
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gs protein peptidomimetics as allosteric modulators of the β2-adrenergic receptor†
Lotte-Emilie
Boyhus
a,
Mia
Danielsen
 a,
Nina Smidt
Bengtson
a,
Micha
Ben Achim Kunze
b,
Xavier
Kubiak
c,
Tjerk J.
Sminia
a,
Jacob Hartvig
Løper
a,
Phuong Thu
Tran
a,
Kresten
Lindorff-Larsen
b,
Søren G. F.
Rasmussen
c,
Jesper Mosolff
Mathiesen‡
*a and
Daniel Sejer
Pedersen‡
*a
a,
Nina Smidt
Bengtson
a,
Micha
Ben Achim Kunze
b,
Xavier
Kubiak
c,
Tjerk J.
Sminia
a,
Jacob Hartvig
Løper
a,
Phuong Thu
Tran
a,
Kresten
Lindorff-Larsen
b,
Søren G. F.
Rasmussen
c,
Jesper Mosolff
Mathiesen‡
*a and
Daniel Sejer
Pedersen‡
*a
aDepartment of Drug Design and Pharmacology, University of Copenhagen, Jagtvej 162, 2100 Copenhagen, Denmark. E-mail: jmm@sund.ku.dk; daniel.pedersen@sund.ku.dk
bStructural Biology and NMR Laboratory, Department of Biology, University of Copenhagen, Ole Maaløes Vej 5, 2200 Copenhagen, Denmark
cDepartment of Neuroscience, University of Copenhagen, Blegdamsvej 3, 2200 Copenhagen, Denmark
First published on 9th January 2018
Abstract
A series of Gs protein peptidomimetics were designed and synthesised based on the published X-ray crystal structure of the active state β2-adrenergic receptor (β2AR) in complex with the Gs protein (PDB 3SN6). We hypothesised that such peptidomimetics may function as allosteric modulators that target the intracellular Gs protein binding site of the β2AR. Peptidomimetics were designed to mimic the 15 residue C-terminal α-helix of the Gs protein and were pre-organised in a helical conformation by (i, i + 4)-stapling using copper catalysed azide alkyne cycloaddition. Linear and stapled peptidomimetics were analysed by circular dichroism (CD) and characterised in a membrane-based cAMP accumulation assay and in a bimane fluorescence assay on purified β2AR. Several peptidomimetics inhibited agonist isoproterenol (ISO) induced cAMP formation by lowering the ISO maximal efficacy up to 61%. Moreover, some peptidomimetics were found to significantly decrease the potency of ISO up to 39-fold. In the bimane fluorescence assay none of the tested peptidomimetics could stabilise an active-like conformation of β2AR. Overall, the obtained pharmacological data suggest that some of the peptidomimetics may be able to compete with the native Gs protein for the intracellular binding site to block ISO-induced cAMP formation, but are unable to stabilise an active-like receptor conformation.
Introduction
The importance of G protein-coupled receptors (GPCRs) within drug discovery is undisputed. It is estimated that >25% of FDA approved drugs act via GPCRs.1 However, only 27% of non-olfactory GPCRs are currently targeted by an approved drug and 15% are currently in clinical trials, leaving 232 non-olfactory GPCRs that remain entirely unexploited as drug targets.2 Despite the central importance of GPCRs, we still have a very rudimentary understanding of the structure and function of this family of membrane-spanning receptors, particularly with respect to how GPCRs interact with intracellular proteins to achieve signal transduction and physiological responses. GPCR ligands generally bind to the extracellular side of the receptor and target the orthosteric or allosteric binding sites, or both as bivalent ligands.3 On the other hand, the intracellular surface of GPCRs has largely been ignored in the development of allosteric modulators. Such allosteric modulators could conceivably be designed to target the receptor surface responsible for recruiting intracellular transducers such as the G proteins and arrestins and thus be useful pharmacological tool compounds for studying GPCR signal transduction and possibly provide a new avenue for drug discovery. Moreover, such compounds could be useful compounds for X-ray crystallography to stabilise GPCRs in their active state conformation, which is particularly difficult to crystallise due to the high degree of receptor flexibility in the receptor active state. Recently, Lefkowitz and co-workers reported the discovery of an intracellular small molecule-like allosteric modulator for the β2-adrenergic receptor (β2AR) using a combinatorial approach with DNA encoded libraries.4 The allosteric ligand was found to bind to the intracellular surface of the receptor and inhibit both G protein and arrestin mediated signalling. Kobilka and co-workers crystallised a closely related analogue of the same ligand in complex with the β2AR-T4 lysozyme fusion protein with the orthosteric inverse agonist carazolol bound. The structure (PDB 5X7D) clearly shows the ligand occupying the G protein binding pocket.5Based on recent bio-structural data of active state GPCRs in complex with GPCR interacting proteins (GIP)6,7 or mimics thereof8–12 we wondered if it would be possible to rationally design peptidomimetics that target the GIP interface. Such allosteric ligands could be beneficial for stabilisation of GPCRs in various conformational states for structural studies, as pharmacological tool compounds, and could possibly provide a new avenue for therapeutic molecules. There have been some reports in the literature on using proteinogenic peptides as GPCR ligands. Hamm and co-workers have published several papers describing that peptides derived from the C-terminus of various G protein α subunits (Gα) are capable of reducing cAMP accumulation by blocking G protein coupling.13–15 Moreover, Scheerer et al. have reported the X-ray crystal structure of rhodopsin in complex with an 11-mer C-terminal peptide from the Gt protein.10 More recently, we reported our efforts to develop a peptidomimetic that mimics the function of nanobody 80 (Nb80) a well-known allosteric modulator of the β2AR that binds at the same site of the receptor as the native Gs protein.9,16
Using the X-ray crystal structure of the β2AR in complex with the Gs protein (β2AR-Gs) as a template (PDB 3SN6)7 we embarked on a project to identify such allosteric modulators for the β2AR by a structure-based design approach.
Results and discussion
Peptidomimetic design
Hamm and co-workers previously reported that a peptide comprised of the last 12 amino acid residues from the Gαs C-terminus (GαsCT12) was capable of inhibiting Gs protein coupling to the β2AR and increased agonist affinity for the receptor.15 However, in our hands the corresponding proteinogenic 15-mer peptide (GαsCT15) did not block agonist induced cAMP formation in β2AR cell membranes, whereas Nb80 significantly inhibited the maximal efficacy of ISO (Fig. 1c). Whereas Hamm and co-workers used saponin-permeabilised C6 glioma cells, all peptides reported herein were evaluated in HEK293 membranes overexpressing the β2AR.17 We selected to work in a cell membrane-based assay setup to render the intracellular surface freely accessible to the ligands and eliminate issues related to cell permeability. Likewise, Rasmussen et al. were not able to observe any effect of the 20-mer GαsCT20 peptide on β2AR receptor function and complex formation with β2AR.7 However, when GαsCT20 was fused to the carboxy terminus of the β2AR and expressed as a fusion protein a 27-fold increase in agonist affinity was observed. Based on the results by Rasmussen et al. we speculate that GαsCT20 mainly adopts a random coil structure in solution rendering binding to the receptor less favourable. This is consistent with our circular dichroism (CD) analysis of GαsCT15 that showed a random coil structure (Fig. 1e). Also, the affinity of a linear GαsCT peptide for β2AR is likely significantly lower than the full Gs protein complex, which has several additional contacts with the receptor. Based on these observations, we hypothesised that it would be necessary to chemically modify the native GαsCT15 peptide to improve binding to the β2AR. In the β2AR-Gs X-ray crystal structure the C-terminus of Gs adopts an α-helix terminated by a 3-residue reverse turn (Fig. 1b).7 Thus, we set out to chemically modify GαsCT15 to pre-organise the peptide in a similar conformation. Peptide stapling is a commonly applied technique for the synthesis of helical peptides.18–20 Among the many available methods for peptide stapling we favour CuAAC-stapling between amino acids with azido- and alkynyl-modified side chains.21,22 This methodology was originally developed by Tornoe et al.23 and later optimised by Cantel et al.24 and has been applied extensively in (i, i + 4)-peptide stapling in recent years.25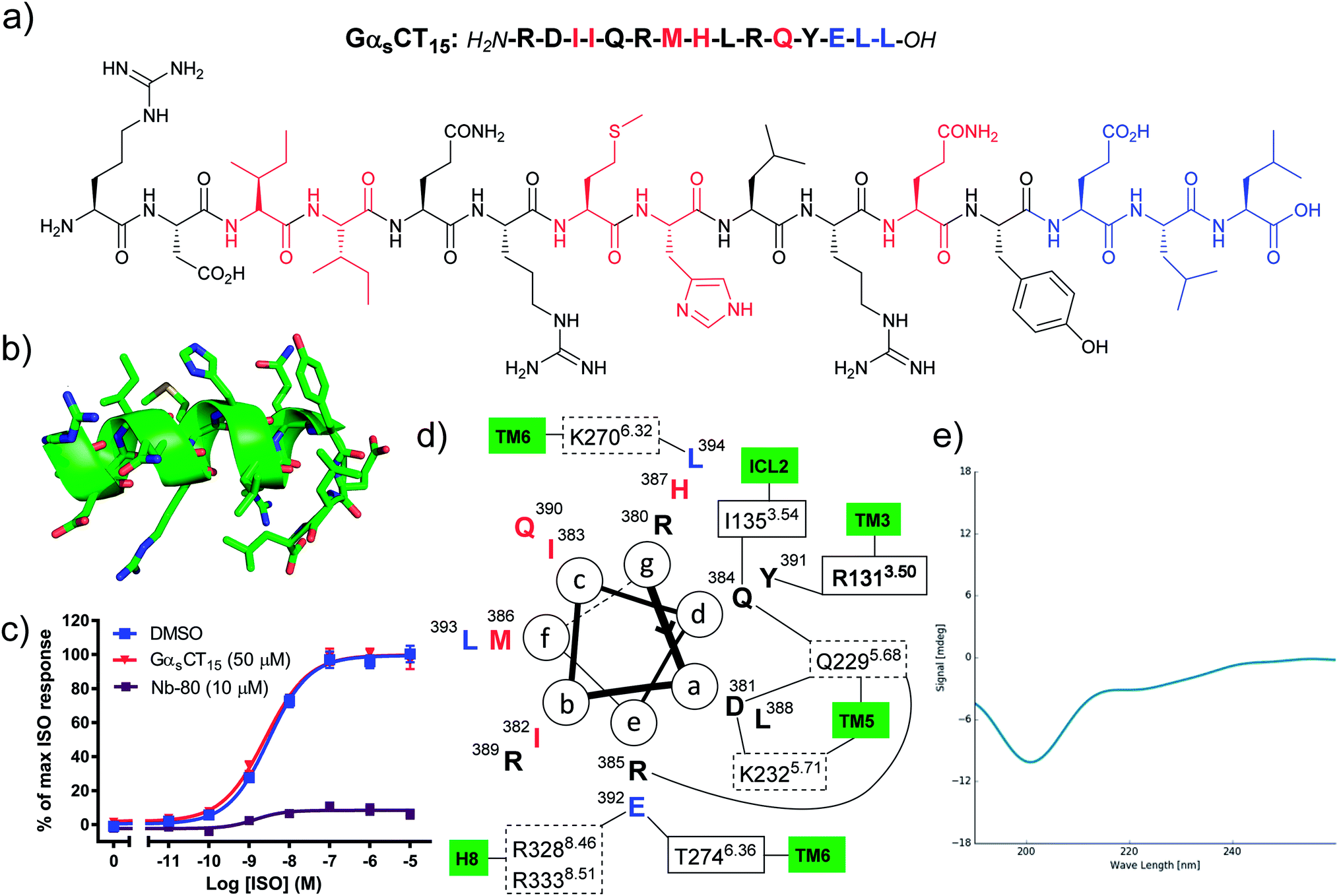 | ||
| Fig. 1 (a) The native GαsCT15 peptide sequences. Red: residues with no or weak receptor contacts. Blue: reverse turn, not helical, (b) the structure of GαsCT15 extracted from PDB entry 3SN6, (c) the GαsCT15 peptide does not inhibit agonist induced cAMP formation of the β2AR. Isoproterenol (ISO) concentration–response curves of cAMP accumulation were generated in the absence and presence of 50 μM peptide using HEK293 cell membranes overexpressing the β2AR. Data represents mean ± SEM from 3–4 independent experiments carried out in duplicates. The known β2AR-interacting nanobody 80 (Nb80) was included for comparison at 10 μM.9 (d) Helical wheel projection showing important contacts determined by MD simulation26 and X-ray crystallography.7 Dashed boxes: polar contacts involving side chains, solid boxes: polar contacts involving backbone carbonyls, bold box: cation–π interaction, (e) CD spectrum of GαsCT15 indicating a random coil structure (at 50 μM in 10 mM NaH2PO4 buffer, pH 6.0). | ||
By visual inspection of the β2AR-Gs X-ray crystal structure we concluded that the 15 C-terminal residues of Gαs participate in important interactions with β2AR. This is consistent with the conclusions drawn by Hildebrand and co-workers based on MD simulations.26 In terms of design, it is important that the staple position does not disrupt binding to the target. By visual inspection and based on the MD simulation study by Hildebrand and co-workers 5 residues were identified as potential staple anchoring points (Fig. 1). These 5 residues are ideally positioned for introduction of (i, i + 4)-staples and four stapling positions would be evaluated in the present study (Fig. 2). Moreover, four different staple designs utilising D- and L-ε-azidolysine (1–2), L-propargyl glycine (3) and O-propargylated L-serine (4) would be explored.
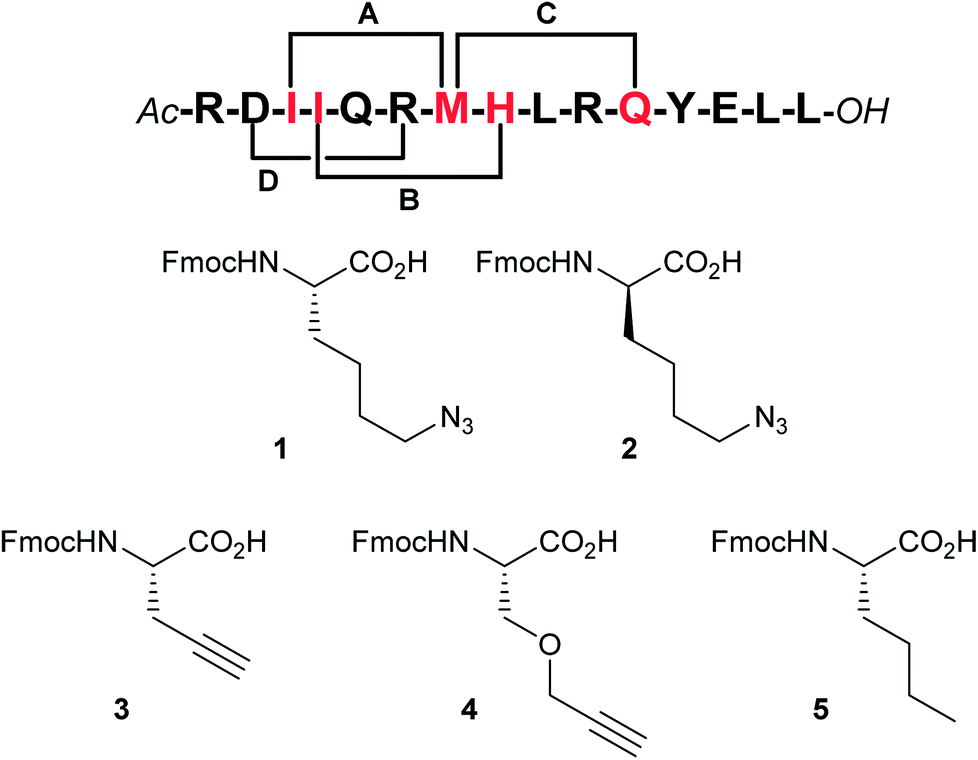 | ||
| Fig. 2 Stapling positions and unnatural amino acid building blocks. According to our analysis the red residues represent the best (i, i + 4)-stapling positions by appropriate substitution with amino acids 1–4 (staples A–C). Staple position D was included to validate the design (a negative control). To circumvent oxidation problems the norleucine building block 5 was employed as a substitute for methionine (stapling positions B and D). | ||
Synthesis
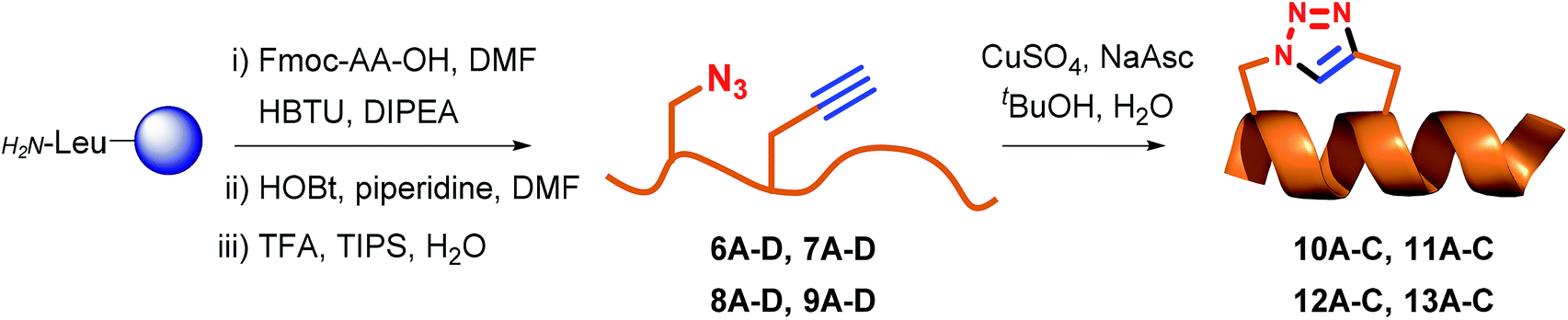
Fmoc-protected propargylglycine (3) is commercially available and the remaining azide and alkyne modified building blocks were synthesised in house. Azides 1–2 were synthesised from the corresponding Fmoc-protected amines as previously reported27 using the diazotransfer reagent imidazole sulfonyl azide.28,29 Alkyne 4 was synthesised in three steps from Boc-protected serine according to the published procedure.27 Finally, for stapling positions B and D commercially available norleucine (5) was employed as a replacement for the oxidation prone methionine residue.30 With amino acids 1–5 in hand the linear peptidomimetics 6–9 were synthesised by standard Fmoc-based solid-phase peptide synthesis (SPPS) on chlorotrityl resin and the N-terminus was acylated (Table 1). The synthesis of linear peptides 6–9A–C was uneventful and they were all purified by standard preparative RP-HPLC. However, the synthesis of peptides 6D–9D where two polar residues (D and R) were replaced proved complicated due to poor solubility after cleavage and deprotection. Because peptides 6D–9D were intended as inactive negative controls we eventually abandoned their purification and stapling due to their poor solubility, which would also translate to problems for pharmacological characterisation. Stapling of purified or crude 6–9A–C (see ESI† for details) was carried out in tBuOH and water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 v/v) using CuSO4·5H2O (1 eq.) and sodium ascorbate (5 eq.) as the in situ reducing agent. In general, the CuAAC reaction was clean and went to completion fast (1–3 h) to give stapled peptidomimetics 10–13A–C that were purified by preparative RP-HPLC to >95% purity in reasonable yields. Full conversion of linear to stapled peptidomimetic was monitored by RP-HPLC by spiking the reaction sample with the linear starting material, and by FT-IR where the azide stretch (at ∼2100 cm−1) is clearly seen to disappear after completion of the stapling reaction (see ESI†).
:2 v/v) using CuSO4·5H2O (1 eq.) and sodium ascorbate (5 eq.) as the in situ reducing agent. In general, the CuAAC reaction was clean and went to completion fast (1–3 h) to give stapled peptidomimetics 10–13A–C that were purified by preparative RP-HPLC to >95% purity in reasonable yields. Full conversion of linear to stapled peptidomimetic was monitored by RP-HPLC by spiking the reaction sample with the linear starting material, and by FT-IR where the azide stretch (at ∼2100 cm−1) is clearly seen to disappear after completion of the stapling reaction (see ESI†).
|
|
||||
|---|---|---|---|---|
| Linear | Amino acid sequence | Stapled | Yielda/NPCb (%) | Reaction time |
| a Yield after preparative HPLC purification to >95% purity (non-NPC corrected). b Net peptide content (NPC) (mass% of peptide, the remainder being constituted by counter ions and water). Determined by qNMR (see ESI for details). c Synthesised from pure linear peptide. d Synthesised from crude linear peptide. Overall yield based on resin loading. e Presumably finished in <3 hours but left overnight for practical reasons. | ||||
| GαsCT15 | H-R-D-I-I-Q-R-M-H-L-R-Q-Y-E-L-L-OH | NA | NA | NA |
| 6A | Ac-R-D-1-I-Q-R-3-H-L-R-Q-Y-E-L-L-OH | 10A | 36c/84 | 15 min |
| 6B | Ac-R-D-I-1-Q-R-5-3-L-R-Q-Y-E-L-L-OH | 10B | 10d/63 | Overnighte |
| 6C | Ac-R-D-I-I-Q-R-1-H-L-R-3-Y-E-L-L-OH | 10C | 10d/45 | 3 h |
| 6D | Ac-R-1-I-I-Q-3-5-H-L-R-Q-Y-E-L-L-OH | NA | NA | NA |
| 7A | Ac-R-D-2-I-Q-R-3-H-L-R-Q-Y-E-L-L-OH | 11A | 56c/77 | 15 min |
| 7B | Ac-R-D-I-2-Q-R-5-3-L-R-Q-Y-E-L-L-OH | 11B | 11d/72 | Overnighte |
| 7C | Ac-R-D-I-I-Q-R-2-H-L-R-3-Y-E-L-L-OH | 11C | 26d/71 | 1 h |
| 7D | Ac-R-2-I-I-Q-3-5-H-L-R-Q-Y-E-L-L-OH | NA | NA | NA |
| 8A | Ac-R-D-1-I-Q-R-4-H-L-R-Q-Y-E-L-L-OH | 12A | 69c/60 | 1 h |
| 8B | Ac-R-D-I-1-Q-R-5-4-L-R-Q-Y-E-L-L-OH | 12B | 15d/74 | 1 h |
| 8C | Ac-R-D-I-I-Q-R-1-H-L-R-4-Y-E-L-L-OH | 12C | 14d/53 | 2 h |
| 8D | Ac-R-1-I-I-Q-4-5-H-L-R-Q-Y-E-L-L-OH | NA | NA | NA |
| 9A | Ac-R-D-2-I-Q-R-4-H-L-R-Q-Y-E-L-L-OH | 13A | 40c/67 | Overnighte |
| 9B | Ac-R-D-I-2-Q-R-5-4-L-R-Q-Y-E-L-L-OH | 13B | 13d/77 | Overnighte |
| 9C | Ac-R-D-I-I-Q-R-2-H-L-R-4-Y-E-L-L-OH | 13C | 11d/60 | 3 h |
| 9D | Ac-R-2-I-I-Q-4-5-H-L-R-Q-Y-E-L-L-OH | NA | NA | NA |
Structural analysis by circular dichroism (CD)
All purified linear and stapled peptides were subjected to structural analysis by CD. Prior to recording CD spectra, the net peptide content (NPC) for all peptidomimetics was determined by quantitative NMR (qNMR, see ESI†).The CD data in Fig. 3 shows that stapling in general leads to peptidomimetics with a more helical structure when compared to the linear counterparts. However, the magnitude of the induced helicity varies significantly. While the position of the staple (A, B or C, Fig. 2) does not have a significant effect on the induced helicity, the employed amino acid residues (1–4) that form the staple have a tremendous effect. Employing residues 1 and 3 clearly increases the helicity of the peptide the most at all three stapling positions. The use of residues 1 and 4 likewise shows strong induction of helicity. In contrast, the use of the D-amino acid 2 in combination with either 3 or 4 lowers the helical induction markedly.
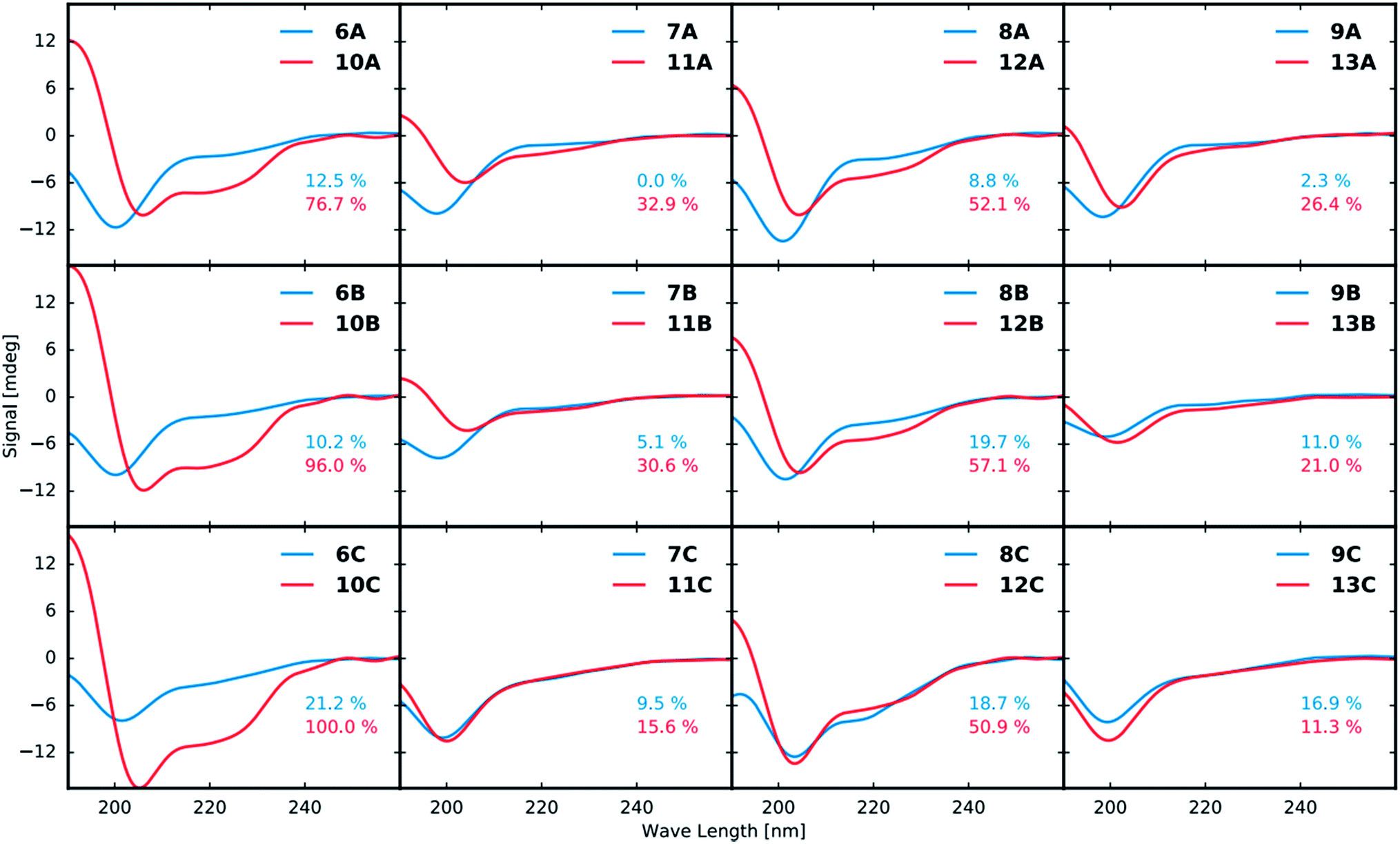 | ||
| Fig. 3 CD spectra of linear (blue curves) and corresponding stapled (red curves) peptidomimetics at a concentration of 50 μM (10 mM NaH2PO4 buffer, pH 6.0). The first row compares stapling at position A (Fig. 2), the second row shows stapling position B and the third row shows a comparison of stapling position C. The columns show a comparison of the different combinations of stapling residues 1 + 3, 2 + 3, 1 + 4 and 2 + 4, respectively. The helicity of the most helical peptidomimetic 10C as determined by the induced minima at 222 nm and maxima at 190 nm was set to 100% and the helicity of all other peptidomimetics were determined relative to that of 10C. | ||
Pharmacology
All peptidomimetics stapled at position A had little to no effect on the maximum efficacy of ISO (Table 2). Several of the peptidomimetics stapled at positions B and C displayed a small effect on the maximum efficacy of ISO; 7B, 8B, 8C and 11C all significantly decreased the maximal efficacy to approximately 80% (Table 2 and Fig. 4). Peptidomimetics 12B and 12C had a more pronounced effect and inhibited the maximal efficacy to ∼73% on average. Finally, peptidomimetic 10C affected the maximal ISO efficacy the most by lowering ISO maximal efficacy to 61%. With respect to peptidomimetic-induced effects on ISO potency, 9B and 13B significantly decreased the potency by 39- and 8-fold, respectively, but not the efficacy of ISO (Fig. 4). No significant effects of the peptidomimetics were observed on the basal cAMP levels or on the Hill-slope of the fitted curves.
| Peptide | Basal level | Maximum efficacy | pEC50 | Hill slope | n |
|---|---|---|---|---|---|
| Vehicle | 0.0 ± 0.0 | 100.0 ± 0.0 | 8.64 ± 0.03 | 0.81 ± 0.05 | 6 |
| Nb80 | −1.2 ± 0.7 | 11.3 ± 0.6* | 8.98 ± 0.16 | 0.88 ± 0.29 | 3 |
| 6A | 0.7 ± 1.1 | 90.8 ± 6.0 | 8.68 ± 0.04 | 0.81 ± 0.07 | 3 |
| 6B | −2.9 ± 4.2 | 81.5 ± 4.5 | 8.48 ± 0.08 | 0.81 ± 0.12 | 3 |
| 6C | 4.6 ± 1.9 | 89.7 ± 1.5 | 8.33 ± 0.02 | 0.83 ± 0.06 | 3 |
| 7A | −0.5 ± 1.5 | 88.8 ± 8.0 | 8.74 ± 0.01 | 0.83 ± 0.10 | 3 |
| 7B | −4.2 ± 1.8 | 79.9 ± 2.0* | 8.58 ± 0.03 | 0.84 ± 0.04 | 3 |
| 7C | 8.4 ± 4.3 | 88.1 ± 3.6 | 8.17 ± 0.11 | 0.87 ± 0.06 | 3 |
| 8A | −1.1 ± 0.7 | 97.7 ± 5.3 | 8.67 ± 0.04 | 0.72 ± 0.02 | 3 |
| 8B | −8.8 ± 2.8 | 77.1 ± 3.9* | 8.46 ± 0.15 | 0.82 ± 0.07 | 3 |
| 8C | −3.7 ± 2.3 | 78.0 ± 5.2* | 8.65 ± 0.06 | 0.71 ± 0.07 | 3 |
| 9A | 0.4 ± 0.7 | 92.7 ± 7.5 | 8.73 ± 0.08 | 0.83 ± 0.04 | 3 |
| 9B | 3.3 ± 4.6 | 84.1 ± 2.4 | 7.05 ± 0.38* | 0.89 ± 0.05 | 3 |
| 9C | 5.7 ± 2.1 | 88.1 ± 4.5 | 8.21 ± 0.33 | 0.92 ± 0.06 | 3 |
| 10A | 3.0 ± 0.4 | 82.8 ± 3.3 | 8.67 ± 0.05 | 0.78 ± 0.03 | 3 |
| 10B | −5.0 ± 2.6 | 80.5 ± 2.2 | 8.45 ± 0.04 | 0.72 ± 0.05 | 3 |
| 10C | −11.0 ± 4.3 | 61.0 ± 4.1* | 7.87 ± 0.01 | 0.63 ± 0.06 | 3 |
| 11A | −1.4 ± 0.7 | 90.7 ± 4.6 | 8.53 ± 0.05 | 0.74 ± 0.03 | 3 |
| 11B | −5.8 ± 3.0 | 88.0 ± 4.1 | 8.46 ± 0.07 | 0.72 ± 0.06 | 3 |
| 11C | −1.9 ± 8.4 | 78.4 ± 2.6* | 8.10 ± 0.12 | 0.85 ± 0.06 | 3 |
| 12A | 0.6 ± 0.4 | 94.5 ± 6.3 | 8.68 ± 0.03 | 0.73 ± 0.04 | 3 |
| 12B | −2.8 ± 4.0 | 74.0 ± 3.3* | 7.87 ± 0.53 | 0.88 ± 0.09 | 3 |
| 12C | −6.0 ± 2.1 | 71.7 ± 3.0* | 8.52 ± 0.05 | 0.90 ± 0.06 | 3 |
| 13A | 2.4 ± 0.3 | 90.0 ± 6.4 | 8.71 ± 0.05 | 0.85 ± 0.03 | 3 |
| 13B | 7.1 ± 5.7 | 85.1 ± 3.0 | 7.72 ± 0.30* | 0.89 ± 0.04 | 3 |
| 13C | 2.3 ± 1.2 | 82.8 ± 3.8 | 8.46 ± 0.43 | 0.80 ± 0.04 | 3 |
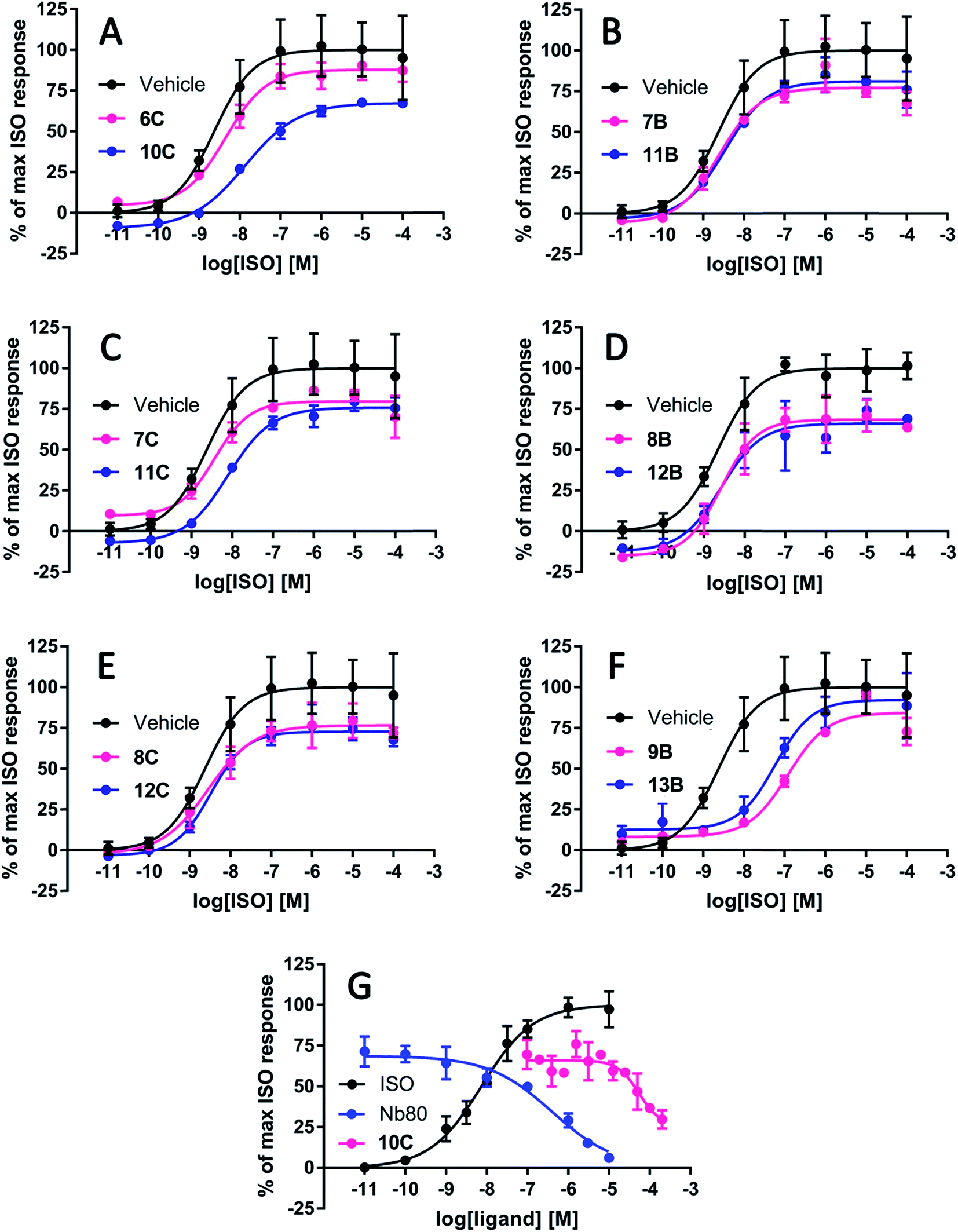 | ||
| Fig. 4 Representative graphs of the most effective peptidomimetics on the cyclic adenosine monophosphate (cAMP) production induced by the full agonist isoproterenol (ISO) at the β2-adrenergic receptor (β2AR). The stapled peptidomimetic 10C inhibited the maximal response of ISO to 61% whereas its linear precursor 6C had no significant effect on the ISO-induced cAMP production (A). The linear 7B (B) and stapled peptidomimetic 11C (C) inhibited ISO efficacy to a smaller degree (∼80%) but significantly. Their stapled (11B) and linear counterparts (7C) had a minor (∼90%) albeit non-statistically significant effects. The linear 8B and stapled peptidomimetic 12B pair decreased the maximum response of ISO to 70–80% (D), which is also the case for the linear and stapled pair 8C and 12C (E). The linear 9B and stapled peptidomimetic 13B, decreased the potency of ISO by 39- and 8-fold, respectively, whereas the efficacy was not affected (F). In presence of ISO corresponding to EC75, the IC50 of 10C was estimated to 55 μM, and the IC50 of Nb80 was estimated to 0.40 μM (G). | ||
To estimate the potency of the most efficacious peptidomimetic 10C, increasing concentrations of 10C up to 200 μM, and Nb80 as a control were applied in the presence of an ISO concentration corresponding to EC75. The IC50 of 10C was estimated to 55 μM whereas the IC50 of Nb80 was estimated to 0.40 μM (Fig. 4).
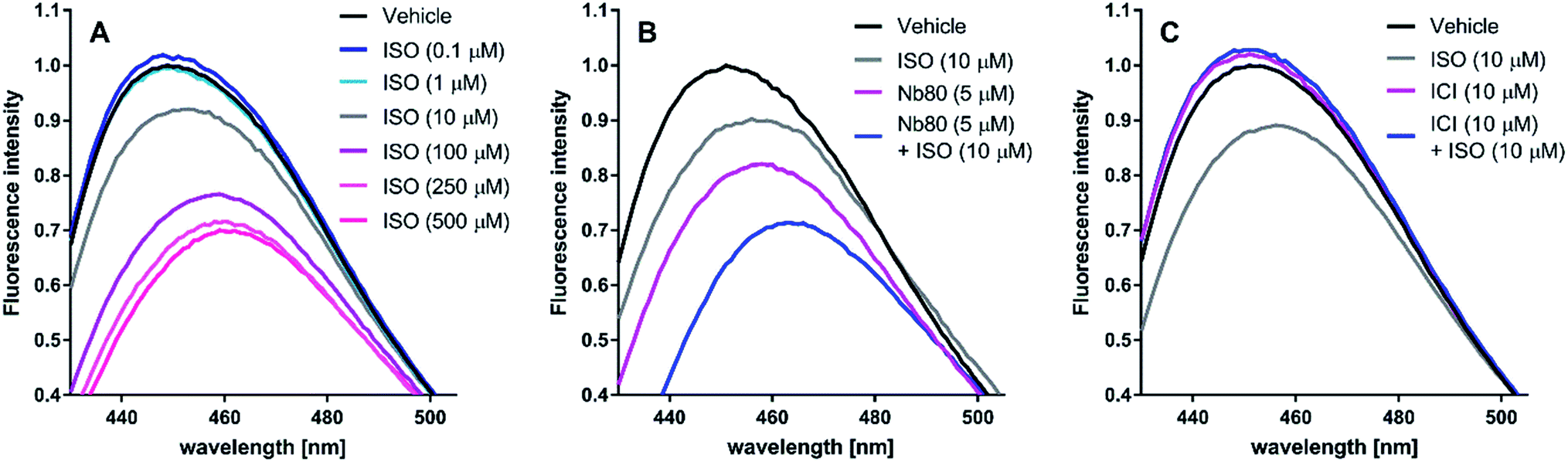 | ||
| Fig. 5 Representative bimane emission spectra of isoproterenol (ISO), nanobody 80 (Nb80) and ICI-118,551 (ICI) at the β2-adrenergic receptor (β2AR). ISO induces an active receptor conformation in a concentration-dependent manner. ISO displays saturating effects at 250 μM and 500 μM (A). Nb80 (5 μM) potentiates the 10 μM ISO-induced bimane-fluorescence response to that of ISO at 250 μM and 500 μM (B). At 10 μM, ICI prevents the receptor activation induced by 10 μM ISO and has a similar effect on the bimane-fluorescence response alone (C). | ||
Based on the results obtained with the cAMP assay, the linear and cyclic peptidomimetic pairs 6C/10C, 7C/11C, 8C/12C, 8B/12B and 9B/13B were tested in the bimane assay at 20 μM (Fig. 6). The peptidomimetics were tested for possible effects on receptor conformation alone and in the presence of 10 μM ISO, which allows detection of peptidomimetic-induced active conformation stabilisation as seen for Nb80. Bimane-fluorescence curves in presence of agonist or peptidomimetic alone or in combination were normalised to that of the receptor alone.
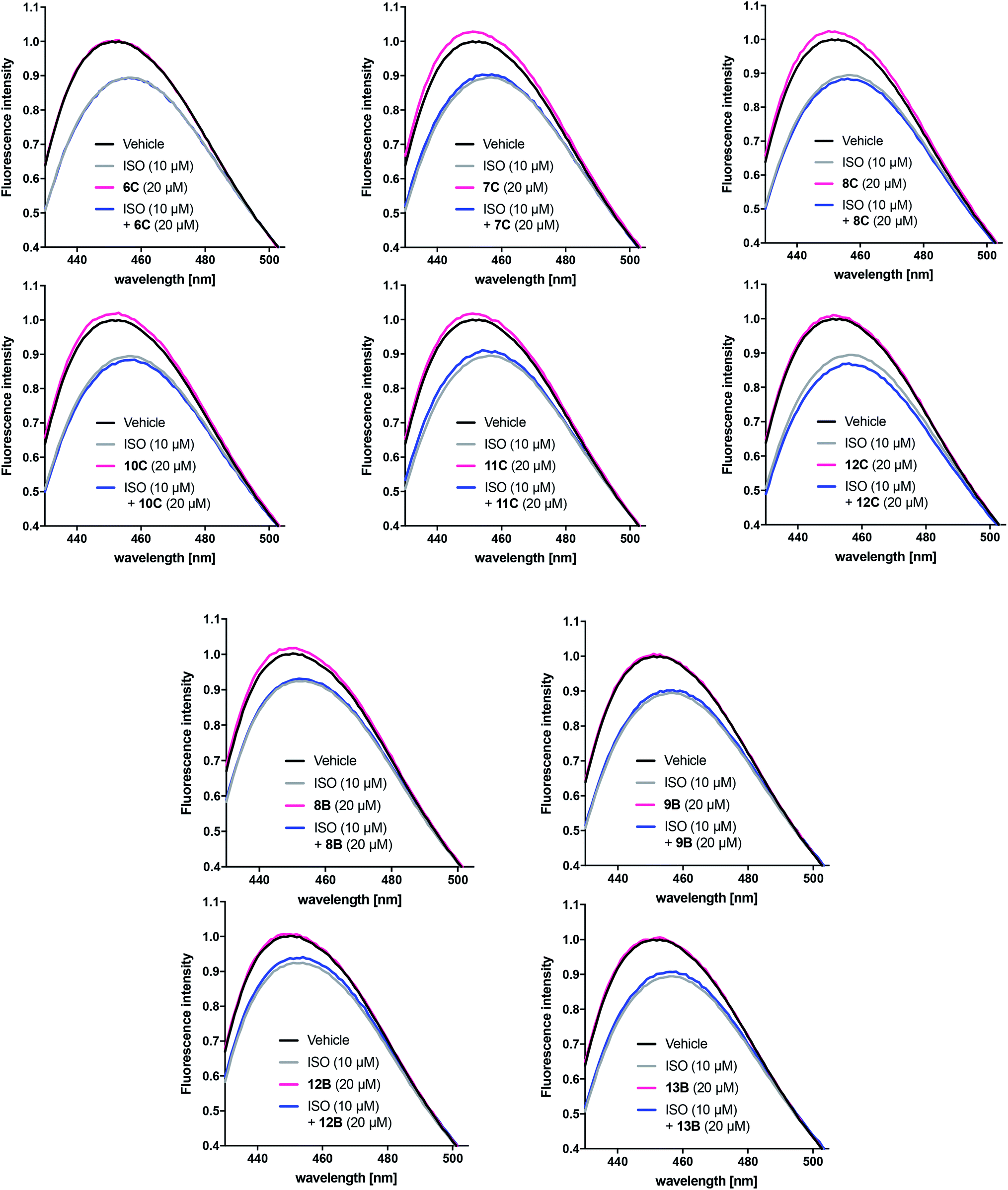 | ||
| Fig. 6 Representative bimane emission spectra of selected peptidomimetics with significant effects in the cAMP assay. The responses of the linear and cyclic peptidomimetic pairs 6C/10C, 7C/11C, 8C/12C, 8B/12B and 9B/13B were normalised to that of the unliganded β2AR alone (black solid line). The peptides were tested at 20 μM (n = 2 of measurements in triplicate) in the absence (pink solid line) and the presence (blue solid line) of ISO 10 μM (grey solid line). | ||
Unlike Nb80, none of the tested peptidomimetic were observed to stabilise an active-like conformation by decreasing FI and red-shifting λmax on their own or by potentiating the response beyond that of 10 μM ISO alone. Although there was a tendency for several peptidomimetics to shift the bimane-fluorescence in the opposite direction (8B, 7–8C and 10–11C), the peptidomimetics did not affect the response of 10 μM ISO alone. Interestingly, with the exception of peptidomimetic 8B only the peptidomimetics stapled at position C closest to the C-terminal were able to increase FI and blue-shift λmax in a similar way to that seen for the inverse agonist ICI (Fig. 5C).
Discussion
As anticipated CD analysis revealed that the stapled peptidomimetics generally had a higher helical content than their linear counterparts and the native Gαs 15-mer. The staples comprised of building blocks 1, 3 and 4 had the highest helical content, whereas the peptidomimetics stapled with D-amino acid 2 and alkynes 3 and 4 contained significantly less helicity. There was no trend regarding the helical content and the stapling positions A–C.The peptidomimetics were evaluated for their ability to block agonist-induced cAMP formation in cell membranes overexpressing the β2AR. Stapling the peptidomimetics at position A was not optimal for blocking cAMP formation by ISO. Thus, both stapled peptidomimetics 12A–13A and linear peptidomimetics 8A–9A were essentially inactive, indicating that this stapling position is not ideal for binding to the receptor using the present chemistry. Moving the staple towards the C-terminus (position B and C) was more favourable. The linear peptidomimetics 7B and 8B had a slight, yet significant effect on the maximal ISO response, lowering the efficacy to approximately 80%. Stapled peptidomimetic 12B had a more pronounced effect on the maximum efficacy with a lowering to 74%. Unexpectedly the 9B/13B pair red-shifted the ISO CRC comparable to that of a competitive antagonist rather than decreasing the maximal agonist efficacy as would be expected for a non-competitive antagonist. The linear 8C and stapled peptidomimetic 11C both lowered the maximum efficacy of ISO to approximately 80%. On the other hand, stapled 12C lowered the efficacy to 72%, whereas stapled 10C lowered the efficacy to approximately 61%. Thus, 10C clearly gave the most significant decrease in the maximum efficacy of ISO. In general, the stapled peptidomimetics gave the highest reduction in efficacy and a small tendency for peptidomimetics with a high helical content to have a greater effect was observed (ESI Fig. S1†). Thus, stapled peptidomimetics 10C, 12B and 12C with a relatively high helical content were found to lower the efficacy the most (61–74%). However, the data also shows that a high degree of helicity on its own is not sufficient to give a notable reduction in the formation of cAMP (e.g. stapled peptidomimetic 10B).
When tested in the conformational bimane fluorescence shift assay none of the peptidomimetics stabilised an active-like conformation similar to that of the G protein mimetic Nb80. However, a small tendency to increase the fluorescence intensity (FI) and blue-shift λmax similar to that for the inverse agonist ICI was seen. The effect was most pronounced for peptidomimetics stapled at position C but no correlation between the degree of helicity and the effect on the FI was observed. It cannot be excluded that the concentration tested in the bimane assay was too low to induce a more significant effect.
One interpretation of the obtained pharmacological data is that some of the peptidomimetics (e.g.10C) are capable of modulating ISO-induced cAMP formation by binding to and thus overlapping with the intracellular binding site of the G protein. However, unlike Nb80, none of the peptidomimetics reported herein stabilise an active-like receptor conformation. Although only small effects of the peptidomimetics were observed it does support the idea of developing intracellular modulators of GPCR signalling derived from hotspot domains of GPCR interacting proteins (e.g. the C-termini of G proteins). However, the native C-terminal peptide sequence (GαsCT15) that was employed as a template herein clearly does not provide potent peptidomimetic analogues despite stapling these in a helical conformation. Thus, to render this class of ligands of use for pharmacological and biophysical studies significant optimisation is required. It should be noted that the present study does not provide data that demonstrates that the peptidomimetics are binding in the intended G protein binding pocket. In principle, the peptidomimetics could be engaging the β2AR elsewhere. Further studies with more potent analogues will be required to determine the mode of action.
Conclusion
In the present study, a series of peptidomimetics that mimic the C-terminal α-helix of the Gαs protein were synthesised as potential allosteric modulators of the β2AR. The peptidomimetics were characterised pharmacologically in a cAMP accumulation assay and bimane fluorescence assay. Several peptidomimetics inhibited agonist ISO induced cAMP formation by lowering the maximal efficacy of ISO up to 61%. For the most potent peptidomimetic 10C the IC50 for blocking ISO induced cAMP formation was determined to 55 μM. Moreover, some peptidomimetics could decrease the potency of ISO significantly (up to 39-fold). In the bimane fluorescence assay none of the tested peptidomimetics could stabilise an active-like β2AR conformation. However, we observed a tendency to shift the bimane assay in the opposite direction for some peptidomimetics. Taken together, the data suggests that some of the peptidomimetics can compete with the native Gs protein for the intracellular binding site, but are unable to stabilise an active-like receptor conformation.To render the peptidomimetics of use as pharmacological tool compounds significant optimisation is required to increase ligand potency. Likewise, to elucidate the mode of action for this class of ligands more potent ligands are required. To this end we are in the process of strengthening ligand–receptor interactions by substitution with natural and unnatural amino acids aided by computational design. The results from these endeavours will be reported elsewhere in due course.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Lundbeck Foundation and the Carlsberg Foundation are gratefully acknowledged for financial support. T. T. P. was supported by the Vietnam International Education Development. M. B. A. K. was supported by a postdoctoral fellowship from the Lundbeck Foundation and K. L. L. was supported by a Hallas-Møller stipend from the Novo Nordisk Foundation. S. G. F. R. was supported by the Lundbeck Foundation (Grant R37-A3457), the Danish Independent Research Council (Grant 0602-02407B), and the UNIK Center for Synthetic Biology.References
- J. P. Overington, B. Al-Lazikani and A. L. Hopkins, How many drug targets are there?, Nat. Rev. Drug Discovery, 2006, 5, 993–996 CrossRef CAS.
- A. S. Hauser, M. Attwood, M. Rask-Andersen, H. B. Schiöth and D. E. Gloriam, Trends in GPCR Drug Discovery – New Targets and Indications, Nat. Rev. Drug Discovery, 2017, 16, 829–842 CrossRef CAS PubMed.
- P. Fronik, B. I. Gaiser and D. S. Pedersen, Bitopic Ligands and Metastable Binding Sites: Opportunities for G Protein-Coupled Receptor (GPCR) Medicinal Chemistry, J. Med. Chem., 2017, 60, 4126–4134 CrossRef CAS.
- S. Ahn, A. W. Kahsai, B. Pani, Q. T. Wang, S. Zhao, A. L. Wall, R. T. Strachan, D. P. Staus, L. M. Wingler, L. D. Sun, J. Sinnaeve, M. Choi, T. Cho, T. T. Xu, G. M. Hansen, M. B. Burnett, J. E. Lamerdin, D. L. Bassoni, B. J. Gavino, G. Husemoen, E. K. Olsen, T. Franch, S. Costanzi, X. Chen and R. J. Lefkowitz, Allosteric β-blocker isolated from a DNA-encoded small molecule library, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1708–1713 CrossRef CAS.
- X. Liu, S. Ahn, A. W. Kahsai, K. C. Meng, N. R. Latorraca, B. Pani, A. J. Venkatakrishnan, A. Masoudi, W. I. Weis, R. O. Dror, X. Chen, R. J. Lefkowitz and B. K. Kobilka, Mechanism of intracellular allosteric β2AR antagonist revealed by X-ray crystal structure, Nature, 2017, 548, 480–484 CrossRef CAS.
- Y. Kang, X. E. Zhou, X. Gao, Y. He, W. Liu, A. Ishchenko, A. Barty, T. A. White, O. Yefanov, G. W. Han, Q. Xu, P. W. de Waal, J. Ke, M. H. Tan, C. Zhang, A. Moeller, G. M. West, B. D. Pascal, E. N. Van, L. N. Caro, S. A. Vishnivetskiy, R. J. Lee, K. M. Suino-Powell, X. Gu, K. Pal, J. Ma, X. Zhi, S. Boutet, G. J. Williams, M. Messerschmidt, C. Gati, N. A. Zatsepin, D. Wang, D. James, S. Basu, S. Roy-Chowdhury, C. E. Conrad, J. Coe, H. Liu, S. Lisova, C. Kupitz, I. Grotjohann, R. Fromme, Y. Jiang, M. Tan, H. Yang, J. Li, M. Wang, Z. Zheng, D. Li, N. Howe, Y. Zhao, J. Standfuss, K. Diederichs, Y. Dong, C. S. Potter, B. Carragher, M. Caffrey, H. Jiang, H. N. Chapman, J. C. Spence, P. Fromme, U. Weierstall, O. P. Ernst, V. Katritch, V. V. Gurevich, P. R. Griffin, W. L. Hubbell, R. C. Stevens, V. Cherezov, K. Melcher and H. E. Xu, Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser, Nature, 2015, 523, 561 CrossRef CAS.
- S. G. F. Rasmussen, B. T. DeVree, Y. Zou, A. C. Kruse, K. Y. Chung, T. S. Kobilka, F. S. Thian, P. S. Chae, E. Pardon, D. Calinski, J. M. Mathiesen, S. T. A. Shah, J. A. Lyons, M. Caffrey, S. H. Gellman, J. Steyaert, G. Skiniotis, W. I. Weis, R. K. Sunahara and B. F. Kobilka, Crystal structure of the β2 adrenergic receptor-Gs protein complex, Nature, 2011, 477, 549 CrossRef CAS.
- B. Carpenter, R. Nehmé, T. Warne, A. G. W. Leslie and C. G. Tate, Structure of the adenosine A2A receptor bound to an engineered G protein, Nature, 2016, 536, 104 CrossRef CAS PubMed.
- S. G. F. Rasmussen, H. J. Choi, J. J. Fung, E. Pardon, P. Casarosa, P. S. Chae, B. T. DeVree, D. M. Rosenbaum, F. S. Thian, T. S. Kobilka, A. Schnapp, I. Konetzki, R. K. Sunahara, S. H. Gellman, A. Pautsch, J. Steyaert, W. I. Weis and B. K. Kobilka, Structure of a nanobody-stabilized active state of the β2 adrenoceptor, Nature, 2011, 469, 175 CrossRef CAS.
- P. Scheerer, J. H. Park, P. W. Hildebrand, Y. J. Kim, N. Krauss, H. W. Choe, K. P. Hofmann and O. P. Ernst, Crystal structure of opsin in its G-protein-interacting conformation, Nature, 2008, 455, 497 CrossRef CAS.
- A. K. Shukla, A. Manglik, A. C. Kruse, K. Xiao, R. I. Reis, W. C. Tseng, D. P. Staus, D. Hilger, S. Uysal, L. Y. Huang, M. Paduch, P. Tripathi-Shukla, A. Koide, S. Koide, W. I. Weis, A. A. Kossiakoff, B. K. Kobilka and R. J. Lefkowitz, Structure of active β-arrestin-1 bound to a G-protein-coupled receptor phosphopeptide, Nature, 2013, 497, 137 CrossRef CAS.
- M. Szczepek, F. Beyrière, K. P. Hofmann, M. Elgeti, R. Kazmin, A. Rose, F. J. Bartl, D. von Stetten, M. Heck, M. E. Sommer, P. W. Hildebrand and P. Scheerer, Crystal structure of a common GPCR-binding interface for G protein and arrestin, Nat. Commun., 2014, 5, 4801 CrossRef CAS PubMed.
- E. L. Martin, S. Rens-Domiano, P. J. Schatz and H. E. Hamm, Potent peptide analogues of a G protein receptor-binding region obtained with a combinatorial library, J. Biol. Chem., 1996, 271, 361 CrossRef CAS PubMed.
- M. R. Mazzoni, S. Taddei, L. Giusti, P. Rovero, C. Galoppini, A. D'Ursi, S. Albrizio, A. Triolo, E. Novellino, G. Greco, A. Lucacchini and H. E. Hamm, A galpha(s) carboxyl-terminal peptide prevents G(s) activation by the A(2A) adenosine receptor, Mol. Pharmacol., 2000, 58, 226 CrossRef CAS.
- M. M. Rasenick, M. Watanabe, M. B. Lazarevic, S. Hatta and H. E. Hamm, Synthetic peptides as probes for G protein function. carboxyl-terminal G alpha s peptides mimic Gs and evoke high affinity agonist binding to beta-adrenergic receptors, J. Biol. Chem., 1994, 269, 21519 CAS.
- C. Martin, S. Moors, M. Danielsen, C. Betti, C. Fabris, D. Sejer Pedersen, E. Pardon, M. Peyressatre, K. Fehér, J. C. Martins, J. Mosolff Mathiesen, M. Morris, N. Devoogdt, V. Caveliers, F. De Proft, J. Steyaert and S. Ballet, Rational Design of Nanobody80 Loop Peptidomimetics: Towards Biased β2 Adrenergic Receptor Ligands, Chem.–Eur. J., 2017, 23, 9632 CrossRef CAS.
- J. M. Mathiesen, L. Vedel and H. Bräuner-Osborne, cAMP biosensors applied in molecular pharmacological studies of G protein-coupled receptors, Methods Enzymol., 2013, 522, 191 CAS.
- T. A. Hill, N. E. Shepherd, F. Diness and D. P. Fairlie, Constraining Cyclic Peptides To Mimic Protein Structure Motifs, Angew. Chem., Int. Ed., 2014, 53, 13020 CrossRef CAS.
- Y. H. Lau, P. de Andrade, Y. Wu and D. R. Spring, Peptide stapling techniques based on different macrocyclisation chemistries, Chem. Soc. Rev., 2015, 44, 91 RSC.
- C. J. White and A. K. Yudin, Contemporary strategies for peptide macrocyclization, Nat. Chem., 2011, 3(7), 509 CrossRef CAS.
- P. T. Tran, C. Ø. Larsen, T. Røndbjerg, M. De Foresta, M. B. A. Kunze, A. Marek, J. H. Løper, L.-E. Boyhus, A. Knuhtsen, K. Lindorff-Larsen and D. S. Pedersen, Diversity-Oriented Peptide Stapling: A Third Generation Copper-Catalysed Azide−Alkyne Cycloaddition Stapling and Functionalisation Strategy, Chem.–Eur. J., 2017, 23, 3490 CrossRef CAS.
- A. D. Pehere, M. Pietsch, M. Gutschow, P. M. Neilsen, D. S. Pedersen, S. Nguyen, O. Zvarec, M. J. Sykes, D. F. Callen and A. D. Abell, Synthesis and Extended Activity of Triazole-Containing Macrocyclic Protease Inhibitors, Chem.–Eur. J., 2013, 19, 7975 CrossRef CAS.
- C. W. Tornoe, C. Christensen and M. Meldal, Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides, J. Org. Chem., 2002, 67, 3057 CrossRef CAS.
- S. Cantel, A. L. C. Isaad, M. Scrima, J. J. Levy, R. D. DiMarchi, P. Rovero, J. A. Halperin, A. M. D'Ursi, A. M. Papini and M. Chorev, Synthesis and conformational analysis of a cyclic peptide obtained via i to i + 4 intramolecular side-chain to side-chain azide – alkyne 1,3-dipolar cycloaddition, J. Org. Chem., 2008, 73, 5663 CrossRef CAS.
- D. S. Pedersen and A. Abell, 1,2,3-Triazoles in Peptidomimetic Chemistry, Eur. J. Org. Chem., 2011, 2399 CrossRef CAS.
- A. S. Rose, M. Elgeti, U. Zachariae, H. Grubmüller, K. P. Hofmann, P. Scheerer and P. W. Hildebrand, Position of Transmembrane Helix 6 Determines Receptor G Protein Coupling Specificity, J. Am. Chem. Soc., 2014, 136, 11244–11247 CrossRef CAS.
- T. J. Sminia and D. S. Pedersen, Azide- and Alkyne-Functionalised α- and β3-Amino Acids, Synlett, 2012, 23, 2643 CrossRef CAS.
- E. D. Goddard-Borger and R. V. Stick, An Efficient, Inexpensive, and Shelf-Stable Diazotransfer Reagent: Imidazole-1-sulfonyl Azide Hydrochloride, Org. Lett., 2007, 9, 3797 CrossRef CAS PubMed.
- H. Johansson and D. S. Pedersen, Azide- and Alkyne-Derivatised α-Amino Acids, Eur. J. Org. Chem., 2012, 4267 CrossRef.
- A. M. Gilles, P. Marlière, T. Rose, R. Sarfati, R. Longin, A. Meier, S. Fermandjian, M. Monnot, G. N. Cohen and O. Bârzu, Conservative replacement of methionine by norleucine in Escherichia coli adenylate kinase, J. Biol. Chem., 1988, 263, 8204 CAS.
- X. J. Yao, G. Vélez-Ruiz, M. R. Whorton, S. G. F. Rasmussen, B. T. DeVree, X. Deupi, R. K. Sunahara and B. Kobilka, The effect of ligand efficacy on the formation and stability of a GPCR-G protein complex, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 9501 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra11713b |
| ‡ These authors contributed equally as principal investigators. |
| This journal is © The Royal Society of Chemistry 2018 |