., 2003, 197, 173–183 CrossRef CAS.
- F. Magalhães, F. C. Moura and R. M. Lago, Desalination, 2011, 276, 266–271 CrossRef.
- Q. Zhu, X. H. Hu, M. S. Stanislaus, N. Zhang, R. D. Xiao, N. Liu and Y. N. Yang, Sci. Total Environ., 2017, 577, 236–244 CrossRef CAS PubMed.
- S. Y. Guo, J. G. Dai and T. J. Zhao, et al., RSC Adv., 2017, 7(58), 36787–36792 RSC.
- G. Li, X. Nie, J. Chen, Q. Jiang, T. An, P. K. Wong, H. Zhang, H. Zhao and H. Yamashita, Water Res., 2015, 86, 17–24 CrossRef CAS PubMed.
- P. Cheng, C. Deng, M. Gu and W. Shangguan, J. Mater. Sci., 2007, 42(22), 9239–9244 CrossRef CAS.
- Z. Yuan and L. Zhang, J. Mater. Chem., 2001, 11(4), 1265–1268 RSC.
- H. Yang, X. Y. Li, Q. D. Zhao, X. Quan and G. H. Chen, Environ. Sci. Technol., 2010, 44(13), 5098–5103 CrossRef PubMed.
- Y. Hou, X. Y. Li and Q. D. Zhao, Adv. Funct. Mater., 2010, 20(13), 2165–2174 CrossRef CAS.
- E. Moreira, L. A. Fraga and M. H. Mendonça, J. Nanopart. Res., 2012, 14(6), 1–10 CrossRef.
- M. Wang, L. Sun and J. Cai, J. Mater. Chem. A, 2013, 1(39), 12082–12087 CAS.
- J. Zhang, X. Wang, X. Wang, J. Song, J. Huang, B. Louangsouphom and J. Zhao, RSC Adv., 2015, 5, 71922–71931 RSC.
- X. Wang, W. Wang, X. Wang, J. Zhang, Z. Gu, L. Zhou and J. Zhao, Appl. Surf. Sci., 2015, 349, 264–271 CrossRef CAS.
- S. Hosseini, S. Borghei, M. Vossoughi and N. Taghavinia, Appl. Catal., B, 2007, 74, 53–62 CrossRef CAS.
- L. C. R. Machado, C. B. Torchia and R. M. Lago, Catal. Commun., 2006, 7, 538–541 CrossRef CAS.
- B. Wang, C. Li, J. Pang, X. Qing, J. Zhai and Q. Li, Appl. Surf. Sci., 2012, 258, 9989–9996 CrossRef CAS.
- J. Song, X. J. Wang, Y. J. Bu, X. Wang, J. Zhang, J. Y. Huang, R. R. Ma and J. F. Zhao, Appl. Surf. Sci., 2017, 391, 236–250 CrossRef CAS.
- A. N. Ökte and D. Karamanis, Appl. Catal., B, 2013, 142, 538–552 CrossRef.
- A. Dagar and A. K. Narula, J. Mater. Sci.: Mater. Electron., 2017, 28(12), 8643–8654 CrossRef CAS.
- Y. Ao, J. Xu and D. Fu, Sep. Purif. Technol., 2008, 61(3), 436–441 CrossRef CAS.
- C. G. Anchieta, A. Cancelier and M. A. Mazutti, et al., Materials, 2014, 7(9), 6281–6290 CrossRef CAS PubMed.
- N. Dhiman, B. P. Singh and A. K. Gathania, J. Nanophotonics, 2012, 6(1), 063511-1–063511-10 CrossRef.
- C. Yang, T. Wang and P. Liu, Curr. Opin. Solid State Mater. Sci., 2009, 13(5), 112–118 CrossRef CAS.
- M. Donohue and G. Aranovich, Adv. Colloid Interface Sci., 1998, 76, 137–152 CrossRef.
- M. Kruk and M. Jaroniec, Chem. Mater., 2001, 13(10), 3169–3183 CrossRef CAS.
- Z. Jing and J. Zhan, Adv. Mater., 2008, 20, 4547–4551 CrossRef CAS.
- S. Delekar, H. Yadav, S. Achary, S. Meena and S. Pawar, Appl. Surf. Sci., 2012, 263, 536–545 CrossRef CAS.
- L. V. Saraf, S. L. Patil and S. B. Ogale, J. Mod. Phys., 1998, 12(25), 2635–2647 CrossRef CAS.
- X. Wang, X. Wang, J. Zhao, J. Chen, J. Zhang, J. Song and J. Huang, RSC Adv., 2015, 5, 66611–66620 RSC.
- T. Watanabe, T. Takirawa and K. Honda, J. Phys. Chem., 1977, 81, 1845–1851 CrossRef CAS.
- E. Moreira, L. A. Fraga and M. H. Mendonça, et al., J. Nanopart. Res., 2012, 14(6), 937 CrossRef.
- I. Kryuko, S. Y. Kuchmii and V. D. Pokhodenko, Theor. Exp. Chem., 2000, 36(2), 63–81 CrossRef.
- L. G. Devi and S. G. Kumar, Appl. Surf. Sci., 2011, 257, 2779–2790 CrossRef CAS.
- H. Wang, X. Yuan, Y. Wu, G. Zeng, X. Chen, L. Leng and H. Li, Appl. Catal., B, 2015, 174, 445–454 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra11055c |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *abc
*abc