
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly dispersed Pd on macroporous SmMn2O5 mullite for low temperature oxidation of CO and C3H8†
Yuning Zhu‡
a,
Chun Du‡a,
Zijian Fenga,
Yongjie Chena,
Hang Lic,
Rong Chenb,
Meiqing Shen c and
Bin Shan*a
c and
Bin Shan*a
aState Key Laboratory of Material Processing and Die and Mould Technology, School of Materials Science and Technology, Huazhong University of Science and Technology, Wuhan 430074, China. E-mail: bshan@mail.hust.edu.cn
bState Key Laboratory of Digital Manufacturing Equipment and Technology, School of Mechanical Science and Engineering, Huazhong University of Science and Technology, Wuhan 430074, China
cSchool of Chemical Engineering and Technology, Tianjin University, 92 Weijin Road, Nankai District, Tianjin 300072, China
First published on 31st January 2018
Abstract
The catalytic behavior of a palladium catalyst supported on macroporous SmMn2O5 mullite (Pd/SMO-EG&M) for CO and C3H8 oxidation was measured under lean-burn conditions. Different analytical techniques including XRD, Raman, BET, CO chemisorption, SEM, FTEM, XPS, TPD, TPR and CO + O2 pulse were undertaken to evaluate its physical and chemical properties. It was concluded that the crystal structure, morphology and specific surface area (SSA) of SmMn2O5 remained unchanged after Pd addition. The Pd/SMO-EG&M exhibited a low complete transformation temperature for CO (105 °C) and C3H8 (350 °C) oxidation. Such remarkable oxidation activity was attributed to high Pd dispersion (38.4%), which improved the reducibility and mobility of oxygen species, as revealed by TPR and TPD measurements. The high activity of oxygen species for Pd/SMO-EG&M above 250 °C accelerated the oxidation capacity as well. In a word, our study indicates that the macroporous Pd–mullite catalyst has potential applications in exhaust purification for gasoline vehicle.
1. Introduction
The noxious exhaust of automobiles has become an urgent and compelling issue across the world. Specifically, because of the low oxidation efficiency of conventional three-way catalysts (TWC) below 300 °C, a large portion (50–80%) of unburned carbon monoxide (CO) and hydrocarbons (HCs) is emitted into the air during the initial cold-start transients, resulting in photochemical smog and the greenhouse effect directly.1,2 To meet the strict current legislation of governments for ultra low-emission or zero-emission vehicles, it is imperative to explore highly efficient catalysts at relatively low temperatures. Among various types of commercial platinum group metals (PGMs), Pd has been regarded as an active component in automotive converters to eliminate CO and other exhaust fumes.3–5 Owing to the interaction between the metal oxide support and the active metal species, the catalytic behavior, durability and dispersion of heterogeneous Pd catalysts are closely associated with the inherent physical and chemical properties of the support.6–9 So far, the simple metal oxides, such as CeO2, Co3O4, TiO2, ZrO2 and MnOx, have been extensively studied for their large specific surface area.10–13 Whereas in practical applications, metal oxide supported catalysts are unstable after redox cycling, which results in the deactivation of active Pd species.In the last decade, perovskite-type mixed oxides have been developed as potential low-cost catalysts for diesel exhaust treatment system.14–16 Due to their unique thermal, mechanical and redox stability and catalytic performance, perovskites could serve as active support to reduce the noble metal loading.17–19 Despite their sufficient catalytic efficiency, the weak hydrothermal resistance and small oxygen storage capacity (OSC) lead to depletion of catalytic activity directly after rapid aging cycles.20–22 In 2012, Wang et al. proposed a new type of mixed-phase oxide catalyst for NO oxidation based on Mn mullite ((Sm, Gd)Mn2O5), which has exhibited favorable thermal and structural stability.23 Compared with the representative LaCoO3 perovskite catalyst, the SmMn2O5 mullite catalyst has higher TWC reactivity, anti-poisoning ability and OSC.24 Hence, it is probable that mullite-supported Pd catalysts should display improved performance and serve as potential candidate as low precious metal loading catalysts. Note that diffusion has been considered as the limit process for the overall performance of bulk catalysts, fabricating a porous structure is desirable to achieve high amount of surface accessible active sites and a better dispersion, ultimately stimulating its catalytic oxidation efficiency.7,25,26 Therefore, it is meaningful to explore Pd on hydrothermally stable porous SmMn2O5 support with high performance.
In this work, a macroporous SmMn2O5 mullite catalyst (SMO) was prepared by the combustion of organic solutions according to our previous study.27 Afterwards, a small fraction (0.5 wt%) of Pd was loaded on the catalyst and its catalytic activities were determined by CO and C3H8 oxidation. The physical and chemical properties of as-prepared Pd catalysts were analysed by XRD, Raman, SSA, SEM, FTEM, ICP, XPS, H2-TPR, O2-TPD and CO + O2 pulse studies. The catalyst exhibits superior catalytic activity and stability for CO and C3H8 oxidation due to a larger number of active sites induced by higher Pd dispersion (38.4%). The adsorbed oxygen species increases dramatically for dispersed Pd/SmMn2O5-EG&M catalyst as revealed by O2-TPD test. The higher catalytic property is attributed to the higher Pd dispersion, inducing stronger ability of oxygen migration and more adsorption site on the macroporous mullite as demonstrated by TPO and CO-TPD test. Meanwhile, the easier activation of available oxygen species bond confirmed by H2-TPR and CO + O2 pulse experiment accelerates the reaction as well.
2. Catalyst characterization
The PANalytical Xpert PRO diffractometer recorded the X-ray diffraction (XRD) spectrum from 10° to 60° in 2θ angle at room temperature using Cu Kα radiation (λ = 1.5406 Å) with a 0.02° step scan. The crystal structures were identified using the JCPDS data bank. Raman spectrum analysis was conducted on a Renishaw InVia Reflex spectrometer from 150 to 800 cm−1. The excitation wavelength of the laser was set at 532 nm with a spectral resolution of 1.5 cm−1. The surface textural parameters of each sample was measured by nitrogen adsorption using Micromeritics ASAP 2020M physisorption analyser at 77 K. Prior to the measurement, the samples were degassed in vacuum oven at 120 °C overnight. The field-emission scanning electron microscopy (FE-SEM) was performed on a Nova NanoSEM 450 microscope, and field-emission transmission electron microscopy (FE-TEM) and high-resolution transmission electron microscopy (HRTEM) images were analysed on a Tecnai G2 30 microscope. Inductively coupled plasma atomic emission spectroscopy (ICPAES) was produced by an Optical 4300DV to determine the actual mass loading of Pd. X-ray photoelectron spectroscopy (XPS) was acquired to study the chemical compositions at 10−7 Pa using an AXIS-ULTRA-DLD spectrometer. The binding energy (BE) of C 1s was calibrated to 284.8 eV. The adsorption properties, oxygen storage capacity (OSC) measurements and catalytic activity measurements were undertaken on an automatic adsorption instrument (FINESORB-3010E, Zhejiang Finetec Co). The detailed experimental process was given in ESI (S2†).3. Results and discussion
3.1. Physical characterization
Fig. 1a displays the XRD profiles of pristine SMO supports (SMO-EG&M and SMO-CP) and Pd loaded catalysts (Pd/SMO-EG&M and Pd/SMO-CP). All samples exhibit characteristic reflections of the single-phase orthorhombic SmMn2O5 structure (SmMn2O5 phase [52-1096]) at 29.1° and 31.2°. Specifically, additional phases of SmMnO3 and Mn2O3 are observed for CP samples around 32.9°, which is similar to our previous work.28,29 For Pd loaded catalysts, due to low content or small crystalline size, no reflections of PdOx or metallic Pd are observed. Additionally, the Pd/SMO catalysts have similar half-height width and intensity of the typical peaks with that of SMO-EG&M and SMO-CP supports, implying negligible changes on crystallization and crystalline size after Pd addition.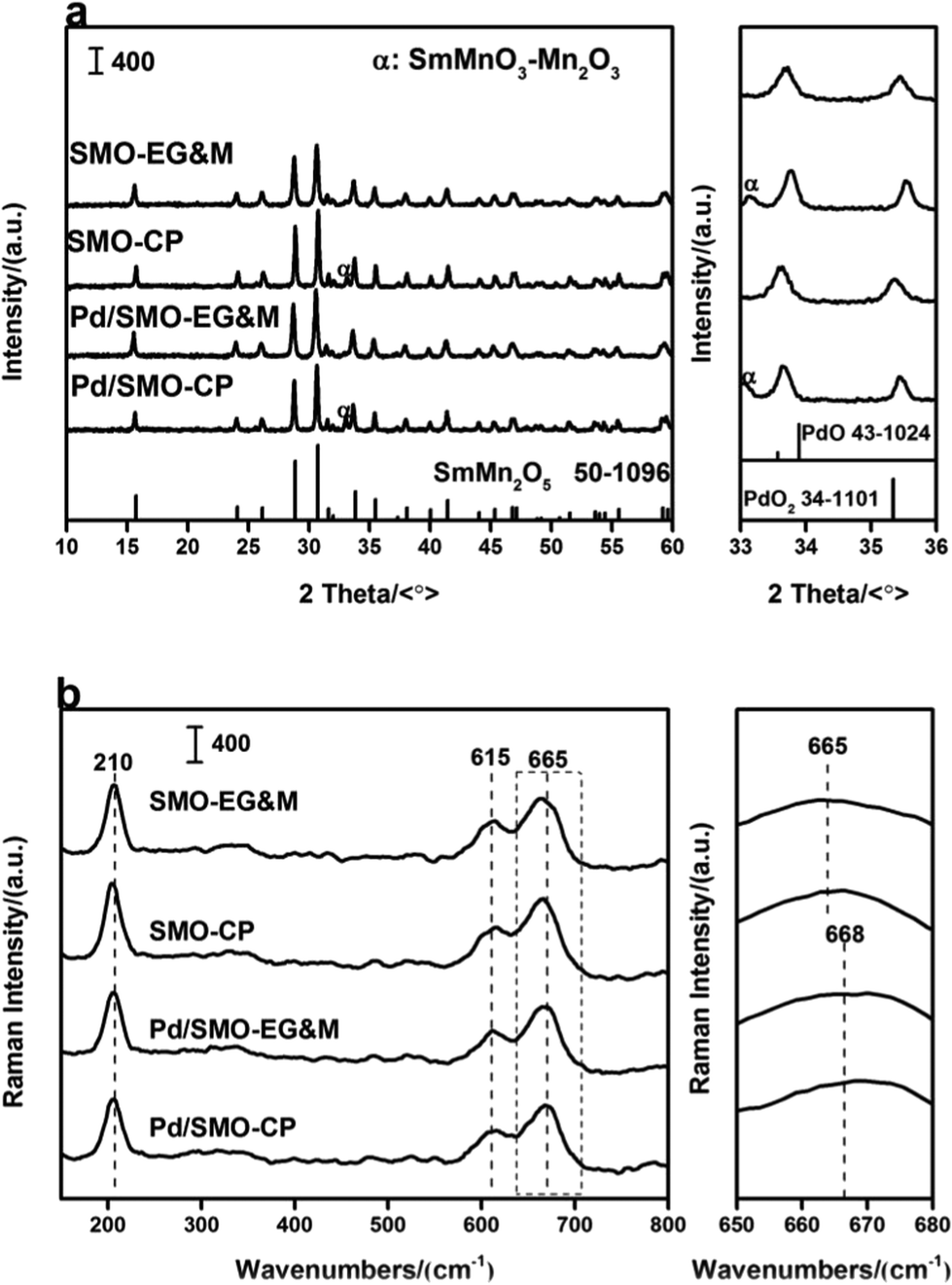 | ||
| Fig. 1 (a) XRD profile; (b) Raman spectra of the SMO and Pd/SMO. | ||
The catalysts were then analysed by Raman spectroscopy to elucidate their structural features. In Fig. 1b, all samples display three characteristic bands at 210, 615 and 665 cm−1, respectively. On the basis of lattice dynamics calculations, the band at 210 cm−1 is associated with Ag mode of Mn–Mn translation motion along the chain direction,26,28,30 while the rest two pronounced bands at higher frequencies (>600 cm−1) correspond to the asymmetric stretching vibration of Mn–O–Mn in Mn4+O6 units.30–33 Moreover, the peak at 665 cm−1 shifts to a higher frequency after Pd addition, implying the generation of new phases. Note that the vibration mode of Pd–O bond at 672 cm−1 has been observed for some Pd-containing samples,34,35 the band at 665 cm−1 for the Pd/SMO can be deduced as a combination of vibration bands from mullite structure and palladium oxides.27 Moreover, the length and angle of Mn–O band might be influenced by the Pd addition since the lattice oxygen of mullite might be shared with Pd species, contributing to the variation of the peak at 665 cm−1 as well.36,37 Thus variation might affect the activity of lattice oxygen.
The morphologies of Pd/SMO samples were investigated by SEM, TEM and HRTEM as shown in Fig. 2. In Fig. 2a, the EG&M sample exhibits a foamlike surface, stacked with agglomerated particles with high porosity. The catalyst shows a macroporous surface structure with aperture size above 50 nm,38,39 and they are ineffective in increasing specific surface area (SSA) as indicated in Table 1. These features are beneficial for exposing Pd nanoparticles as active sites and improving metal dispersion.27 Fig. 2b shows the morphology of the CP sample where agglomerated nanoparticles with uniform size are formed. Interestingly, the supported samples maintain their initial morphologies of pristine SMO samples (Fig. S3†). In TEM images of Fig. 2c and d, one can observe that the particle size of the catalysts was 50–60 nm, approximately four times larger than the value (13 nm) estimated from XRD analysis (Table 1). The pore size and volume tends to decrease as shown in Table 1, implying that the pore was partially occupied by the Pd species after the impregnation procedure.40 Thus, the larger pore size is beneficial to exposing superficial active site of Pd particles. As expected, the CO chemisorption analysis (Fig. S4†) illustrates a higher dispersion of Pd for the EG&M sample (38.4%) compared with that of the SMO-CP sample (16.6%). Using the eqn S(3),† the metallic Pd particle size was in the ranges of 2.7–3.0 nm for Pd/SMO-EG&M and 5.9–7.5 nm for Pd/SMO-CP, respectively. This suggests the suppression of Pd aggregation on SMO-EG&M support.26,38 In HRTEM images of Fig. 2e and f, the PdOx particles are clearly observed and segregated on the mullite surface with a mean particle size of 3 nm and 6 nm for EG&M and CP samples, which is well consistent with the estimated value. The observed inter-planar distance of the particles in each figure is 0.223 and 0.254 nm approximately, corresponding to the (2 0 0) and (1 1 0) plane of tetragonal PdO2.41,42 In addition, the presence of well-crystallized mullite-type SmMn2O5 is confirmed by its typical d-spacing (d2 1 0 = 0.293 nm and d1 1 2 = 0.253 nm).23,28
 | ||
| Fig. 2 SEM, TEM and HRTEM images of (a), (c) and (e) Pd/SMO-EG&M and (b), (d) and (f) Pd/SMO-CP. | ||
| Catalysts | Pda (wt %) | Daveb (nm) | SSAc (m2 g−1) | Pore sizeave (nm) | Pore volumed (cm3 g−1) | Surface coveragee (μmol gcat−1) | Pd dispersione (%) | Pd sizee (nm) |
|---|---|---|---|---|---|---|---|---|
| a ICP-OES.b Debyb–Scherrer formula.c BET method.d t-Plot method.e CO-chemisorption. | ||||||||
| SMO-EG&M | — | 12.56 | 16 | 41.6 | 0.11 | — | — | — |
| SMO-CP | — | 13.26 | 15 | 15.1 | 0.05 | — | — | — |
| Pd/SMO-EG&M | 0.51 | 13.04 | 12 | 37.2 | 0.08 | 18.0 ± 1.0 | 38.4 ± 2.2 | 2.7–3.0 |
| Pd/SMO-CP | 0.53 | 13.01 | 10 | 14.6 | 0.04 | 7.9 ± 0.9 | 16.6 ± 2.0 | 5.9–7.5 |
3.2. XPS
The surface composition and chemical electronic states of the samples was examined by XPS. High resolution XPS spectrum of Pd 3d is included in Fig. 3b. Briefly, the components Pd 3d5/2 at 337.7 eV and Pd 3d3/2 at 342.7 eV are observed, herein the BE of the Pd 3d5/2 is higher than that of Pd2+ ion (336.1–336.9 eV), indicating the formation of highly oxidized cationic of palladium species (Pd3+ or Pd4+).19,43–45 This behavior is in good agreement with the FTEM image. Recent reports have shown that the higher oxidized Pd species, such as PdO2, displays a high thermal stability and reactivity toward CO oxidation at room temperature,42,46,47 thus we speculate that the performance of CO oxidation for mullite could be enhanced after Pd addition. Fig. 3c illustrates the Mn 2p XPS spectrum with Mn 2p3/2 at 641.9 eV and Mn 2p1/2 at 652.9 eV, respectively. Herein, the lower asymmetrical Mn 2p3/2 peak can be fitted into two components with a BE of 641.3–641.7 eV and 642.2–643.0 eV, referring to the Mn3+ and Mn4+ species, respectively.24,27–29 The surface Mn4+/Mn3+ ratio of 0.72 deviates from the theoretical value of 1.0 (Table S2†), which suggests the surface enrichment of Mn3+.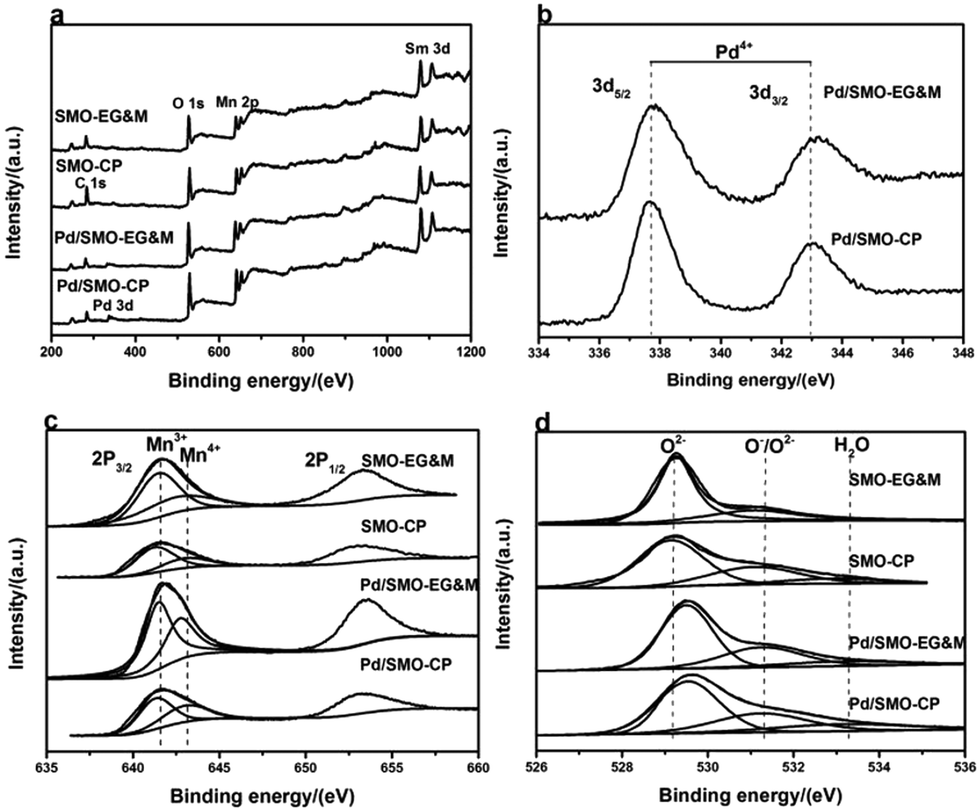 | ||
| Fig. 3 XPS spectrum of SMO and Pd/SMO: (a) survey spectrum; (b) Pd 3d; (c) Mn 2p; (d) O 1s. | ||
Fig. 3d is the fined scanned XPS spectrum of the O 1s. Three types of signals at low BE (529.3–529.9 eV), medium BE (531.0–531.4 eV) and high BE (above 533.2 eV) are associated with lattice oxygen (Olatt: O2−), surface adsorbed oxygen (Oads: O2−, O− or OH groups) and adsorbed molecular water, respectively.26–28,48 For the pristine EG&M sample, the BE values for lattice oxygen and surface adsorbed oxygen species are 529.1 eV and 531.2 eV, respectively. However, the values shift to 529.7 eV and 531.4 eV after Pd addition, and a similar behavior is found for the CP sample. Induced by defects of palladium oxide, the presence of a larger amount of oxygen vacancies is demonstrated by the increase of BE, which illustrates higher capacity of electronic capture and release, further accelerating the mobility and redox properties of reactive oxygen species.28,49 Consistent with our previous work,27 the SMO-EG&M sample possesses a larger Oads/Olatt ratio than that of SMO-CP (Table S2†). After Pd addition, the values increase to 0.54 and 0.49 for Pd/SMO-EG&M and Pd/SMO-CP, respectively. The larger Oads concentration for the supported samples is attributed to the well dispersed Pd species, which facilitates the adsorption of dissociative oxygen in light of the oxygen spillover effect.43–46 Moreover, considerable interface would be formed between mullite and Pd species, with the formation of defect and oxygen vacancies, which explains the increased oxygen binding energy and Oads concentration. Since a higher Oads concentration is beneficial for oxidative reaction, the Pd/SMO-EG&M exhibits higher catalytic activity. Besides, the reducibility and mobility of Oads are crucial for CO and C3H8 oxidation as Oads should be activated at the first step,47,49 which will be investigated by the following O2-TPD and H2-TPR measurements.
3.3. O2-TPD and H2-TPR
The oxygen desorption behavior for the prepared samples was investigated by O2-TPD as shown in Fig. 4a. The amount of desorbed oxygen was estimated and listed in Table 2. Overall, the desorption profile is categorized into three regions with the demarcation intervals of 350 °C and 650 °C, where the desorbed oxygen species are denoted as α-O, β-O and γ-O, respectively.27 α-O is known to represent the weakly bonded oxygen species on the surface (O2− and O− species), which is responsible for oxidative reactions at relatively low temperature.50–54 β-O and γ-O, showing correlated response to methane oxidation, are ascribed as the release of equivalent oxygen in surface lattice and bulk mullite skeleton, respectively.27,51,53–56 Note that the temperature window of CO and C3H8 oxidation was mainly encompassed with α-O, its concentration and mobility would largely affect the catalytic performance. In the enlarged figure of the desorption region, we see that the signal of α-O shows a weak plateau-like peak with its concentration in the order of Pd/SMO-EG&M (38.5 μmol g−1) > Pd/SMO-CP (27.9 μmol g−1) > SMO-EG&M (24.9 μmol g−1) > SMO-CP (20.4 μmol g−1), speculating that the generation of surface oxygen species is more facile for the Pd/SMO-EG&M, which is well correlated with the highest ratio of Oads/Olatt for the Pd/SMO-EG&M as attained from XPS results. The higher concentration of α-O for Pd/SMO-EG&M is attributed to the stronger oxygen spillover effect between Pd species and EG&M support (Fig. S6†). In addition, the higher mobility of active oxygen species are crucial for CO oxidation and suggested by the lower desorption temperature. We see the desorption temperatures of α-O follow the sequence of Pd/SMO-CP (67 °C) ≈ Pd/SMO-EG&M (70 °C) < SMO-EG&M (110 °C) < SMO-CP (128 °C), suggesting the stronger mobility of Oads after Pd addition. The high mobility indicates an ease of oxygen migration, which provides sufficient available oxygen species and is beneficial for activation of reactants.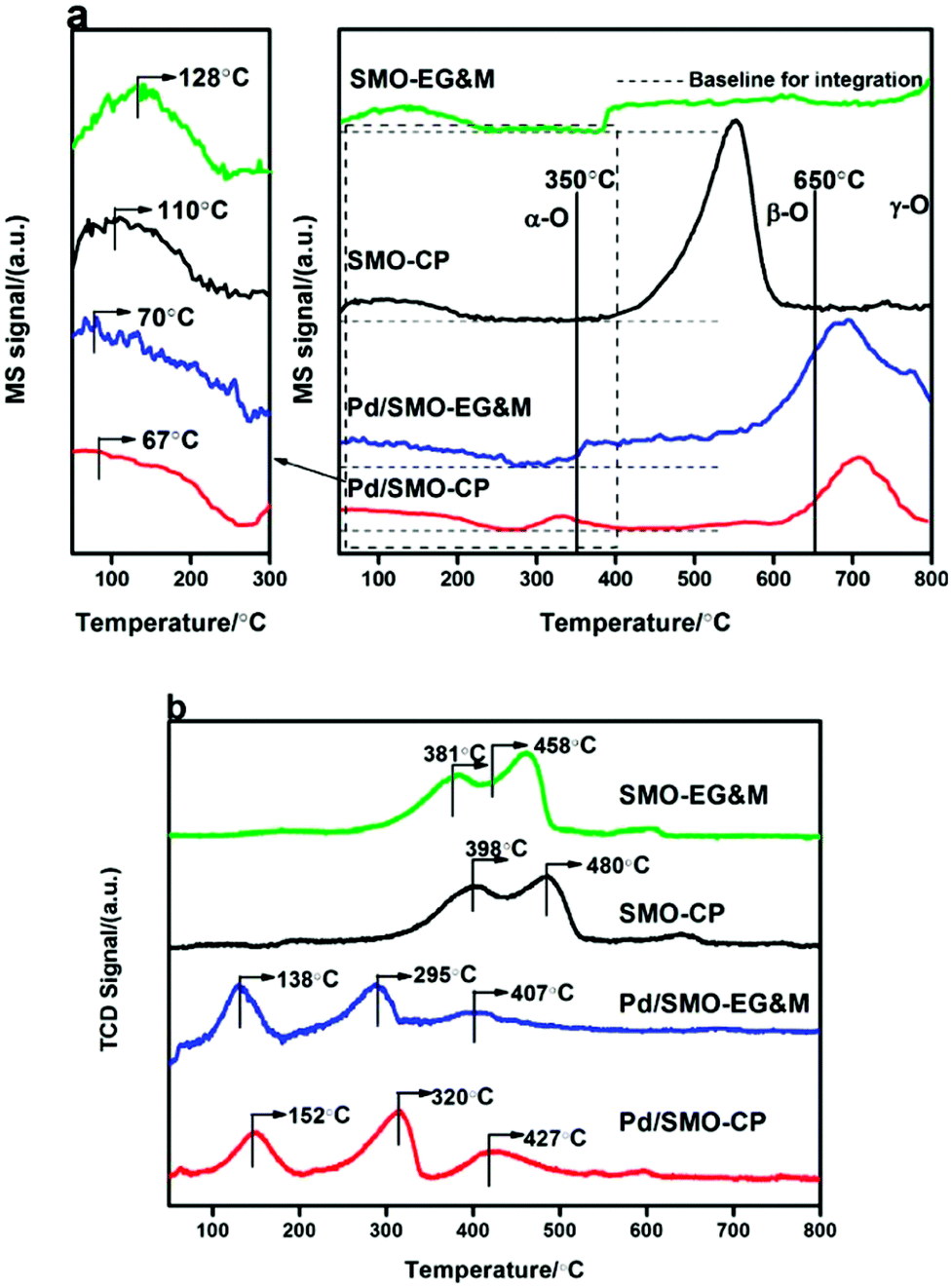 | ||
| Fig. 4 (a) O2-TPD and (b) H2-TPR profiles of the SMO and Pd/SMO. | ||
| Catalysts | O2 desorption amounta (μmol g−1) | CO adsorption amountb (μmol g−1) | CO2 adsorption amountc (μmol g−1) | ||||
|---|---|---|---|---|---|---|---|
| α-O (<350 °C) | β-O (350–650 °C) | γ-O (>650 °C) | <350 °C | 350–800 °C | <450 °C | 450–800 °C | |
| a Desorbed concentration of O2 during O-TPD (Fig. 4).b Desorbed concentration of CO2 during CO-TPD (Fig. S5).c Desorbed concentration of CO2 during CO2-TPD (Fig. S6). | |||||||
| SMO-EG&M | 24.9 | 59.1 | 33.8 | 100.5 | 139.6 | 42.6 | 7.1 |
| SMO-CP | 20.4 | 135.2 | 0.7 | 83.7 | 81.0 | 39.9 | 2.7 |
| Pd/SMO-EG&M | 38.5 | 75.8 | 97.4 | 151.7 | 145.6 | 60.6 | 13.5 |
| Pd/SMO-CP | 27.9 | 13.6 | 51.0 | 129.7 | 231.6 | 50.3 | 13.6 |
The reduction behavior of the catalysts was studied by H2-TPR. As shown Fig. 4b, the TPR profiles of the pristine EG&M sample present three reduction peaks, which indicate the multiple reductive processes for SMO mullite. The first two consecutive peaks at 381 °C and 458 °C result from the consumption of adsorbed oxygen and reduction of Mn4+ to Mn3+.24,27,28 After the deposition of Pd, the release of the surface oxygen species for EG&M sample is greatly promoted as evidenced by the decline of reduction peak to 138 °C. In addition, the reduction temperature of Mn4+ and Mn3+ decreases to 295 °C and 407 °C, suggesting the facile reduction of adsorbed oxygen and Mn ion by loading palladium species. The reason for the decrease of reduction temperatures is attributed to the hydrogen spillover effect, because hydrogen is easily dissociated at the reduced Pd surface and spilled to the SMO support.57–59 In addition, the enhanced activity of oxygen species (certified by CO + O2 pulse hereafter) contributes as well.28,37 As the Pd dispersion is higher for the Pd/SMO-EG&M compared with other samples, its surface Pd site was highly exposed and could be reduced easier. The lower temperature suggests higher reducibility of the Pd/SMO-EG&M in the whole range, suggesting that the reduction of mobile oxygen and surface adsorbed oxygen species is more facile. The superior reducibility for the Pd/SMO-EG&M indicates a stronger Pd-interaction, and it is beneficial for exhaust removal.
3.4. CO + O2 pulse test
The oxygen storage capacity (OSC) has been commonly applied as a critical factor to quantify the amount of active oxygen available for chemical reactions. Following the thermodynamic and kinetic laws, the diffusion of lattice oxygen is largely accelerated with rised temperatures, which enhances the release of oxygen for all samples.24,60–62 In Fig. 5, we see that all samples show similar OSC below 250 °C, while the desorbed oxygen species that include motivated surface adsorbed oxygen and lattice oxygen increase rapidly above 250 °C for the supporting samples. Based on CO-TPR profile (Fig. S6†), we find that the reduction of adsorbed oxygen and highly oxidized manganese ion occurs in this region, which leads to higher CO consumption in reductive pulse. In oxidation pulse, the Pd species could accomplish a quick oxygen migration and accelerate the reoxidation of reduced manganese ion via the oxygen spillover effect (S10†). Hence, better redox capacity of oxygen species and manganese ion lead to higher CO2 production for all supported samples. Moreover, we see that Pd/SMO-EG&M shows a lower reduction temperature compared with the Pd/SMO-CP from the CO-TPR (Fig. S6†). Indeed, the Pd/SMO-EG&M sample possesses a higher amount of activated oxygen species than that of the Pd/SMO-CP in the whole temperature range, which is in agreement with the results of O2-TPD and CO2-TPD (Fig. S6†). The origin of easier activation of O2 is attributed to a larger amount of adsorption sites on EG&M support, which contributes to easier accessibility of active sites and activation of reactants (CO, C3H8 and O2). Note that the oxidation temperature for C3H8 exceeds 250 °C, we conclude that Pd/SMO-EG&M provides sufficient active oxygen species and displays the superior catalytic performance.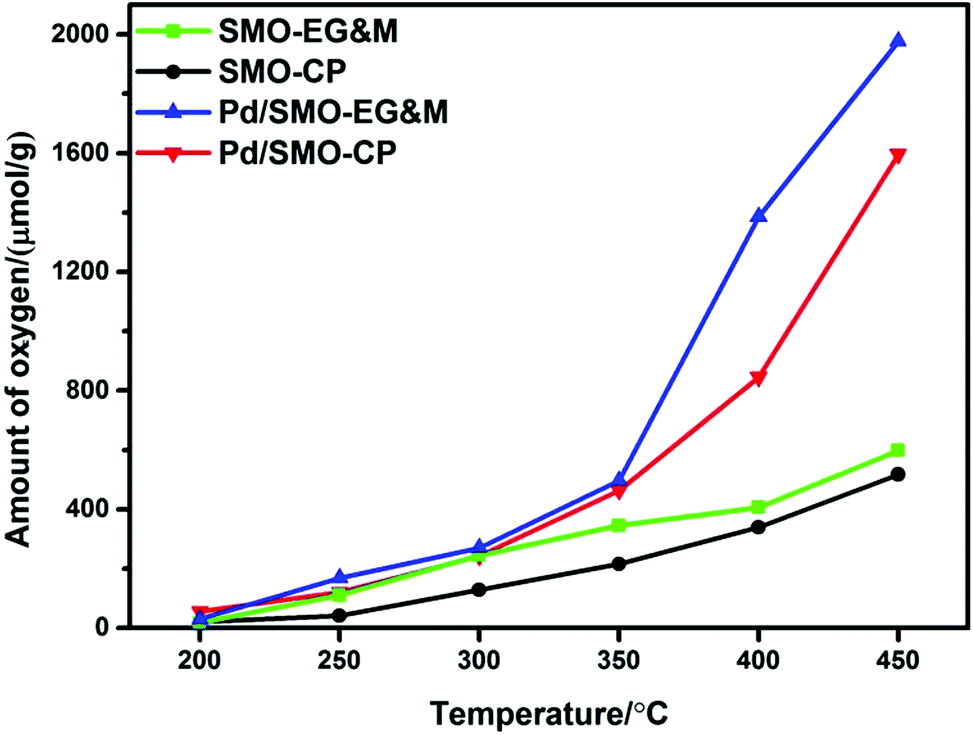 | ||
| Fig. 5 OSC of the SMO and Pd/SMO. | ||
3.5. Performance evaluation
The combustion activity of CO and C3H8 for were given in Fig. 6. In panel (a), the SMO-EG&M achieves a significant conversion of CO at 160 °C, comparable to that of supported Pd/CeO2 and Pd/LMO conventional catalysts. Through deposition of 0.5 wt% Pd over the catalyst, the corresponding temperatures of light-off curve drop by 75 °C relative to the pristine SMO-EG&M sample. Similarly, considerable catalytic performance is identified below 250 °C and complete conversion of C3H8 is obtained at 320 °C after Pd addition, as seen in Fig. 6b. The similar improved catalytic efficiency is observed for CP sample as well, whereas its combustion activity is lower compared with EG&M. All Pd/SMO catalysts display better catalytic performance than the SMPO reference catalysts (Fig. S8†). In addition, we extensively compared the catalytic activity of Pd/SMO with some other Pd supported catalysts reported in literature, the results were listed in Table 3. It is found that the Pd/SMO-EG&M catalyst exhibits an optimal catalytic activity for CO and C3H8 oxidation with lower T50 values, lower Pd loading and higher gas hourly space velocity. The CeO2–Co3O4 and CeO2–CuO catalysts have shown better C3H8 oxidation catalytic activity, mainly due to the lower gas hourly space velocity and higher BET surface area.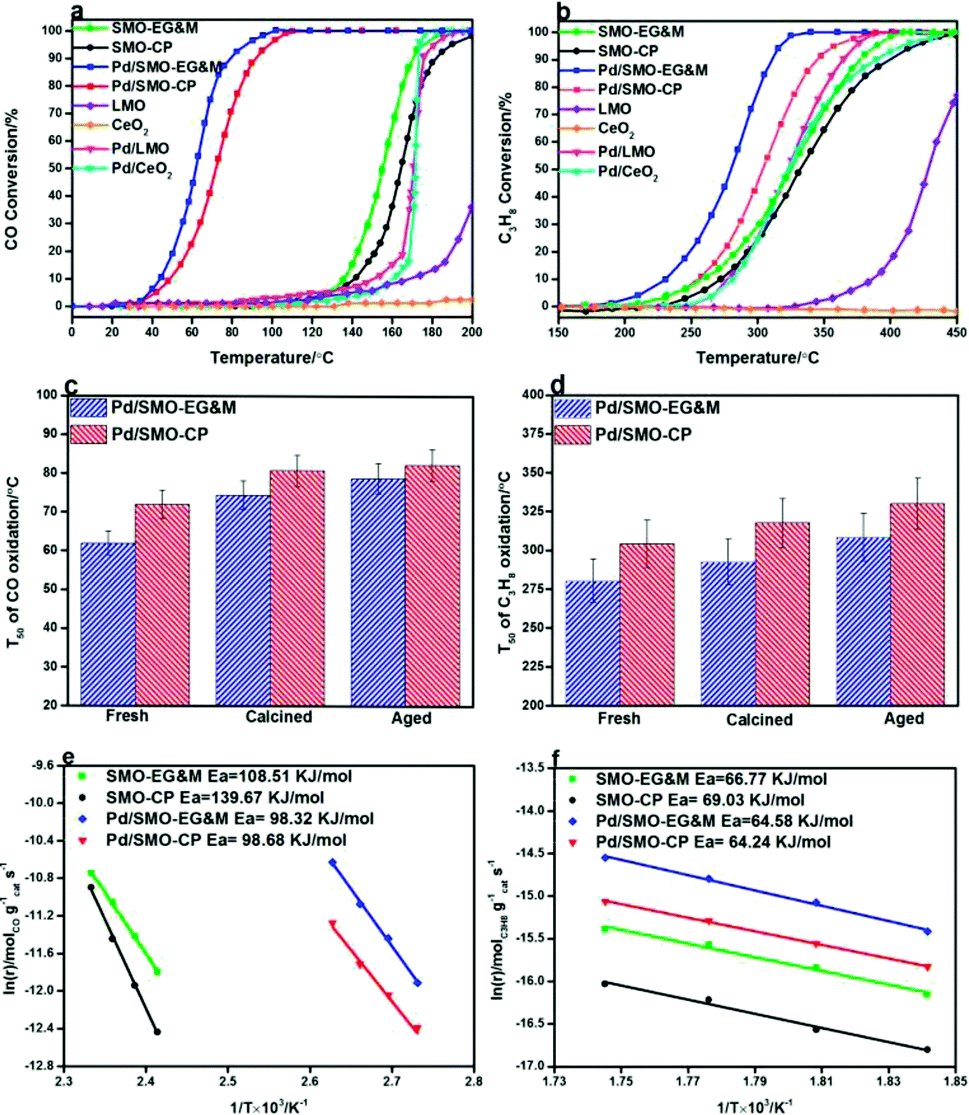 | ||
| Fig. 6 (a) Catalytic activities of CO oxidation. Feed: 1% CO, 10% O2 and N2 as balance; (b) catalytic activities of C3H8 oxidation. Feed: 500 ppm C3H8, 10% O2 and N2 as balance; (c) T50 of CO oxidation curve catalyzed by Pd supported catalysts after calcination of hydrothermal aging treatment; (d) T50 of C3H8 oxidation curve catalyzed by Pd supported catalysts after calcination of hydrothermal aging treatment; (e) Arrhenius plots for CO oxidation; (f) Arrhenius plots for C3H8 oxidation. | ||
| Supports | Pd content (wt%) | T50 (°C) | Reaction conditions | Ref. | |
|---|---|---|---|---|---|
| CO | C3H8 | ||||
| SMO-EG&M | 0.5 | 61.9 | 280.2 | 1% CO/500 ppm C3H8, 10% O2, N2 balance, 150 mL min−1, 50 mg | This work |
| SMO-CP | 71.9 | 304.1 | |||
| CeO2–Co3O4 | 0.5 | 70.0 | 231.4 | 1% CO/0.5% C3H8, 5% O2, N2 balance, WHSV = 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mL g−1 h−1, 600 mg 000 mL g−1 h−1, 600 mg |
1 |
| CeO2–NiOx | 99.0 | 298.4 | |||
| CeO2–MnOx | <40.0 | 240.6 | |||
| CeO2–CuO | 66.0 | 289.7 | |||
| CeO2–R | 1.0 | — | 401.0 | 1% CO, 20% O2, Ar balance, 50 mL min−1, 10 mg/0.2% C3H8, 2% O2, Ar balance, 100 mL min−1, 20 mg | 63 |
| CeO2–C | — | 336.0 | |||
| CeO2–O | — | 268.1 | |||
| LaCoO3 | 2.1 | 210.1 | 352.1 | 6% CO, 0.2% C3H8, 4.4% O2, Ar balance, 50 cm3 STP per min, 25 mg | 64 |
| LaCoO3-reduced | 162.3 | 303.5 | |||
| La2O3 | 1.0 | 210.1 | — | 1% CO, 21% O2, N2 balance, 30 mL min−1, WHSV = 72000 mL g−1 h−1 |
65 |
| SnO2 | 162.3 | — | |||
| Al2O3 | 157.2 | — | |||
| CeZr/Al2O3 | 2.0 | — | 348.2 | 0.3% C3H8, 3% O2, N2 balance, GHSV = 30000 h−1, 600 mg |
66 |
| CeZr–Y/Al2O3 | — | 312.8 | |||
| ZSM-5 | 1.5 | — | 327.1 | 2000 ppm C3H8, 2% O2, N2 balance, 100 mL min−1, 200 mg | 67 |
A high temperature of 900 °C is usually encountered in automobile exhaust where catalysts are easily sintered and deactivated.68 To examine the industrial applications, the resistance of thermal and hydrothermal aging of the samples were measured. The T50 of CO and C3H8 oxidation is plotted in Fig. 6c and d, respectively. Despite a severe agglomeration of Pd and SMO support (Fig. S13 and S14†), both samples maintain their good CO oxidation activity after calcination or aging treatment. For C3H8 oxidation, the Pd/SMO-EG&M sample still shows a T50 below 320 °C, which indicates most of active sites are maintained under harsh conditions. The observations demonstrate the Pd/SMO-EG&M sample with higher Pd dispersion exhibits the higher activity and stability, with great promise in purification of hazardous emissions.
Arrhenius plots of lnr versus 1/T for CO and C3H8 oxidation are displayed in Fig. 6e and f, respectively. For CO oxidation dynamic test, we can see that the reaction rate of EG&M sample is higher compared to that of CP sample at the whole reaction region in Fig. 6e, which is consistent with the result of catalytic performance results. The apparent activation energy (Ea) for all samples was calculated and ranked in the order SMO-CP (139.67 kJ mol−1) > SMO-EG&M (109.51 kJ mol−1) > Pd/SMO-CP (98.68 kJ mol−1) ≈ Pd/SMO-EG&M (98.32 kJ mol−1). In Fig. 6f, a similar sequence of reaction rate and Ea are observed for C3H8 oxidation. The higher reaction rate and lower activation energy for Pd/SMO-EG&M indicate that CO and C3H8 oxidation proceed more easily with macroporous SMO support. The Pd supported catalysts show similar TOFPd values for CO oxidation at 100 °C and for C3H8 oxidation at 300 °C (Table S3†), suggesting an identical active center for CO and C3H8 oxidation. Normally, TOFPd represents the converted number of the reactant on per Pd site. Thus, with an identical TOFPd value, a larger number of active sites over EG&M support would give rise to the higher reaction rate and activity.
Scheme 1 illustrates the detailed reaction pathway of Pd/SMO catalysts for CO oxidation. In brief, CO molecule is chemisorbed on the surface of the catalyst (Step 1) and reacts with surface active oxygen species to form bidentate carbonate, followed by its decomposition into CO2 (Step 2) to regenerate oxygen vacancy.1,37 Finally, gas-phase oxygen is replenished onto the oxygen vacancies to accomplish the redox cycle (Step 3). During the whole procedure, the activation of surface oxygen species are supposed to play a rate-determination step since the reaction occurs at low temperatures where little oxygen species are motivated in this temperature range as illustrated in OSC test. Thus, fast oxygen activation plays a critical role in CO oxidation. H2-TPR test has revealed the easily reducible surface Oads for the Pd/SMO-EG&M, and its mobility is higher by the stronger spillover effect as demonstrated in TPO test. Most of active sites for CO oxidation are at the interface between transition metal oxide and the supported species where spilled oxygen are transmitted via support.1,36,37 In this way, the oxygen species of PdOx on EG&M support could be motivated at lower temperatures and spilled to the surface of catalyst, and such activated oxygen atom could preferentially react with the adsorbed CO and form bidentate carbonate via spillover, giving rise to the improved catalytic performance. Moreover, due to a larger number of exposed Pd sites, the Pd/SMO-EG&M is beneficial to CO chemisorption (Fig. S5†), which leads to the improved CO oxidation activity as well.
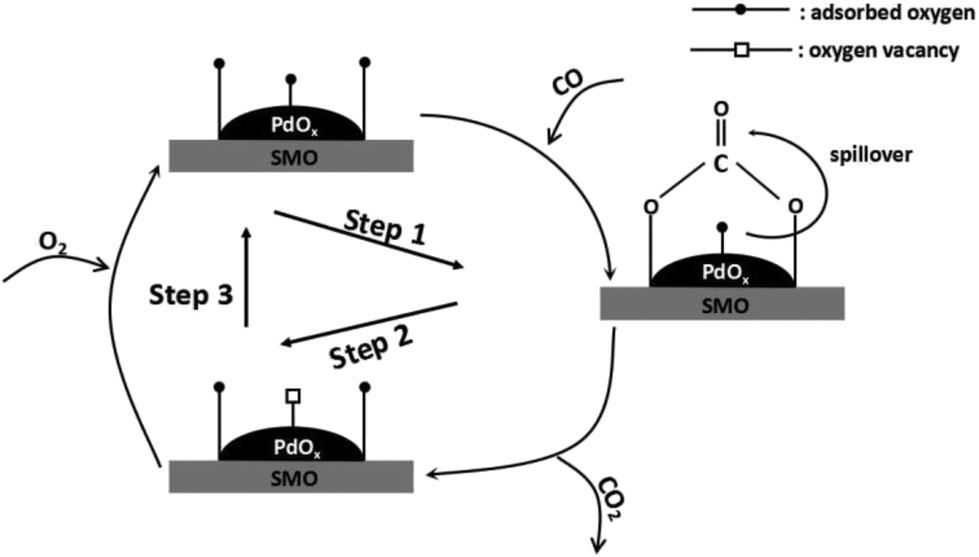 | ||
| Scheme 1 The reaction pathway for CO oxidation of the Pd/SMO catalysts. | ||
When it comes to C3H8 oxidation, the oxygen activation is largely accelerated in this temperature region, the fast oxygen activation is no longer regarded as the rate-limiting step. Note that the light-off temperature for the saturated conversion is closely associated with the strength of C–H bond, the reaction rate is decided by the abstraction of H atom in C3H8 molecule.1,58,59 After abstracting C–H bond, the active oxygen species available for hydrocarbons oxidation becomes a dominate factor.1,69 A larger number of basic centers of Pd/SMO-EG&M has been confirmed by CO2-TPD measurement (Fig. S5†) and demonstrates an easier activation of C–H bond. The O2-TPD and CO + O2 pulse tests display higher mobility and OSC for Pd/SMO-EG&M above 250 °C, confirming higher amount of available oxygen species for C3H8 activation. Above all, the higher dispersion of Pd species on EG&M support suggests the presence of larger amount of Pd active sites,26,38 leading to stronger synergistic effect and serving superior catalytic performance of CO and C3H8 oxidation. In a word, this catalyst has great potential in exhaust purification for gasoline vehicle for it shows a high catalytic efficiency at low temperatures even after calcination of hydrothermal aging treatment for a long time.
4. Conclusions
Pd-decorated SMO mullite catalysts have been synthesized via an incipient-wetness impregnation method. The Pd/SMO-EG&M sample with excellent CO and C3H8 oxidation catalytic activity possesses a higher dispersion and smaller particle size of Pd species than that of CP sample. The higher Pd dispersion on EG&M support is beneficial to exposing active sites and enhancing the reducibility of Oads, which accelerates the oxygen spillover effect and promotes its CO oxidation capability. The higher oxygen mobility for Pd/SMO-EG&M means a facile oxygen transfer and helps the activation of available oxygen species above 250 °C, improving the activation of C–H bond and further resulting in optical C3H8 oxidation performance. It is concluded that SMO support functions well with outstanding thermal and structural stability, which exhibits great potential to ravel out the existing problems associated with metal oxide/Pd catalysts.Conflicts of interest
The authors declare that there are no conflicts to declare.Acknowledgements
We thank the financial support of the National Natural Science Foundation of China (51575217, 51572097) and the Hubei Province Funds for Distinguished Young scientists (2015CFA034). Rong Chen acknowledges the Thousand Young Talents Plan, the Recruitment Program of Global Experts and the Program for Changjiang Scholars and Innovative Research Team in University (IRT13017). The authors are also grateful to the technology support of the Flexible Electronics Research Center and Analytic Testing Center of HUST.Notes and references
- J. Y. Luo, M. Meng, J. S. Yang, X. G. Li, Y. Q. Zha, X. T. Wang and T. Y. Zhang, Appl. Catal., B, 2009, 87, 92 CrossRef CAS.
- W. P. Shan and H. Song, Catal. Sci. Technol., 1996, 30, 3529 Search PubMed.
- J. Y. Luo, D. Kisinger, A. Abedi and W. S. Epling, Appl. Catal., A, 2010, 383, 182 CrossRef CAS.
- H. Sharma and A. Mhadeshwar, Appl. Catal., B, 2012, 127, 190 CrossRef CAS.
- F. Y. Zhou, X. X. Du, J. Yu, D. S. Mao and G. Z. Lu, RSC Adv., 2016, 6, 66653 Search PubMed.
- C. S. Deng, B. Li, L. H. Dong, F. Y. Zhang, M. G. Fan, G. Z. Jin, J. B. Gao, L. W. Hao and X. P. Zhou, Phys. Chem. Chem. Phys., 2015, 17, 16092 RSC.
- M. Haneda, M. Todo, Y. Nakamura and M. Hattori, Catal. Today, 2017, 281, 447 CrossRef CAS.
- P. Weerachawanasak, O. Mekasuwandumrong, M. Arai, S.-I. Fujita, P. Praserthdam and J. Panpranot, J. Catal., 2009, 262, 199 CrossRef CAS.
- J. Liu, B. Sun, J. Y. Hu, Y. Pei, H. X. Li and M. H. Qiao, J. Catal., 2010, 274, 287 CrossRef CAS.
- J. J. Lovón-Quintanaa, J. B. O. Santos, A. S. P. Lovóna and N. La-Salviaa, J. Mol. Catal. A: Chem., 2016, 411, 117–127 CrossRef.
- M. S. Kumar, A. Eyssler, P. Hug, N. van Vegten, A. Baiker, A. Weidenkaff and D. Ferri, Appl. Catal., B, 2013, 94, 77–84 CrossRef.
- Y. Sekine, H. Takamatsu, S. Aramaki, K. Ichishima, M. Takada, M. Matsukata and E. Kikuchi, Appl. Catal., A, 2009, 352, 214 CrossRef CAS.
- G. Zhang, Z. Zhao, J. Liu, G. Liang, A. Duan, J. Zheng, S. Chen and R. Zhou, Chem. Commun., 2010, 46, 457–459 RSC.
- C. H. Kim, G. S. Qi, K. Dahlberg and W. Li, Science, 2010, 327, 1624 CrossRef CAS PubMed.
- C. Zhou, X. Liu, C. Z. Wu, Y. W. Wen, Y. J. Xue, R. Chen, Z. L. Zhang, B. Shan, H. F. Yin and W. G. Wang, Phys. Chem. Chem. Phys., 2014, 16, 5106 RSC.
- N. Li, A. Boreave, J. Deloume and F. Gaillard, Solid State Ionics, 2008, 197, 1396 CrossRef.
- M. A. Pen and L. G. Fierro, Chem. Rev., 2011, 101, 1981–2017 CrossRef.
- Y. Nishihata, J. Mizuki, T. Akao, H. Tanaka, M. Uenishi, M. Kimura, T. Okamoto and N. Hamada, Nature, 2002, 418, 164 CrossRef CAS PubMed.
- J.-M. Giraudon, A. Elhachimi, F. Wyrwalski, S. Siffert, A. Aboukais, J.-F. Lamonier and G. Leclercq, Appl. Catal., B, 2007, 75, 157 CrossRef CAS.
- S. Royer, H. Alamdari, D. Duprez and S. Kaliaguine, Appl. Catal., B, 2005, 58, 273 CrossRef CAS.
- R. Ran, X. D. Wu, D. Weng and J. Fan, J. Alloys Compd., 2013, 577, 288 CrossRef CAS.
- M. Uenishi, H. Tanaka, M. Taniguchi, I. Tan, Y. Sakamoto, S. -i. Matsunaga, K. Yokota and T. Kobayashi, Appl. Catal., A, 2005, 295, 114 CrossRef.
- W. Wang, G. McCool, N. Kapur, G. Yuan, B. Shan, M. Nguyen, U. M. Graham, B. H. Davis, G. Jacobs, K. Cho and X. Hao, Science, 2012, 337, 832 CrossRef CAS PubMed.
- P. F. Zhao, P. F. Yu, Z. J. Feng, R. Chen, L. W. Jia, J. Q. Wang, M. Q. Shen and B. Shan, RSC Adv., 2016, 6, 65950–65959 RSC.
- M. Sadakane, T. Horiuchi, N. Kato, C. Takahashi and W. Ueda, Chem. Mater., 2007, 19, 5779–5785 CrossRef CAS.
- Y. X. Liu, H. X. Dai, J. G. Deng, L. Zhang, B. Z. Gao, Y. Wang, X. W. Li, S. H. Xie and G. S. Guo, Appl. Catal., B, 2013, 140–141, 317–326 CrossRef CAS.
- Z. J. Feng, J. Q. Wang, X. Liu, Y. W. Wen, R. Chen, H. F. Yin, M. Q. Shen and B. Shan, Catal. Sci. Technol., 2016, 6, 5580–5589 CAS.
- Z. J. Feng, Q. H. Liu, Y. J. Chen, P. F. Zhao, Q. Peng, K. Cao, R. Chen, M. Q. Shen and B. Shan, Catal. Sci. Technol., 2017, 7, 838–847 CAS.
- S. Thampy, V. Ibarra, Y.-J. Lee, G. McCool, J. W. P. Hsu and K. Cho, Appl. Surf. Sci., 2016, 385, 490–497 CrossRef CAS.
- S. Thampy, Synthesis and characterization of transition metal–mullite catalysts for nitric oxide (NO) oxidation, PhD thesis, The University of Texas at Dallas, USA, 2014.
- A. F. García-Flores, E. Granado, H. Martinho, R. R. Urbano, C. Rettori, E. I. Golovenchits, V. A. Sanina, S. B. Oseroff, S. Park and S.-W. Cheong, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 104411 CrossRef.
- A. Grzechnik, M. Tolkiehn, W. Morgenroth and K. Friese, J. Phys.: Condens. Matter, 2010, 22, 275401 CrossRef PubMed.
- D. K. Shukla, R. Kumar, S. Mollah, R. J. Choudhary, P. Thakur, S. K. Sharma, N. B. Brookes and M. Knobel, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 73, 174432 CrossRef.
- L. Meng, A. P. Jia, J. Q. Lu, L. F. Luo, W. X. Huang and M. F. Luo, J. Phys. Chem. C, 2011, 115, 19789–19796 CAS.
- X. Liu, Y. W. Wen, Y. T. Tang, Y. Lang, B. Shan and R. Chen, Catal. Sci. Technol., 2017, 7, 4462 CAS.
- Z. Q. Zou, M. Ming and Y. Q. Zha, J. Phys. Chem. C, 2010, 114, 468–477 CAS.
- J. Y. Luo, M. Meng, Y. Q. Zha and L. H. Guo, J. Phys. Chem. C, 2008, 112, 8694–8701 CAS.
- X. W. Li, H. X. Dai, J. G. Deng, Y. X. Liu, S. H. Xie, Z. X Zhao and Y. Wang, Chem. Eng. J., 2013, 228, 965–975 CrossRef CAS.
- Y. X. Liu, H. X. Dai, Y. C. Du, J. G. Deng, L. Zhang and Z. X. Zhao, Appl. Catal., B, 2012, 119–120, 20–31 CrossRef CAS.
- J. Morère, M. J. Tenorio, M. J. Torralvo, C. Pando, J. A. R. Renuncio and A. Cabanas, J. Supercrit. Fluids, 2011, 56, 213–222 CrossRef.
- G. S. Guo, K. Lian, F. B. Gu, D. M. Han and Z. H. Wang, Chem. Commun., 2014, 50, 13575 RSC.
- J. Y. Ye and C. J. Liu, Chem. Commun., 2011, 47, 2167–2169 RSC.
- J. C. Ding, H. Y. Li, Z. X. Cai, X. X. Wang and X. Guo, Sens. Actuators, B, 2016, 22, 517–524 CrossRef.
- H. Gabasch, A. Knop-Gericke, R. Schlőgl, M. Borasio, C. Weilach, G. Rupprechter, S. Penner, B. Jenewein, K. Hayeka and B. Klőtzer, Phys. Chem. Chem. Phys., 2007, 9, 533–540 RSC.
- J.-M. Giraudon, A. Elhachimi and G. Leclercq, Appl. Catal., B, 2008, 84, 251–261 CrossRef CAS.
- F. P. Leisenberger, G. Koller, M. Sock, S. Surnev, M. G. Ramsey, F. P. Netzer, B. Klőtzer and K. Hayek, Surf. Sci., 2000, 445, 380–393 CrossRef CAS.
- L. S. Kibis, A. I. Stadnichenko, S. V. Koscheev, V. I. Zaikovskii and A. I. Boronin, J. Phys. Chem. C, 2012, 116, 19342–19348 CAS.
- G. N. Li, L. Li, Y. S. Lia and J. L. Shi, New J. Chem., 2015, 39, 1742–1748 RSC.
- B. P. Barbero, J. A. Gamboa and L. E. Cadús, Appl. Catal., B, 2006, 65, 21–30 CrossRef CAS.
- R. Westerholm, A. Christensen and A. Rosén, Atmos. Environ., 1996, 30, 3529 CrossRef CAS.
- P. Doggali, Y. Teraoka, P. Mungse, I. K. Shah, S. Rayalu and N. Labhsetwar, J. Mol. Catal. A: Chem., 2012, 358, 23–30 CrossRef CAS.
- Y. Wei, J. Liu, Z. Zhao, Y. Chen, C. Xu, A. Duan, G. Jiang and H. He, Angew. Chem., Int. Ed., 2011, 50, 2326–2329 CrossRef CAS PubMed.
- C. Zhang, Y. Guo, Y. Guo, G. Lu, A. Boreave, L. Retailleau, A. Baylet and A. Giroir-Fendler, Appl. Catal., B, 2014, 148–149, 490–498 CrossRef CAS.
- D. D. Song, X. Z. Shao, M. L. Yuan, L. Wang, W. C. Zhan, Y. L. Guo, Y. Guo and G. Z. Lu, RSC Adv., 2016, 6, 88117–88125 RSC.
- C. L. Li, B. S. Jiang, W. L. Fanchiang and Y. C. Lin, Catal. Commun., 2011, 16, 165–169 CrossRef CAS.
- D. Fino, N. Russo, G. Saracco and V. Specchia, J. Catal., 2003, 217, 367–375 CrossRef CAS.
- H. Q. Zhu, Z. F. Qin, W. J. Shan, W. J. Shen and J. G. Wang, J. Catal., 2004, 225, 267 CrossRef CAS.
- M. Y. Ni and Z. Zeng, J. Mol. Struct., 2009, 910, 14–19 CrossRef CAS.
- J. Roques, C. Lacaze-Dufaure and C. Mijoule, J. Chem. Theory Comput., 2007, 3, 878–884 CrossRef CAS PubMed.
- J. Fan, X. D. Wu, X. D. Wu, Q. Liang, R. Ran and D. Weng, Appl. Catal., B, 2008, 81, 38–48 CrossRef CAS.
- L. Meng, J. J. Lin, Z. Y. Pu, L. F. Luo, A. P. Jia, W. X. Huang, M. F. Luo and J. Q. Lu, Appl. Catal., B, 2012, 119–120, 117–122 CrossRef CAS.
- J. Wang, Y. G. Su, X. Q. Wang, J. H. Chen, Z. Zhao and M. Q. Shen, Catal. Commun., 2012, 25, 106–109 CrossRef CAS.
- Z. Hu, X. F. Liu, D. M. Meng, Y. Guo, Y. L. Guo and G. Z. Lu, ACS Catal., 2016, 6, 2265–2279 CrossRef CAS.
- S. Sartipi, A. A. Khodadadi and Y. Mortazavi, Appl. Catal., B, 2008, 83, 214–220 CrossRef CAS.
- J. S. Tian, H. G. Peng, X. L. Xu, W. M. Liu, Y. H. Ma, X. Wang and X. J. Yang, Catal. Sci. Technol., 2015, 5, 2270 CAS.
- G. Wang, R. You and M. Meng, Fuel, 2013, 103, 799–804 CrossRef CAS.
- Z. Z. Zhu, G. Z. Lu, Z. G. Zhang, Y. Guo, Y. L. Guo and Y. Q. Wang, ACS Catal., 2013, 3, 1154–1164 CrossRef CAS.
- Q. Xu, K. C. Kharas, B. J. Croley and A. K. Datye, ChemCatChem, 2011, 3, 1004–1014 CrossRef CAS.
- S. H. Liang, T. G. Xu, F. Teng, R. L. Zong and Y. F. Zhu, Appl. Catal., B, 2010, 96, 267–275 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed CO-pulse test conditions, catalytic performance of individual oxides, the research of synthetic effect, detailed experiment process, SEM images of pristine SmMn2O5 mullite, etc. See DOI: 10.1039/c7ra11551b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |