
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInhibition of the expression of oncogene SRSF3 by blocking an exonic splicing suppressor with antisense oligonucleotides
Jihua Guo†
ab,
Xiaoxuan Che†a,
Xiaole Wanga and
Rong Jia *a
*a
aHubei-MOST KLOS & KLOBME, School & Hospital of Stomatology, Wuhan University, 237 Luoyu Road, Wuhan, 430079, PR China. E-mail: jiarong@whu.edu.cn; Tel: +86 87686268
bDepartment of Endodontics, School & Hospital of Stomatology, Wuhan University, Wuhan, PR China
First published on 14th February 2018
Abstract
Antisense oligonucleotides (ASOs) have been widely used to regulate alternative splicing of pre-mRNA by targeting splice sites, branch points, or exonic splice enhancers to increase exon skipping or intron retention. So far, few studies have used ASOs to block exonic splicing suppressor (ESS) and increase exon inclusion. Previously, we demonstrated that serine and arginine rich splicing factor 3 (SRSF3) (also called SRp20) is an oncogene. The inclusion of its alternative exon 4 down-regulates its expression. An ESS motif is responsible for the skipping of alternative exon 4. Here, we used an economical method to screen effective anti-ESS ASO. We discovered that an ASO targeting the ESS motif can promote the inclusion of exon 4, reduce SRSF3 expression, and inhibit cell growth in oral cancer cells. Our results suggested that using anti-ESS ASOs can efficiently increase exon inclusion and be used as a potential anti-cancer drug.
Introduction
Serine and arginine rich splicing factor 3 (SRSF3, also called SRp20) belongs to the serine/arginine (SR)-rich family, and plays important roles in constant and alternative splicing of pre-mRNA. In addition, SRSF3 was also reported to regulate alternative RNA polyadenylation,1 RNA export,2,3 DNA repair,4 and protein translation.5 Like other SR protein family members, SRSF3 protein contains an N-terminal RNA recognition motif (RRM) and a C-terminal arginine/serine-rich domain (RS).6SRSF3 is an oncogene and frequently overexpressed in cancer cells.7,8 Knockdown of SRSF3 significantly reduced cancer cell proliferation.5,9–12 SRSF3 promotes the expression of oncogenes, including FoxM1,7 and PKM2,13 and inhibits the expression of tumor suppressor genes, including PDCD4,14 and p53.15 Previously, we have found the expression of SRSF3 is autoregulated by the inclusion of its alternative exon 4, which contains a pre-mature stop codon. Only transcript without exon 4 is able to encode full-length functional SRSF3. Inclusion of exon 4 may encode a truncated SRSF3 or cause degradation of transcript.16
We have also demonstrated that an exonic splicing suppressor (ESS) is responsible for the skipping of alternative exon 4.16 PTBP1 and PTBP2 are potential oncogenic proteins.17 These two splicing factors bind to exon 4 ESS motif and prevent the inclusion of exon 4. In cancer cells, increased expression of PTBP1 and PTBP2 impaired the autoregulation mechanism and promoted the expression of SRSF3 protein.16 Interference of the interaction between ESS motif and PTBP proteins may increase the inclusion of exon 4 and decrease the expression of oncogenic SRSF3.
Misregulated pre-mRNA alternative splicing can be corrected by antisense oligonucleotides (ASOs).18 ASOs bind to splice sites and block the usage of splice site and cause exon skipping. ASOs can also bind to exonic or intronic splicing enhancers, and then inhibit the usage of splice sites. So far, few study has used ASOs to block ESS and increase exon inclusion. In this study, we established an economical method to screen effective anti-ESS ASO. We successfully found that an anti-ESS ASO was able to efficiently increase the inclusion of exon 4, and then inhibit SRSF3 expression and cancer cell growth.
Experimental procedures
Cells and plasmid
CAL 27 and FaDu are oral squamous carcinoma cell lines. HEK 293 is a human embryonic kidney cell line. All cell lines were grown and maintained in Dulbecco's modified Eagle medium (DMEM, Hyclone, USA) supplemented with 10% fetal bovine serum (FBS, Gibco, USA). A plasmid carrying SRSF3 minigene of genomic DNA sequence from exon 3 to exon 5 was shown in Fig. 1A.16 An ATG was added to 5′ of exon 3 to allow the expression of exon 3, exon 5 and GFP fusion protein. Inclusion of exon 4 will include a premature stop codon and block the expression of GFP.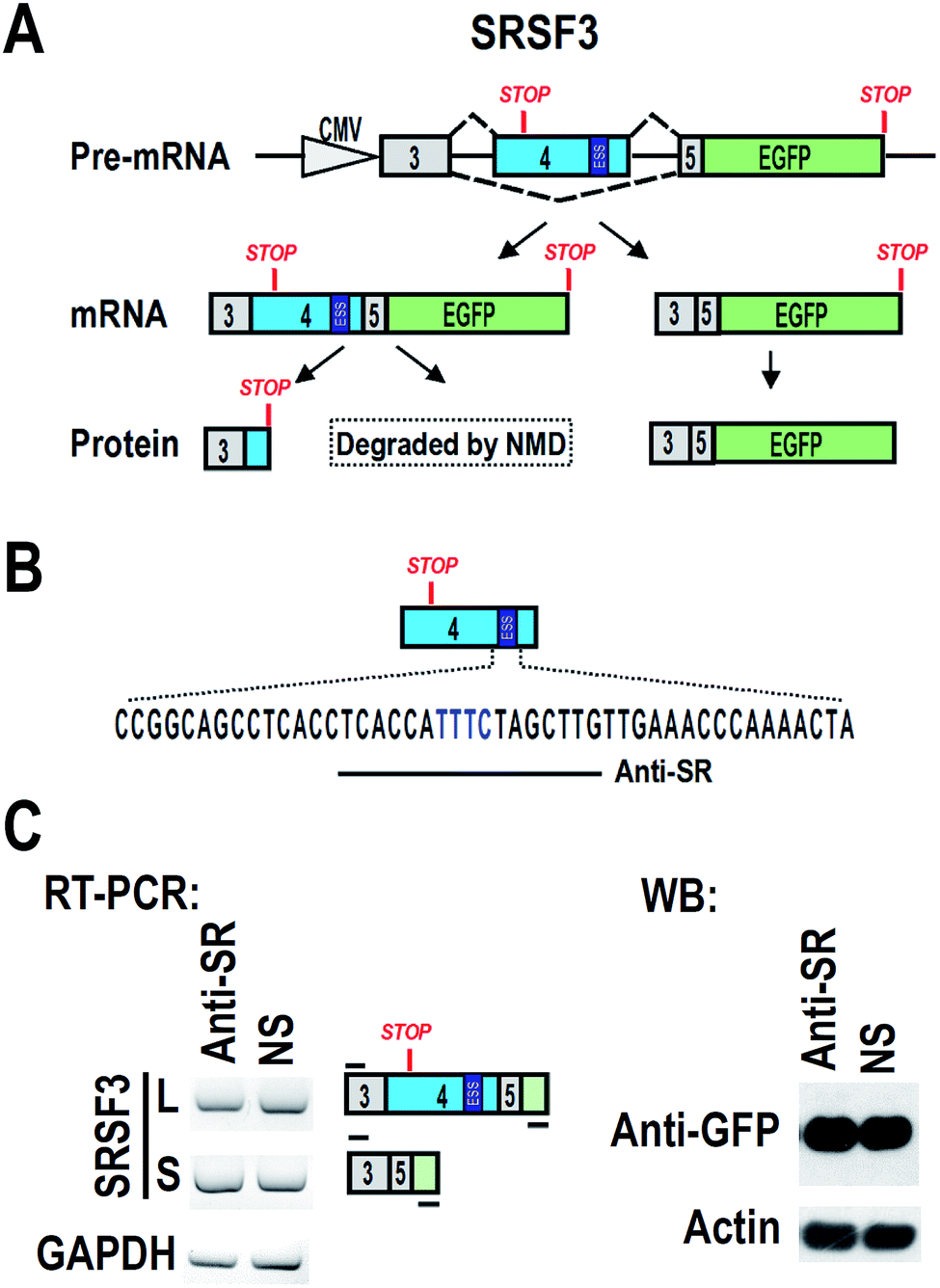 | ||
| Fig. 1 The design of antisense oligonucleotide to block ESS motif in SRSF3 exon 4. (A) Schematic diagram of SRSF3 minigene. Genomic sequence of SRSF3 (exon 3 to exon 5) were cloned into pEGFP-N1. ATG was added to 5′ end of minigene. GFP is allowed to be translated in the absence of exon 4. Inclusion of exon 4 will block the expression of GFP or cause degradation by nonsense-medicated decay (NMD). An exonic splicing suppressor (ESS) is highlighted in 3′ part of exon 4. (B) An antisense oligonucleotide (Anti-SR, the line below the sequence) was designed to base-pair to ESS region (highlighted in blue). (C) HEK 293 cells were co-transfected with SRSF3 minigene and 20 nM antisense oligonucleotide Anti-SR. Alternative splicing of exon 4 were analyzed by RT-PCR. GAPDH served as loading control. The expression of GFP was detected by western blot. Actin served as loading control. | ||
Antisense oligonucleotide transfection
Antisense oligonucleotides modified by phosphorothioate or 2′-O-methyl-phosphorothioate were synthesized in Sangon Biotech (China). For cell transfection, 40 pmol ASOs were diluted in 25 μL Opti-MEM medium (ThermoFisher, USA), and then mixed with 1.5 μL Lipofectamine 3000 (ThermoFisher, USA) diluted in 25 μL Opti-MEM medium. After incubation for 15 minutes in room temperature, the transfection complex was added to cell culture medium. The final concentration of ASOs is 20 nM. A non-specific ASO (5′ ACTCTATCTGCACGCTGACT 3′) was used as control. Forty eight hours later, total RNA and protein were collected.RNA purification and RT-PCR
Total RNA was purified from cells by using AxyPrep Multisource Total RNA Miniprep Kit (Axygen, USA). One microgram total RNA was treated with DNase I (Thermo fisher scientific, USA), and then reversely transcribed with Moloney Murine Leukemia Virus (MMLV) reverse transcriptase (Promega, USA) and random primers (hexadeoxynucleotides) (Promega, USA). One microliter of the cDNA was subjected to PCR with Taq DNA polymerase (Takara, Japan) and following primers:oGJH209 5′ GGAGTCCTCCACCTCGTCGCA 3′ and oGJH101 5′ GCTCCTCGCCCTTGCTCACCA 3′ for detecting the alternative splicing of exogenous exon 4 in the minigene; oGJH759 5′ CATGTGAAACGACACCAGCCAAGC 3′ and oGJH211 5′ CTCCCTCTTGGGGTCGTCGC 3′ for detecting exon 4-included endogenous SRSF3; oGJH213 5′ CCATAGAGAATTACACCTTTGTGTCACTG 3′ (exon 7) and oGJH761 5′ AGTCCTCCACCTCGTCGCAGATCTC 3′ (exon 3–5 junction primer) for detecting exon 4-skipped endogenous SRSF3; 5′ GTCATCAATGGAAATCCCATCACC 3′ and 5′ TGAGTCCTTCCACGATACCAAA 3′ for internal control GAPDH.
Western blot
Total protein samples were collected by adding 2× SDS sampling buffer to cells. Samples were denatured for 5 minutes at 95 degree and separated in a 10% SDS-PAGE gel, followed by being transferred to a nitrocellulose membrane. Specific proteins were detected by anti-GFP antibody (Santa cruz, USA), anti-SRSF3 antibody (Thermo Fisher Scientific, USA), or horseradish peroxidase-labeled mouse anti-β-actin antibody (Sigma-Aldrich, USA).Cell proliferation analysis
Cell proliferation was analyzed by trypan blue exclusion method or MTS assay. Trypan blue exclusion method was performed with a 0.4% trypan blue solution (Thermo Fisher Scientific, USA). MTS assay was performed by using the celltiter 96 aqueous one solution cell proliferation assay kit (Promega, USA) according to the manufacturer's instructions. Absorbance values at 490 nm were obtained using powerWave XS2 plate reader (BioTek, USA).Statistical analyses
All two-group statistical comparisons of means were determined using Student's t test (Excel, Microsoft).Results and discussion
Blocking of ESS with antisense oligonucleotide
We hypothesized that blocking of ESS with ASO may increase the inclusion of exon 4, and then down-regulate the expression of full-length SRSF3. We designed an ASO, named “Anti-SR”, according to the ESS and nearby sequence (Fig. 1B). Anti-SR was modified by 2′-O-methyl-phosphorothioate to increase stability and reduce RNase H activity in cells.19 Anti-SR was co-transfected with SRSF3 minigene plasmid into 293 cells. After 24 hours, the alternative splicing of SRSF3 was analyzed by RT-PCR. We found that Anti-SR could not increase the inclusion of exon 4, indicating that this ASO is not suitable for blocking ESS. Hybridization with target mRNA is the necessary characteristic for effective ASO.20 Anti-SR may be not able to efficiently bind to the ESS of exon 4 due to the RNA secondary structure.Screening of effective antisense oligonucleotide
To find effective ASO to block the ESS motif, we need to screen a serial of ASOs based on the ESS motif and nearby sequence (Fig. 2A). However, 2′-O-methyl-phosphorothioate modification is quite expensive. Unmodified ASOs are much cheaper than 2′-O-methyl-phosphorothioate modification ASOs, but they are generally unstable in cells. In comparison, phosphorothioate modification of oligonucleotides are stable in cells and also much cheaper than 2′-O-methyl-phosphorothioate modification (at least 10 fold less expenditure). Therefore, we used phosphorothioate ASOs for screening experiment. In general, phosphorothioate ASOs cannot be used to modify alternative splicing, because binding of phosphorothioate ASOs to target mRNA will induce RNase H-mediated degradation of target mRNA. However, significant degradation may correlated with the efficient binding of ASO to target mRNA.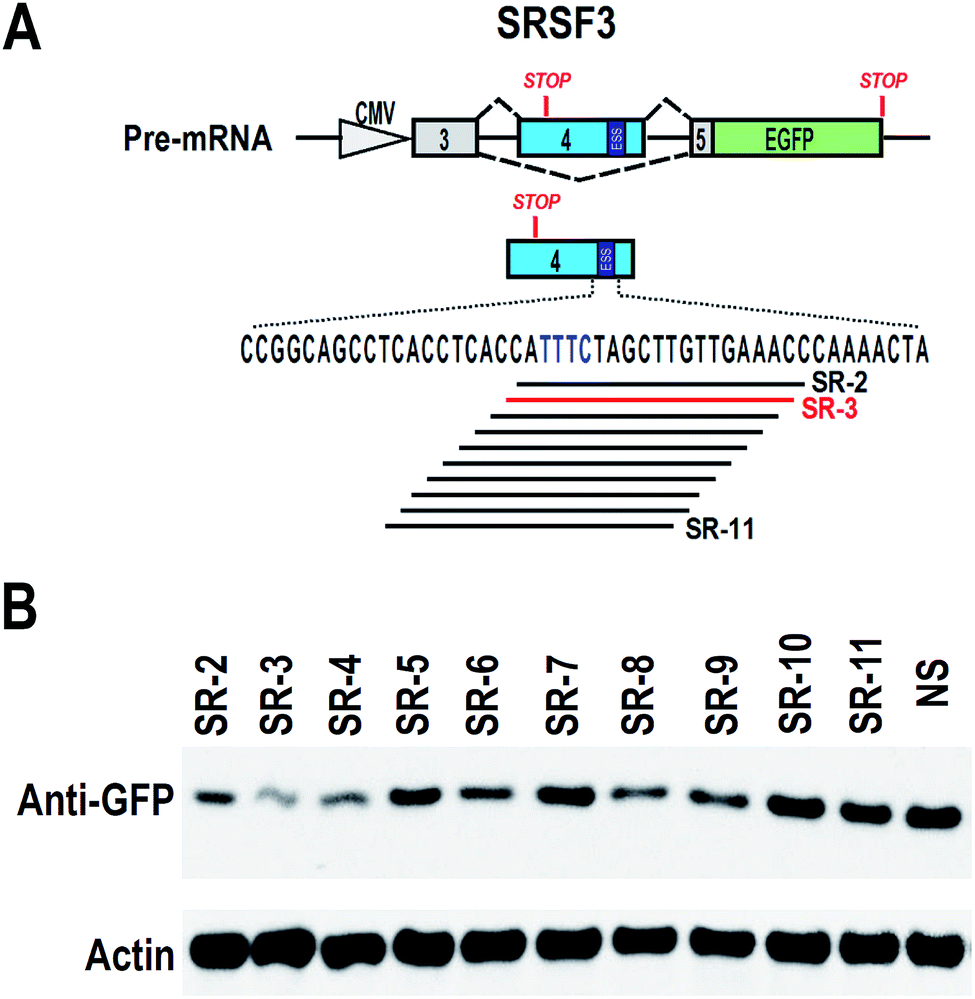 | ||
| Fig. 2 Screening of effective antisense oligonucleotides against ESS in SRSF3 exon 4. (A) Mapping the ASOs against ESS (the lines below the sequence). (B) HEK 293 cells were co-transfected with SRSF3 minigene and 20 nM ASOs. The expression of GFP was detected by western blot. Actin served as loading control. Inclusion of exon 4 will block GFP expression or cause degradation of transcripts. | ||
We synthesized a serial of ASOs modified by phosphorothioate and based on the ESS motif and nearby sequence (Fig. 2A). These ASOs were also co-transfected with SRSF3 minigene into 293 cells. If ASOs are able to bind to SRSF3-GFP fusion transcripts, these transcripts will be degraded and the expression of SRSF3-GFP fusion protein will be reduced. Indeed, we successfully observed that several ASOs reduced the expression of SRSF3-GFP fusion protein compared with non-specific ASO. The most significant down-regulation of GFP expression was found in ASO SR-3 (Fig. 2B). We speculated that SR-3 may be also effective on modification of alternative splicing of SRSF3 exon 4.
To test the effect of SR-3 on alternative splicing of SRSF3 exon 4, we synthesized 2′-O-methyl-phosphorothioate modified SR-3 and performed transfection in two oral cancer cell lines, CAL 27 and FaDu. RT-PCR results showed that SR-3 was able to significantly increase the inclusion of SRSF3 exon 4 (Fig. 3). This result demonstrated that the strategy of screening effective ASO against ESS by using phosphorothioate ASOs is successful. It is useful and economical in studies of alternative splicing modification by ASOs.
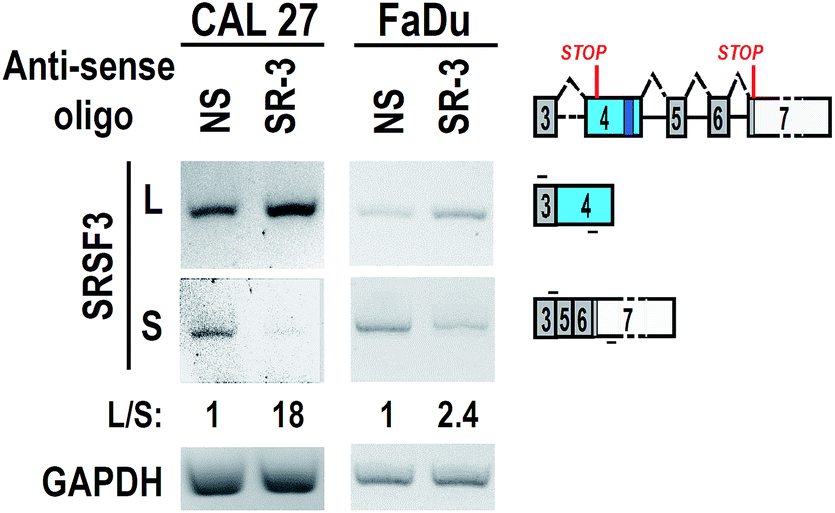 | ||
| Fig. 3 Antisense oligonucleotide promotes the inclusion of exon 4. CAL 27 or FaDu oral cancer cells were transfected with 20 nM 2′-O-methyl-phosphorothioate ASO SR-3 or NS (non-specific oligonucleotide). Alternative splicing of exon 4 were analyzed by RT-PCR. GAPDH served as loading control. | ||
Antisense oligonucleotides have been widely used to regulate alternative splicing of pre-mRNA.19,21,22 Most of ASOs were designed to target splice site, branch point, or exonic splice enhancer to increase exon skipping or intron retention, and then reduce the expression of oncogenic proteins. For example, MDM4 is an oncogene and contains an alternative exon 6. Dewaele et al. designed an ASO to target 5′-donor site and SRSF3-binding motif, and promote exon 6 skipping. Consequently, the expression level of functional and full-length MDM4 decreased, and melanoma cell growth was impaired.23 Shchelkunova et al. designed ASOs to target splice sites of oncogene STAT5B intron 18, and increased intron 18 retention, which converts oncogenic full-length STAT5B protein into truncated STAT5B protein with tumor suppressor characteristics.24 Theoretically, blocking of ESS with ASOs will promote exon inclusion. Increased inclusion of novel exon may also interfere with the expression of functional oncoproteins. However, to the best of our knowledge, few study has used ASOs to block ESS and increase exon inclusion. Our results demonstrated that 2′-O-methyl-phosphorothioate modified anti-ESS ASOs can efficiently increase exon inclusion.
Blocking of ESS to suppress SRSF3 expression and the growth of cancer cells
To further study the function of SR-3 ASO in cancer cells, we analyzed the effect of SR-3 on the expression level of SRSF3 protein, and cell growth using trypan blue exclusion method or MTS assay. We found that SRSF3 protein level significantly decreased in SR-3 treated cells compared with non-specific ASO (Fig. 4C). SR-3 treatment significantly retarded the growth of both CAL 27 and FaDu cells compared with non-specific ASO (Fig. 4A and B). Moreover, repeated treatment with 20 nM SR-3 ASO significantly prevented cell growth without the development of resistance in both CAL 27 and FaDu cells (Fig. 5). These results demonstrated that blocking of ESS with ASO is a novel and efficient method to inhibit the expression of oncogene SRSF3.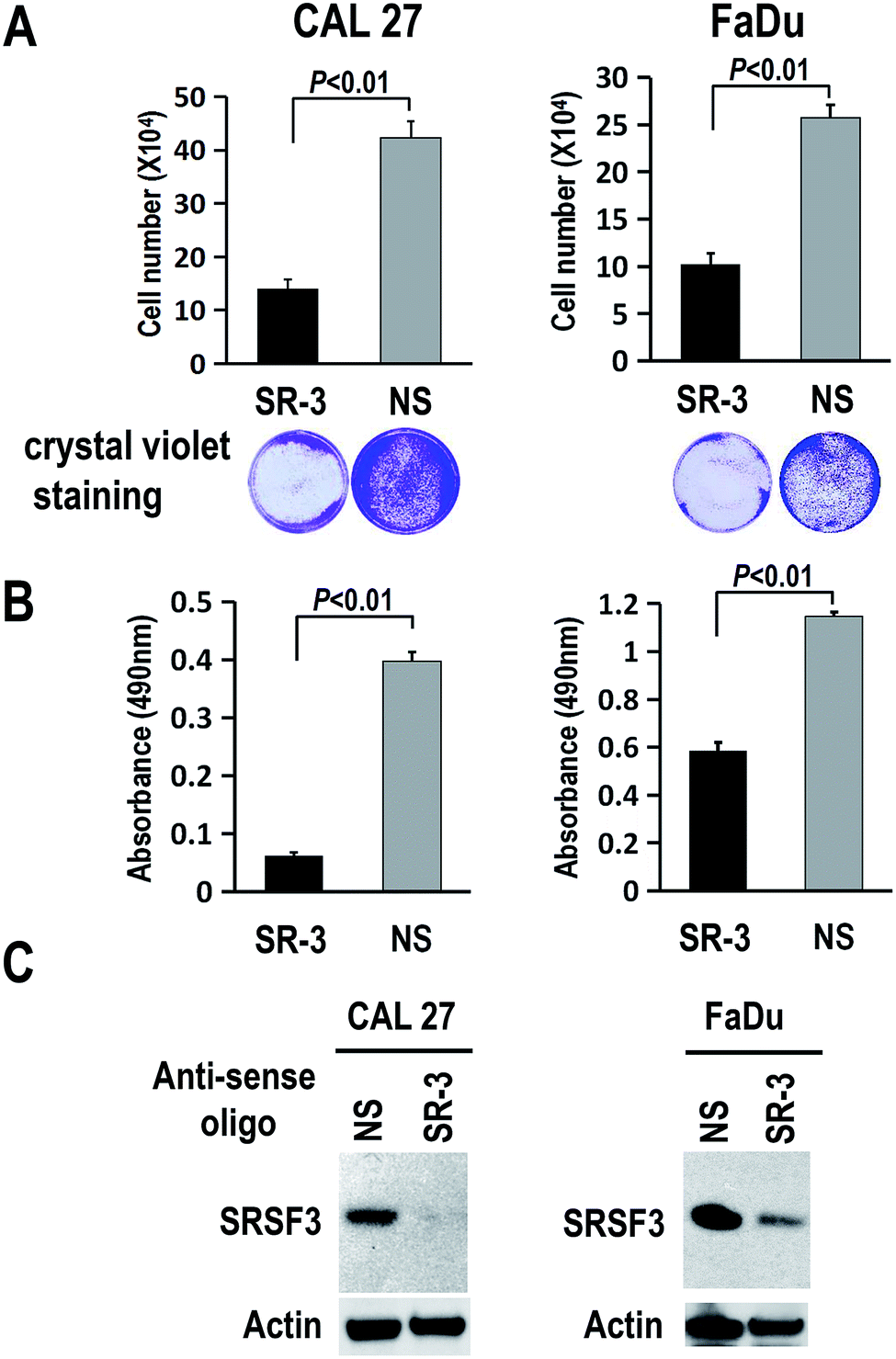 | ||
| Fig. 4 Antisense oligonucleotide SR-3 decreases the expression of SRSF3 and inhibits the growth of CAL 27 or FaDu oral cancer cell. (A and B) CAL 27 or FaDu oral cancer cells were transfected with 20 nM 2′-O-methyl-phosphorothioate antisense oligonucleotide SR-3 or NS control. Three days after transfection, cell proliferation was tested by using trypan blue exclusion method (A) or MTS assay (B). The transfected cells stained with crystal violet were shown at the bottom of panel A. The difference of means between SR-3 and NS group was determined using Student's t test. (C) Western blot of SRSF3. Actin served as loading control. | ||
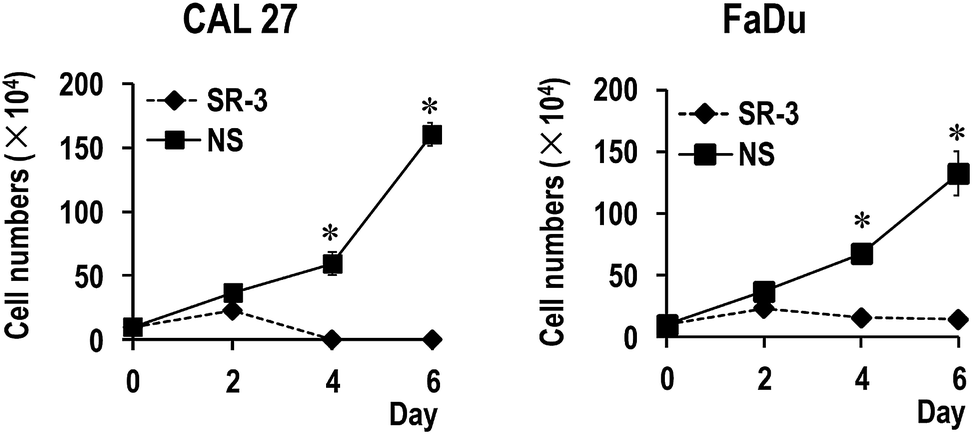 | ||
| Fig. 5 Repeated treatment with SR-3 significantly prevented cancer cell growth. CAL 27 or FaDu cells were treated with 20 nM 2′-O-methyl-phosphorothioate antisense oligonucleotide SR-3 or NS control three times in a 48 hour interval. * p < 0.01. The difference of means between SR-3 and NS group was determined using Student's t test. | ||
SRSF3 has been demonstrated to be overexpressed in multiple cancers, including cervical cancer,7 ovarian cancer,10 colon cancer,25 and so on. Repression of SRSF3 expression decreased cell proliferation,26 and metastasis.27 We found that SR-3 ASO is able to inhibit SRSF3 expression and significantly suppress the growth of cancer cells. Therefore, we consider that SR-3 ASO may provide a new method to block the expression of oncogene SRSF3 in oral cancer. Co-administration of SR-3 ASO with standard anti-cancer drugs may also enhance the efficacy and reduce dosage of standard drugs. Currently, antisense oligonucleotides have attracted extensive attention. For example, Nusinersen, an antisense oligonucleotide drug, was recently approved by FDA for spinal muscular atrophy (SMA) and showed efficiency in treating SMA in a phase III clinical trial study.28 For future translational application, we will try to increase the stability of SR-3 ASO and evaluate its anti-cancer efficiency in vivo.
Autoregulation of pre-mRNA alternative splicing is a common mechanism in splicing factors to maintain their relative stable expression levels in cells. For example, oncogene SRSF1 has alternative intron 3 and exon 4. Exogenous overexpression of SRSF1 significantly reduced endogenous expression of functional SRSF1 by promoting the inclusion of intron 3 or partial deletion of exon 4.29 SRSF2 has alternative exon and intron in 3′-UTR region. Overexpression of SRSF2 significantly increased an exon inclusion and an intron excision in 3′-UTR region. The resulted SRSF2 transcript is unstable, which then causes a decrease in the SRSF2 protein level.30 Both SRSF1 (ref. 31) and SRSF2 (ref. 32) are oncogenic proteins and potential therapeutic targets in cancers. Our results suggested that 2′-O-methyl-phosphorothioate modified ASOs may be useful to regulate autoregulation of SRSF1 or SRSF2 pre-mRNA and inhibit their expression in cancers.
Conclusion
In summary, we discovered an effective antisense oligonucleotide targeting the ESS motif to promote inclusion of SRSF3 exon 4. This strategy is useful to design novel anti-cancer drugs.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by Hubei Provincial Natural Science Foundation of China, 2016CFA067. This work was also supported by Grant 81271143, 81470741 and 81571024 from the National Science Foundation of China.References
- H. Lou, K. M. Neugebauer, R. F. Gagel and S. M. Berget, Mol. Cell. Biol., 1998, 18, 4977–4985 CrossRef CAS PubMed.
- Y. Huang, R. Gattoni, J. Stevenin and J. A. Steitz, Mol. Cell, 2003, 11, 837–843 CrossRef CAS PubMed.
- G. M. Hautbergue, M. L. Hung, A. P. Golovanov, L. Y. Lian and S. A. Wilson, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 5154–5159 CrossRef CAS PubMed.
- X. He and P. Zhang, Mol. Cancer, 2015, 14, 158 CrossRef PubMed.
- J. Kim, R. Y. Park, J. K. Chen, J. Kim, S. Jeong and T. Ohn, Cell Death Differ., 2014, 21, 481–490 CrossRef CAS PubMed.
- P. J. Shepard and K. J. Hertel, Genome Biol., 2009, 10, 242 CrossRef PubMed.
- R. Jia, C. Li, J. P. McCoy, C. X. Deng and Z. M. Zheng, Int. J. Biol. Sci., 2010, 6, 806–826 CrossRef CAS PubMed.
- C. Corbo, S. Orru and F. Salvatore, Biochem. Biophys. Res. Commun., 2013, 436, 1–5 CrossRef CAS PubMed.
- L. Peiqi, G. Zhaozhong, Y. Yaotian, J. Jun, G. Jihua and J. Rong, Int. J. Biol. Sci., 2016, 13, 533–539 Search PubMed.
- X. He, A. D. Arslan, M. D. Pool, T. T. Ho, K. M. Darcy, J. S. Coon and W. T. Beck, Oncogene, 2011, 30, 356–365 CrossRef CAS PubMed.
- H. R. Kim, C. H. Shin, H. Lee, K. H. Choi, D. H. Nam, T. Ohn and H. H. Kim, Oncotarget, 2017, 8, 8342–8355 Search PubMed.
- K. Kurokawa, Y. Akaike, K. Masuda, Y. Kuwano, K. Nishida, N. Yamagishi, K. Kajita, T. Tanahashi and K. Rokutan, Oncogene, 2014, 33, 1407–1417 CrossRef CAS PubMed.
- Z. Wang, D. Chatterjee, H. Y. Jeon, M. Akerman, H. M. Vander, L. C. Cantley and A. R. Krainer, J. Mol. Cell Biol., 2012, 4, 79–87 CrossRef CAS PubMed.
- S. K. Park and S. Jeong, Biochem. Biophys. Res. Commun., 2016, 470, 431–438 CrossRef CAS PubMed.
- Y. Tang, I. Horikawa, M. Ajiro, A. I. Robles, K. Fujita, A. M. Mondal, J. K. Stauffer, Z. M. Zheng and C. C. Harris, Oncogene, 2013, 32, 2792–2798 CrossRef CAS PubMed.
- J. Guo, J. Jia and R. Jia, Sci. Rep., 2015, 5, 14548 CrossRef CAS PubMed.
- H. C. Cheung, T. Hai, W. Zhu, K. A. Baggerly, S. Tsavachidis, R. Krahe and G. J. Cote, Brain, 2009, 132, 2277–2288 CrossRef PubMed.
- S. M. Hammond and M. J. Wood, Trends Genet., 2011, 27, 196–205 CrossRef CAS PubMed.
- D. R. Mercatante and R. Kole, Biochim. Biophys. Acta, 2002, 1587, 126–132 CrossRef CAS.
- R. B. Runyan, C. C. Wendler, L. A. Romano, A. S. Boyer, J. M. Dagle and D. L. Weeks, Methods, 1999, 18, 316–321 CrossRef CAS PubMed.
- K. Siva, G. Covello and M. A. Denti, Nucleic Acid Ther., 2014, 24, 69–86 CrossRef CAS PubMed.
- P. Spitali and A. Aartsma-Rus, Cell, 2012, 148, 1085–1088 CrossRef CAS PubMed.
- M. Dewaele, T. Tabaglio, K. Willekens, M. Bezzi, S. X. Teo, D. H. Low, C. M. Koh, F. Rambow, M. Fiers, A. Rogiers, E. Radaelli, M. Al-Haddawi, S. Y. Tan, E. Hermans, F. Amant, H. Yan, M. Lakshmanan, R. C. Koumar, S. T. Lim, F. A. Derheimer, R. M. Campbell, Z. Bonday, V. Tergaonkar, M. Shackleton, C. Blattner, J. C. Marine and E. Guccione, J. Clin. Invest., 2016, 126, 68–84 Search PubMed.
- A. Shchelkunova, B. Ermolinsky, M. Boyle, I. Mendez, M. Lehker, K. S. Martirosyan and A. V. Kazansky, Int. J. Biol. Sci., 2013, 9, 45–54 CrossRef CAS PubMed.
- M. Ajiro, R. Jia, Y. Yang, J. Zhu and Z. M. Zheng, Nucleic Acids Res., 2016, 44, 1854–1870 CrossRef PubMed.
- H. N. Jang, M. Lee, T. J. Loh, S. W. Choi, H. K. Oh, H. Moon, S. Cho, S. E. Hong, D. H. Kim, Z. Sheng, M. R. Green, D. Park, X. Zheng and H. Shen, Biochim. Biophys. Acta, 2014, 1839, 25–32 CrossRef CAS PubMed.
- H. R. Kim, S. J. Hwang, C. H. Shin, K. H. Choi, T. Ohn and H. H. Kim, Exp. Cell Res., 2017, 358, 161–170 CrossRef CAS PubMed.
- R. S. Finkel, E. Mercuri, B. T. Darras, A. M. Connolly, N. L. Kuntz, J. Kirschner, C. A. Chiriboga, K. Saito, L. Servais, E. Tizzano, H. Topaloglu, M. Tulinius, J. Montes, A. M. Glanzman, K. Bishop, Z. J. Zhong, S. Gheuens, C. F. Bennett, E. Schneider, W. Farwell and D. C. De Vivo, N. Engl. J. Med., 2017, 377, 1723–1732 CrossRef CAS PubMed.
- S. Sun, Z. Zhang, R. Sinha, R. Karni and A. R. Krainer, Nat. Struct. Mol. Biol., 2010, 17, 306–312 CAS.
- A. Sureau, R. Gattoni, Y. Dooghe, J. Stevenin and J. Soret, EMBO J., 2001, 20, 1785–1796 CrossRef CAS PubMed.
- R. Karni, E. de Stanchina, S. W. Lowe, R. Sinha, D. Mu and A. R. Krainer, Nat. Struct. Mol. Biol., 2007, 14, 185–193 CAS.
- C. Luo, Y. Cheng, Y. Liu, L. Chen, L. Liu, N. Wei, Z. Xie, W. Wu and Y. Feng, Cancer Res., 2017, 77, 1168–1178 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |