
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Natural 6-hydroxy-chromanols and -chromenols: structural diversity, biosynthetic pathways and health implications†
Marc Birringer *a,
Karsten Siemsb,
Alexander Maxonesa,
Jan Frankc and
Stefan Lorkowskide
*a,
Karsten Siemsb,
Alexander Maxonesa,
Jan Frankc and
Stefan Lorkowskide
aDepartment of Nutritional, Food and Consumer Sciences, Fulda University of Applied Sciences, Leipziger Straße 123, 36037 Fulda, Germany. E-mail: marc.birringer@oe.hs-fulda.de
bAnalytiCon Discovery GmbH, Hermannswerder Haus 17, 14473 Potsdam, Germany. E-mail: k.siems@ac-discovery.com
cInstitute of Biological Chemistry and Nutrition, University of Hohenheim, Garbenstr. 28, 70599 Stuttgart, Germany. E-mail: jan.frank@nutres.de
dInstitute of Nutrition, Friedrich Schiller University Jena, Dornburger Str. 25, 07743 Jena, Germany. E-mail: stefan.lorkowski@uni-jena.de
eCompetence Cluster for Nutrition and Cardiovascular Health (nutriCARD), Halle-Jena-Leipzig, Germany
First published on 26th January 2018
Abstract
We present the first comprehensive and systematic review on the structurally diverse toco-chromanols and -chromenols found in photosynthetic organisms, including marine organisms, and as metabolic intermediates in animals. The focus of this work is on the structural diversity of chromanols and chromenols that result from various side chain modifications. We describe more than 230 structures that derive from a 6-hydroxy-chromanol- and 6-hydroxy-chromenol core, respectively, and comprise di-, sesqui-, mono- and hemiterpenes. We assort the compounds into a structure–activity relationship with special emphasis on anti-inflammatory and anti-carcinogenic activities of the congeners. This review covers the literature published from 1970 to 2017.
1. Introduction
In 1922, Bishop and Evans discovered α-tocopherol as an essential lipid-soluble factor that promotes the gestation of rat fetuses.1 Since then, numerous structurally related 6-hydroxy-chromanols and -chromenols have been discovered. Tocochromanols of the vitamin E class represent the most widely distributed and predominant chromanols in nature. However, only photosynthetic organisms, such as plants, algae, and cyanobacteria as well as fungi, corals, sponges and tunicates, are able to perform the biosynthetic steps leading to a chromanol ring system. However, mammals, including humans, rely on these resources (esp. plant oils), since vitamin E is essential for a wide range of higher organisms.2The term vitamin E is traditionally used for the eight structurally related vitamers α-, β-, γ-, δ-tocopherol, and α-, β-, γ-, δ-tocotrienol, with α-tocopherol being the compound with the highest vitamin activity.3
Tocochromanols belong to the family of prenylquinones that also include plastochromanol-8, phylloquinones (vitamin K), and ubiquinones (coenzyme Q10). Due to its unique 6-hydroxy-chromanol structure, the vitamin E forms may act as antioxidants that prevent lipid peroxidation in cellular membranes and quench harmful reactive oxygen species (ROS) in plants and animals (including humans). The proton of the 6-hydroxy group can quench a reactive radical, in turn leading to a tocopheryl radical that, depending on the substitution pattern of the ring system, remains stable, with a half-life of several seconds, and can be subsequently recycled by vitamin C. The review does not aim to discuss the complex antioxidant and redox chemistry of tocopherols forming corresponding radicals, quinones, dimers or polymers. These issues have already been discussed in several excellent reviews.4,5 Further, biosynthesis, bioactivity and chemical properties of tocopherols and tocotrienols are summarized in several outstanding reviews,6 and will be discussed here only briefly. This work focuses on the structural diversity of chromanols due to side chain modifications and attempts to merge structural aspects with biological activity.
In general, 6-hydroxy-chromanols derive from the parent structure 2-methyl-3,4-dihydro-2H-chromen-6-ol (1) and 6-hydroxy-chromenols derive from 2-methyl-2H-chromen-6-ol (2) that comprise a class of bicyclic heterocycles formed by cyclisation of substituted 1,4-benzoquinones (Fig. 1).
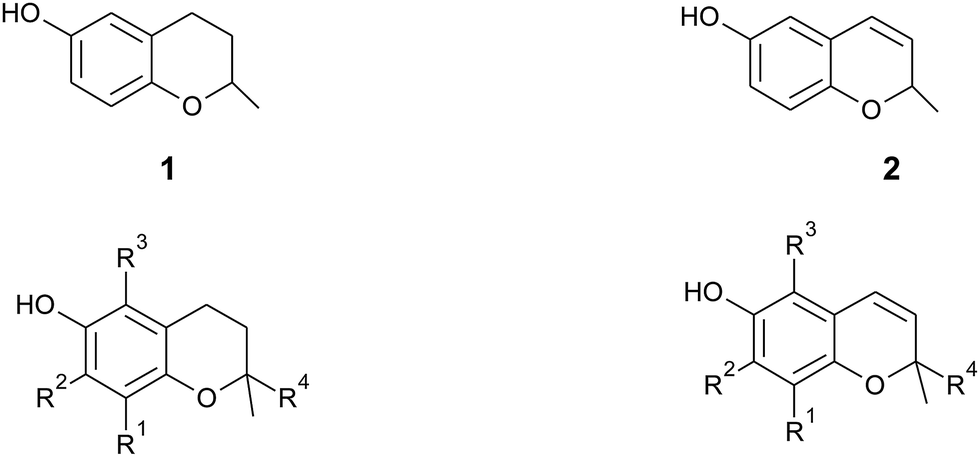 | ||
| Fig. 1 Core structures 2-methyl-3,4-dihydro-2H-chromen-6-ol (1) and 2-methyl-2H-chromen-6-ol (2) with substitution patterns of the chromanol and chromenol ring systems. | ||
Besides the methylation pattern (R1–R3) of the chromanol ring system, side chain modifications (at R4) show the highest structural variability. In particular tocotrienols are prone to (partial) reduction of the double bonds or oxidation of the methyl groups by cytochrome P450-dependent hydroxylases and oxidases, which ultimately results in the formation of oxidation products, such as alcohols, ketones, aldehydes, carboxylic acids, and truncations of the side chain. Furthermore, intramolecular cyclisation and/or rearrangements of the isoprene units can build up mono-, bi-, and tri-cyclic ring systems. These modifications are well known for compounds in marine organisms, especially in brown algae and sponges (see below), but have been found also in higher plant species. Along with side chain modifications, increased bioactivity is observed for many of these structures in vitro and in vivo.
The following chapters describe these compounds, sorted by the length of the carbon skeleton, following the order (mero)-diterpenes, -sesquiterpenes, -monoterpenes and -hemiterpenes.
2. Methods
Chromanols and chromenols presented here were selected by a chemical substructure search of 1 and 2, respectively, within several databases. We received 307 matches from the Dictionary of Natural Products and 128 matches from the Dictionary of Marine Natural Products (both at Chemnet BASE). We included a PubChem substructure search and PubMed keyword searches for “tocochromanol*”, “tocochromenol*” and “mero(di)terpenoid*”. Patents were searched by chemical names at the website of the European Patent Office.7 Finally, we performed a reference-related snowball sampling and deleted all doublets. All identified meroterpenoids were sorted by the length of their carbon-skeleton and number of prenyl units, respectively. Numbering of the carbon skeleton of metabolites was conducted in analogy to IUPAC rules, however for better clearness, side chain numbers were primed (e.g. 13′, see Fig. 3). Metabolites were further classified by their occurrence in the above-mentioned species (including animal metabolism) and not by structural matching. Within each species, metabolites were sorted by functionalization of the side chain (e.g. saturated, unsaturated and oxidized). Physio-chemical properties were predicted by Molinspiration WebME editor version 1.16 (http://www.molinspiration.com).With respect of the extent of the review, we excluded corresponding oxidized 1,4-benzoquinones or dimeric (and polymeric) structures that derive from natural or chemical oxidation processes that may occur during work-up procedures.
Many natural products with phenolic hydroxy groups, e.g. flavonoids, cumarins, caffeic acids, anthraquinones, or xanthones, bear a prenyl or to a minor extent geranyl or farnesyl residues in ortho position to the phenol. In some cases, this phenol forms a six-membered chromene ring by addition to the double bond of the prenyl (geranyl, farnesyl) residue. These mainly plant-derived compounds are not related to tocopherol biosynthesis and usually do not have the substructure of the 6-OH-chromanol. These chromanols are also not covered by this review.
3. Meroditerpenes
3.1 Plants
In the last decades, hundreds of publications referring to tocopherols and tocotrienols have been published, covering chemical, physical and biological properties of vitamin E as well as analytical procedures to detect the vitamers from biological origin.The main sources of the fat-soluble vitamin E are plant oils. To understand the structural variability of tocochromanols in plants and other photosynthetic organisms, a brief introduction into their biosynthesis is presented. The biosynthesis of tocochromanols was primarily investigated in the leaves of green plants, however all photosynthetic organisms as well as apicomplexa parasites such as Plasmodium falciparum8 are capable of the necessary biosynthetic steps. The biosynthetic pathways of tocotrienols, tocomonoenols, tocopherols and plastochromanol-8 are depicted in Fig. 2 and consist of five main steps. First, the transformation of p-hydroxyphenylpyruvate (HPP) to homogentisic acid (HGA), which is catalyzed by hydroxyphenylpyruvate dioxygenase (HPPD). Second, the synthesis of the isoprenoid side chain that originates from the 1-deoxy-D-xylulose-5-phosphate (DOXP) pathway in plastids. Here, geranylgeraniol reductase (GG-reductase) determines the degree of side chain saturation that leads to dihydro-geranylgeraniol diphosphate (DHGG-DP), tetrahydro-geranylgeraniol diphosphate (THGG-DP) and phytyl diphosphate, respectively. The reduction of the double bonds between C-3′–C-4′ and C-7′–C-8′ results in two R-configurated stereogenic centers at C-4′ and C-8′ of the later tocopherols (Fig. 3).
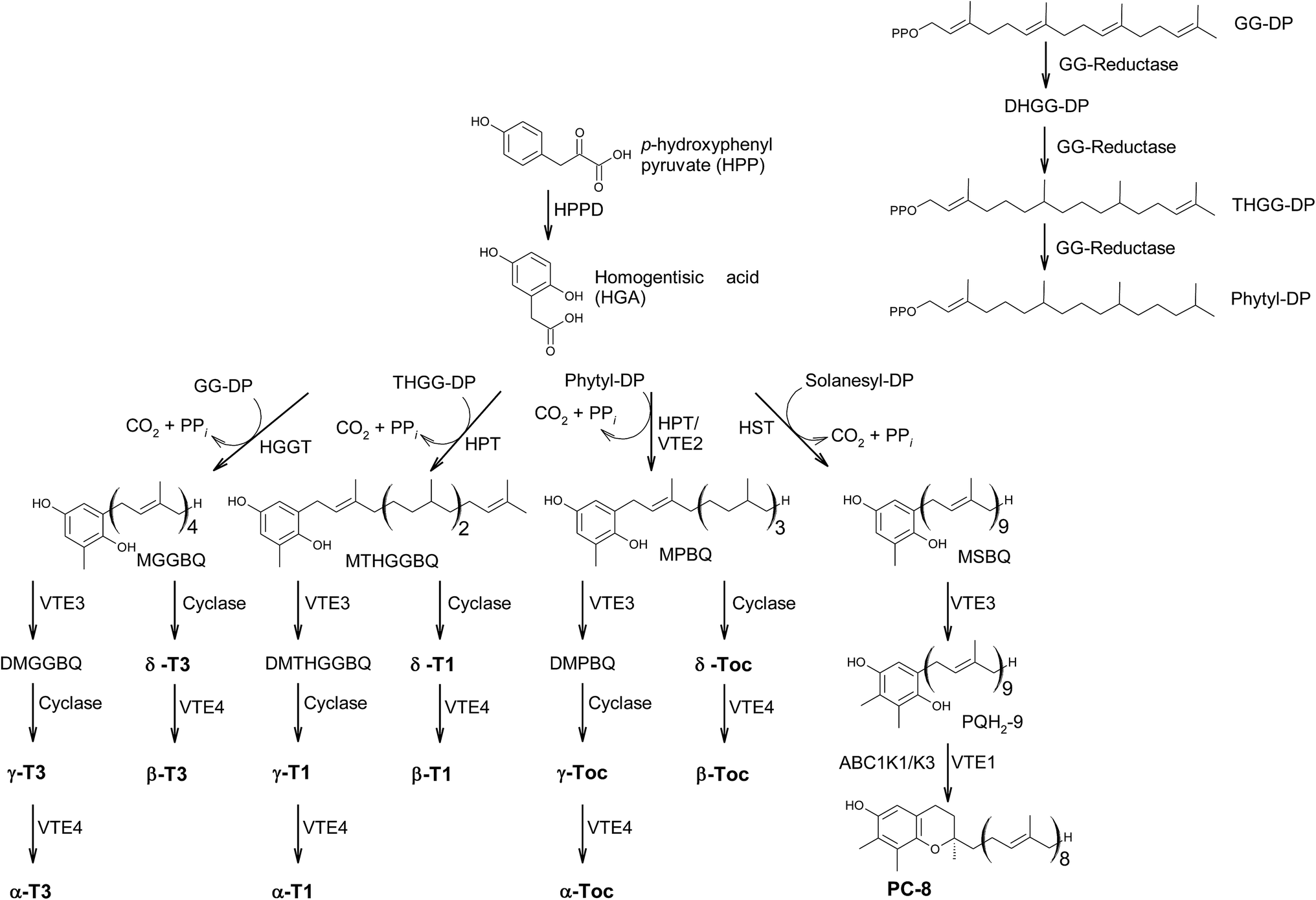 | ||
| Fig. 2 Biosynthetic pathway towards the formation of chromanols within photosynthetic organisms. Abr.: HPP: p-hydroxyphenylpyruvate; HGA: homogentisic acid; HPPD hydroxyphenylpyruvate dioxygenase; DOXP: 1-deoxy-D-xylulose-5-phosphate; GG-reductase: geranylgeraniol reductase; DHGG-DP: dihydro-geranylgeraniol diphosphate; THGG-DP: tetrahydro-geranylgeraniol diphosphate; MGGBQ: methyl-geranygeraniol benzoquinol; MTHGGBQ: methyl-tetrahydro-geranylgeraniol benzoquinol; MPBQ: methyl-phytyl benzoquinol; MSBQ: methyl-solanesyl benzoquinol. | ||
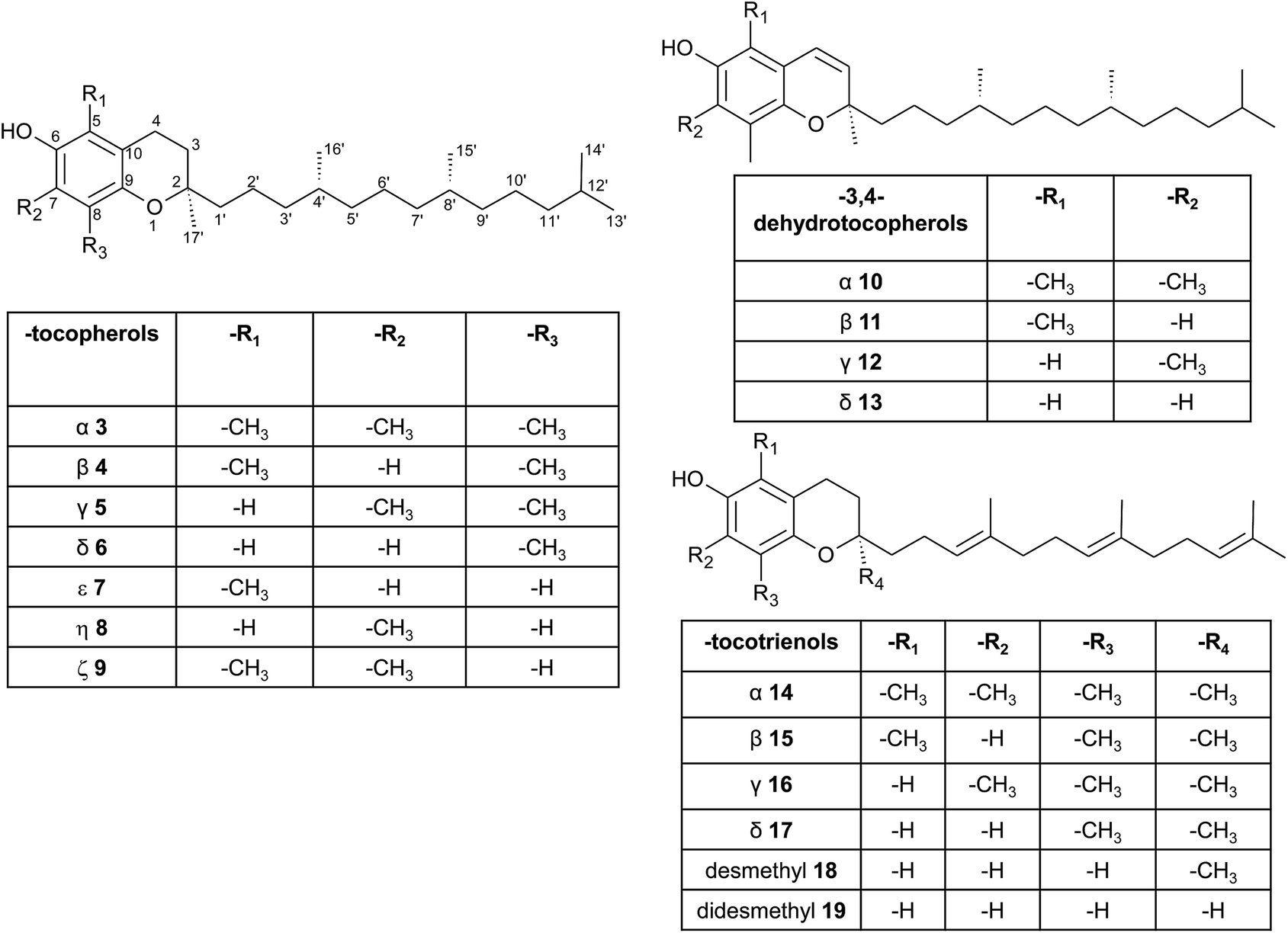 | ||
| Fig. 3 Structures and substitution patterns of tocopherols (3 to 9), dehydrotocopherols (10 to 13) and tocotrienols (14 to 19). | ||
In addition, solanesyl diphosphate, containing nine isoprene units, is formed by solanesyl diphosphate synthase.9 The above-mentioned diphosphates serve as substrates for transferases, which catalyze the alkylation of HGA, leading to benzoquinol derivatives, such as methyl-geranygeraniol benzoquinol (MGGBQ), methyl-tetrahydro-geranylgeraniol benzoquinol (MTHGGBQ), methyl-phytyl benzoquinol (MPBQ), and methyl-solanesyl benzoquinol (MSBQ), respectively (step 3).
The methylation pattern of tocochromanols depends on the next steps (step 4 and 5) of the biosynthesis. δ- and β-tocochromanols are formed by immediate cyclization, followed by S-adenosyl methionine-dependent methylation of the chromanol ring, whereas γ-tocochromanols are build by methylation followed by cyclization. Finally, α-tocochromanols results from methylation of γ-tocochromanols. The cyclization of the prenylated quinones to chromanols by tocopherol cyclase occurs within plastoglobules. The latter biosynthetic step yields R-configuration at C-2 atom and thus seems to be unique for plant species. For in-depth details of the biosynthetic pathways, the reader is referred to previously published excellent reviews.10,11
According to the methylation pattern of the 6-hydroxy-chromanol ring system, tocopherols are divided into the most prominent vitamers α(5,7,8-trimethyl)-tocopherol (3), β(5,8-dimethyl)-tocopherol (4), γ(7,8-dimethyl)-tocopherol (5) and δ(8-methyl)-tocopherol (6), respectively (Fig. 3). The tocopherols are ubiquitously found in most plant oils, whereas tocotrienols occur only in non-photosynthetic organs of higher plants, mainly eudicots and monocots.11,12
Alternative methylations of the chromanol ring lead to ε-tocopherol (5-methyltocol) (7), η-tocopherol (7-methyltocol) (8) and ζ-tocopherol (5,7-dimethyltocol) (9), which are found in trace amounts in rice bran.13 The latter congeners have not been described in recent literature and therefore their existence seems to be questionable and may have been the result of analytical artifacts.
In the past, all tocopherols and tocotrienols were studied in gestation-fetal resorption assays, with RRR-α-tocopherol (3) being the most potent vitamer.14
Although described in textbooks, primary literature on tocopherylesters at C-6 is scarce.15,16 Acylesters of saturated fatty acids (C12:0, C14:0, C16:0 and C18:0) and tocopherols were found in Nuphar luteum and Nymphea alba15 and in the pulp of yellow bell pepper (Capsicum annuum).16 In the last decade, the occurrence, angiogenic and vasculogenic properties of α-tocopheryl-phosphate were intensively studied by Azzi et al.17,18 This molecule was found in food but only in low amounts.18
Dehydrotocopherols derive from the biochemical elimination between C-3 and C-4 of the chromanol ring and were first isolated as α-, β- and γ-dehydrotocopherols (10, 11, 12) from wheat germ oil19 and from various Stemona species, such as Korean Stemonae Radix (Fig. 3) (Stemona tuberosa).20,21 γ-Dehydrotocopherol shows proliferative effects on mouse NIH 3T3 fibroblasts and a potential use as wound healing agent has been suggested.21
As mentioned above, α-, β-, γ- and δ-tocotrienols (14, 15, 16, 17) usually occur as trace vitamers, however, several plant tissues and oils accumulate higher amounts of tocotrienols. For example, α-tocotrienol was found in barley (76 mg/100 g), γ- and δ-tocotrienol in palm oil (36 mg and 8 mg/100 g, respectively). The distribution of tocotrienols in plant has been reviewed in detail elsewhere.6,12,22
In contrast to tocopherols, tocotrienols exhibit higher bioactivity in vertebrates. Ashan et al. recently reviewed the bioactivity of tocotrienols, which may act as anti-cancer, anti-diabetic, anti-inflammatory, antioxidant, immune-stimulatory, cardio-, neuro-, hepato- and nephro-protective molecules.23
Alternation in the methylation pattern has been also described for tocotrienols. Qureshi et al. found desmethyltocotrienol (3,4-dihydro-2-methyl-2-(4,8,12-trimethyltrideca-3′(E),7′(E),11′-trienyl)-2H-1-benzopyran-6-ol) (18) and didesmethyltocotrienol (3,4-dihydro-2-(4,8,12-trimethyltrideca-3′(E),7′(E),11′-trienyl)-2H-1-benzopyran-6-ol) (19) in rice bran24 (Fig. 3). Most interestingly, the latter compounds show cholesterol lowering activity in chicken, most likely by inhibition of 3-hydroxy-3-methylglutaryl-CoA reductase, which catalayzes the rate-limiting step of the cholesterol biosynthesis pathway. The compounds reduced total serum cholesterol by 26% and 31% relative to a control diet and reduced LDL cholesterol by 41% and 48%, respectively. Similar to tocotrienols, both compounds suppress proliferation B16 melanoma cells24,25 (Table 3). Interestingly, oxidation of the aromatic methyl groups are rare in nature. Two unusual formyl-derivatives (at C-5 (20) and C-7 (21), respectively) of δ-tocotrienol have been isolated in trace amounts from Garcinia virgata (Fig. 4).26
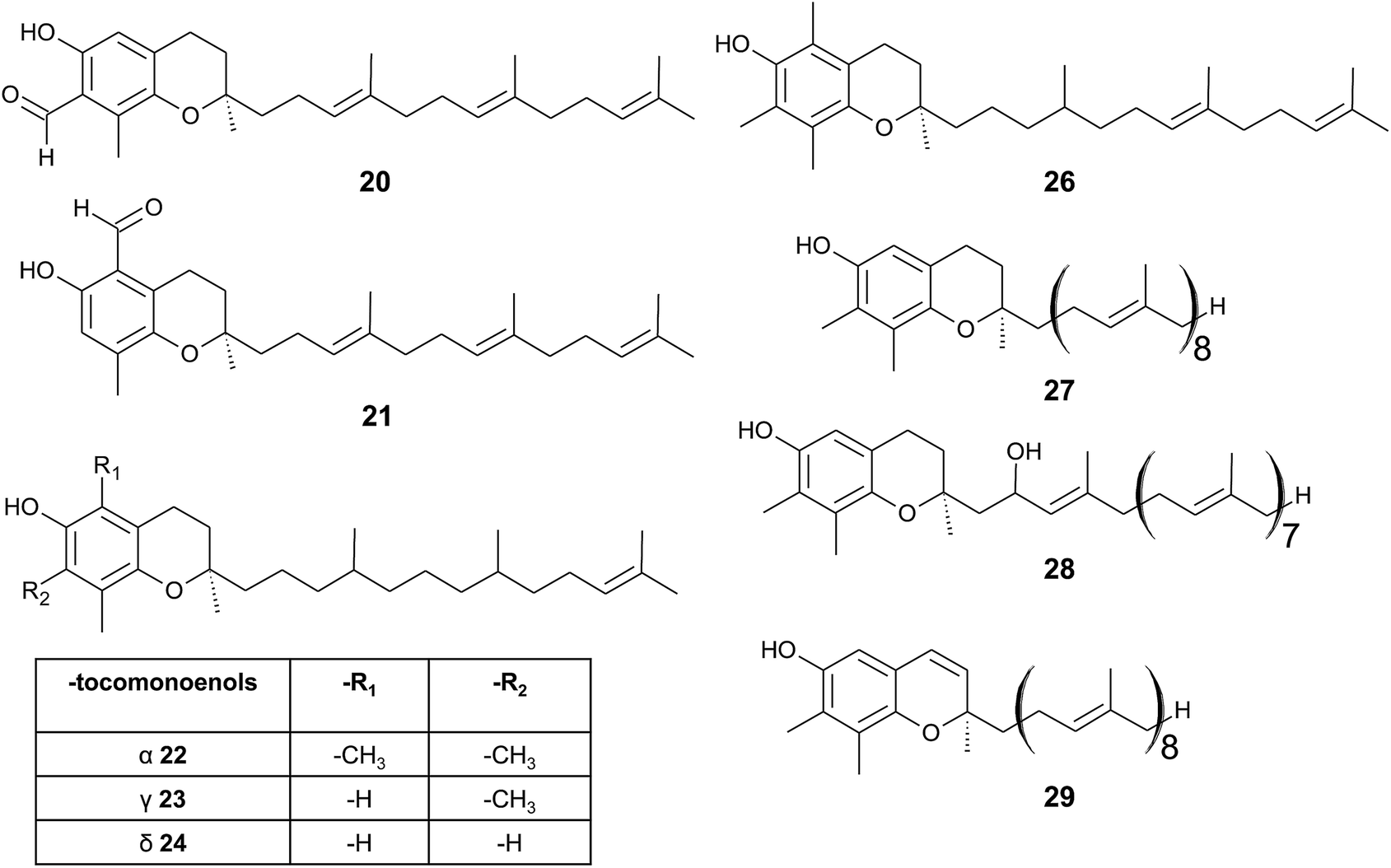 | ||
| Fig. 4 Structures and substitution patterns of formyl derivatives (20) and (21), tocomonoenols (22 to 24), tocodienol (26) and plastochromanols (27 to 29). | ||
As a result of a partial reduction of the prenyl side chain by geranylgeraniol reductase during the synthesis of tocopherols, tocodienols and tocomonoenols were found in several plant species (Fig. 4).11 α-Tocomonoenol (22) was isolated in palm seed and pumpkin seed oils,27,28 γ-tocomonoenol (23) in pumpkin seed oil as well as green leaves and etiolated beans of Kalanchoe daigremontiana and Phaseolus coccineus,11,28 and δ-tocomonoenol (24) was found in kiwi fruits (Actinidia chinensis).29 Of note, marine-derived α-tocomonoenol (25) (MDT) is a structural isomer of the above mentioned α-tocomonoenol with a terminal double bond at C-13′ (see section on Phytoplankton (3.3.2)).
α-Tocodienol (26) has recently been discovered as a trace compound (0.2% of the total vitamin E content) in palm oil.30 Interestingly to note is a recent publication by Hammann et al., who have tentatively identified 170 unsaturated tocochromanol compounds in palm oil by GC-MS, which were most likely produced (in trace amounts) by the thermal oil refining process and are thus unlikely genuine natural products.31
Beside tocopherols and tocotrienols, some plant species produce plastochromanol-8 (27), a γ-tocochromanol with eight isoprenoid units in the side chain. The biosynthesis of the polyterpene follows that of tocotrienols except of the use of solanesyl-diphosphate synthase to form the elongated side chain of plastochromanol-8 (Fig. 4).32 Plastochromanol-8 was first discovered in leaves of the rubber tree (Hevea brasiliensis) and since then in many higher plants, where it acts as a fat-soluble antioxidant.32–34 Nutritional sources, such as rapeseed and linseed oil, accumulate between 5.57 and 18.47 mg/100 g, respectively.34 Nutritional or physiological effects of plastochromanol-8 in animals or humans have not been described so far. As a result of the non-enzymatic oxidation of plastochromanol-8 by singlet oxygen, hydroxy-plastochromanol (28) was identified in Arabidopsis leaves.35 Solanachromene (29) (plastochromenol-8) contains a double bond in the chromanol ring and was found in relatively high amounts (0.05% of dry weight) in aged flue-cured tobacco leaves.33,36
δ-Garcinoic acid (30) (E-13′-carboxy-δ-tocotrienol, δ-garcinoic acid), an oxidation product of δ-tocotrienol, is probably the most investigated plant tocotrienol with side chain modification, so far (Fig. 5).37 δ-Garcinoic acid was first isolated from Clusia grandiflora by Delle Monache et al. and later by Terashima et al. from the African bitter nut Garcinia kola and was further characterized for its chemical and physiological properties.37–42 It has been detected in different amounts within the Clusiaceae family including Tovomitopsis psychotriifolia, Clusia obdeltifolia, Clusia burlemarxii, Clusia pernambucensis, Garcinia kola and together with γ-garcinoic acid (31) in the bark of Garcinia amplexicaulis.43,44 Recently, γ-garcinoic acid was isolated in small amounts from the Algerian conifer Cedrus atlantica (Pinaceae).45 A mixture of 2(Z)-δ-garcinoic acid and 2(E)-δ-garcinoic acid was isolated from the stem of Clusia obdeltifolia.46
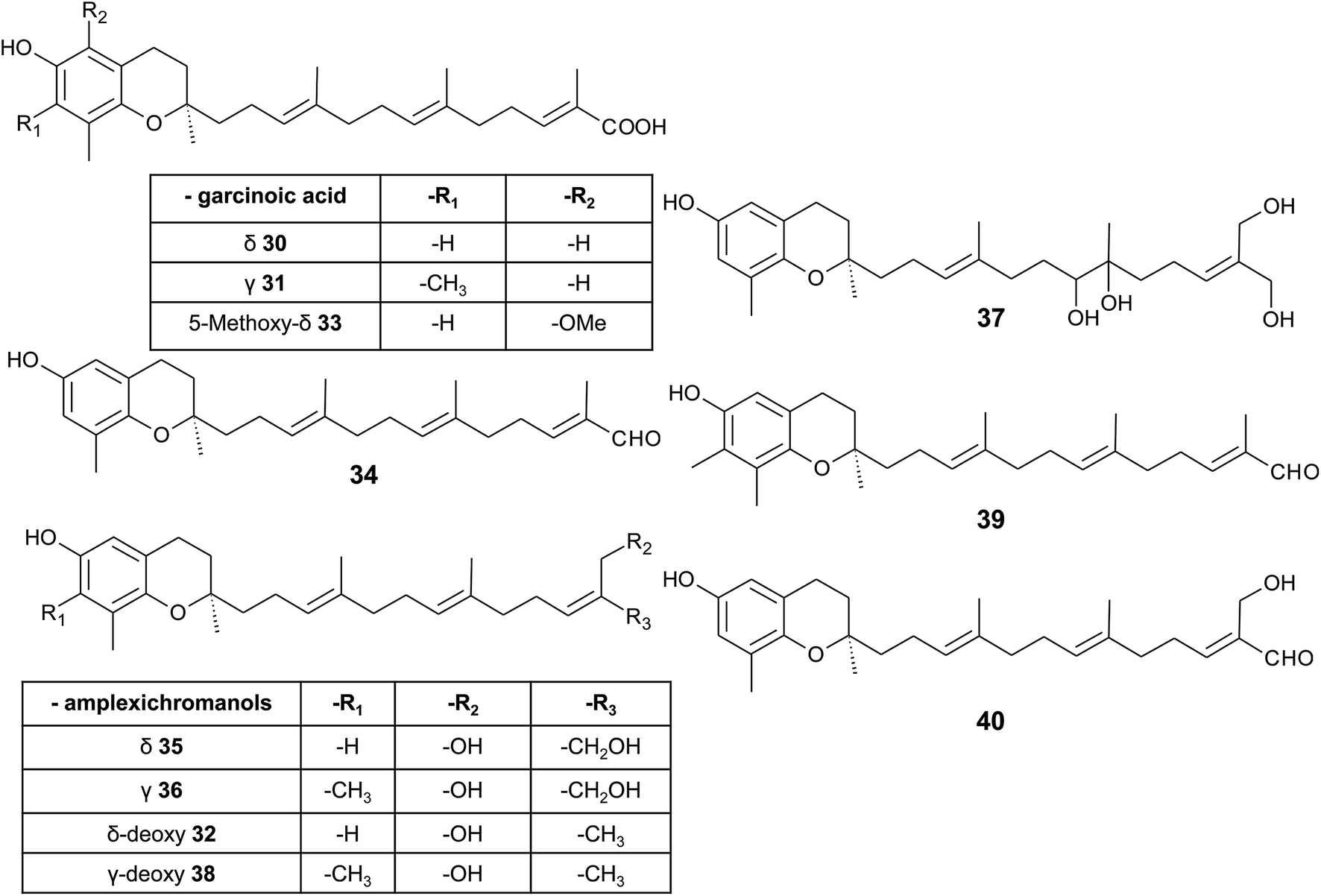 | ||
| Fig. 5 Structures and substitution patterns of garcinoic acids (30 to 33) and meroditerpenes (34 to -40) from Garcinia amplexicaulis. | ||
δ-Garcinoic acid exerts potent anti-inflammatory, anti-proliferatory and antibacterial properties (see corresponding sections). As a possible target for its anti-inflammatory action, microsomal prostaglandin E2 synthase has been identified recently.44 The two natural (δ- and γ-garcinoic acid) isoforms as well as semi-synthesized β- and α-garcinoic acid inhibited the enzyme with IC50 values of 6.7, 2.0, 2.8 and 7.8 μM, respectively.
δ-Garcinoic acid reduced the growth of C6 cells and RAW264.7 mouse macrophages with an EC50 of 10 μM and 5 μM, respectively.37,38 As demonstrated by Maloney and Hecht, δ-garcinoic acid inhibits DNA polymerase β with an IC50 of about 4 μM.47 Whether this inhibition is a useful approach to prevent growth of cancer cells needs to be elucidated.
As mentioned above, γ-garcinoic acid and δ-(E)-deoxy-amplexichromanol (32) (see below) were isolated together with a 5-methoxy-δ-garcinoic acid derivative (33) from Cedrus atlantica (Fig. 5).45 All compounds showed only moderate anti-bacterial activity against different bacterial strains (see Table 3).
As a by-product of the isolation of garcinoic acid, garcinal (34) (δ-(E)-garcinal), with a terminal aldehyde group, was found in the G. kola nut.41 So far, the bioactive properties of garcinal are unknown.
Another interesting group of side chain-modified compounds with large structural variability has been isolated from the bark of Garcinia amplexicaulis, an endemic shrub from New Caledonia. δ- and γ-amplexichromanol (35) and (36) are terminal-hydroxylated δ- and γ-tocotrienols, respectively, carrying two hydroxy-groups at carbon-13′ and -14′ (Fig. 5).43 Both compounds inhibited capillary formation of VEGF-induced human primary endothelial cells at 25 nM concentration. Interestingly, only δ-amplexichromanol decreased the adhesion of VEGF-induced human primary endothelial cells whereas γ-amplexichromanol had no significant effect, suggesting different modes of action. δ-Dihydroxy-amplexichromanol (37) results from dihydroxylation of the double bond between C-7′ and C-8′. Besides γ-(Z)- and γ-(E)-deoxy-amplexichromanol (38) as well as δ-(Z)- and δ-(E)-deoxy-amplexichromanol, two aldehydes, namely γ-(E)-deoxy-amplexichromanal (39) (which is identical to γ-(E)-garcinal) and δ-(E)-amplexichromanal (40) were isolated from Garcinia amplexicaulis.43,48 δ-(E)-Deoxy-amplexichromanol (32) has also been described in Cedrus atlantica.45 δ-(Z)-Deoxy-amplexichromanol was earlier described by Teixeira et al. in Clusia obdeltifolia.46 In addition, dimeric oxidation and condensation products of amplexichromanols have been characterized.43
From the methanolic extract of leaves of Litchi chinensis (Sapindaceae), several δ-tocotrienol derivatives with side chain and chromanol modifications were isolated and investigated for their anti-cancerogenic potential.49 Litchtocotrienols A–G (41–47) are hydroxylated at C-11′ with R-configuration and E-F (45, 46) contain a ketone group at C-11′ (Fig. 6). An additional methoxy-group is introduced at position C-5 of the chromane ring for litchocotrienols B, D, F and G, respectively. Position C-12′ is hydroxylated for A, B, G or methoxylated for C and D. Macrolitchtocotrienol A (48) derives from an intramolecular condensation between C-12′ and C-6 to form an ansa-chromane. The structural motive is similar to the smenochromene sesquiterpenes. Finally, cyclolitchtocotrienol A (49) with a cyclohexene ring within the side chain was isolated. The latter compound is a structural isomer of walsurol (50) with related biosynthesis (Fig. 7). Litchtocotrienols presumably derive from the precursor 11′-12′-epoxide that undergoes nucleophilic ring opening and further modifications. Litchtocotrienols A–G and macrolitchtocotrienol A showed moderate cytotoxicity in HepG2 liver cells and gastric epithelial cells (AGS), with IC50 values ranging from 10–50 μM (Table 2).
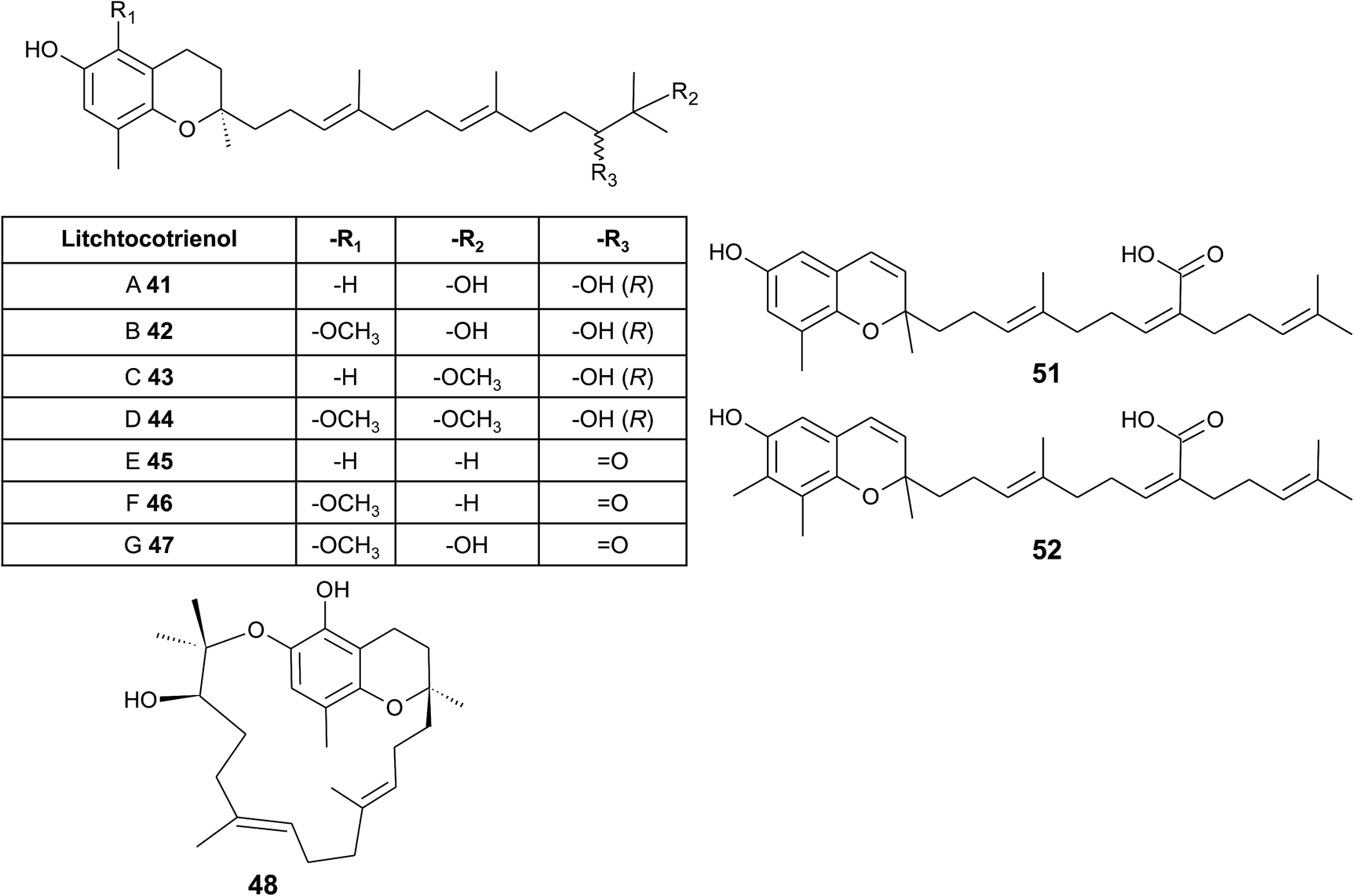 | ||
| Fig. 6 Structures and substitution patterns of litchtocotrienols (41 to 48) from Litchi chinensis, sargachromenols (51) and (52). | ||
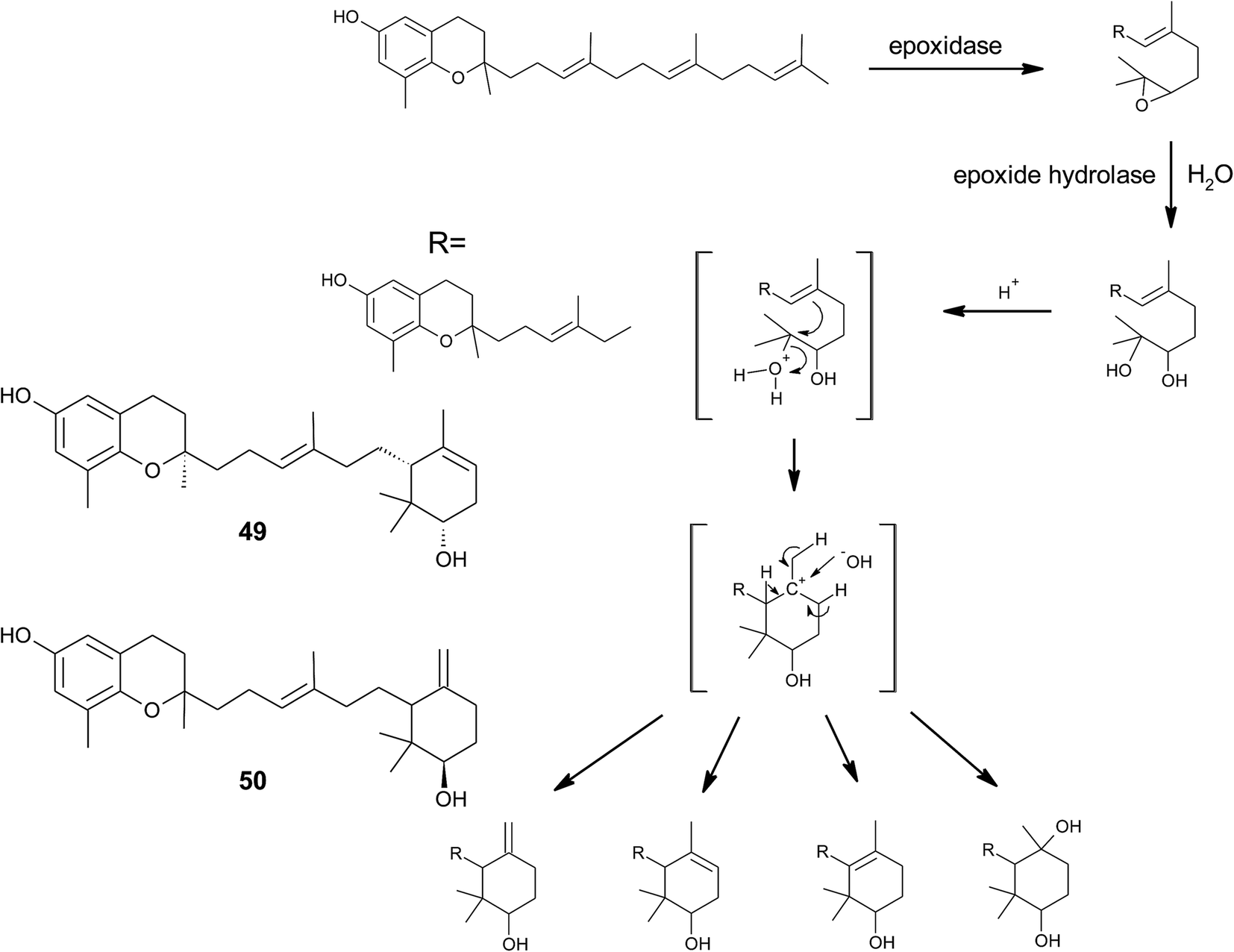 | ||
| Fig. 7 Scheme of an acid-catalyzed cyclization cascade including final cyclization of the chromane ring according to Etse et al.61 Examples are cyclolitchtocotrienol (49) and walsurol (50). | ||
All isolated compounds from Garcinia amplexicaulis and Litchi chinensis show high structural similarity to tocochromanols from Saragassum species (see section on Algae). In conclusion, Garcinia amplexicaulis and Litchi chinensis present the highest degree of structural variability among angiosperms.
Side chain-modified tocochromanols have been found in the fruits of the Amazonian Myristicaceae Iryanthera juruensis and Iryanthera grandis,50–52 and in vegetal parts of the Mexican Asteraceae Roldana barba-johannis.53 Iryanthera leaves were used by the indigenous population to treat infected wounds and cuts, and the latex of the bark was used against infections.52 δ-Sargachromenol (51) was found in all the above-mentioned plants and was obtained in 0.4% and 0.8% yield (dry mass) from Roldana and Iryanthera species, respectively. δ-Sargachromenol is a δ-dehydrotocotrienol derivative with a carboxyl group located at C-15′ of the side chain and is thus a structurally related form of δ-garcinoic acid (30) (Fig. 5). Sargachromenol was named after the brown algae Sargassum serratifolium, from which it was first isolated by Kusumi et al.54 For a detailed description of the biological properties, please see the section on Algae.
Besides δ-sargachromenol, 7-methyl-sargachromenol (52) (γ-sargachromenol) was isolated from the fruits of Iryanthera juruensis by Silva et al.50
To the best of our knowledge, besides cyclolitchtocotrienol A (49), walsurol (50) obtained from the bark of the Yunnan tree Walsura yunnanensis (Meliaceae) is the only meroditerpene in higher plants that forms a 6-membered ring structure within the side chain.55 Interestingly, walsurol was obtained as the main lipid constituent from powdered bark (0.08% yield). Here, the authors discussed a possible mechanism that leads to cyclization reactions in the side chain. Epoxidation of the terminal double bonds in isoprenoid structures are well described for squalene and also for tocotrienols.56,57 Nucleophilic ring-opening results in a 11′,12′-diol structure that has also been described for algae.58–60 Etse et al. proposed an acid-catalyzed rearrangement that leads to a variety of cyclic structures formed from the diol. Elimination of water and ring closure between carbon 7′ and 12′ forms endo- (e.g. (49).) and exo-double bonds (e.g. (50)), respectively (Fig. 7).61 The metabolic pathway described here also applies to the formation of chromarols (see section on sponges).
3.2 Fungi
Although mushrooms and fungi produce a large number and variety of meroterpenoids,62,63 our database search found only scarce information on long-chain or cyclic 6-hydroxy-chromanols or -chromenes. The occurrence of α-, β-, γ-, and δ-tocopherols has been summarized in a review by Ferreira et al.64 Interestingly, no tocotrienols have been found in fungi so far. Several meroterpenoid structures were described with a 5-hydroxy-chromene ring, which originated from orsellinic acid as the aromatic precursor.623.3 Marine organisms
Since 1960, more than 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 distinct chemical compounds were discovered from marine organisms.65 Of these, algae and sponges form two third of all natural marine products found from 1965 to 2007.66 Marine natural products (MNP) with isoprenoid structures account for almost 60% of all natural products found in marine organisms.67 Several excellent reviews have summarized meroterpene structures from marine fungi,68 invertebrates,69 and algae.67,70,71 Tocopherols are well known to be produced by algae as well as marine invertebrates and microorganisms.69,72 Most interestingly, δ-tocotrienol (17) is widely distributed (especially in algae and sponges) and appears as the lead structure of most of the diverse compounds described in this review. Among them, sargachromanols, sargachromenols, cystoseira metabolites, chromarols, epitaondiols, smenochromenes and strongylophorines constitute the largest and best studied groups. Anti-bacterial, anti-viral, anti-inflammatory and cytotoxic properties were attributed to these compounds, making them potential lead structures for drug development.73
000 distinct chemical compounds were discovered from marine organisms.65 Of these, algae and sponges form two third of all natural marine products found from 1965 to 2007.66 Marine natural products (MNP) with isoprenoid structures account for almost 60% of all natural products found in marine organisms.67 Several excellent reviews have summarized meroterpene structures from marine fungi,68 invertebrates,69 and algae.67,70,71 Tocopherols are well known to be produced by algae as well as marine invertebrates and microorganisms.69,72 Most interestingly, δ-tocotrienol (17) is widely distributed (especially in algae and sponges) and appears as the lead structure of most of the diverse compounds described in this review. Among them, sargachromanols, sargachromenols, cystoseira metabolites, chromarols, epitaondiols, smenochromenes and strongylophorines constitute the largest and best studied groups. Anti-bacterial, anti-viral, anti-inflammatory and cytotoxic properties were attributed to these compounds, making them potential lead structures for drug development.73
There is increasing interest in and knowledge about the isolation, and structural elucidation of meroditerpenes and their quinone precursors from brown algae. Recently, Culioli and colleagues described the analytical procedure for the extraction, chromatographic isolation and structural determination by sophisticated one- and two-dimensional nuclear magnetic resonance spectroscopic methods.74
As mentioned above, sargachromanols and sargachromenols show the highest structural diversity among all meroditerpenes. They derive from the common precursor geranylgeranyltoluquinol and subsequently from δ-tocotrienol and δ-dehydro-tocotrienol, respectively. δ-Tocotrienol-11′-12′-epoxide (53) was one of the first sargachromanols discovered in brown algae by Kato et al. in 1975.57 The activation of the terminal double bond leads to hydroxyl-, oxo-, and cyclic derivatives, respectively. However, the sequence of the chemical reactions leading to cyclic derivatives remains elusive (see also Fig. 7). Observational studies showed that an extract of Sargassum tortile induced the settling of swimming larvae of the hydrozoa Coryne uchidai, thus obviously acting as an intercellular signaling molecule.75 The epoxide was found by bioactivity-guided fractionation of the lipid extract.
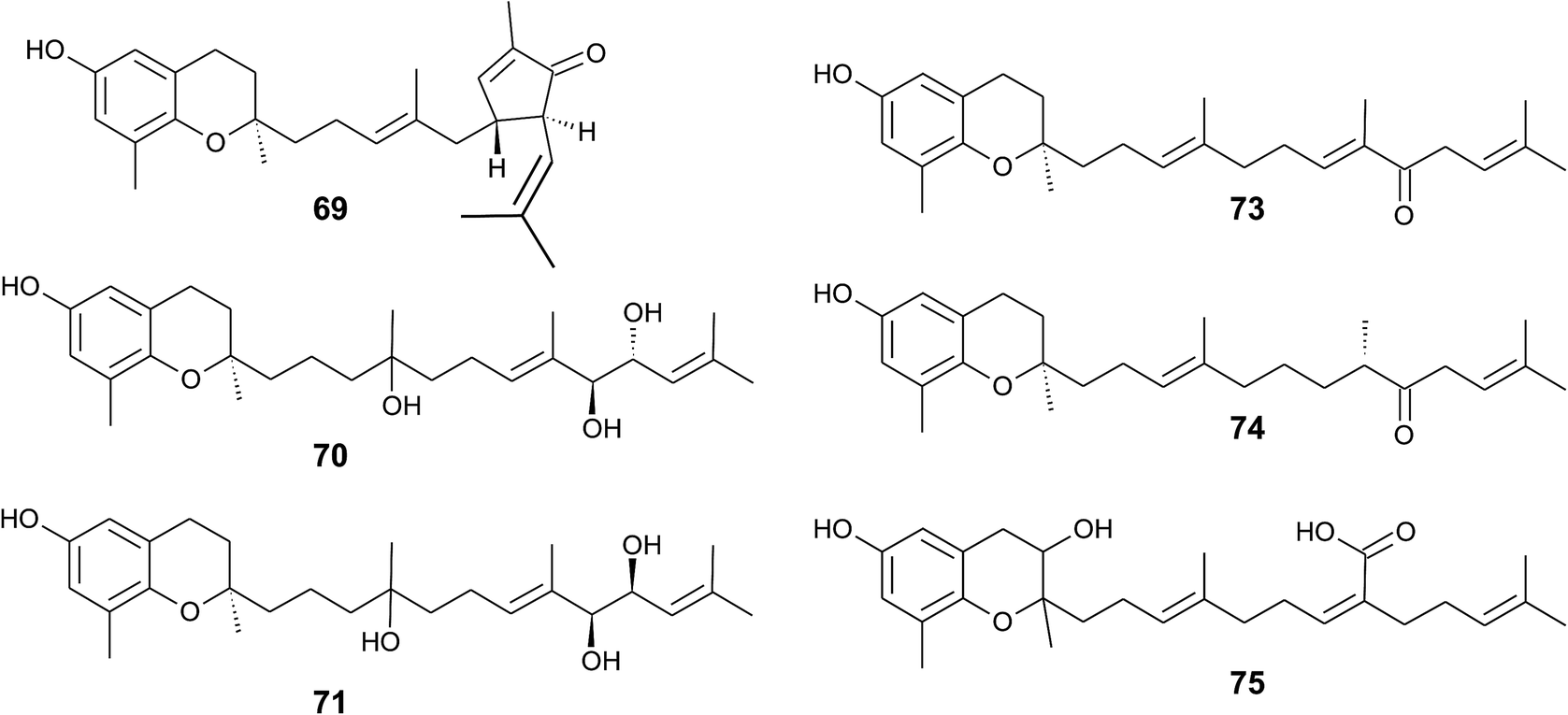
In 2005, Jang et al. isolated a series of sargachromanols (A to P) (54–69) from Sargassum siliquastrum and characterized them by extensive two-dimensional nuclear magnetic resonance experiments.76 Later, Lee et al. isolated the structures Q to S (70–72) from the same species.77 Since sargachromanols A, B and S are sesquiterpenes, they are described here in the corresponding section.
Sargachromanol C (56) contains a 9′-hydroxyl group with R-configuration in the δ-tocotrienol side chain. The two diols, sargachromanols D (57) and E (58) possess hydroxyl groups at C-9′ and C-10′ and are diastereomers of each other. Sargachromanol F (59) has a methoxy group at C-9′ and a hydroxyl group with R-configuration at C-10′. Sargachromanols G to J (60–63) share similar side chain modifications consisting of a C-9′ carbonyl and a C-10′ hydroxyl group. They differ in the numer and type of saturation of the double bonds between C-7′ and C-8′ (I, J) (62, 63) and between C-11′ and C-12′ (H, J) (61, 63), respectively, and a double bond shift from C-7′ to C-6′ (H) (61). Sargachromanol K (64) is an isomer of sargachromanol G, where the carbonyl and hydroxyl groups are shifted to C-10′ and C-9′, respectively. Two-dimensional nuclear magnetic resonance experiments revealed that sargachromanols L to P underwent carbon skeleton rearrangements of the terminal prenyl group. Thus, the C-8′–C-9′ bond is rearranged to C-8′–C-10′. Sargachromanol L (65) contains a hydroxyl group at C-9′, whereas sargachromanols M (66) and N (67) are structural cis/trans-isomers containing an aldehyde group at C-9′. In addition, the double bond from C-7′ migrated to C-8′–C-10′. Further oxidation of sargachromanol L leads to sargachromanol O (68), which bears a carboxyl group at C-9′ und is thus a structural isomer of δ-garcinoic acid (30) (Fig. 8). The highest isolation yield was obtained for sargachromanols G (60) and I (62) (0.062 and 0.04%, respectively).76
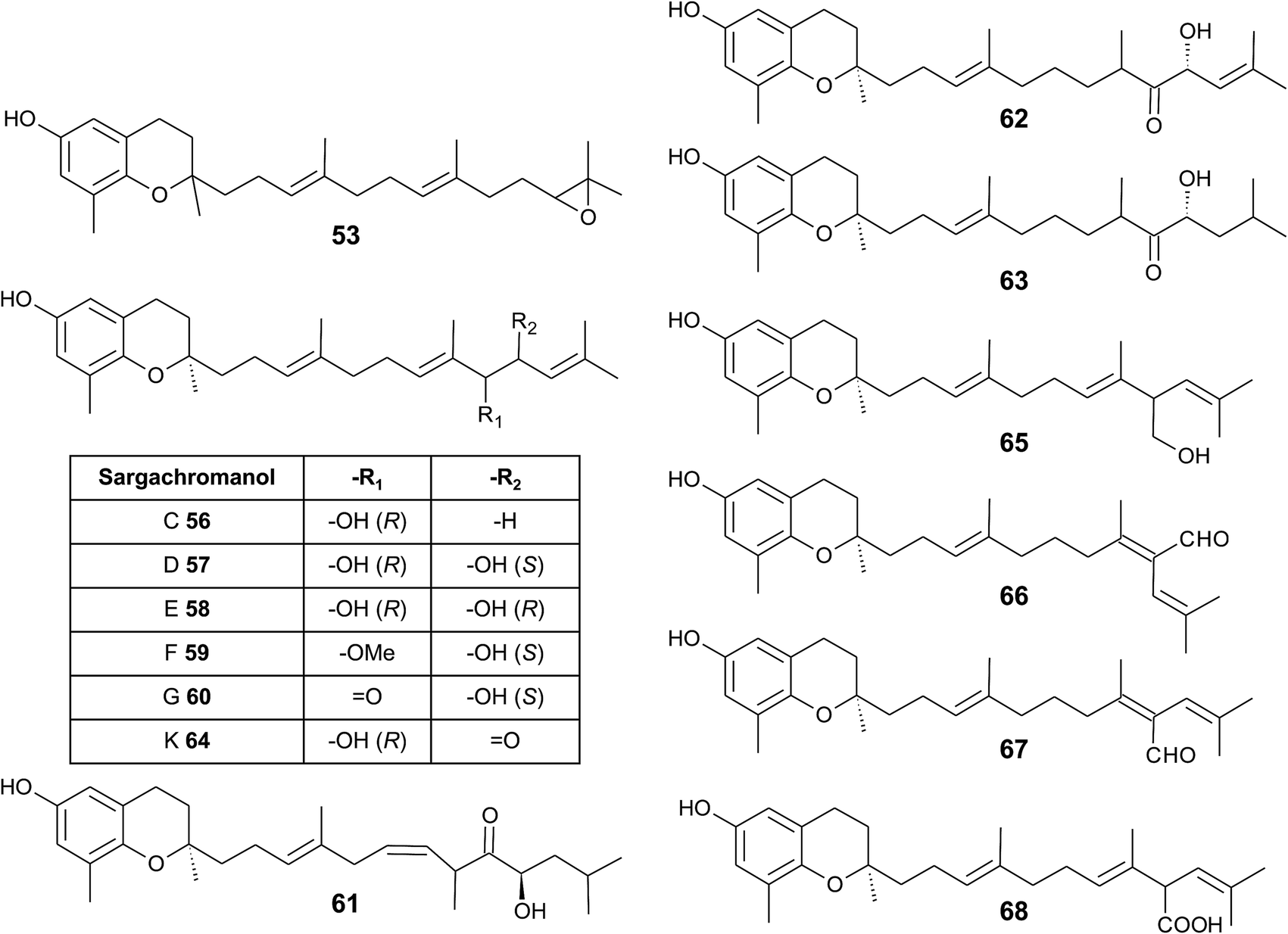 | ||
| Fig. 8 Structures and substitution patterns of sargachromanols (56 to 68) from Sargassum species. | ||
The formation of an α,β-unsaturated cyclopentenone within the side chain leads to sargachromanol P (69). Sargachromanols Q (70) and R (71) share high structural similarity to sargachromanols D and E, respectively, but bear an additional tert-hydroxyl group at the saturated C-4′ (Fig. 9).77
 | ||
| Fig. 9 Structures and substitution patterns of sargachromanols (69 to 75) from Sargassum species. | ||
Sargachromanols D, F, H and L are strong Na+/K+-ATPase ion pump inhibitors, with IC50 values of 3.6, 6.0, 4.6 and 7.0 μM, respectively.78 The study revealed that the hydroxyl groups at C-9′ and/or C-10′ are important for this inhibitory activity. Bioassay-guided fractionation of Sargassum siliquastrum extracts revealed anti-inflammatory action of sargachromanol D.79 The compound reduced lipopolysaccharide (LPS)-induced production of nitric oxide and prostaglandin (PG) E2 in murine RAW 264.7 macrophages and inhibited the expression of the pro-inflammatory enzymes inducible nitric oxide synthetase (iNOS) and COX-2. In addition, the production of the pro-inflammatory cytokines TNF-α, interleukin-1β (IL)-1β and IL-6 was reduced by sargachromanol D.79 Recently, sargachromanol D was suggested as an anti-hypertensive agent, since it showed dual antagonistic activity towards an L-type Ca2+-channel and endothelin A/B2 receptor (Table 3).80 The use of sargachromanols is protected by several patents.81
Sargassum siliquastrum was also used as a natural source of sargachromanol E and G for bioactivity studies.82–87 Both compounds inhibited the expression of pro-inflammatory cytokines in LPS-stimulated murine RAW 264.7 macrophages.83,84,86 In addition, sargachromanol E induced apoptosis via caspase-3 activation in promyelocytic HL-60 leukemia cells82 and inhibited ultraviolet A-induced ageing of human dermal fibroblasts.88 Sargachromanol G showed anti-osteoclastogenic effects on the expression of IL-1β-induced osteoclastogenic factors in the human osteoblast cell line MG-63 and suppressed the activation of nuclear factor κB (NF-κB) and mitogen-activated protein kinase (MAPK) in receptor activator of NF-κB ligand (RANKL)-induced RAW264.7 cells.85,86
Besides sargachromanol I and K, another two sargachromanols, (2R)-9′-oxo-δ-tocotrienol (73) and (2R)-7′-8′-dihydro-9′-oxo-δ-tocotrienol (74) were isolated from Sargassum micracanthum, however, in very low yield (Fig. 9).89
Seo et al. isolated a racemic mixture of thunbergol A (75) from Sargassum thunbergii. The compound features a 3-hydroxyhydrobenzopyran structure with a 15′-carboxy group and thus presumably derives from sargachromenol (Fig. 9).90
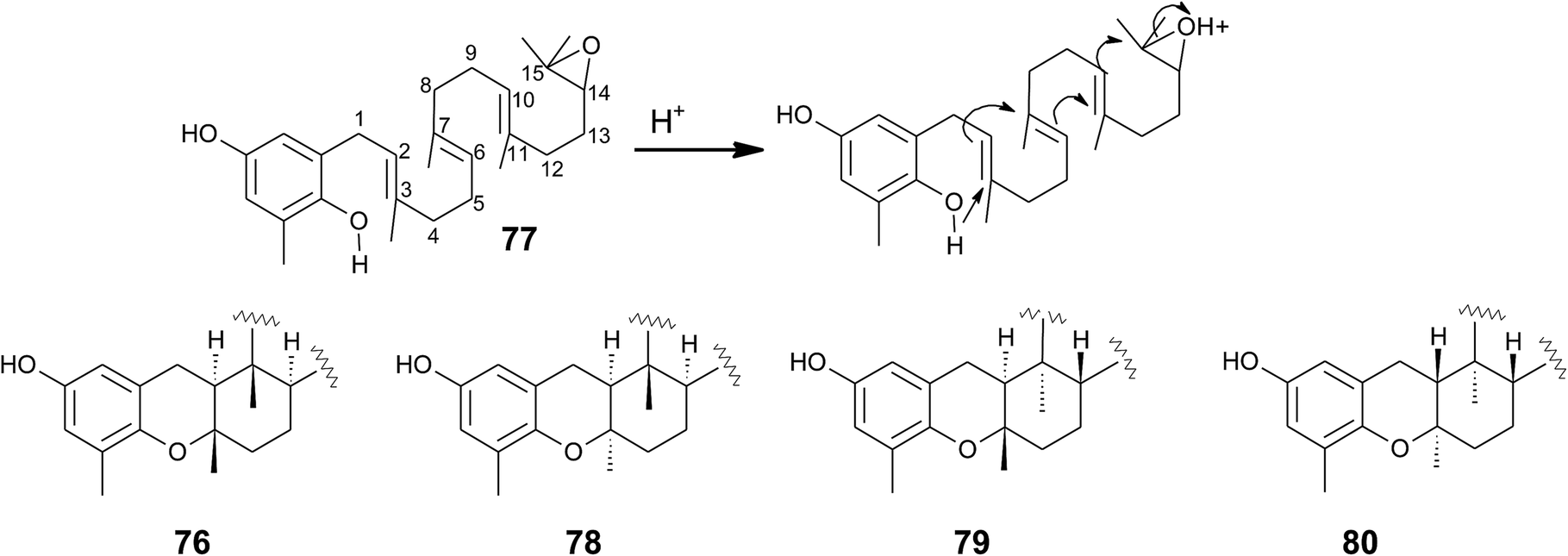
Cyclic sargachromanols are widely distributed in brown algae. Taondiol (76) (Fig. 10) was the first cyclic side chain-derivative of tocotrienol that was isolated in 0.05% yield from Taonia atomaria (order Dictyotales).91 The authors proposed an enzyme-initiated synchronous cyclization cascade of the prenylated 1,4-hydroquinone leading to the tetracyclic ring system. We and others propose an alternative cyclization mechanism starting from 1,4-hydroquinone-14-15-epoxide (77), analogous to lanosterol synthesis92,93 (Fig. 10). The protonation of 77 via an epoxide-hydrolase enzyme would increase the susceptibility of intramolecular attacks of the C-2–C-3 and C-6–C-7 double bonds. The stereochemistry of the possible isomers of taondiol at C-2 and C-3 and C-6 and C-7 has been a matter of debate. Recently, Areche et al. assigned the stereochemistry of isoepitaondiol (78) isolated from Stypopodium flabelliforme to the formerly described isotaondiol.94 By now, the structures of taondiol, isoepitaondiol, epitaondiol (79) and 2β,3α-epitaondiol (80) (Fig. 10) have been unambiguously assigned.94–96
 | ||
| Fig. 10 Proposed enzyme-catalyzed cyclization leading to cyclic sargachromanols (76 to 80). | ||
Epitaondiol (79) was isolated from Stypopodium zonale and Stypopodium flabelliforme (both species are members of the order Dictyotales) and its bioactivity was intensively studied.93,95–102 The polycyclic compound shows ichthyotoxic, anti-herpes and anti-human metapneumovirus (HMPV) activity and acts as an anti-inflammatory agent in vitro and in vivo (see Tables 1–3).98,99,102 Further, epitaondiol inhibited cell proliferation of human colon adenocarcinoma (Caco-2), human neuroblastoma (SH-SY5Y), rat basophilic leukemia (RBL-2H3) cells, and murine macrophages (RAW.267), but not of non-cancer Chinese hamster fibroblasts (V79) (Table 2).100 2β,3α-Epitaondiol (80) exhibited moderate neurotoxicity towards mouse neuro-2a cells with LC50 values of 2 μM.92 Epitaondiol was effective in the prevention of HCl/ethanol-induced gastric lesions in mice at an ED50 value of 40 mg kg−1 bodyweight.101,103 Anti-insecticide activity was found against Spodoptera frugiperda.96 Finally, the compound induced the settlement of the mussel Perna perna.98
| Compounds | Test system | Effective concentrationsb | References |
|---|---|---|---|
| a Abbreviations: thromboxane B2 (TXB2), leukotriene B4 (LTB4), cyclooxygenase (COX)-1 and -2, 3α-hydroxysteroid dehydrogenase (3α-HSD), lipoxygenase (LOX), xanthine oxidase (XO), horseradish peroxidase (HRP).b Estimated from original publication. | |||
| Meroditerpenes | |||
| α-Tocopherol (3) | IL-1β-stimulated A549 cells | 8% nitric oxide inhibition at 10 μM | 279 |
| 25% iNOS inhibition at 10 μM | |||
| IC50 PGE2 inhibition: > 50 μM (A549 cells) | |||
| γ-Tocopherol (5) | IL-1β-stimulated A549 cells | IC50 PGE2 inhibition: 7.5 μM (RAW264.7) | 279 |
| IC50 5-LOX inhibition: > 50 μM | 245,257 | ||
| IC50 COX-2 (A549 cells) inhibition: > 50 μM | |||
| δ-Tocopherol (6) | IL-1β-stimulated A549 cells | IC50 COX-2 (A549 cells)) inhibition: > 50 μM | 257 |
| IC50 5-LOX inhibition: > 50 μM | |||
| IC50 PGE2 inhibition: 3 μM (A549 cells) | |||
| α-Tocotrienol (14) | LPS-induced RAW 264.7 cells | 5% NO inhibition at 33 μM | 280 |
| δ-Tocotrienol (17) | 31% NO inhibition at 26 μM | ||
| γ-Tocotrienol (16) | 19% NO inhibition at 30 μM | ||
| IL-1β-stimulated A549 cells | IC50 PGE2 inhibition: 1 μM (A549 cells) | 245 | |
| 13′-carboxy-α-tocopherol (205) (α-13′-COOH) | LPS-induced RAW 264.7 cells | Total inhibition at 2.7 μM | 255 |
| IC50 NO production: 0.2–0.5 μMb | |||
| LPS-induced RAW 264.7 cells | 88% NO inhibition at 5 μM | 256 | |
| 100% iNOS inhibition at 5 μM | |||
| 13′-carboxy-δ-tocopherol (229) (δ-13′-COOH) | Inhibition of COX-1 and COX-2 | IC50 COX-1 (bovine) inhibition: 5.0 μM | 245 |
| IC50 COX-2 (human) inhibition: 4.0 μM | 245 | ||
| IC50 COX-2 (A549 cells)) inhibition: 4.0 μM | 257 | ||
| Inhibition of human recombinant 5-LOX | IC50 5-LOX inhibition: 0.5–1.0 μM | 253 | |
| IC50 (LTB4) generation: 4–7 μM | |||
| Neutrophils and promyelocytic HL-60 | |||
| Leukemia cells generated LTB4 | 79% NO inhibition at 5 μM | ||
| LPS-induced RAW 264.7 cells | 56% iNOS inhibition at 5 μM | 256 | |
| 13′-Hydroxy-α-tocopherol (204) (α-13′-OH) | LPS-induced RAW 264.7 cells | 49% COX-2 inhibition at 10 μM | 241,256 |
| 53–60% iNOS inhibition at 10 μM | |||
| 54% PGE2 inhibition at 10 μM | |||
| 44–69% NO inhibition at 10 μM | |||
| 13′-Hydroxy-δ-tocopherol (231) (δ-13′-OH) | LPS-induced RAW 264.7 cells | 49% NO inhibition at 10 μM | 256 |
| 53% iNOS inhibition at 10 μM | |||
| δ-Garcinoic acid (30) | Inhibition of COX-2 and 5-LOX | IC50 COX-2 inhibition: 9.8 μM | 257 |
| IC50 5-LOX inhibition: 1.0 μM | |||
| Inhibition of LPS-stimulated NO production in RAW 264.7 macrophages | IC50 NO production: 1.0 μMb | 260 | |
| α-, β-,< γ-,< δ-Garcinoic acid (209, 232, 231, 230) | Inhibition of PGE2-synthase (PGES-1) | IC50 7.8 (α-), 2.8 (β-), 2.0 (γ-), 6.7 (δ-) garcinoic acid | 44 |
| δ-Sargachromenol (51) | TPA-induced mouse ear edema | IC50 edema reduction: 0.36 mg per ear | 53 |
| Inhibition of COX-1 and -2 | 98% COX-1 inhibition at 100 ppm | 52 | |
| 84% COX-2 inhibition at 100 ppm | |||
| LPS-induced RAW 264.7 and BV-2 cells | IC50 NO production: 82 μM (RAW264.7) | 129 | |
| IC50 PGE2 inhibition: 30.2 μM (RAW264.7) | |||
| IC50 NO production: 1.3–2.7 μM (BV-2) | 134,261 | ||
| Sargachromanol D (57) | LPS-induced RAW 264.7 cells | IC50 NO production: 40 μMb (RAW264.7) | 79 |
| IC50 PGE2 inhibition: 15 μMb (RAW264.7) | |||
| Sargachromanol E (58) | LPS-induced RAW 264.7 cells | IC50 NO production: 16.3 μM | 83 |
| Sargachromanol G (60) | LPS-induced RAW264.7 cells | IC50 NO production: Ca. 15 μMb | 84 |
| Chromarols (A–D) (113–116) cyclic | Inhibitors of 12- and 15-LOX | 15-hLO IC50: 0.6(A), 4.0(B), 0.7(C), 1.1 μM(D) | 154 |
| 12-hLO IC50: all >100 μM | |||
| Epitaondiol (79) cyclic | TPA-induced mouse ear edema | IC50 edema reduction: 20.7 μg per ear | 99 |
| IC50 myeloperoxidase activity: 17.8 μg per ear | |||
| Eicosanoid inhibition | IC50 (TXB2) generation: 3.8 μM | 99 | |
| IC50 (LTB4) generation: 30.1 μM | |||
|
|||
| Merosesquiterpenes | |||
| Capillobenzopyranol (172) cyclic | LPS-induced RAW 264.7 cells | 36.7% NO inhibition at 10 μM | 202 |
| 9′-Carboxy-δ-tocopherol (206) (δ-9′-COOH) | Inhibition of COX-1, -2 | IC50 COX-1 (bovine) inhibition: >20 μM | 245 |
| IC50 COX-2 (human) inhibition: >20 μM | |||
| IC50 COX-2 (A549 cells)) inhibition: 6.0 μM | |||
|
|||
| Monoterpenes | |||
| Cordiachromene A (173) | Inhibition of PGI2 biosynthesis | IC50 8.2 μM | 216 |
| Carrageenan induced rat paw endema | IC50 18.9 μM | 211 | |
| Inhibitor of 15-LOX | IC50: 0.82 μM | 214 | |
| IC50: 2 μM | |||
| α-CMBHC (207) | IC50 COX-1 (bovine) inhibition: 160 μM | 245 | |
| IC50 COX-2 (human) inhibition: 140 μM | |||
| α-CEHC (208) | TNFα-stimulated NO and PGE2 production in RAEC cells | IC50 PGE2 inhibition: 59 μM | 244 |
| IC50 NO production: 56 μM | |||
| γ-CEHC (233) | IC50 COX-1 (bovine) inhibition: 300 μM | 245 | |
| IC50 COX-2 (human) inhibition: 450 μM | |||
| IC50 COX-2 (A549 cells)) inhibition: 35–70 μM | |||
| IC50 PGE2 inhibition: 30.0 μM (RAW 264.7) | 279 | ||
|
|||
| Hemiterpenes | |||
| Quercinol (199) | In vitro cytokine inhibition | IC50 COX-1 inhibition: 4.7 μM | 235 |
| IC50 COX-2 inhibition: 0.63 μM | |||
| IC50 3α-HSD inhibition: 114 μM | |||
| IC50 XO inhibition: 21 μM | |||
| IC50 HRP: 68 μM | |||
| Compound | Isolated from | Cell line/organism | Effective concentrationc | Reference |
|---|---|---|---|---|
| a Abbreviation: human gastric adenocarcinoma cells (AGS), human microvascular endothelial cells (HMEC), hepatocellular carcinoma cells (SMMC-7721), human lung epithelial cells (A549), human hepatocellular carcinoma cells (HepG2), B16 melanoma cells, vascular endothelial growth factor (VEGF), human umbilical vein endothelial cells (HUVEC).b Estimated from original publication.c For better comparison μg ml−1 were converted to μM. | ||||
| Meroditerpenes | ||||
| α-Tocopherol (3) | Plant oils | HepG2 cells, | >100 μM | 39 |
| MDA-MB-231, MCF7 cells | Not achieved | 281 | ||
| γ-Tocopherol (5) | Plant oils | Jurkat, HBTII, MCF7, MCF7-C3 cells | >50 μM | 267 |
| δ-Tocopherol (6) | Plant oils | Jurkat, HBTII, MCF7, MCF7-C3 cells | >50 μM | 267 |
| α-Tocotrienol (14) | Palm oil | MDA-MB-435, MCF7, B16 cells | IC50: 210 μM, 14 μM, 110 μM | 25,273 |
| MDA-MB-231, MCF7 cells | IC50: 24 μM, 26 μM | 281 | ||
| γ-Tocotrienol (16) | Jurkat, HBTII, MCF7, MCF7-C3 cells | ∼50%, 35%, 30%, 35% at 50 μM | 267 | |
| MDA-MB-231, MCF7 cells | IC50: 11 μM, 15.6 μM | 281 | ||
| SKBR3 cells, BT474 cells | IC50: 4.1 μM, 4.4 μM | 275 | ||
| δ-Tocotrienol (17) | MCF7, B16 cells | IC50: 15 μM, 10 μM | 25,276 | |
| MDA-MB-231, MCF7 cells | IC50: 17 μM, 17 μM | 281 | ||
| Sargaol (95) | Stypopodium flabelliforme | Human epithelial gastric cells | IC50: 18 μM | 103 |
| Human fibroblasts | IC50: 12 μM | |||
| Sargassum tortile | P338 leukemia cells | EC50: 52 μM | 120 | |
| Desmethyltocotrienol (18) (P21-tocotrienol) | Rice bran | B16 cells (suppression of proliferation) | IC50 > 1 μMb | 24 |
| Didesmethyltocotrienol (19) (P25-tocotrienol) | IC50: 0.9 μM | 25 | ||
| 13′-Carboxy-δ-tocopherol (229) | Semisynthetic from Garcinia kola, human metabolites | Glioma C6 cells | n.d. | 38 |
| HepG2 cells | EC50: 6.5 μM (δ-13-COOH) | 39 | ||
| THP-1 macrophages | EC50: 11.1 μM (δ-13-COOH) | 277 | ||
| HCT-116 cells | EC50: 8.9 μM (δ-13-COOH) | 257 | ||
| HT-29 | EC50: 8.9 μM (δ-13-COOH) | |||
| 13′-Hydroxy-α-tocopherol (204) | HepG2 cells | EC50 > 100 μM | 39 | |
| THP-1 macrophages | EC50 > 100 μM | 277 | ||
| 13′-Carboxy-α-tocopherol (205) | HepG2 cells | EC50: 13.5 μM (α-13-COOH) | 39 | |
| THP-1 macrophages | EC50: 7.4 μM (α-13-COOH) | 277 | ||
| δ-Garcinoic acid (30) | Garcinia kola | Glioma C6 cells | EC50: 10 μM | 38 |
| RAW264.7 macrophages | EC50: 5.5 μM | 255 | ||
| HCT-116 cells | EC50: 16 μM (δ-garcinoic acid) | 257 | ||
| HT-29 | EC50: 17 μM (δ-garcinoic acid) | |||
| Inhibition of DNA polymerase β | IC50: 4 μM | 47 | ||
| δ-Sargachromenol (51) | Sargassum sagamiamum | Caspase-3 induced apoptosis in HaCaT cells | EC60: 11.8 μMb | 126 |
| Fallachromenoic acid (105) | Sargassum fallax | P338 leukemia cells | IC50 > 27–29 μM | 133 |
| δ-Amplexichromanol (35) | Garcinia amplexicaulis | Antiangiogenicity in VEGF-induced HUVECs | Effective at 25 nM and 2.5 μM | 43 |
| γ-Amplexichromanol (36) | ||||
| Litchtocotrienol A-G (41–47) | Litchi chinensis | HepG2 cells | IC50: 11.1 (A), 14.2 (B), 22.7 (C), 10.7 (E), 12.3 (F), 34.1 (G) μM | 49 |
| AGS cells | IC50: 10.9 (A), 32 (B), 24.2 (C), 26.8 (D), 27.4 (E), 49.2 (F) 43.2 (G) μM | |||
| Crassumtocopherol A (134) | Lobophytum crissum | P338 leukemia cells | IC50: 6.7 μM | 169 |
| IC50: 5.2 μM | ||||
| Crassumtocopherol B (135) | Cytotoxicity in HT-29 cells | IC50: 7.5 μM | ||
| Sargachromanol E (58) | Sargassum siliquastrum | Caspase-3 induced apoptosis in promyelocytic HL-60 leukemia cells | EC50: 20 μMb | 82 |
| Sargatriol (98) | Sargassum tortile | P-338 leukemia cells | EC50: 42 μM | 118 |
| Sargadiol-I (96) | EC50: 34 μM | 120 | ||
| Sargadiol-II (97) | EC50: 41 μM | |||
| Sargadiol I (96) | Desmaretia menziesii | Artemia salina | EC50: 233 μM | 123 |
| Epitaondiol (79) cyclic | Stypopodium flabelliforme | Human epithelial gastric cells | IC50: 29 μM | 103 |
| Human fibroblasts | IC50: 19 μM | |||
| RAW 264.7 | IC50: 12.7 μM | 100 | ||
| Isoepitaondiol (78) cyclic | Human epithelial gastric cells | IC50: 42 μM | 103 | |
| Human fibroblasts | IC50: 65 μM | |||
| Neuro-2a cell line | LC50: 2 μM | 92 | ||
| NCI-H460 | LC50: 24 μM | |||
| Macrolitchtocotrienol A (48) cyclic | Litchi chinensis | HepG2 cells | IC50: 16.5 μM | 49 |
| Cystoseirol A (86) cyclic | Cystoseira mediterranea | Crown-gall potato bioassay | 73% tumor inhibition at 10−2 M | 110 |
| Demethoxy cystoketal chromane (89) cyclic | Cystoseira amentacea | Cytotoxicity in HepG2 cells | IC50: 35 μM | 113 |
| Cystoseira tamariscifolia | ||||
| Strongylophorine 2 (117) | Petrosia corticata | Cytotoxicity in HeLa cells | IC50: >100 μM | 163 |
| Strongylophorine 3 (118) | IC50: 45.2 μM | |||
| Strongylophorine 4 (119) | IC50: 50.5 μM | |||
| Strongylophorine 22 (128) | IC50: 26.6 μM | |||
| Strongylophorine 23 (129) | IC50: 62.0 μM | |||
| Strongylophorine 24 (130) | IC50: >100 μM | |||
| All cyclic | ||||
|
||||
| Merosesquiterpenes | ||||
| Riccardiphenol C (146) cyclic | Riccardia crassa | P338 leukemia cells | IC50 > 80 μM | 177 |
| Aureol (155) cyclic | Smenospongia aurea | A549, | IC50: 13.6 μM | 189 |
| HT-29, | IC50: 14.9 μM | |||
| EL-4 | IC50: 31.5 μM | |||
| Panicein A2 (160) cyclic | Reniera mucosa | P388, A549, MEL20, HT29 | EC50: 14.8 μM | 195 |
| Panicein F2 (161) cyclic | EC50: 14.2 μM | |||
|
||||
| Monoterpenes | ||||
| Cordiachromene A (173) | Aplidium antillense | Human carcinoma KB | CI50: 14.3 μM | 212 |
| Murine leukemic P388 | CI50: 20.5 μM | |||
| Aplidium aff. densum | Lymphoblastic leukemia CEM-WT cells | IC50: 30 μM | 213 | |
| Didehydroconicol (185) | Aplidium aff. densum | Lymphoblastic leukemia CEM-WT cells | IC50: > 10 mM | 213 |
| Epiconicol (184) | IC50: 60 mM | |||
| Didehydroconicol (185) | Aplidium aff. densum | Sea urchin eggs of P. lividus and S. granularis | IC50: >25 μM and 9.8 μM | 218 |
| Epiconicol (184) | IC50: >11.3 μM and >25 μM | |||
| Chaetopyranin (188) | Chaetomium globosum | HMEC, SMMC-7721, A549 | IC50: 49 μM, 90 μM, 124 μM | 220 |
| α-CEHC (208) | Human metabolites | Growth inhibition at 50 μM | 282 | |
| γ-CEHC (233) | Prostate cancer PC-3 | 42% (α-), 83% (γ-) | ||
| HTB-82 | 34% (α-), 58% (γ-) | |||
| HECV | 9% (α-), 19% (γ-) | |||
| Sargasal-I (176) | Sargassum tortile | P-338 leukemia cells | EC50: 20.3 μM | 118 |
| Sargasal-II (177) | EC50: 21 μM | 120 | ||
|
||||
| Hemiterpenes | ||||
| Mollugin (189) | Rubica cordifolia | MCF-7/adriamycin | IC50 of doxorubicin decreased from 60 to 7.5 μg ml−1 at 10 μM mollugin | 226 |
A series of cyclic meroditerpenes was isolated from different Cystoseira species collected along the Mediterranean and contiguous Atlantic coasts.104 According to AlgaeBase,105 more than 289 species (and infraspecific) names were found, of which 42 have been marked as currently accepted taxonomically.
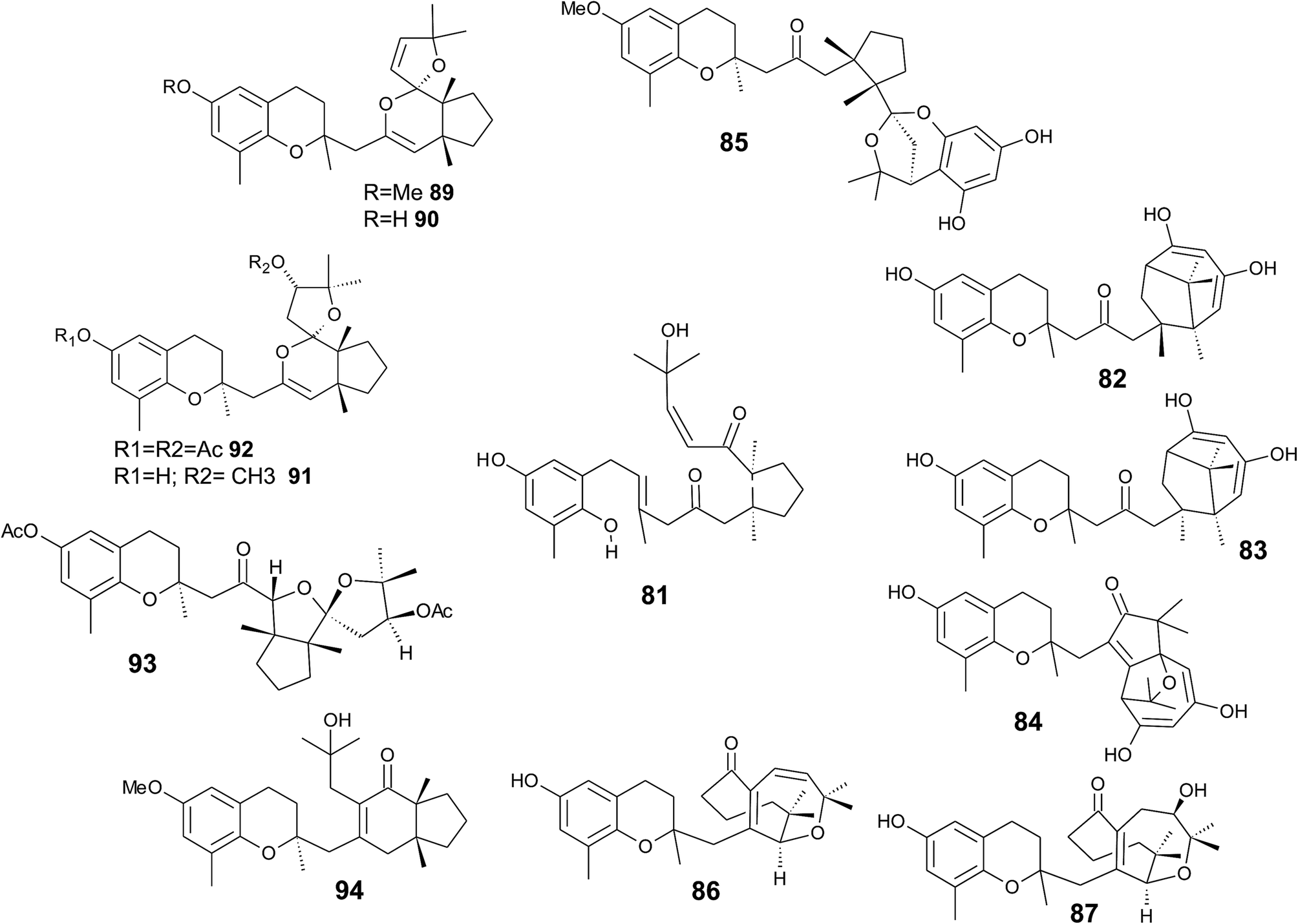
It was suggested that the following cyclic diterpenes origin from a common biosynthetic precursor, namely bifurcarenone (81) (Fig. 11). Among them, mediterraneols C (82), D (83), and E (84) have been isolated as their trimethoxy-derivatives from Cystoseira mediterranea in high yield (0.11, 0.14 and 2.0% from dry weight algae, respectively).106,107 Mediterraneols C and D are stereoisomers at C-4′ and compromise a bridged cyclooctane structure with two dienol moieties. Mediterraneol E (84) is a tricyclic oxygen-bridged diterpene with antineoplastic activity.107 So far, the biosynthesis of mediterraneols is largely unknown.106,108 Mediterraneols have been found to inhibit the mobility of sea urchin sperm and the mitotic cell division (ED50 values of 2 μg ml−1) of fertilized urchin eggs.106
 | ||
| Fig. 11 Structures of mediterraneols (82 to 84), cystophloroketal E (85), cystoseirols (86 to 88), chromanes (90 to 92), (94) and cystoseirone (93) from Cystoseira species. Bifurcarenone (81) as the common biosynthetic precursor is depicted in the center of the figure. | ||
Recently, cystophloroketal E (85), a meroditerpene with a 2,7-dioxabicylo[3.2.1]octane core was isolated from Cystoseira tamariscifolia.108 The authors assumed that ketal formation was preceded by a Michael addition of phloroglucinol onto the unsaturated carbonyl of 4-methoxy-bifurcarenone. The compound showed anti-bacterial, anti-microalgal and anti-invertebrate activity (Table 3).
| Compound | Activity | Species | Effective concentration | reference |
|---|---|---|---|---|
| a Abbreviations: herpes simplex virus 1 and 2 (HSV-1 and -2), minimum concentration that inhibits (MIC) bacterial growth, 2-aminoanthracene (2AN), ethyl methanesulfonate (EMS).b Estimated from original publication. | ||||
| Meroditerpenes | ||||
| γ-Dehydrotocopherol (12) | Proliferation, wound healing | 21 | ||
| Desmethyltocotrienol (18) (P21-tocotrienol) | Hypocholesterolemic activity | Chicken | Reduction of total cholesterol (mmol L−1) vs. control diet: 26% and 31% | 24 |
| Didesmethyltocotrienol (19) (P25-tocotrienol) | Reduction of LDL cholesterol (mmol L−1) vs. control diet: 41% and 48% | |||
| δ-Garcinoic acid (30) | Anti-bacterial | Staphylococcus aureus, Bacillus cereus, Pseudomonas aeruginosa | 1 mm zone of inhibition | 42 |
| Sargaol (95) | Gastroprotective against HCl/ethanol-induced gastric lessions | Mice | 30 mg kg−1 | 103 |
| Taondiol (76) | ||||
| Sargachromanol D, F, H, L (57, 59, 61, 65) | Ion pump inhibitor Na+,K+-ATPase | In vitro | IC50: 3.6 μM, 6.0 μM, 4.6 μM, 7.0 μM | 78 |
| Epitaondiol (79) | Anti-viral | HSV-1 | EC50: 1.34 M | 98 |
| Strongylophorine 3 (118) | Insecticidal activity | Spodoptera littoralis | EC50: 60 ppm | 157 |
| Bifurcarenone chromane (94) | Photoprotection | Human fibroblasts | IC50: 5–20 μg ml−1 | 116 |
| Bifurcarenone chromene (107) | Anti-macroalgal | Sargassum muticum | IC50: 2.5 μg ml−1 | 143 |
| Sargadiol-I (96) | Anthelmintic | Nippostrongylus brasiliensis | EC50: 307 μg ml−1 | 123 |
| 11′,12′-dihydroxy-3,4-dehydro-δ-tocotrienol (102) | Anti-viral | Human cytomegalovirus | IC50 virus absorption: 0.2 μM | 60 |
| IC50 antiviral activity: 0.49 μM | 58 | |||
| HSV-1 | 2.8 μM | |||
| HSV-2 | 2.6 μM | |||
| Mumps virus | 7.6 μM | |||
| Measles virus | 2.7 μM | |||
| Adeno virus | 14 μM | |||
| Influenza virus | 4.4 μM | |||
| Poliovirus | 9.0 μM | |||
| Coxsackievirus | 6.8 μM | |||
| 11′,12′-dihydroxy-3,4-dehydro-δ-tocotrienol (102) | Inhibition of bone resorption | Osteoclast-like cells (OCLs) | IC50: ∼8 μMb | 59 |
| Sarcochromenol sulfate A (110) | Ion pump inhibitor Na+,K+-ATPase | IC50: 1.6 μM | 152 | |
|
||||
| Merosesquiterpenes | ||||
| Chromazonarol (154) | Algicidal activity | Heterosigma akashiwo | Mean mortality after 4h at 1 μg ml−1: | 182 |
| Chattonella antiqua | 78% | |||
| Heterocapsa circularisquama | 42% | |||
| 93% | ||||
|
||||
| Monoterpenes | ||||
| Cordiachromene A (173) | Anti-bacterial | Staphylococcus aureus | 2–64 μg ml−1 | 212 |
| Streptococcus faecalis | 1–64 μg ml−1 | |||
| Escherichia coli | MIC > 2 mmol | 213 | ||
| Micrococcus luteus | MIC > 0.51 mmol | |||
| Didehydroconicol (185) | Anti-bacterial | Escherichia coli, Micrococcus luteus | MIC > 2 mmol, MIC > 0.51 mmol | 213 |
| Epiconicol (184) | MIC > 2 mmol, MIC > 0.13 mmol | |||
| Cymobarbatol (181) and 4-isocymobarbatol (182) | Antimutagenic inhibition of 2AN and EMS mutagenicity towards Salmonella thyphimurium | T-98 stain | 75, 150 and 300 μg/plate | 208 |
| T-100 stain | (EMS): 32.5–300 μg/plate | |||
|
||||
| Hemiterpenes | ||||
| Precocene 2 (194) | Anti juvenile hormone | Induction of precocious metamorphosis in milkweed bug | 0.7 μg cm−2 90% precocious adults | 230 |
| Daedalin A (199) | Inhibition of melanin synthesis | Tyrosinase inhibition | IC50: 194 μM | 234 |
Another group of complex bicyclic compounds was isolated from Cystoseira stricta, Cystoseira mediterranea and Cystoseira tamariscifolia. Cystoseirols A (86), B (87) and C (88) possess a oxabicyclo[5:4:1]dodecane ring that results from a single methyl group displacement, supplementary bridges and ring fissions.107,109,110 Cystoseirol A (86) inhibited plant tumor formation in a crown-gall potato bioassay (73% at 10 μM).110
Amico et al. isolated cystoketal chromane (89) from the Sicilian brown alga Cystoseira balearica. Structural elucidation revealed a tricyclic ring system within the side chain and an epimeric mixture at C-2 (Fig. 11). Thus, the authors proposed cystoketal chromane to be an artefact of the extraction process.111 Later, demethoxy cystoketal chromane (90) was isolated from Mediterranean Cystoseira amentacea and recently from Cystoseira tamariscifolia.112,113 Demethoxy cystoketal chromane showed cytotoxic activity with high selectively towards HepG2 cells (IC50 = 14.77 μg ml−1).113 A screening of 55 Cystoseira species found several bicyclic meroditerpenes, namely 14-methoxyamentol chromane (91), amentolchromane and cystoseirone, respectively.114 The latter two compounds were unstable and were therefore isolated as their acetate derivatives. Interestingly, isolated amentolchromane acetate (92) could be transferred to cystoseirone acetate (93) by chemical oxidation with meta-chloroperoxybenzoic acid in methylenchloride. Again, it was suggested that amentols and cystoseirone have a common biosynthetic precursor, bifurcarenone (81) bearing the typical cis orientation for the bridgehead methyls.
Finally, bifurcarenone chromane (94), the cyclization product of 81, was found in Cystoseira baccata,104,115 and Sargassum muticum,116 from which it was isolated as epimeric mixture at C-2 (Fig. 11). The mixture showed anti-leishmanial activity at IC50 values of 44.9 μM and decreased the intracellular infection index (IC50 value of 25.0 μM).117
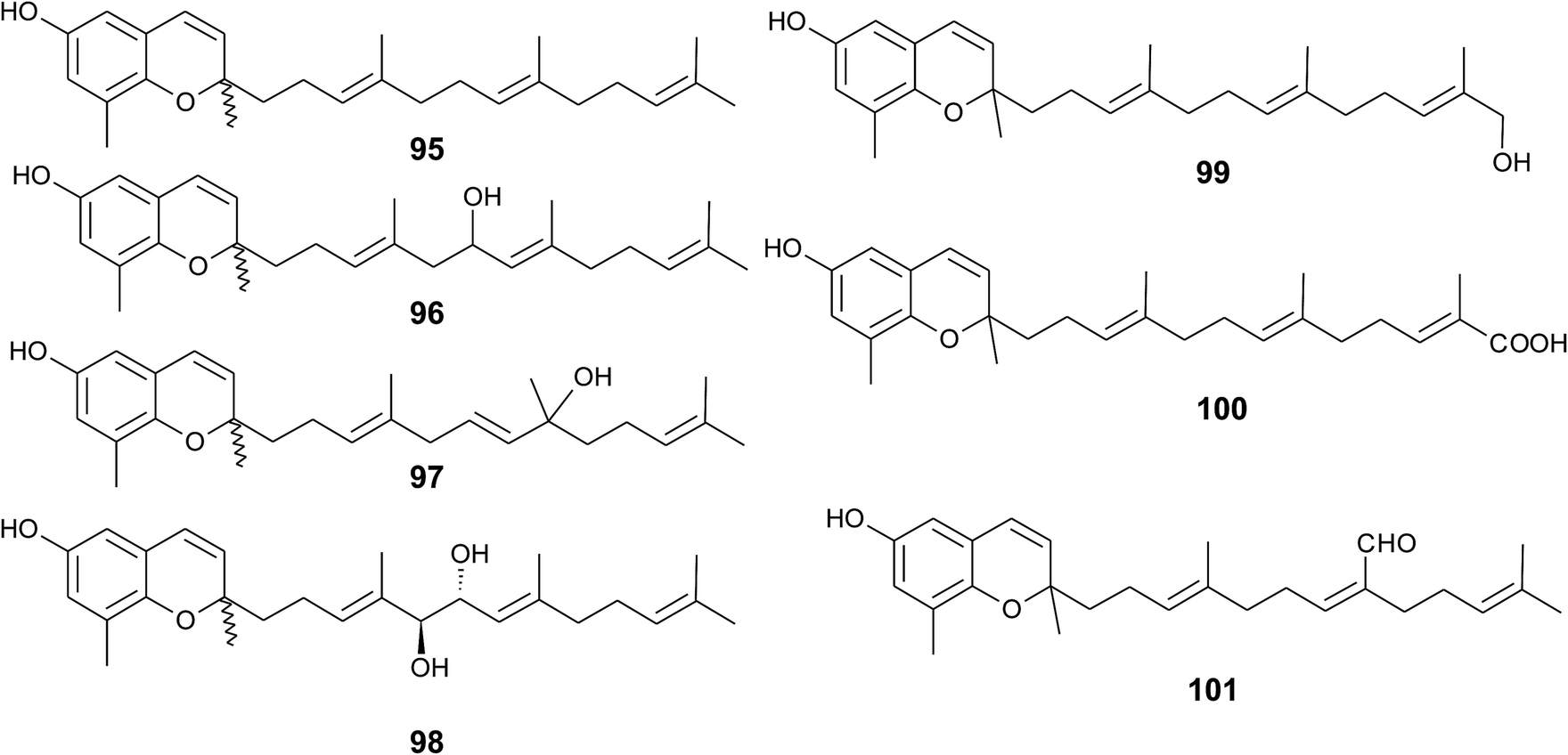
Sargaol (95) or dehydro-δ-tocotrienol is the potential biosynthetic precursor for most of the chromenols found in brown algae. It was originally isolated from Sargassum tortile collected at the Japanese Tanabe Bay. A lipid extract of the algae exhibited high cytotoxic activity and was used as a skin-lightening agent.118,119 Fractionation of the extract resulted in the isolation of sargaol (95), sargadiols-I (96) and -II (97), and sargatriol (98) (Fig. 12).120,121 All compounds were moderately cytotoxic towards murine P-388 leukemia cells with ED50 values of 52, 34, 41 and 42 μM, respectively (Table 2).118,120 Sargadiols (96) and (97) bear a hydroxyl group at C-6′ and C-8′, respectively, and sargatriol has two hydroxyl groups at C-5′ and C-6′. All compounds were suggested to be artefacts of the isolation since epimers at C-2 were found in all cases. In addition, heating of the corresponding 1,4-hydroquinones in organic solvents led to the epimeric chromenes described in this paragraph.
 | ||
| Fig. 12 Structures of sargachromenols (95 to 101) from Sargassum species. | ||
Two chromenols were isolated as minor compounds from Desmarestia menziesii collected from the Antarctic King George Island, one bearing a hydroxy group at C-13′ (99) and the other a carboxy group at C-13′ (100). The latter is a structural isomer of δ-sargachromenol (51) (see below) and shares structural similarity with garcinoic acid (30).122,123 Again, no optical activity was found for the two chromenes suggesting an epimeric center at C-2. However, the authors suggested a non-enzymatic ring closure within the living algae since no corresponding 1,4-benzoquinone was found as a potential precursor.
A C-15′-aldehyde-bearing chromenol (101) with anti-leishmanial activity was found as minor compound in the Southern Australian brown alga Sargassum paradoxum and the Japanese algae Sargassum yamadae.124
δ-Sargachromenol is one the most investigated meroditerpenoid obtained from marine organisms. As mentioned above, its unique structure resembles a δ-chromenol ring system with an unsaturated side chain containing a carboxy group at C-15′. δ-Sargachromenol is widely distributed in Sargassum species such as Sargassum sagamianum,73,125,126 Sargassum serratifolium,54,127,128 Sargassum micracanthum,129 Sargassum horneri,130 Sargassum macrocarpum,131,132 and Sargassum fallax.133 The latter species contains δ-sargachromenol as high as 0.13% of the dry weight. It has also been isolated from Myagropsis myagroides (Sargassaceae),134 from the tunicate Botryllus tuberatus135 and other algae.134
Kusumi et al. claimed 51 to be an artefact that is produced from sargaquinoic acid during the clean-up procedure. Although there is an asymmetric carbon center at C-2, the authors found no optical rotation. Literature data on the stereochemistry of sargachromenol are inconsistent. δ-Sargachromenol isolated from plant species showed optical rotation with an [α]D of +5°,53 whereas δ-sargachromenol isolated from Sargassum fallax showed an [α]D of -23.7°.133 Choi et al. isolated a racemic mixture from Botryllus tuberatus and separated δ-sargachromenol stereoisomers by chiral HPLC coupled with circular dichroism spectroscopy. They determined the absolute configuration for R-sargachromenol with an [α]D of -68° and S-sargachromenol with an [α]D of +88°.135 It is yet not clear whether sargachromenol should be considered as an artefact of the work-up procedure or as a natural product.136
Sargachromenol received attention in drug research since it has inhibitory activity against enzymes related to Alzheimer's disease, strong anti-inflammatory activity and anti-hyperproliferative properties in skin cells (Tables 1–3). Several patents are pending on the use of sargachromenol as drug candidate.137
Choi et al. found acetylcholinesterase- and butyrylcholinesterase-inhibitory activity with IC50 values of 32.7 and 7.3 μM, respectively.125 Recently, Seong et al. repeated the enzyme assays and determined slightly higher IC50 values (97.3 and 9.4 μM, respectively) for these enzymes. In addition, the authors found that 51 is a non-peptidic, noncompetitive inhibitor of β-site amyloid precursor protein-cleaving enzyme 1 (BACE1) with an IC50 value of 7.0 μM and a Ki value of 2.9 μM.127 Molecular docking experiments revealed that sargachromenol interacts with the allosteric side of BACE1.127 In line with these results, sargachromenol promotes neurite outgrowth and survival of rat PC12D pheochromocytoma cells via activating phosphatidylinositol-3 kinase.131 Based on its lipid-solubility and low molecular weight (<500 Dalton), sargachromenol should be able to cross the blood brain barrier, making δ-sargachromenol an interesting drug candidate for treating Alzheimer's disease and other neurodegenerative diseases.
Similar to δ-garcinoic acid, sargachromenol is a potent anti-inflammatory compound that prevented TPA-induced ear edema in mice with an IC50 value of 0.36 mg per ear.53 In addition, δ-sargachromenol inhibits lipoxygenase (LOX) (76% at 100 ppm) and cyclooxygenase (COX)-1 and -2 (98% and 84% at 100 ppm; Table 1).52
Sargachromenol inhibited LPS-induced inflammation markers in murine RAW 264.7 macrophages. Production of PGE2 and nitric oxide was inhibited (IC50 values of 30.2 and 82 μM, respectively) accompanied by a reduced protein expression of iNOS and COX-2.129 Kim et al. reported the inhibition of nitric oxide formation in LPS-stimulated murine microglial BV-2 cells with an EC50 value of 1.14 μg ml−1 (2.7 μM). These effects are accompanied by a suppression of the release of TNF-α, IL-1β, and IL-6.134 Several markers of vascular inflammation were also decreased in primary endothelial cells by δ-sargachromenol, namely TNF-α induced ICAM-1 and VCAM-1 expression, adhesion of monocytes to HUVEC and decreased production of monocyte chemoattractant protein-1 and matrix metalloproteinase-9 (MMP-9).128 Both epimers of sargachromenol bind to human farnesoid X receptor and inhibit its transactivation (IC50 values of 9.0 μM (R-epimer) and 17.0 μM (S-epimer), respectively). It is known that farnesoid X receptor agonists decrease plasma triacylglycerides and increase HDL cholesterol by regulating the expression of apolipoprotein C-I and C-IV.135 Summarizing the evidence (also from plant species), δ-sargachromenol (51) clearly is a candidate for an anti-atherogenic drug.
Sargachromenol has also been suggested as a drug for skin health, since it induced apoptosis in hyperproliferative human keratinocyte HaCaT cells and suppressed MMP-1, -2 and -9.126,130 Finally, insecticidal activity was found against the larvae of Spodoptera frugiperda with a LD50 value of 2.94 μg ml−1.136
Iwashima et al. synthesized a dihydroxylation product of sargachromenol from the corresponding plastoquinone precursor that had been isolated from Sargassum micracanthum.58 To the best of our knowledge, 11′-,12′-dihydroxy-sargachromenol (102) (Fig. 13) has never been isolated as a natural product from algae before. However, the compound has been investigated for its anti-viral activity against human cytomegalovirus,58,60 its anti-ulcer activity in ethanol-induced gastric lesions in rats,138 and inhibitory activity in osteoclastogenesis (bone resorption), thus suggesting that this compound is an interesting pharmacological lead structure.59
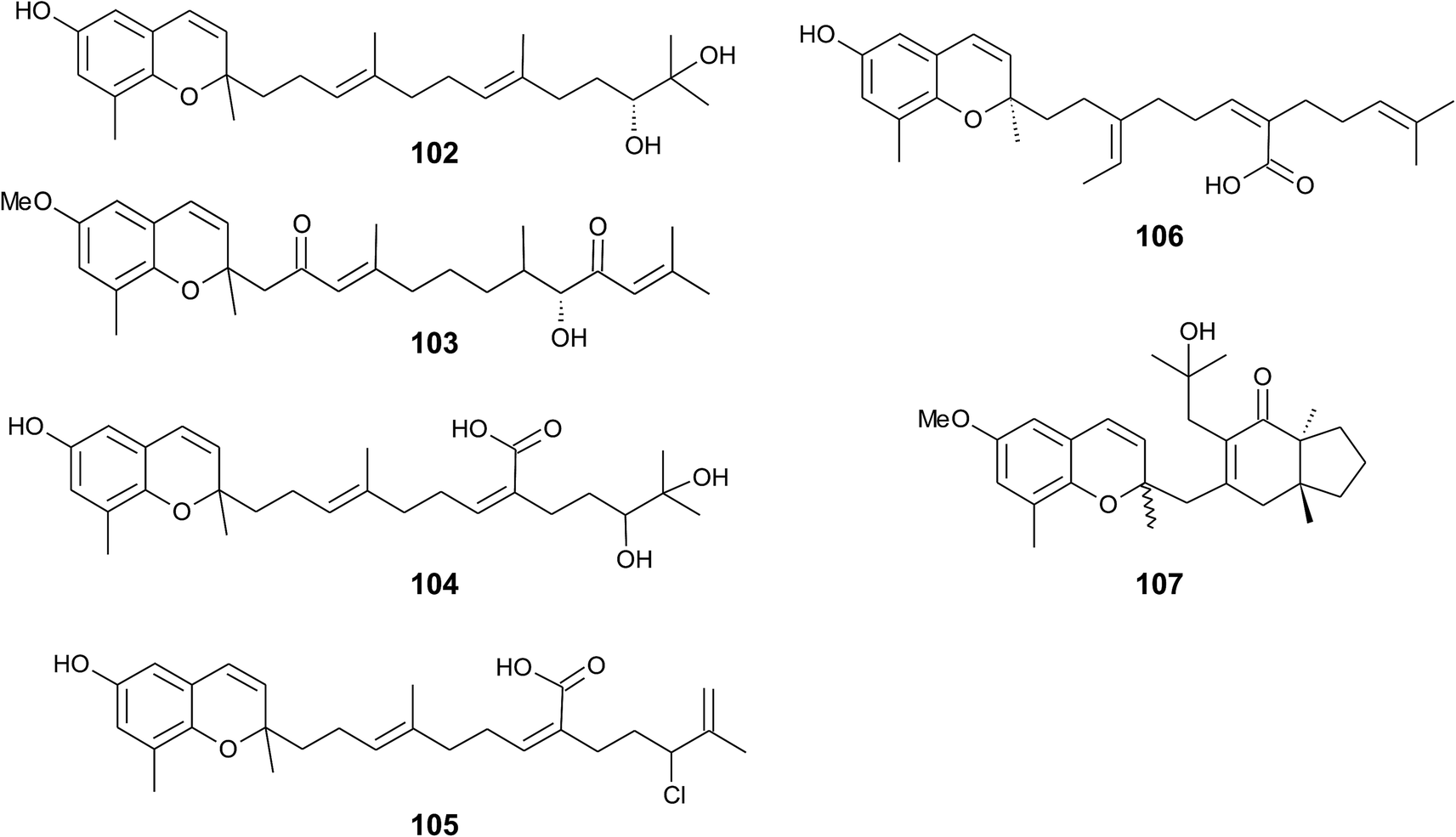 | ||
| Fig. 13 Structures of sargachromenols (102 to 107) from Sargassum species. | ||
Multiple biosynthetic oxidation steps lead to a highly oxidized chromane (103), which was found in Halidrys siliquosa (Sargassaceae) from the French Atlantic coast.139 Two keto groups at positions C-2′ and C-10′ and a hydroxyl group at C-9′ with R-configuration could be assigned by two-dimensional NMR spectroscopy.
Natural derivatives of δ-sargachromenol 52 have been isolated from different algae species. Besides δ-sargachromenol, sargothunbergol A (104), a sargachromenol with two additional hydroxyl groups at C-11′ and C-12′, was isolated as a minor compound from Sargassum thunbergii, collected from the shore of the Korean Youngdo Island.66,140 Fallachromenoic acid (105) from the Australian alga Sargassum fallax is an interesting variation as it bears a chlorine atom at C-11′ and a terminal double bond (Fig. 13).133,141 Fallachromenoic acid was isolated in 0.06% yield (dry mass) and exhibited moderate anti-tumor activity in the murine leukemia P388 cell assay (IC50 value of 29 μM).
Along with the sargachromanols described by Jang et al.,76 mojabanchromanol (106) has been isolated from Sargassum siliquastrum,142 showing a rearranged carbon skeleton at C-3′ of the side chain.
Only two chromenols with cyclic side chain modifications were found in the literature. A 3,4-unsaturated analogue of bifurcarenone chromane (107) was identified in Cystoseira amentacea collected from the French Riviera and an unsaturated analogue of compound 107 from Cystoseira baccata.143
As reported by Yamamoto et al., cold water fish contains a substantial amount of marine-derived tocopherol (25) (MDT), an α-tocomonoenol with a terminal double bond between C-12′ and C-13′ (Fig. 14).147 Since tocochromanols are only synthesized by photoactive organisms, the authors suggested a dietary source for MDT in fish. In fact, phytoplankton contains up to 21% (of total tocopherol) MDT. Also Antarctic krill (Euphasia superba) contains up to 8% (of total tocopherols) MDT.148 The biosynthesis of 25 is largely unknown; however, the authors suggested that the terminal double bond is introduced by side chain desaturation of α-tocopherol, similar to that of fatty acids.
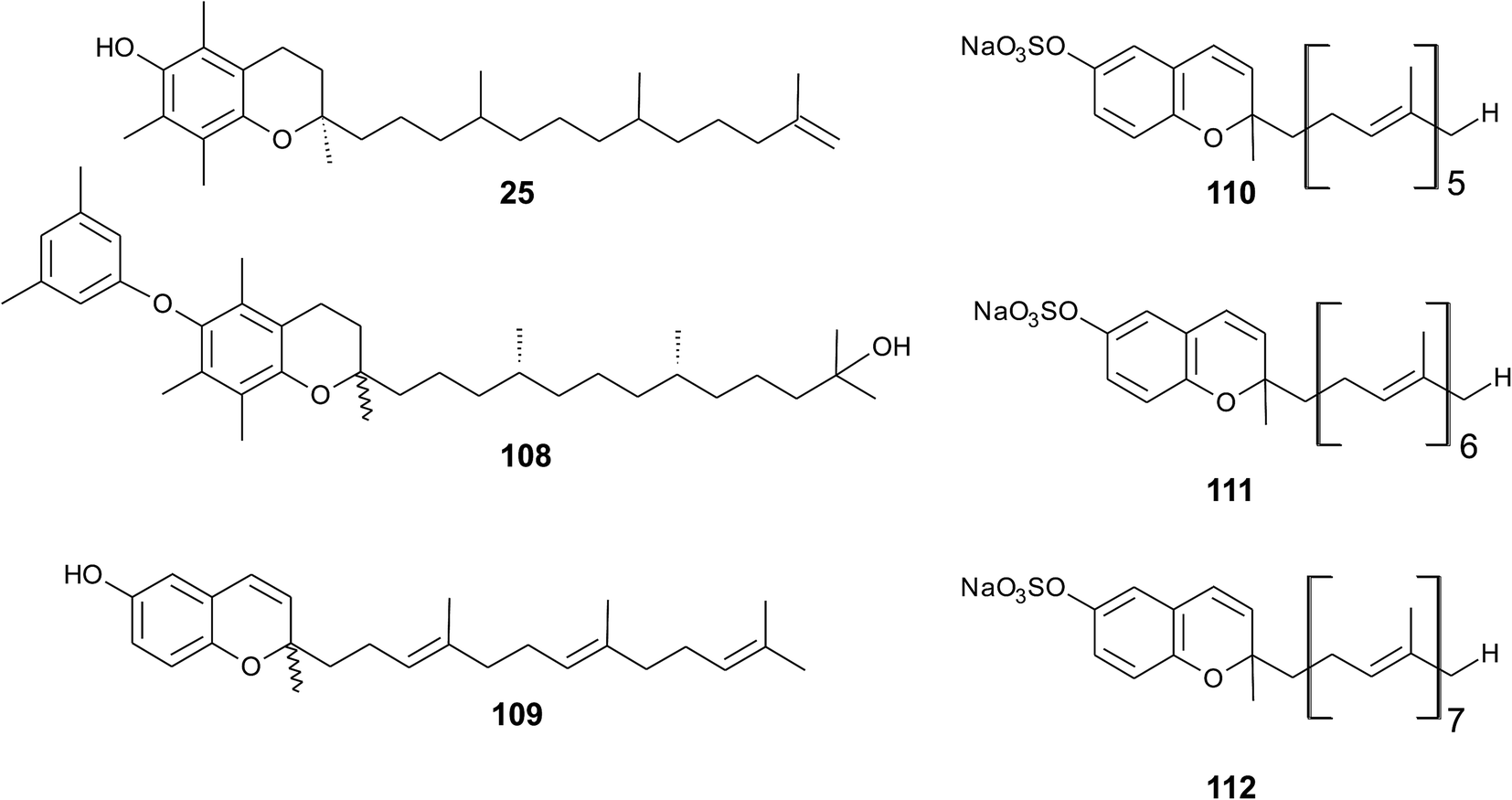 | ||
| Fig. 14 Structures of meroditerpenoids (25) and (108) from phytoplankton and sarcochromenols (110 to 112) from Sarcotragus spinulosus. | ||
Recently, an unusual α-tocopheroid, α-tocoxylenoxy (108), containing a 3,5-dimethylphenoxyl moiety was isolated from the seaweed Caulerpa racemosa,149 taxonomically also belonging to the green algae (Chlorophyta).
A hypothetic biosynthetic precursor of the chromene structure was found in the Western Australian sponge Fasciospongia species (order of Dictyoceratida, family of Thorectidae).151 Fascioquinol F (109) is a demethylated 3-4-dehydro-tocotrienol that might undergo cyclization to form complex ring systems in analogy to taondiols (see the section on Brown algae). The structure is similar to sargaol (95), but lacks the methyl group at C-8 (Fig. 14). Fascioquinol F revealed moderate antibacterial activity against Staphylococcus aureus and Bacillus subtilis (IC50 values of 13 and 30 μM, respectively).
Sarcochromenols A (110), B (111) and C (112) are a group of long-chain tocochromenols with five, six and seven isoprene units, respectively (Fig. 14). They were isolated from the Pacific Ocean sponge Sarcotragus spinulosus (Schmidt) (family of Thorectidae) and showed Na+/K+-ATPase inhibitory activity similar to that of the sargachromanols D, F, H and L (IC50 value for sarcochromenol A of 1.6 μM).78,152 The compounds have also been isolated from the Indian sponge Ircinia fasciculate (Spongillidae).153 In addition, an un-sulfated form of sarcachromenol B was isolated in 0.1% yield.
A screening for selective human 15-LOX inhibitors from an extract of the Papua New Guinean sponge Psammocinia (order of Dictyoceratida, family of Irciniidae) revealed chromarols A to D (113 to 116; Fig. 15).154 The IC50 values for chromarols A to D were 0.6, 4.0, 0.7 and 1.1 μM, respectively. The authors found high selectivity since the IC50 values for 12-LOX were above 100 μM. The biosynthesis of the cyclohexene ring system in the side chain of chromarols presumably derives from an acid-catalyzed cyclization.
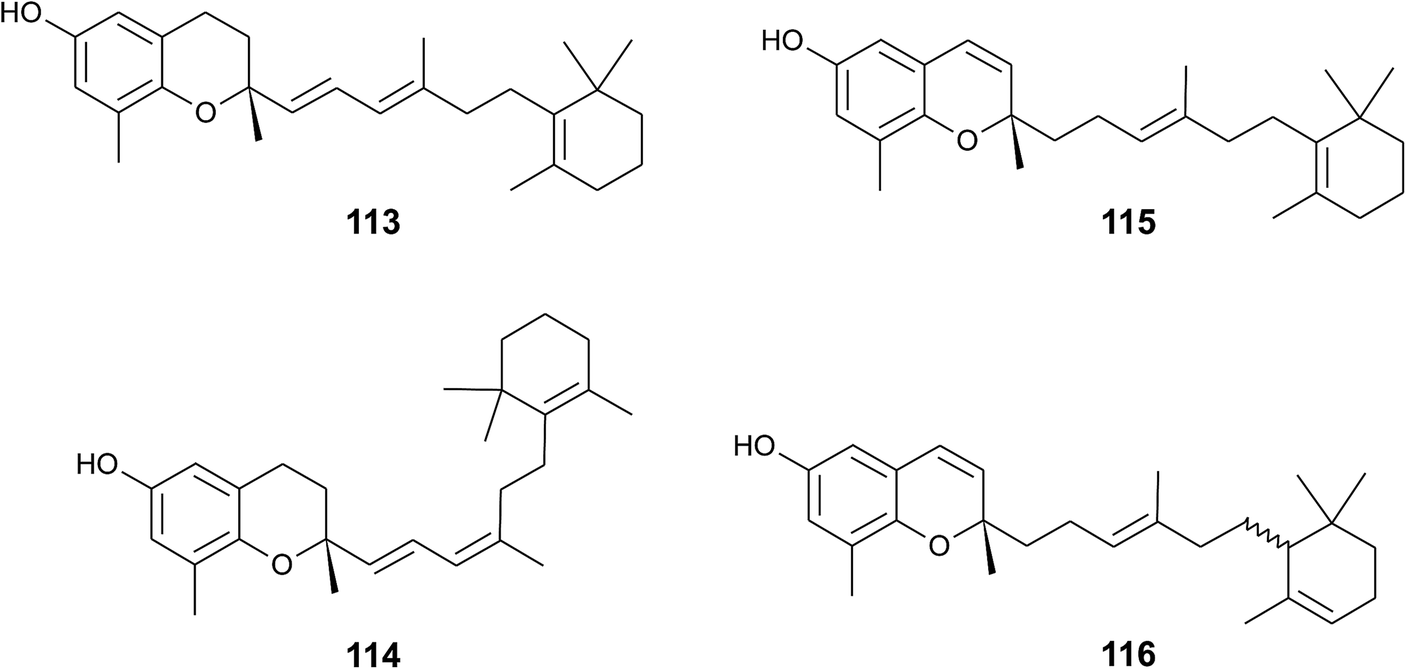 | ||
| Fig. 15 Structures of chromarols (113 to 116) from Psammocinia species. | ||
Several sponges produce a group of eight polycyclic strongylophorines that resemble taondiol structural motives (Fig. 16). They contain a demethylated aromatic ring and modifications at the methyl groups at C-13′ and/or C-15′. They were discovered by Braekman et al. because of their ichthyotoxic activity.155 The biosynthesis follows that of taondiol and is an enzyme-catalyzed cyclization cascade (see Fig. 10). Strongylophorines 2 (117), 3 (118), 4 (119), and 5 (120) were isolated from Strongylophora durissima from Maricabiin Island, Philippines,156 and a different, as yet undescribed Strongylophora species from Ilocos Sur, Philippines.157 These molecules contain a cyclic lactone, a carboxy, an aldehyde or a hydroxyl group moiety at C-13′, respectively. Strongylophorine 3, bearing a terminal carboxy group, was isolated with 0.1% yield (dry weight).156 Furthermore, the known strongylophorines 3, 9 (121) and 11 (122) were isolated from a Taiwanese species of Strongylophora durissima. The 6-methoxy (121) and 6-acetyl (122) derivatives are structurally related to strongylophorine 2, which contains a cyclic lactone moiety.158 Liu et al. isolated the strongylophorines 15 (26R) (123) and 16 (26S) (124), respectively, from the Okinawan sponge Strongylophora strongylata as epimers at the hemiacetal carbon.159 Biosynthetic O-methylation and O-ethylation gave the acetals 26-O-methoxystrongylophorine 16 (125) and 26-O-ethoxystrongylophorine 16 (126), respectively.160,161 Noda et al. found a mixture of strongylophorines 15 and 16 to be strong inhibitors of the proteasome with IC50 values of 3.6 μM.160 The same study compared the proteasome-inhibitory activity of structurally related strongylophorines and found the following order: hemiacetal > acetal ∼ carboxy > lactone > no modification.
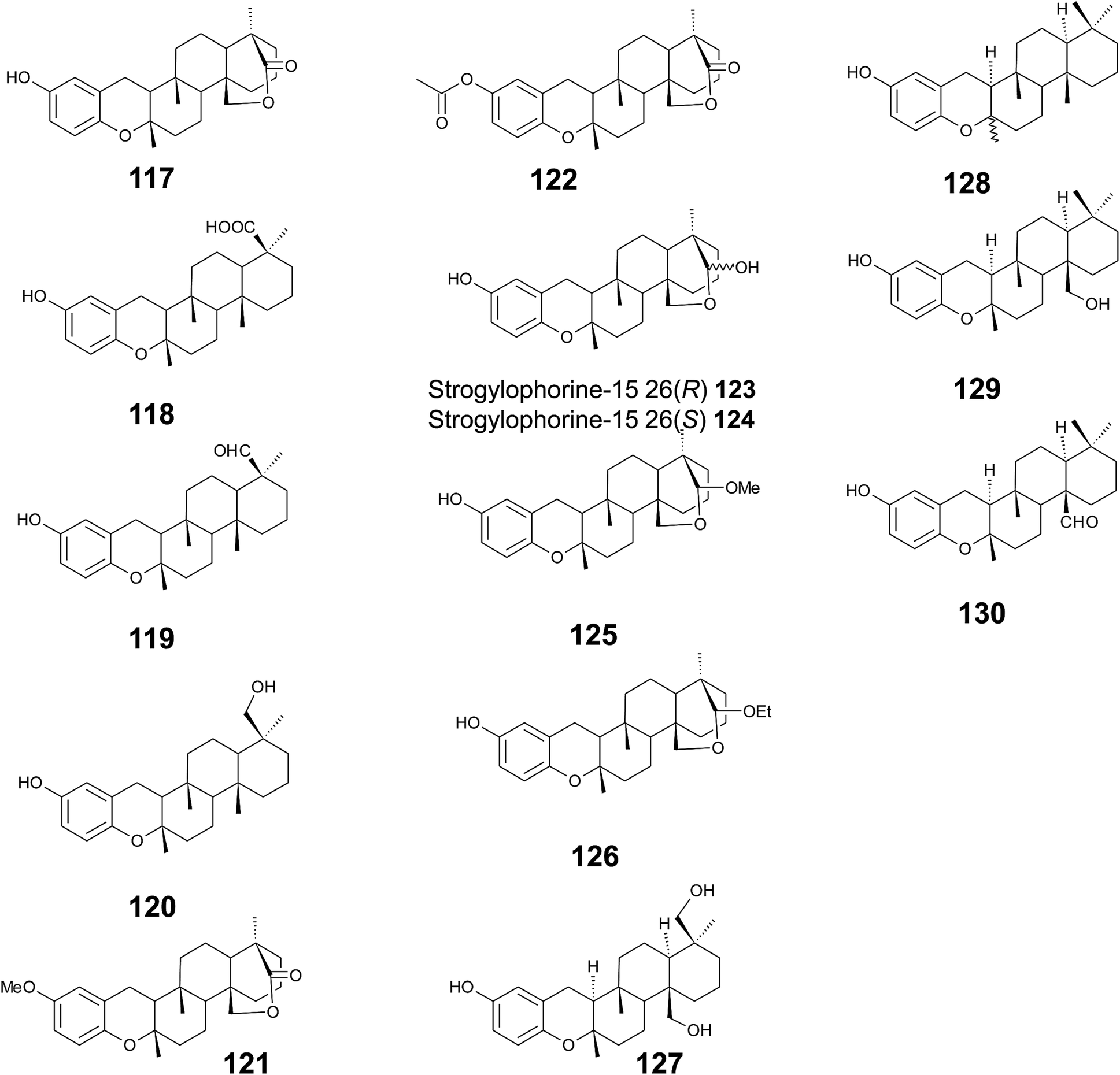 | ||
| Fig. 16 Structures of strongylophorines (117 to 130) from Strongylophora species. | ||
On their search for inhibitors of protein tyrosine phosphatase 1B, an enzyme that plays a crucial role in the regulation of insulin and leptin signalling, Lee et al. found inhibitory activity for 125, 117, 118, 123, and strongylophorine 17 (127) with IC50 values of 8.5, 24.4, 9.0, 11.9, and 14.8 μM, respectively.161 Strongylophorines 2 and 3 also inhibited hypoxia-inducible factor-1-dependent luciferase expression in engineered U251-HRE glioma cells with EC50 values of 8 and 13 μM.162 Strongylophorines 22 (128), 23 (129), 24 (130), and 17 (127) were isolated from the Okinawan sponge Petrosia corticata and displayed moderate cytotoxic activity against uman cervical carcinoma epithelial (HeLa) cells (Table 2).163 All strongylophorines exhibited ichthyotoxic, insecticidal, anti-bacterial, fungicidal, and cytotoxic properties. Strongylophorine 22 and fascioquinol D are epimers at C-2 and were isolated from Fasciospongia sp.151 The latter compounds displayed anti-microbial activity against Staphylococcus aureus and Bacillus subtilis with IC50 values of 25 and 2.3 μM (for strongylophorine 22) and 7.8 and 2.8 μM (for fascioquinol D), respectively.
Recently, Yu et al. presented the first semi-synthesis of strongylophorine 2 starting from isocupressic acid.164
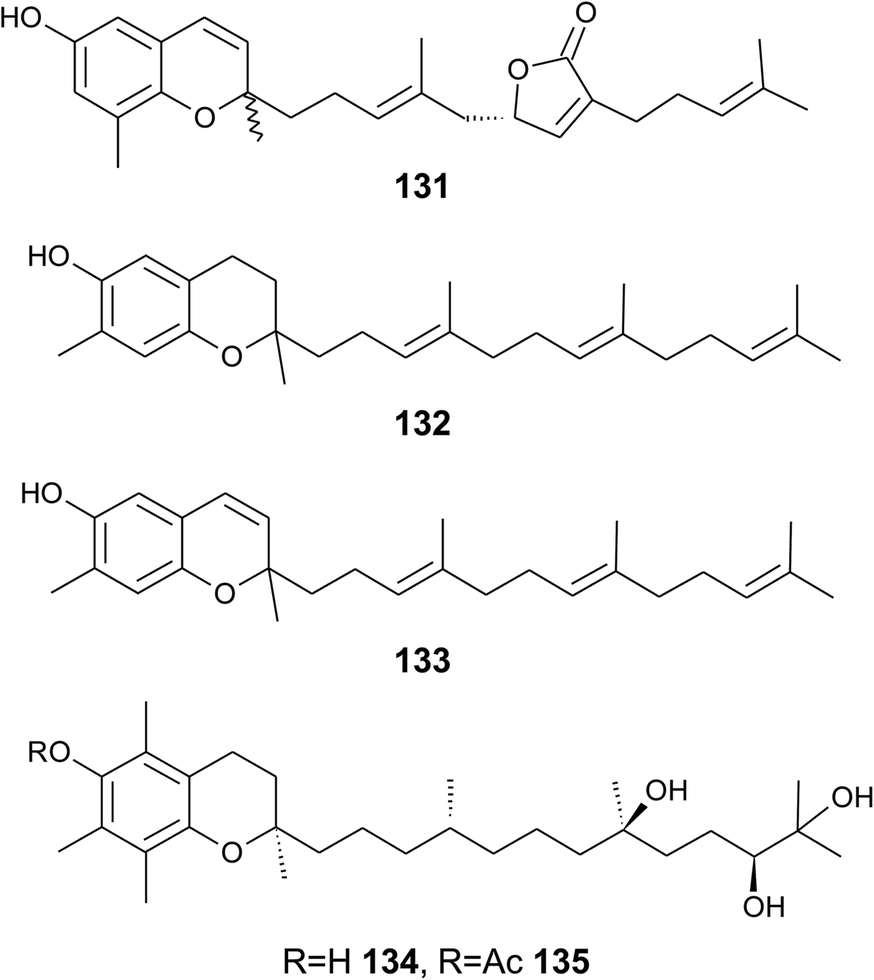 | ||
| Fig. 17 Structures of tuberatolide (131) from the tunicate Botryllus tuberatus and meroditerpenoids (132 to 135) from soft corals. | ||
Bowden et al. isolated tocotrichromenol (132), an isomer of sargaol (95), and its dihydro derivative (133) from an unknown Australian Nephthea species.168 The precursor quinone was also isolated, but did not convert into the chromenol under the work-up conditions; however, no optical activity was found at C-2.
Two α-tocopherol derivatives with three hydroxyl groups at C-8′, C-11′, and C-12′, respectively, were isolated from Lobophytum crissum (Fig. 17). Crassumtocopherol A (134) (R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) H) and B (135) (Racetyl) showed moderate cytotoxicity against murine P-388 leukemia cells with IC50 values of 6.7 and 5.2 μM, respectively.169 Compound 135 also showed cytotoxicity against the human colon adenoma cell line HT-29 with an IC50 value of 7.5 μM.
H) and B (135) (Racetyl) showed moderate cytotoxicity against murine P-388 leukemia cells with IC50 values of 6.7 and 5.2 μM, respectively.169 Compound 135 also showed cytotoxicity against the human colon adenoma cell line HT-29 with an IC50 value of 7.5 μM.
4. Merosesquiterpenes
4.1 Plants
The biosynthesis of chroma(e)nols with sesqui-, mono- and hemi-terpene moieties within the plant kingdom is only poorly understood. These molecules most likely derive from homogentisate condensed with farnesyl-, geranyl- and isoprenyldiphosphates, respectively.Oligandrol (136), a sesquiterpenechromane with an unsaturated side chain, was isolated together with methoxy-oligandrol (137) from the bark of the Australian tree Beilschmiedia oligandra (Lauraceae),170 and from the leaves of the genus Pseuduvaria indochinensis Merr, an Annonaceae variety from the Yunnan province in China (Fig. 18).171 Cytotoxic assessment revealed no activity against promyelocytic HL-60 leukemia cells and human SMMC-7721 hepatocarcinoma cells.171
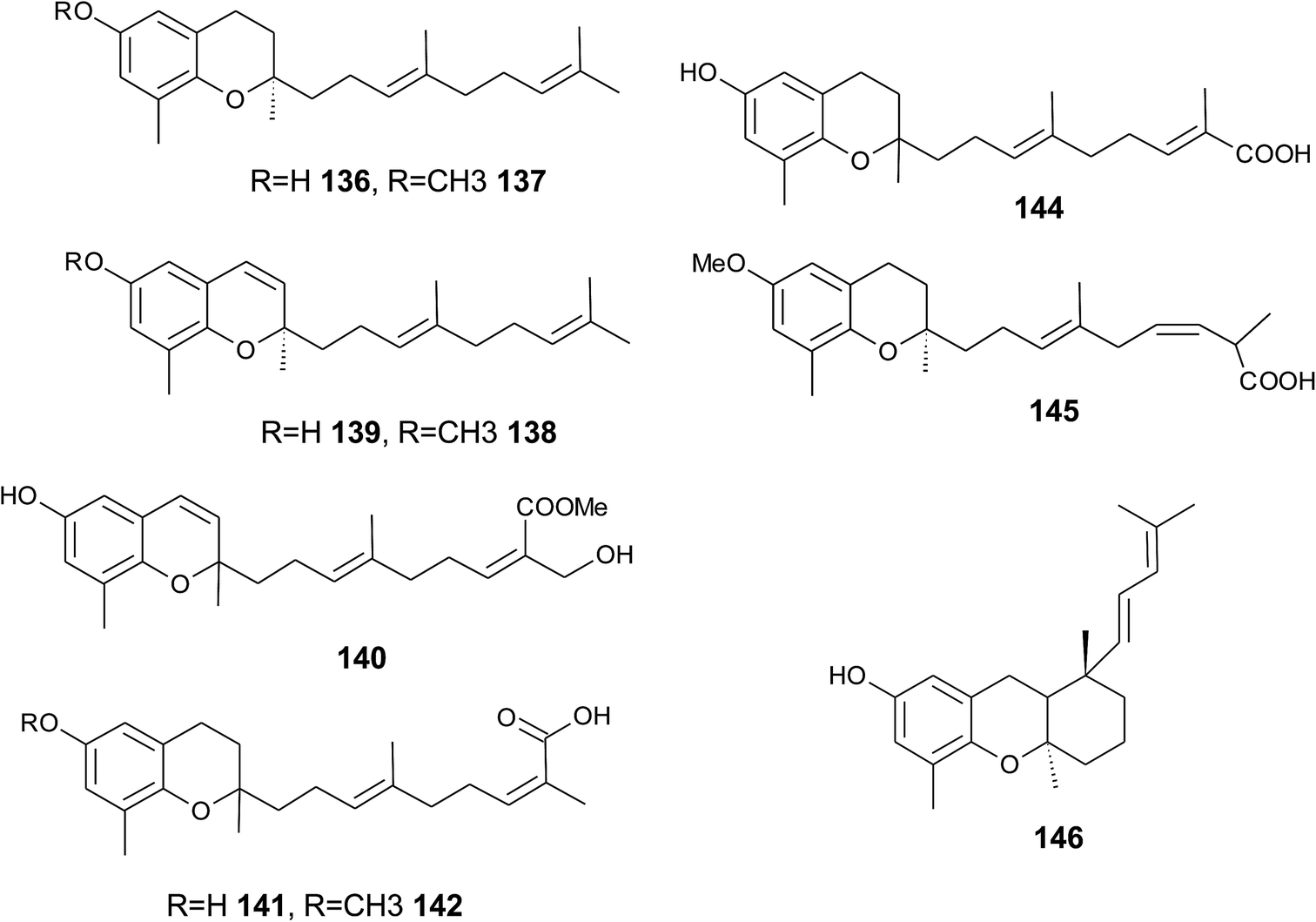 | ||
| Fig. 18 Structures of plant sesquiterpenes (136 to 146). | ||
The methoxy-derivative of dehydrooligandrol (138) was obtained from the root of Beilschmiedia erythrophloia172 and the free dehydrooligandrol (139) from the leaves of Seseli farreynii (Umbelliferae). However, the latter was suggested to be an artefact from the work-up procedure.173 Zhao et al. isolated a dehydrooligandrol with a terminal (Z)-carboxy and a 13′-hydroxy group, respectively, which the authors named pseudindochin (140).171
Polycerasoidol (141), an oligandrol derivative with a terminal (Z)-carboxy-group and its 6-methoxy-derivative polycerasoidin (142) were found in the stem bark of the Papua New Guinean Polyalthia cerasoides.174 Later, the methyl ester of polycerasoidin (143) and the E-isomer of polycerasoidol, termed isopolycerasoidol (144), were identified in the same species.175 Polycerasoidin was isolated at 0.13% yield (dry weight). Polyalthidin (145), a structural isomer of polycerasoidin with a double bond shift from C-7′–C-8′ to C-6′–C-7′ was isolated from P. cerasoides at a yield of 0.09% (Fig. 18).176 Polycerasoidol, polycerasoidin and polyalthidin were found to be inhibitors of the mitochondrial electron transfer chain that block NADPH oxidase activity with IC50 values of 37, 11 and 4.4 μM, respectively.176
Riccardiphenol C (146), a sesquiterpene from the New Zealand liverwort Riccardia crassa, is an example of a chromanol that undergoes intramolecular cyclization to form a condensed ring system. Purification of the crude extract yielded riccardiphenol C in 4 mg g−1 of dry liverwort (Fig. 18). The compound showed cytotoxicity against African green monkey BSC-1 kidney cells and inhibited the growth of Bacillus subtilis.177
4.2 Fungi
A sesquiterpene chromene (147) with a truncated tocochromene-like structure was isolated from Chroogomphus rutilus.178 The mushroom is also known as brown simecap and lives ectomycorrhizally with Pinus species. The compound shows R-configuration at the chiral center C-2 (Fig. 19).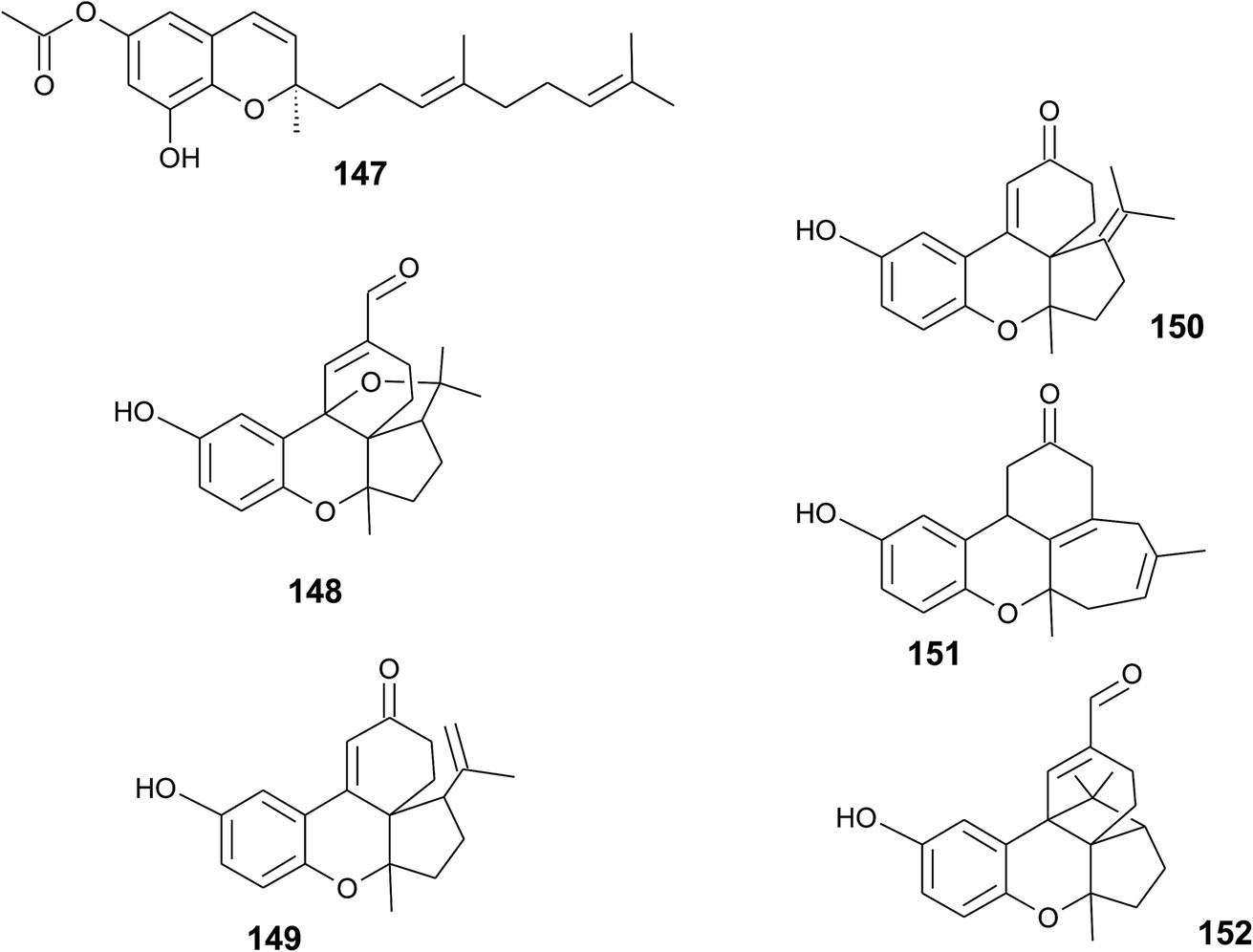 | ||
| Fig. 19 Structures of sesquiterpenes (147 to 152) from fungi species. | ||
Polycyclic sesquiterpenes were isolated from the fruiting bodies of the tropical rot fungus Ganoderma cochlear.179 Ganocin A to C (148–150) possess a spiro[4,5]decane ring, and ganocin D (151) has an eight-membered ring system. As a biosynthetic key step, the authors suggest a Diels–Alder reaction of fornicin C to build up the polycycles. In the same fungus, Dou et al. found cochlearol B (152) with an unusual 4/5/6/6/6 polycyclic ring system (Fig. 19). The compound is a strong inhibitor of the TGF-β/Smad signaling pathway.180
4.3 Marine organisms
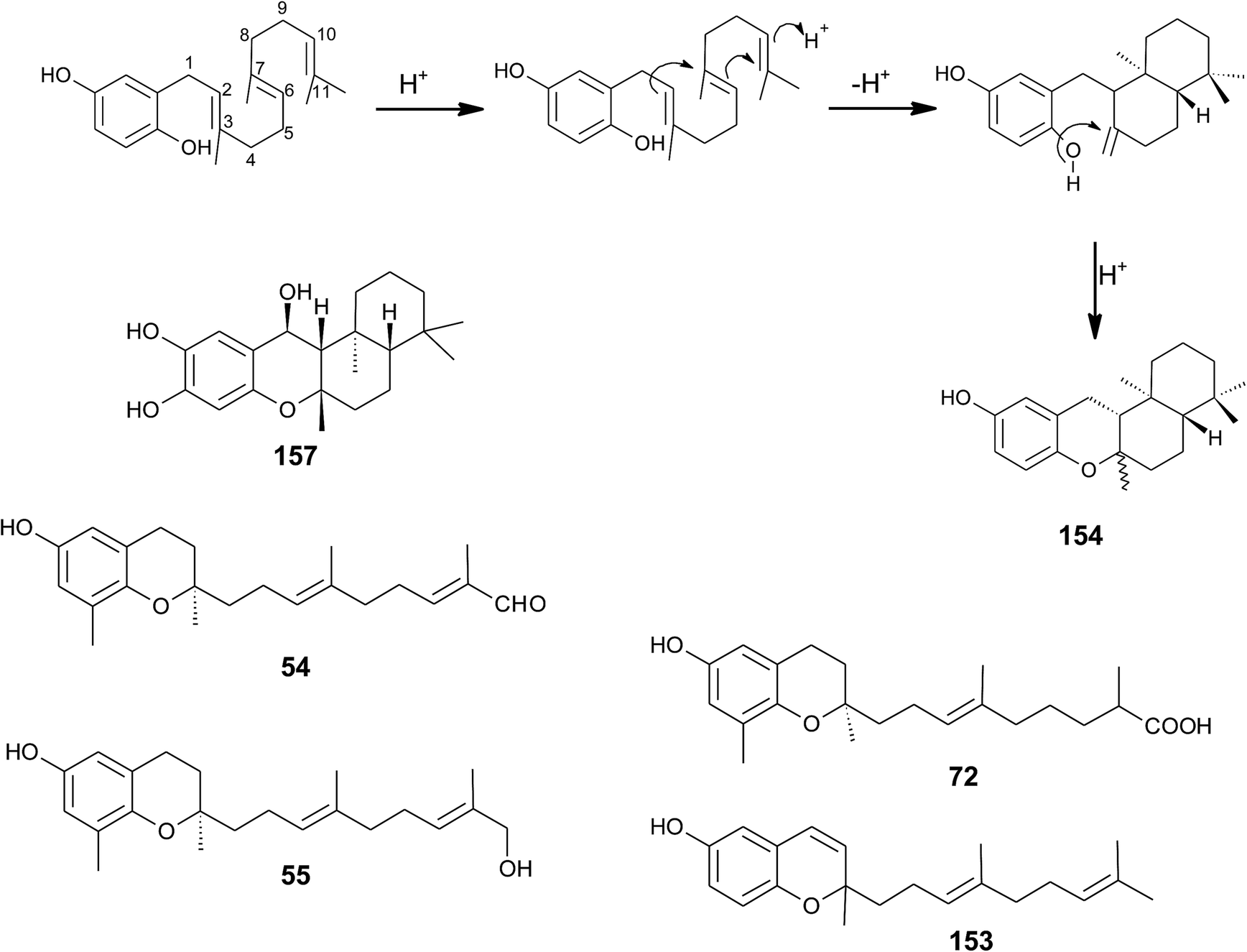 | ||
| Fig. 20 Scheme of multiple acid-catalyzed cyclizations of farnesyl hydroquinone towards chromazoranol (154) as suggested by Kurata et al.186 Structures of cyclic (157) and linear sesquiterpenoids (54, 55, 72 and 153). | ||
Dictyochromenol (153) and its cyclization product chromazonarol (154) were both isolated from the Japanese brown alga Dictyopteris undulata.181–183 Dictyochromenol is comprised of a demethylated chromanol ring which is attached to an unsaturated sesquiterpene moiety. A chemical synthesis route of dictyochromenol was described by Aoki et al.184,185 Kurata et al. suggested an acid-catalyzed cyclization of farnesyl hydroquinone towards zonarol (1,4-hydroquinone) followed by a second acid-catalyzed formation of the epimeric center at C-2 of chromazonarol (Fig. 20).186 Chromazonarol showed algicidal activity towards Heterosigma and Chattonella species.182
Two epimeric sesquiterpene chromenols, named cyclorenierin A and B (158), were found in Haliclona sp., an Indo-Pacific sponge from Vanuatu.192 The biosynthesis of the cyclohexenone ring system seems to follow that of walsurol (Fig. 6).
Panicein B2 (159) bears a chromene ring and an aromatic ring system in the side chain (Fig. 21). It was first isolated by Cimino et al. from Haliclona panacea and later from the Mediterranean sponge Reniera fulva.193,194 Panicein B2 was also found in Reniera mucosa along with panicein A2 (160) and F2 (161).195 It has been suggested that the aromatic group of the side chain is formed from cyclorenierin A/B by a 1,2-methyl migration and subsequent oxidation.193 All paniceins show racemic carbon centers at C-2 suggesting that these compounds may be artefacts from the work-up procedure.
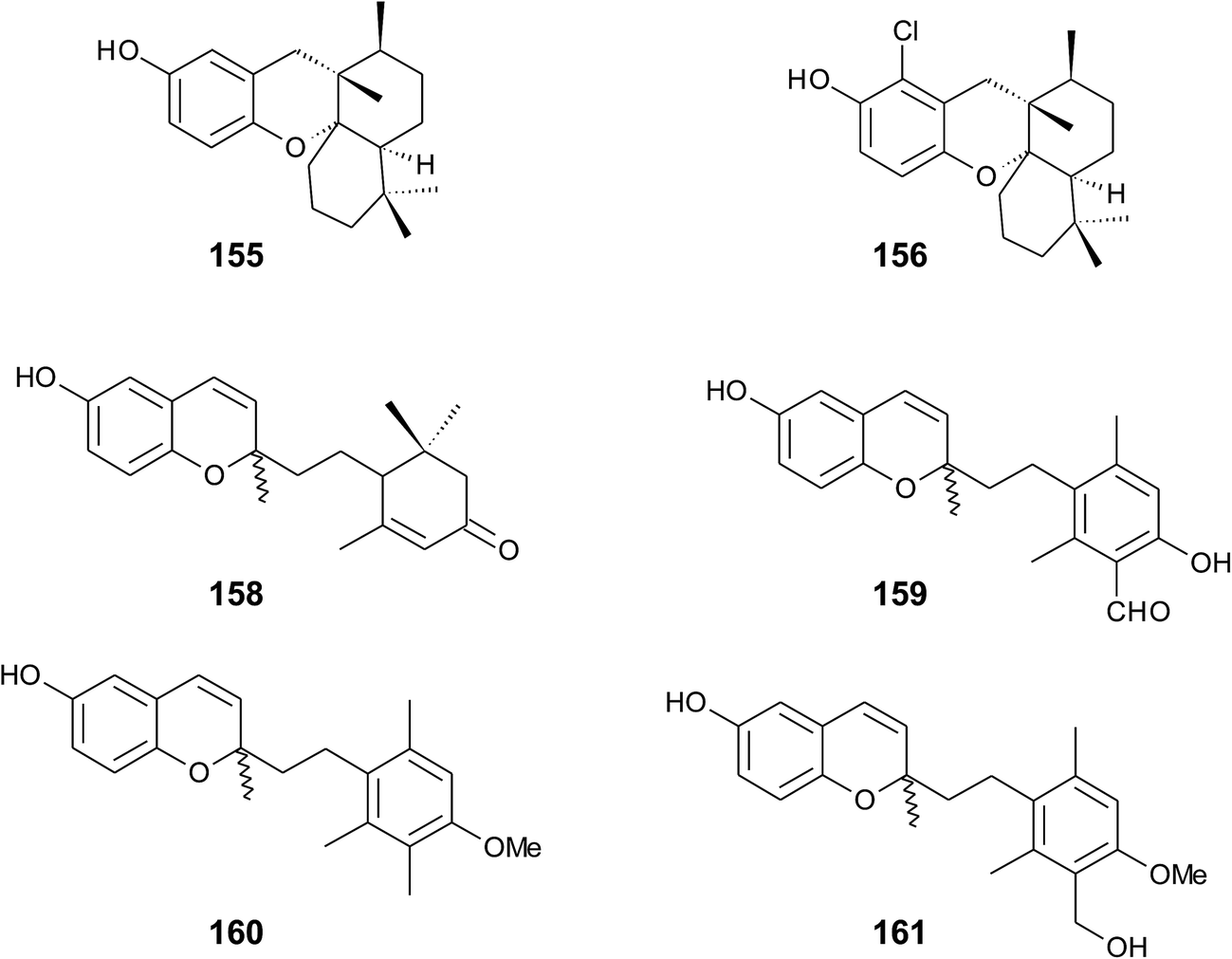 | ||
| Fig. 21 Structures of sesquiterpenes (155 to 161) from sponge species. | ||
Faulkner et al. isolated a series of unusual ansa chromene macrocycles from Smenospongia sp., a sponge from the Seychelles.196 Smenochromes A to D (162–165) were isolated with 0.26% yield (dry weight) for A and 0.037% for B, C and D, respectively (Fig. 22). The compounds showed no optical activity and thus occurred as racemic mixtures. The structurally related likonides A (166) and B (167) were isolated from the Kenyan sponge Haytella sp. with 0.06 and 0.04% yield.197 The biosynthesis of ansa chromenes presumably starts from a farnesylated hydroquinone followed by alkylation at C-5 of the activated hydroquinone ring or alternatively by O-alkylation of the terminal double bond.197
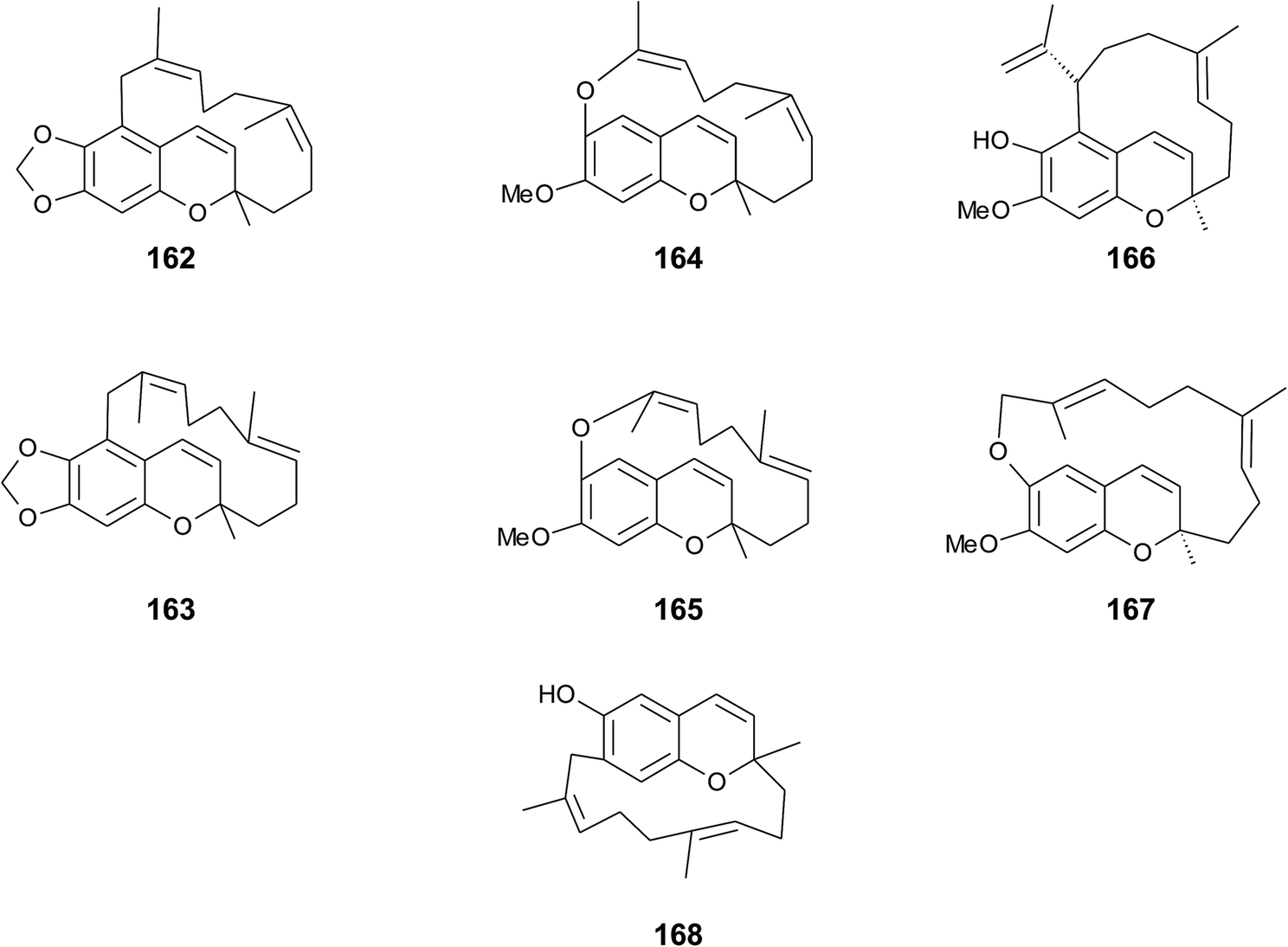 | ||
| Fig. 22 Structures of ansa chromane macrocycles (162 to 167) from Smenospongia species and (168) from tunicate. | ||
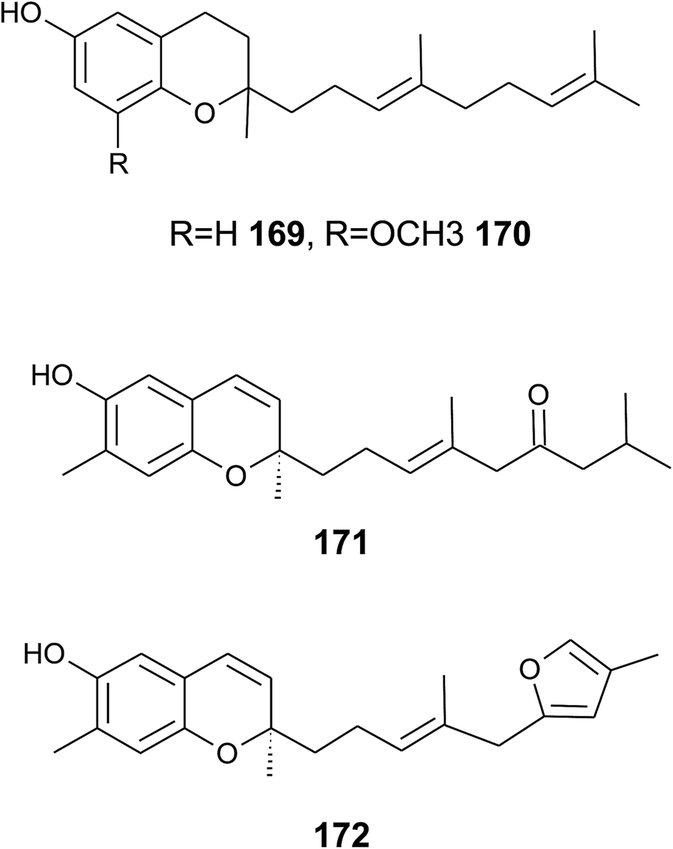 | ||
| Fig. 23 Structures of sesquiterpenes (169–171) from marine nudibranchs and soft coral (172). | ||
5. Monoterpenes
Monoterpenes from plant origin have been used since ancient times to treat certain diseases, such as inflammation or cancer. De Sousa and colleagues summarized the anti-cancer and anti-inflammatory activities of monoterpenes in an outstanding recent review.2045.1 Plants
The monoterpene cordiachromene A (173) was isolated from the heartwood of the tropical American tree Cordia alliodora (Boraginaceae) by Manners et al.205 The authors proposed geranyl benzoquinol as the biogenic precursor of the compound. Interestingly, the woods of Cordia alliodora are recognized for their durability in marine uses. Cordiachromene A was also isolated from the extract of different tunicates and was further tested for its bioactivity (see section on Tunicates (5.2)).As part of the investigation of Garcinia amplexicaulis (see section on Diterpenes), a short-chain chromane (175) with a truncated C-9 carbon skeleton was found (Fig. 24).48
 | ||
| Fig. 24 Structures of monoterpenes (173 to 185) from plants, marine algae and tunicates. | ||
5.2 Marine organisms
Cordiachromene A showed anti-bacterial activity against methicillin resistant Staphylococcus aureus and Streptococcus faecalis,212 but weak activity against Micrococcus luteus (the minimum inhibitory concentration was 0.51 mmol L−1).213 Cytotoxic activity was found against a panel of cancer cell lines, such as murine leukemia P388 cells, human adenocarcinomic A549 alveolar basal epithelial cells, human colon adenocarcinoma HT-29 cells, and African green monkey CV-1 kidney fibroblasts, and drug-sensitive human leukemic lymphoblasts (IC50 value of 30 μM).213,217
So far, three cyclization products of cordiachromene A were found; conical (184), a mixture of C-3, C-4 epimers called epiconicol, and didehydroconicol (185) with a condensed aromatic ring system (Fig. 24).213,215,217,218 All compounds showed cytotoxic and weak anti-bacterial activity.
Two optically active cordiachromenes were isolated from the Australian tunicae Aplidium solidum, one with an additional 2′–3′ double bond (186), the other with a saturated side chain and a 2′-ketone group (187; Fig. 25).219
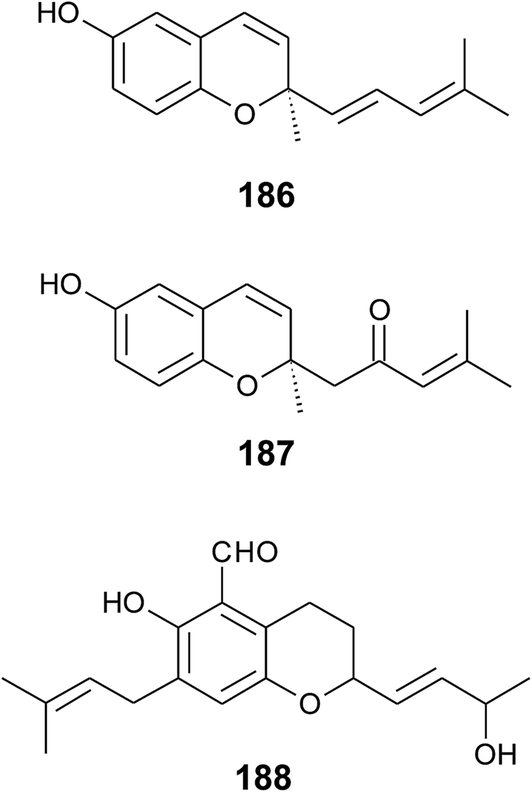 | ||
| Fig. 25 Structures of monoterpenes (186 to 188) from tunicates and algal derived endophytic fungi. | ||
6. Hemiterpenes
6.1 Plants
The following hemiterpenes exhibit interesting biological and pharmacological activities, among them mollugin (189) (methyl 2,2-dimethyl-6-hydroxy-2H-naphtho[1,2-b]pyran-5-carboxylat) from the Chinese medicinal plant Rubia cordifolia. Biogenetically, mollugin was formed by a cyclisation of a prenylated naphthoquinone and is not related to the biosynthetic pathway of tocopherols (Fig. 2). Mollugin has been first reported by Schildknecht et al. as a pigment from the rhizomes of Galium mollugo221 and was further investigated for pharmacological effects, such as anti-platelet aggregation activity and anti-viral activity against hepatitis B virus.222 Mollugin has been shown to induce apoptosis in different types of cancer cells. It exhibited IC50 values of 12.3 μM, 23 μM and 60.2 μM on human colon cancer cells (Col2),223 human Jurkat T-cells224 and murine NIH 3T3-L1 preadipocytes,225 respectively. In the presence of 10 μM mollugin, a human multidrug-resistant breast cancer cell line (MCF-7/adr) was more susceptible to doxorubicin (IC50 decreased from 60 to 7.5 μg ml−1).226 Several patents describe the use of mollugin for different applications (Fig. 26).227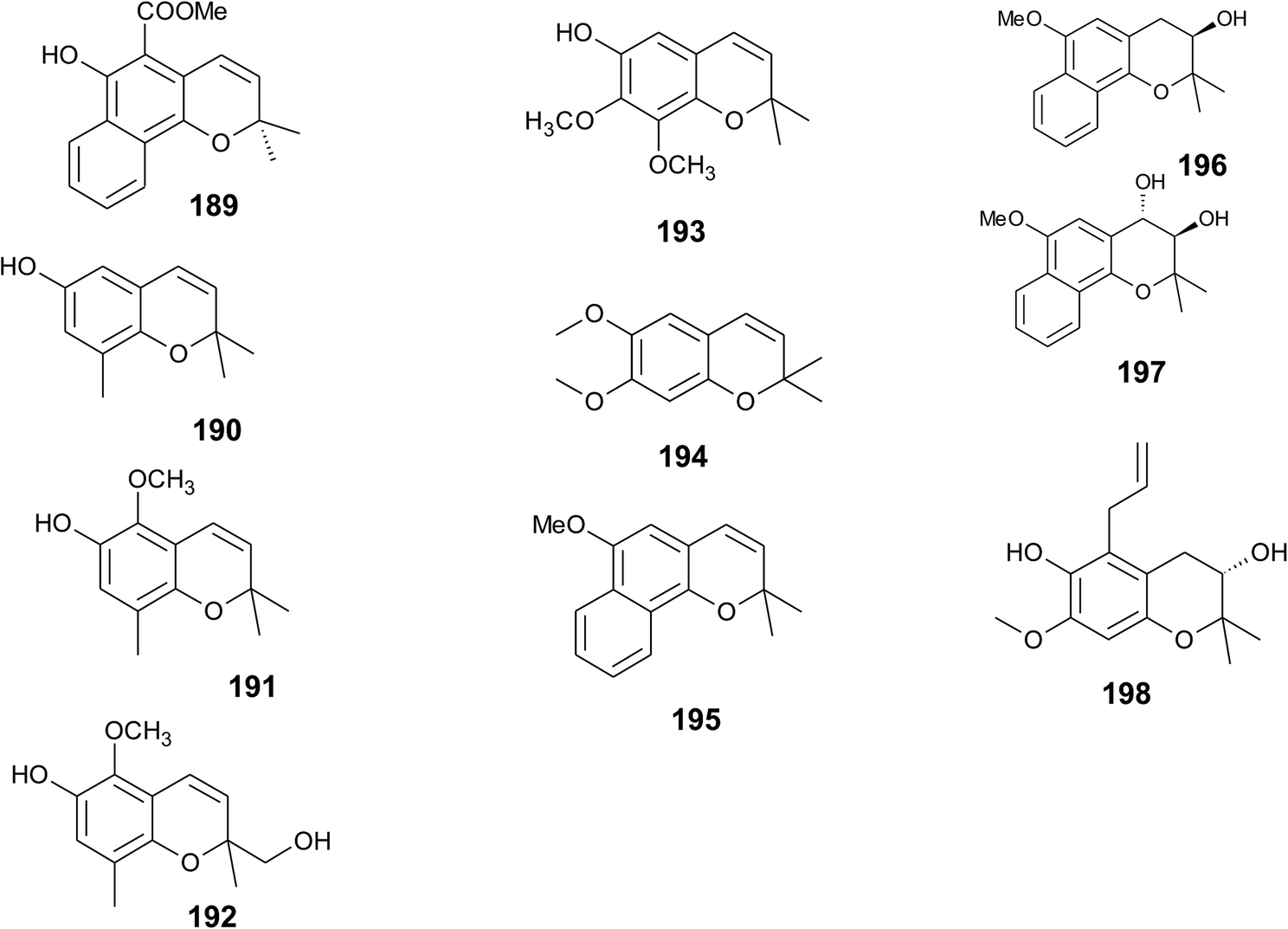 | ||
| Fig. 26 Structures of hemiterpenes (189 to 198) from plant species. | ||
Several low molecular weight chromenes, such as pterochromenes L1 (190), L2 (191) and L4 (192), were isolated from Taiwanese Pteris lingipinna228. 6-Hydroxyeupatoriochromene B (193) was obtained from Ageratina riparia (Asteraceae)229 and finally, one of the first insect anti-juvenile hormones, precocene 2 (194) (6,7-dimethoxy-2,2-dimethylchromene) was found in Ageratum houstonianum (Asteraceae).230 Lapachenol (195), a naphthalene derivative, was isolated from the heartwoods Paratecoma peroba, Tabebuia chrysantha and Tabebuia heptaphylla.231,232 3-Hydroxy (196) and 3-,4-dihydroxy-chromanes (197) were isolated from a trunkwood extract of Tabebuia heptaphylla.232
Zhuang et al. isolated illihenryipyranol A (198) from roots of Illicium henryi in minor amounts.233
6.2 Fungi
Deadalin A (199), also called quercinol, was independently discovered by Morimura et al. and Gebhardt et al. from the mycelial culture broth of Daedalea dickinsii and Daedalea quercina, respectively.234,235Later, 5-methoxy-deadalin A (200), 6-methoxy-deadalin A (201), and 9-deoxy-deadalin A (202) were isolated from Daedalea dickinsii by Morimura and colleagues (Fig. 27).236 Deadalin A has been shown to have anti-tyrosinase activity (IC50 of 194 μM) and thus to inhibit melanin synthesis in a three-dimensional human skin model.
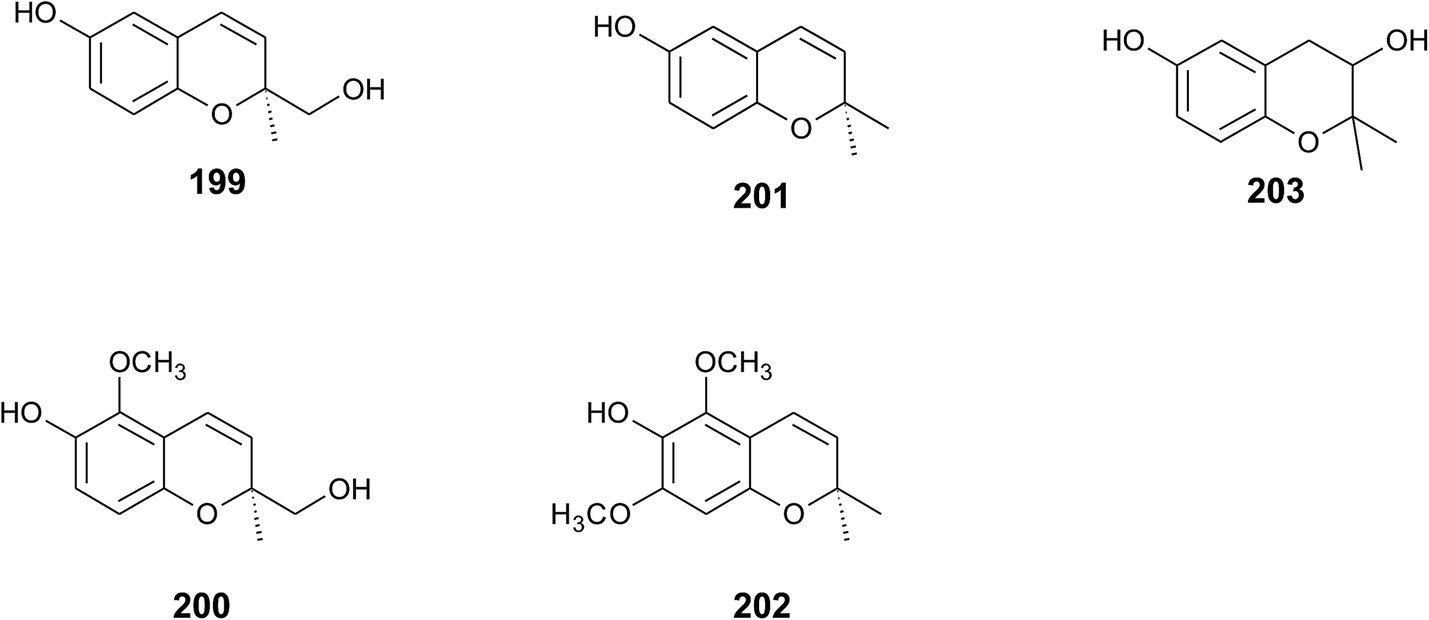 | ||
| Fig. 27 Structures of hemiterpenes (199 to 202) from plants and fungi. | ||
Tanaka et al. isolated a 3-hydroxy-chromane (203) from Acremonium murorum,237 a hyaline phialide.
All hemiterpenoid chromanols from fungi are derived from simple prenylated phenols and not related to the biosynthetic pathway of tocopherols.
7. Animal tocochromanol metabolism
For a complete overview of side chain-modified 6-hydroxy-chromanols, we present in the following animal and human vitamin E metabolites. In recent years, these metabolites have been intensively studied for anti-inflammatory and cytotoxic activity (see also sections below) and were discovered as novel regulatory and signaling molecules. Studies on vitamin E metabolism were summarized in several outstanding reviews;17,238,239 we therefore describe here only briefly the formation and activities of these metabolites.The hepatic metabolism of tocopherols follows the classical activation of branched chain hydrocarbons by cytochrome P450 enzymes (most likely CYP4F2) within the endoplasmic reticulum.240 ω-Hydroxylation of α-tocopherol forms α-13′-hydroxy-tocopherol (204) (13′-OH) with subsequent oxidation to α-13′-carboxy-tocopherol (205) (13′-COOH) by aldehyde dehydrogenase (Fig. 28). Both metabolites were detected in human plasma and show anti-inflammatory and cytotoxic activity in in vitro- and in vivo systems (see corresponding Section 8 and 9).241,242
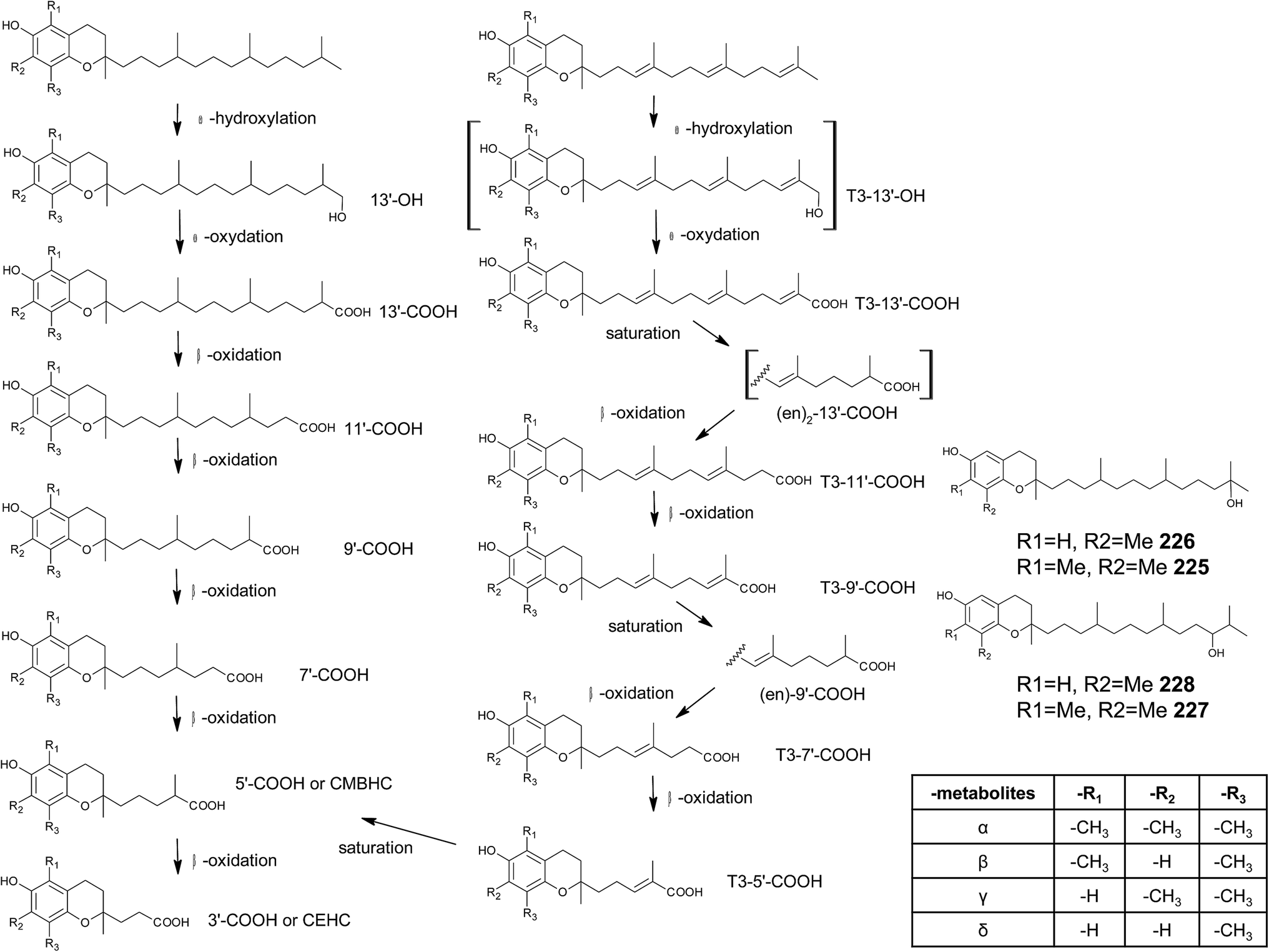 | ||
| Fig. 28 Mammalian metabolism of tocopherols and tocotrienols. α-TOH: α-tocopherol; α-tocotrienol: α-tocotrienol; 13′-OH: 13′-hydroxy-chromanol; 13′-COOH: 13′-carboxy-chromanol; CDMD(en)2HC: carboxy-dimethyl-decadienyl-hydroxy-chromanol; CDMOenHC: carboxy-dimethyl-octenyl-hydroxy-chromanol; CDMHenHC: carboxy-methyl-hexenyl-hydroxy-chromanol; CMBenHC: carboxy-methyl-butadienyl-hydroxy-chromanol; CDMOHC: carboxy-methyl-octyl-hydroxy-chromanol; CDMHHC: carboxy-methyl-hexyl-hydroxy-chromanol; CMBHC: carboxy-methyl-butyl-hydroxy-chromanol; CEHC: carboxy-ethyl-hydroxy-chromanols. | ||
Further degradation of the long-chain metabolites (LCM) occurs like that of methyl branched-chain fatty acids by β-oxidation, subsequently cutting out 2- and 3-carbon units, respectively. The β-oxidation takes place in the peroxisomes and results in 11′-COOH and 9′-COOH LCM.240 Mustacich et al. suggested that further degradation occurs within the mitochondrial matrix where intermediate-chain metabolites (7′-COOH and 5′-COOH) and short-chain metabolites 3′-COOH (or carboxy-ethyl-hydroxy-chromanol (CEHC)) were detected.240 CEHCs were the first metabolites identified in human and animal (rats and mice) studies, since they are secreted in urine.243 δ-9′-COOH (206), α-5′-COOH (also known as α-carboxy-methylbutyl-hydroxy-chromanol, α-CMBHC; 207), and α-3′-COOH (α-CEHC; 208) were investigated for their anti-inflammatory properties in vitro.244,245 Interestingly, the degradation of tocotrienols results in the formation of CEHC, suggesting a similar degradation mechanism as seen for tocopherols. Indeed, analogous catabolic steps were found for tocotrienol in vitro and in vivo242,246 (Fig. 28). Initial ω-hydroxylation followed by five cycles of β-oxidation follows in principle that of tocopherols; however, the double bonds between C-4′–C-5′, C-7′–C-8′ and C-11′–C-12′ undergo a saturation step, which is catalyzed by 2,4-dienoyl-CoA reductase and 3,2-enoyl-CoA isomerase (Fig. 28). The following metabolites of tocotrienol were identified by in vitro experiments in hepatic adenoma HepG2 cells and in mice feces, respectively:242,246 13′-carboxy-trienols (α-, γ-, δ-tocotrienol-13′-COOH; 209, 31, 30) which are identical with the naturally occurring garcinoic acids, e.g. α-, γ-, and δ-garcinoic acid, carboxy-dimethyl-decadienyl-hydroxy-chromanol (α-, γ-, δ-CDMD(en)2HC or α-, γ-, δ-(en)2-11′-COOH, (210, 211, 212)), carboxy-dimethyl-octadienyl-hydroxy-chromanols (α-, γ-, δ-CDMO(en)2HC or α-, γ-, δ-(en)2-9′-COOH, (213, 214, 215)), as well as carboxy-dimethyl-octenyl-hydroxy-chromanols (α-, γ-, δ-CDMOenHC or α-, γ-, δ-(en)-9′-COOH, 216, 217, 218), carboxy-methyl-hexenyl-hydroxy-chromanol (α-, γ-, δ-CMHenHC or α-, γ-, δ-(en)-7′-COOH, (219, 220, 221)), and carboxy-methyl-butadienyl-hydroxy-chromanol (α-, γ-, δ-CMBenHC or α-, γ-, δ-(en)-5′-COOH (222, 223, 224),). Tocotrienols are metabolized with a higher rate than α-tocopherol; thus, depending on dietary tocotrienol intake, the plasma concentrations exceed that of α-tocopherol metabolites. Except for the 13′-carboxy-trienols (see next section), biological properties of tocotrienol metabolites are largely unknown.
All tocopherol and tocotrienol metabolites can occur in free form or as phase II conjugates, such as sulfates or glucuronides.242,247
Bardowell et al. investigated the role of the murine Cyp4f14 gene, an orthologue of the human CYP4F2 gene, in vitamin E metabolism in Cyp4f14-knockout mice and found two new metabolites, namely 12′-hydroxy-tocopherol (12′-OH: γ- and δ-12′-OH, (225 and 226)) and 11′-hydroxy-tocopherol (11′-OH: γ- and δ-11′-OH, (227 and 228)) in fecal pellets of mice fed a diet rich in γ-tocopherol.248,249 The metabolites derive from ω-1 and ω-2-hydroxylation and were excreted via bile into feces of mice and humans.
8. Anti-inflammatory activity of toco-chromanols and -chromenols
Many diseases, including atherosclerosis, diabetes or even cancer, are related to inflammatory processes. A decreased grade of inflammation could lead to a reduced risk for these diseases. In the past, human clinical trials with α-tocopherol as an anti-inflammatory agent revealed contradictory results.17,239,250,251 We here like to broaden the view to structurally related chromanols and chromenols and compare their anti-inflammatory in vitro activity.The anti-inflammatory activities of tocopherols and tocotrienols from the human diet are well known and are compiled in Table 1.239,245,252 In general, the chromanols with saturated and unsaturated side chains showed good to moderate inhibitory activity depending on the anti-inflammatory marker measured and the in vitro system used.253,254 For example, Jiang et al. investigated the inhibition of COX-2 catalyzed PGE2 synthesis in IL-1β stimulated human lung epithelial A549 cells of a series of tocopherols and tocotrienols, respectively.245 The inhibitory activities reached from IC50 > 50 μM for α-tocopherol to IC50 = 1–3 μM for δ-tocopherol and γ-tocotrienol.
The LCM of tocopherols and tocotrienols with a terminal C-13′-carboxy and -hydroxyl group, respectively, were found in nanomolar concentration in human plasma and intensively studied as anti-inflammatory agents. The research on the LCM was promoted by the facile semi-syntheses from garcinoic acid that can be efficiently isolated from Garcinia kola.38,39
α-13′-Carboxy-tocopherol (205) (α-13′-COOH) inhibited the expression of iNOS by 100% at 5 μM and the formation of nitric oxide by 100% at 2.7 μM, respectively.255,256 A recent investigation on the anti-inflammatory activity of α-13′-COOH showed strong inhibition of recombinant 5-LOX and only moderate inhibition of COX-1, leukotriene (LT) C4 synthase, PGES-1 and epoxide hydrolase.254 Human recombinant COX-2 was not inhibited by α-13′-COOH at low concentrations.
The δ-tocopherol metabolite δ-13′-carboxy-tocopherol (229) (δ-13′-COOH) showed a slightly reduced activity, and inhibited iNOS protein synthesis by 56% at 5 μM and nitric oxide formation by 79% at 5 μM, respectively, in LPS-activated murine RAW264.7 macrophages. δ-13′-COOH further inhibited the LPS-induced upregulation of COX-2 expression in the same cells with IC50 values of 4–5 μM.245,257 5-LOX activity was inhibited in the range from 0.5–2.0 μM, depending on the assay used.253
The 13′-Hydroxy-tocopherols of α-, and δ-tocopherol (α-13′-OH (204) and δ-13′-OH (231)) are synthetically available and have been therefore intensively studied. α-13′-OH reduced COX-2 expression (49%) and PGE2 synthesis (54%) and both alcohols inhibited iNOS expression by 53–60% at 10 μM in LPS-induced murine RAW264.7 macrophages.241,256 Both alcohols can be biochemically converted by mammalian cells to the corresponding acids, thus making it difficult to distinguish between the activity of 13′-OH and 13′-COOH (unpublished results).
As described in the section on human metabolism, the metabolic truncation of the LCM leads to several medium- and short-chain metabolites, such as 9′-carboxy-tocopherols (9′-COOH), CMBHC and CEHC, respectively. In general, the anti-inflammatory activities of the medium- and short-chain metabolites seem to decrease with the decreasing lengths of the side chains, resulting in higher IC50 values (Table 1).245
In summary, although the number of in vitro studies ist still limited and different markers of inflammation cannot be compared directly, we roughly estimate the anti-inflammatory activity of tocopherols, tocotrienols and their metabolites as follows: α-tocopherol < non-α-tocopherol ∼ tocotrienols ≪ 13′-OH ∼ 13′-COOH ≫ 9′-COOH > CMBHC ∼ CEHC. However, it must be kept in mind that the molecular modes of action of these molecules seem to be quite different. It is obvious that the impact of the metabolites depends on individual metabolism rates (pharmacokinetics) of the tocochromanols from the diet. Grebenstein et al. proposed that the affinity of vitamers towards the α-tocopherol transfer protein (α-TTP) may predict their degradation by cytochrome P450 enzymes.258 α-TTP has the strongest affinity for α-tocopherol with Kd of 25 nM and much higher Kd values for the other vitamin E forms, depending on their methylation pattern and side chain saturation.239,259 Accordingly, the catabolism of non-α-tocopherol vitamers into the corresponding LCM may occur much faster than that of α-tocopherol, thus generating more anti-inflammatory metabolites. As a result, α-tocopherol per se is less active than all other vitamers following the order: δ-tocopherol ∼ γ-tocotrienol > γ-tocopherol ≫ α-tocopherol.239
δ-Garcinoic acid (30) is the main constituent of several Garcinia species, which are known for their anti-inflammatory properties in African ethnomedicine.37 δ-Garcinoic acid was reported to inhibit COX-2 (IC50 = 10 μM) and, even stronger, 5-LOX with IC50 ranging from 0.04 to 1.0 μM.257 δ-Garcinoic acid down-regulated the LPS-induced expression of pro-inflammatory cytokines, such as TNF-α, IL-6, IL-1β, COX-2 and iNOS in macrophages and reduced production of nitric oxide (IC50 value of 1 μM).255,260 A direct comparison of several carboxy-tocotrienols (tocotrienol-13′-COOH metabolites) from plant origin as inhibitors of microsomal PGE2 synthase-1 revealed the following order of activity: γ-garcinoic acid (31) > β-garcinoic acid (232) > δ-garcinoic acid (30) > α-garcinoic acid (209); however the methylation pattern had only moderate impact.44
Structurally related forms of δ-garcinoic acid, such as δ-sargachromenol (51) with a 15′-COOH group and a chromene ring system, showed only moderate inhibitory activity on in LPS-stimulated production nitric oxide and PGE2 in murine RAW 264.7 macrophages (IC50 values of 82 μM and 30.2 μM, respectively);129 however, much higher activity was observed in BV-2 microglial cells (IC50 value for inhibition of nitric oxide production of 1.3–2.7 μM).134,261
As described above, 13′-OH metabolites have a similar anti-inflammatory potential than the corresponding 13′-COOH. Thus, natural products such as sargachromanols D (57), E (58) and G (60), respectively, with hydroxyl-groups at C-9′ and C-10′ are interesting intermediates. They all showed moderate inhibitory activity on nitric oxide production in LPS-stimulated murine RAW 264.7 cells (IC50 values of 15–40 μM).79,83,84
Cyclic meroditerpenes such as epitaondiol (79) and the chromarols A to D (113 to 116) exhibited anti-inflammatory activity in vitro and in vivo (Table 1).99,154 The four chromarols A to D inhibited 15-LOX with IC50 = 0.6(113), 4.0(114), 0.7(115) and 1.1 μM (116), respectively, but not 12-LOX. Epitaondiol was effective in a TPA-induced mouse ear edema study (IC50 = 20.7 μg per ear) and inhibited eicosanoid synthesis with an IC50 of 3.8 μM for thromboxane B2 (TXB2) and an IC50 of 30.1 μM for LTB4.
The cyclic sesquiterpenes capillobenzopyranol (172) only moderately inhibited nitric oxide production in LPS-stimulated macrophages by 37% at 10 μM.202 The monoterpene cordiachromene A (173) inhibited soybean 15-LOX with an IC50 of 0.82 μM and lipid peroxidation with an IC50 of 2 μM.214
Only moderate anti-inflammatory activity was observed for the hemiterpene quercinol (199), whereas it inhibited COX-2 expression with an IC50 of 0.63 μM.235
In Fig. 29 we postulate the structural motives that are essential for the anti-inflammatory activity based on the structures and properties discussed above. The most effective compounds described are the diterpenes 13′-COOH, 13′-OH, garcinoic acid and δ-sargachromenol, respectively, with strong potential as anti-inflammatory drug candidates. A recent SAR study revealed that the effects of human LCM depend on the presence of the chromanol ring and modifications in the side chain and less on the substitution pattern at the aromatic ring.256 This study is in line with the observation of Silva et al. with δ-sargachromenol (51) and its precursor 1,4-benzoquinone sargaquinoic acid; the latter had less inhibitory activity towards LOX- and COX-enzymes.52 In addition to the natural compounds described above, the anti-inflammatory and anti-diabetic drug troglitazone exhibits a 6-hydroxy-chromane ring system. Troglitazone was used a PPAR-γ-receptor agonist but was withdrawn from the market since it caused hepatotoxicity.262 Obviously, anti-inflammatory activity is enhanced by the occurrence of a 6-hydroxy-chromane and -chromene moiety, respectively.
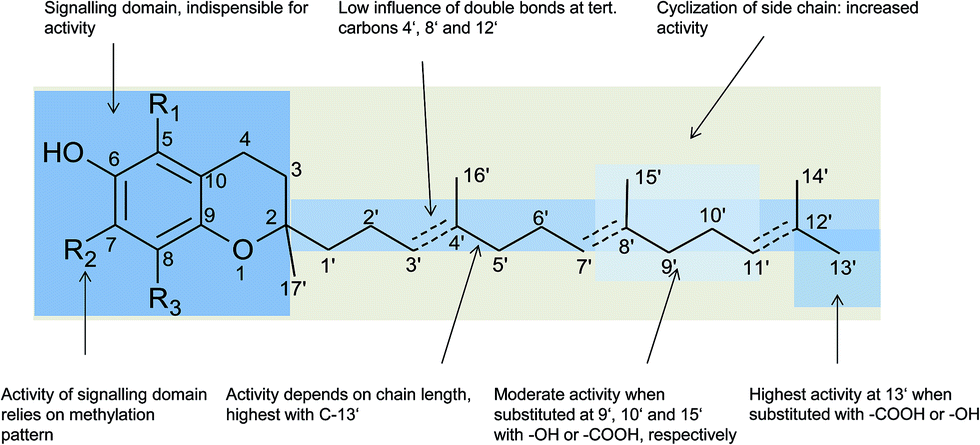 | ||
| Fig. 29 General domains modulating the anti-inflammatory activity of meroterpenoids. | ||
In conclusion, meroditerpenoids with a functional group (COOH, OH) at the side chain have much higher anti-inflammatory activity than the parent chromanols and chromenols, respectively.
9. Anti-proliferative and cytotoxic activity of chromanols and chromenols
Dietary tocopherols and tocotrienols have been extensively investigated for their cancer-preventive potential in several human intervention trials (reviewed in263 and6), but widely failed to prove beneficial effects.264 However, in vitro studies with tocopherols and tocotrienols in cell cultures and in vivo studies have shown pronounced anti-neoplastic and anti-carcinogenic effects.265,266The susceptibility of the cell lines tested for anti-carcinogenic activity varied tremendously and makes thus it difficult to compare the compounds discussed in this section. For example, HepG2 liver cells exhibit greater resistance to drugs and toxins compared to other cells lines, since they actively express phase I and II enzymes. As a result, higher IC50 values are expected for drug resistant cell lines, such as HepG2.
Structure–activity relationship studies revealed that chemical modifications at C-6 of the aromatic ring (ethers or esters) magnified the cytotoxic potential of vitamin E compounds.266,267 In general, drug candidates that were ‘redox-silent’ at C-6, such as tocopherol-succinate, showed promising results in animal studies.268 Although most of the redox-silent compounds were chemically synthesized, distinct structure–activity relationships have been derived from these experiments.266 The studies revealed the importance of three major domains of the chromanols tested: first, the functional domain (I) that needs to be ‘redox silent’ to exert the cytotoxic properties. Second, the signaling domain (II) modified by the methylation pattern of the chromanol ring system. Third, the hydrophobic domain (III) that is mostly covered by saturated and unsaturated side chains.267 Reviewing the structural features of the molecules presented here, we further specify the domains that are relevant for cytotoxicity.
Tocopherols seem to have low or moderate anti-cancer activity in the different cell model systems.267,269 α-Tocopherol (3) in particular is well-tolerated by adenoma and cancer cells in supra-physiological concentrations (above 100 μM) (see Table 2).269 γ-Tocopherol (5) at 25 μM, however, had anti-proliferative activity on colon carcinoma (CaCo-2), androgen-sensitive (LNCaP) and androgen-resistant (PC-3) prostate, lung adenocarcinoma (A549), and osteosarcoma (SaOs-2) cells, but did not induce apoptosis in these cell lines.269,270 Accordingly, we postulate the following order of activity for tocopherols: γ-tocopherol (5) > δ-tocopherol (6) ≫ α-tocopherol (3).
Tocotrienols showed anti-proliferative and pro-apoptotic effects in vitro and in vivo and are in general more potent in the prevention of cancer than tocopherols.271 Several molecular targets were identified for γ- and δ-tocotrienols (16) and (17) (γ- and δ-tocotrienol), respectively (summarized by272). Induction of mitochondrial apoptosis, demonstrated by activation of caspase-3 and -9, along with modulation of apoptogenic genes such as Bcl-2, Bcl-xl and Bax, respectively, has been observed for most of the tocotrienols tested.
α-Tocotrienol (14) showed low to moderate pro-apoptotic activity against different breast cancer (MDA-MB-435, MDA-MB-231, and MCF7) and melanoma (B16) cells, respectively (Table 2).25,273,274 In contrast, γ- (16) and δ-tocotrienol (17) induced apoptosis at low micromolar concentrations in most of the cell lines tested.25,267,275,276 Sargaol (95) (3-4-dehydro-δ-tocotrienol), has been tested with moderate activity in human gastric epithelial cells, human fibroblasts and murine lymphocytic leukemia P338 cells.103,120 Interestingly, fascioquinol F (109) (3-4-dehydro-desmethyl-tocotrienol) showed no inhibitory activity against human gastric adenoma (AGS) and human neuroblastoma (SH-SY5Y) cells, respectively.151 In contrast, desmethyl- and didesmethyl-tocotrienol (18) and (19) strongly induced apoptosis in B16 melanoma cells (IC50 values of about 1 μM).24,25 In conclusion, the activity order is determined by the methylation pattern of the chromanol ring: desmethyl-tocotrienol (18) ∼ didesmethyl-tocotrienol (19) > γ-tocotrienol (16) ∼ δ-tocotrienol (17) ∼ 3-4-dehydro-δ-tocotrienol (95) ≫ α-tocotrienol (14).
Only recently, tocopherol- and tocotrienol-metabolites were investigated for their anti-carcinogenic activity. 13′-Carboxylic acids, including garcinoic acid, induced apoptosis in the lower micromolar range with slight differences depending on their methylation pattern and double bonds in the side chain, respectively. The tocopherol metabolites 13′-carboxy-α-tocopherol (205) and 13′-carboxy-δ-tocopherol (229) induced caspase-3-dependent apoptosis in human HepG2 liver cells (IC50 values of 13.5 μM and 6.5 μM, respectively).39 Similar activities were observed in human THP-1 macrophages, glioma C6, colon carcinoma HCT-116 and colon adenocarcinoma HT-29 cells (Table 2).38,257,277 The natural product and tocotrienol metabolite garcinoic acid (30) showed similar activities. δ-Sargachromenol (51) and fallachromenoic acid (105) were both active in the lower micromolar range.126,133 Thus, the shift of the carboxylic group at C-15′ does not affect the pro-apoptotic activity.
The introduction of hydroxyl group(s) within the side chain is associated with pro-apoptotic effects. Crassumtocopherols A (134) and B (135) (C-8′–C-11′–C-12′-triols) showed strong inhibitory activity towards murine P338 leukemia and human colon adenocarcinoma HT-29 cells (IC50 values of 5.2–7.5 μM).169 Somewhat lower activities were found for diols such as δ- and γ-amplexichromanols (35) and (36) (C-13′–C-14′-diols), litchtocotrienol A (41) (C-11′–C-12′-diol), and sargachromanol E (58) (C-9′–C-10′-diol) (Table 2).43,49,82 Sargatriol (98) (C-5′–C-6′-diol), as well as sargadiol-I (96) (C-6′–OH) and -II (97) (C-8′–OH) are weak inhibitors in P338 leukemia cells with IC50 values between 30 and 40 μM.118,120 All compounds with mono-hydroxy-substituted side chains showed low to no cytotoxic activity. Based on the hydroxylation pattern of the side chain, we estimate the following activity order: C-8′–C-11′–C-12′-triol > C-13′–C-14′-diol ∼ C-11′–C-12′-diol ∼ C-9′–C-10′-diol > C-8′–OH ∼ C-5′–C-6′-diol ∼ C-6′–OH ≫ C-13′–OH.
Cyclizations of the tocotrienol side chain lead to epitaondiols, strongylophorines and bifurcarenone-derived chromanols. All compounds tested showed moderate to weak anti-cancerogenic activities (Table 2)
Unfortunately, only few data exist for the anti-cancer activities of sesquiterpenes. Paniceins A2 (160) and F2 (161) both inhibited growth of P330, lung adenocarcinoma A549 cells, uveal melanoma MEL20 cells, and colon adenocarcinoma HT-29 cells with IC50 values of around 15 μM (ref. 195) and riccardiphenol C (146) was not active.177
Monoterpenes and hemiterpenes both demonstrated medium to low inhibitory activity towards cancer cells (Table 2).
In conclusion, meroditerpenoids exhibited the strongest inhibitory activity towards cancer cells among all meroterpenoids described, especially when a carboxy or more than one hydroxyl group is present at the terminal end of the side chain (Fig. 30).
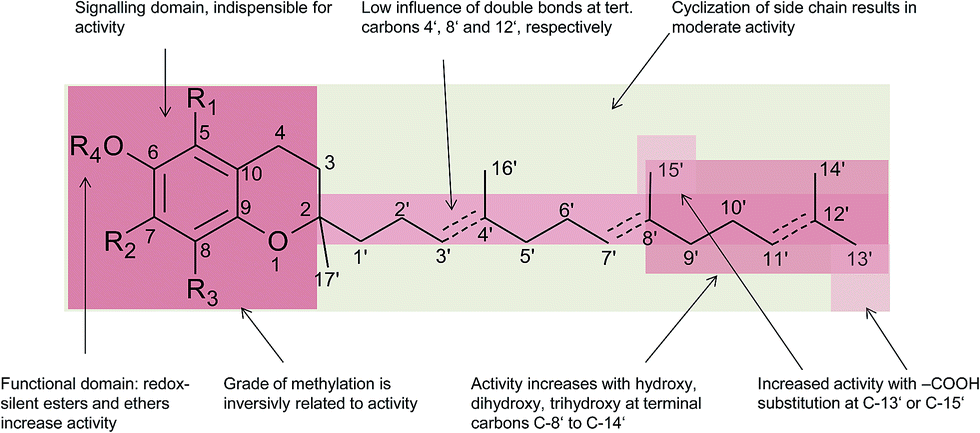 | ||
| Fig. 30 General structural motives important for the anti-cancer activities of meroterpenoids. | ||
10. Discussion
This review describes more than 230 6-hydroxy-chromanols and -chromenols, respectively that were found in terrestrial and marine organisms. Fig. 31a highlights the distribution of meroterpenes within different phylae. Marine organisms, led by brown algae (Phaeophyceae), cover two thirds of the molecules presented in this review, followed by plants and fungi. Interestingly, sponges (porifera) produce 18% of the natural products presented here, mainly cyclic di- and sesqui-terpenes.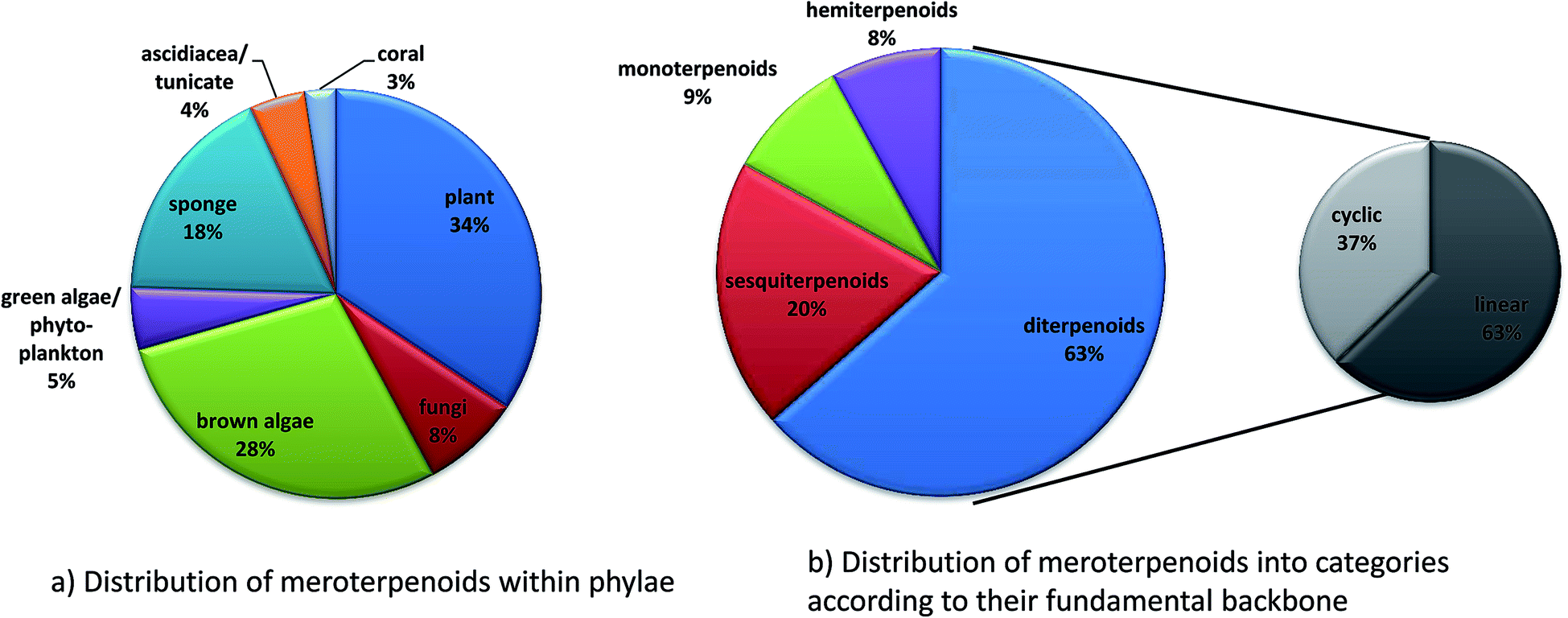 | ||
| Fig. 31 Statistical distribution of meroterpenoids. | ||
Meroditerpenes represent almost two thirds of all compounds discussed and are divided into 63% with linear and 37% with cyclic side chains, respectively (Fig. 31b). The occurrence of sesquiterpenes was dominant in sponges, whereas hemiterpenes were only found in plants and fungi.
During the course of this compilation, the question arose whether or not the stereo-controlled cyclization of toluquinols to a chromane or chromene ring with R-configuration at C-2 occurs exclusively in terrestrial species. The evidence for this process in plants is well documented and the isolation of several cyclases substantiates the biosynthetic step. Marine-derived meroterpenes were often isolated as mixtures of stereoisomers at C-2 and several authors debated the isolation of chrom(e)anols as artefacts of the work-up procedures or as non-enzymatic reaction products within the organism. In addition, the monocyclic 1,4-benzoquinone precursors were isolated in most cases with high yields, whereas toluquinols in plant species occur only in trace amounts and were rarely described. From the 49 diterpenes isolated from plants, 46 (94%) were described with R-configuration. A statistical analysis of chromanols and chromenols from marine species revealed that 73% of chromanols were isolated as R-enantiomers, whereas only 26% of all chromenols show optical activity with R-configuration. In conclusion, we postulate that marine organisms most likely produce chromanols via enzyme-catalyzed cyclization, whereas chromenols may mostly originate from non-enzymatically cyclization or as an artefact during sample work-up.
The structural variability of the compounds described in this review is remarkable. Side chain modifications by oxidation and/or cyclization occur widely, especially in marine organisms. Cytochrome P450 enzymes are most likely responsible for the initial oxidation to epoxy-, hydroxy- and carboxy-derivatives, respectively, although the corresponding enzymes were studied only in animal vitamin E metabolism and are not fully understood yet.248,249
Cyclization of the prenylated side-chain occurs via different pathways. The first pathway begins with an acid-catalyzed cyclization cascade between C-2–C-7, C-6–C-11 and C-10–C-15 of the sesquiterpenes and diterpene backbone, respectively, that leads to di- or tricyclic 1,4-hydroquinones. This is followed by a second acid-catalyzed formation of the chromane ring as described by Kurata et al (Fig. 20).186 Several examples for this cyclization, such as chromazoranol (154) or strongylophorines (117–130), are described above.
The second cyclization pathway occurs via an epoxidation of the terminal double bond, followed by an acid-catalyzed cyclization cascade with a final cyclization of the chromane ring, as first described by Etse et al.61,92,93 (see also Fig. 7 and 10). It remains unclear, if these mechanisms occur simultaneously or sequentially. Walsurol (50), cyclolitchtocotrienol A (49) or the taondiols (76–80) are examples of the second pathway.
A third cyclization pathway occurs via the formation of the 1,4-hydroquinone precursor bifurcarenone (81) by an acid-catalyzed anti-Markovnikoff cyclization between C-7 and C-11.278 Subsequent cyclization reactions lead to cystoketal chromane (89), mediterraneols (82–84) and cystoseirols (86, 87).
Only three meroterpenes with a cyclic side chain have been described in plants, namely walsurol (50), cyclolitchtocotrienol A (49) (Fig. 7) and riccardiphenol C (146) (Fig. 19).
With some exceptions, all higher plants produce side chain-saturated tocopherols with the typical methylation pattern α-, β-, γ-, and δ-, respectively. Next, several algae have the ability to produce tocopherols, although in low yields. 8-Methyl- or desmethyl-tocotrienol moieties were found in most of the structures described from marine organisms. Only three tocopherol-derivatives with a full methylation pattern (α-) were identified in marine organisms, namely marine-derived tocopherol (25) from phytoplankton, α-tocoxylenoxy (108) from the green alga Caulerpa racemosa and chrassumtocopherol from the soft coral Lobophytum crissum.
The primary biological function of the side chain modifications remains unclear. On the one hand, cytotoxicity, algicidal and anti-macroalgal activity was found for several metabolites. On the other hand, the settling of sea urchins and perna eggs was induced by several compounds. Thus, side chain-modified metabolites are presumably used as chemical protectants or as signalling molecules for intercellular communication or both.
Recent advances in the research on human vitamin E metabolites led us to a comprehensive search for chromanol- and chromenol-structures with anti-inflammatory and cytotoxic properties (see Tables 1 and 2). The number of structurally related compounds exceeded our expectations. We therefore merged the available information on over 30 compounds and identified structural motives that correspond to high anti-inflammatory activities (Table 1). Most of the compounds described here affected arachidonic acid metabolism and also the synthesis of pro-inflammatory cytokines. Inhibition of COX-1 and COX-2 expression, respectively, reduced prostaglandin metabolite formation and inhibition of 5- and/or 12-LOX blocked leukotriene synthesis. Further studies will have to reveal if meroterpenoids have the potential to be developed into anti-inflammatory drug candidates.
Cytotoxicity data of approximately 50 compounds were collected (Table 2). Like the anti-inflammatory activities of meroterpenoids, diterpenes showed the strongest activity, led by side chain-modified chromanols. Anti-proliferative and cytotoxic properties were modulated by the presence of hydroxyl and carboxy groups. Activation of caspases-3 and -9, respectively, suggested that most of these compounds induce a mitochondrial death pathway.
Rangasany et al. evaluated the drug-likeness of several natural products isolated from algae and found δ-sargachromenol (51) and epitaondiol (79) as good fits to Lipinski's ‘Rule of Five’. This rule estimates the potential of a drug candidate based on physio-chemical properties, such as molecular weight, number of hydrogen bond acceptors and donors, and distribution coefficient (logP).73,141 We screened a series of compounds described in this review (ESI Table 1†) and found many with good predicted oral bioavailability, based on these calculations which were conducted via Molinspiration WebME editor version 1.16 (http://www.molinspiration.com).
We and others tested several vitamin E metabolites for their biological activity in vitro and in vivo and found them to have anti-bacterial, anti-viral, anti-inflammatory and cytotoxic properties (Tables 1 to 3). In general, any modification of the prenyl side chain increased their biological activity.
In this review, we thoroughly described the class of 6-hydroxy-chromanols and -chromenols within living nature and summarize their biological properties, in particular their anti-inflammatory and anti-carcinogenic potential. Based on the presented evidence, we conclude that the presence of a hydroxyl or carboxy group in the side chain enhances the anti-inflammatory activity of natural chromanols and chromenols. With respect to anti-proliferative and anti-cancer activities, we conclude that, among all meroterpenoids described, meroditerpenoids have the strongest inhibitory activity towards cancer cells, in particular when, again, bearing a carboxy or more than one hydroxyl group at the terminal end of the side chain. We therefore propose that the presence of a terminal hydroxyl or carboxy group in the side chain of the long-chain vitamin E metabolites warrants further investigation and might help us to unravel the as yet unknown essential biological function(s) and modes of action of vitamin E in animals.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge support by the Open Access Publishing Fund of Hochschule Fulda – University of Applied Sciences. The work of Marc Birringer is supportet by grants from the Internal Research Support of Hochschule Fulda – University of Applied Sciences. The work of Stefan Lorkowski is supported by grants from the Federal Ministry of Education and Research (01EA1411A), the Deutsche Forschungsgemeinschaft (DFG; RTG 1715), the German Ministry of Economics and Technology (AiF 16642 BR) via AiF (the German Federation of Industrial Research Associations) and FEI (the Research Association of the German Food Industry), and by the Free State of Thuringia and the European Social Fund (2016 FGR 0045).References
- H. M. Evans and K. S. Bishop, Science, 1922, 56, 650–651 CAS.
- R. Brigelius-Flohé and M. G. Traber, FASEB J., 1999, 13, 1145–1155 CrossRef.
- H. M. Evans, O. H. Emerson and G. A. Emerson, J. Biol. Chem., 1936, 113, 319–332 CAS.
- D. C. Liebler, J. A. Burr, L. Philips and A. J. Ham, Anal. Biochem., 1996, 236, 27–34 CrossRef CAS PubMed.
- L. Gille, T. Rosenau, A. V. Kozlov and W. Gregor, Biochem. Pharmacol., 2008, 76, 289–302 CrossRef CAS PubMed.
- H. Y. Peh, W. S. D. Tan, W. Liao and W. S. F. Wong, Pharmacol. Ther., 2016, 162, 152–169 CrossRef CAS PubMed.
- European Patent Office, https://www.epo.org/.
- R. A. C. Sussmann, W. L. Fotoran, E. A. Kimura and A. M. Katzin, Parasites Vectors, 2017, 10, 461 CrossRef PubMed.
- (a) A. Block, R. Fristedt, S. Rogers, J. Kumar, B. Barnes, J. Barnes, C. G. Elowsky, Y. Wamboldt, S. A. Mackenzie, K. Redding, S. S. Merchant and G. J. Basset, J. Biol. Chem., 2013, 288, 27594–27606 CrossRef CAS PubMed; (b) N. Yan, Y. Liu, H. Zhang, Y. Du, X. Liu and Z. Zhang, Molecules, 2017, 22, pii: E510 CrossRef PubMed.
- (a) L. Spicher and F. Kessler, Curr. Opin. Plant Biol., 2015, 25, 123–129 CrossRef CAS PubMed; (b) J. Falk and S. Munné-Bosch, J. Exp. Bot., 2010, 61, 1549–1566 CrossRef CAS PubMed; (c) P. Dörmann, Planta, 2007, 225, 269–276 CrossRef PubMed; (d) R. Szymańska and J. Kruk, Plant Physiol. Biochem., 2018, 122, 1–9 CrossRef PubMed.
- J. Kruk, A. Pisarski and R. Szymańska, J. Plant Physiol., 2011, 168, 2021–2027 CrossRef CAS PubMed.
- G. Horvath, L. Wessjohann, J. Bigirimana, M. Jansen, Y. Guisez, R. Caubergs and N. Horemans, Phytochemistry, 2006, 67, 1185–1195 CrossRef CAS PubMed.
- (a) J. Green, D. McHale, S. Marcinkiewicz, P. Mamalis and P. R. Watt, J. Chem. Soc., 1959, 3362–3373 RSC; (b) J. Green and S. Marcinkiewicz, Nature, 1956, 177, 86–87 CrossRef CAS PubMed.
- (a) J. Bunyan, D. McHale, J. Green and S. Marcinkiewicz, Br. J. Nutr., 1961, 15, 253–257 CrossRef CAS PubMed; (b) J. Bunyan, Nature, 1961, 181, 1237 Search PubMed.
- G. Klink, A. Buchs and F. O. Gülacar, Phytochemistry, 1994, 36, 813–814 CrossRef CAS.
- S. Krauß, S. Hammann and W. Vetter, J. Agric. Food Chem., 2016, 64, 6306–6311 CrossRef PubMed.
- F. Galli, A. Azzi, M. Birringer, J. M. Cook-Mills, M. Eggersdorfer, J. Frank, G. Cruciani, S. Lorkowski and N. K. Özer, Free Radical Biol. Med., 2017, 102, 16–36 CrossRef CAS PubMed.
- J.-M. Zingg, M. Meydani and A. Azzi, BioFactors, 2012, 38, 24–33 CrossRef CAS PubMed.
- W. Müller-Mulot, G. Rohrer, G. Oesterhelt, K. Schmidt, L. Allemann and R. Maurer, Fette, Seifen, Anstrichm., 1983, 85, 66–71 CrossRef.
- B. Brem, C. Seger, T. Pacher, M. Hartl, F. Hadacek, O. Hofer, S. Vajrodaya and H. Greger, Phytochemistry, 2004, 65, 2719–2729 CrossRef CAS PubMed.
- Y.-S. Kil, J. Park, A.-R. Han, H. A. Woo and E.-K. Seo, Molecules, 2015, 20, 5965–5974 CrossRef CAS PubMed.
- F. Shahidi and A. C. deCamargo, Int. J. Mol. Sci., 2016, 17, 1745–1774 CrossRef PubMed.
- H. Ashan, A. Ahad, J. Iqbal and W. A. Siddiqui, Nutr. Metab., 2014, 52, 1–22 Search PubMed.
- A. A. Qureshi, H. Mo, L. Packer and D. M. Peterson, J. Agric. Food Chem., 2000, 48, 3130–3140 CrossRef CAS PubMed.
- L. He, H. Mo, S. Hadisusilo, A. A. Qureshi and C. E. Elson, J. Nutr., 1997, 127, 668–674 CAS.
- J. Merza, M.-C. Aumond, D. Rondeau, V. Dumontet, A.-M. Le Ray, D. Séraphin and P. Richomme, Phytochemistry, 2004, 65, 2915–2920 CrossRef CAS PubMed.
- A. Irías-Mata, W. Stuetz, N. Sus, S. Hammann, K. Gralla, A. Cordero-Solano, W. Vetter and J. Frank, J. Agric. Food Chem., 2017, 65, 7476–7482 CrossRef PubMed.
- B. Butinar, M. Bučar-Miklavčič, C. Mariani and P. Raspor, Food Chem., 2011, 128, 505–512 CrossRef CAS PubMed.
- A. Fiorentino, C. Mastellone, B. D'Abrosca, S. Pacifico, M. Scognamiglio, G. Cefarelli, R. Caputo and P. Monaco, Food Chem., 2009, 115, 187–192 CrossRef CAS.
- P. T. Gee, C. Y. Liew, M. C. Thong and M. Gay, Food Chem., 2016, 196, 367–373 CrossRef CAS PubMed.
- S. Hammann, A. Kröpfl and W. Vetter, J. Chromatogr. A, 2016, 1476, 77–87 CrossRef CAS PubMed.
- J. Kruk, R. Szymańska, J. Cela and S. Munne-Bosch, Phytochemistry, 2014, 108, 9–16 CrossRef CAS PubMed.
- K. J. Whittle, P. J. Dunphy and J. F. Pennock, Biochem. J., 1965, 96, 17C–19C CrossRef CAS PubMed.
- A. Siger, P. Kachlicki, J. Czubiński, D. Polcyn, K. Dwiecki and M. Nogala-Kalucka, Eur. J. Lipid Sci. Technol., 2014, 116, 413–422 CrossRef CAS.
- R. Szymańska and J. Kruk, Acta Biochim. Pol., 2010, 57, 105–108 Search PubMed.
- R. L. Rowland, J. Am. Chem. Soc., 1958, 80, 6130–6133 CrossRef CAS.
- Studies in Natural Products Chemistry. Chapter 9-Garcinoic Acid: A Promising Bioactive Natural Product for Better Understanding the Physiological Functions of Tocopherol Metabolites, ed. S. Kluge, M. Schubert, L. Schmölz, M. Birringer, M. Wallert and S. Lorkowski, Elsevier B.V., 2016, vol. 51 Search PubMed.
- F. Mazzini, M. Betti, T. Netscher, F. Galli and P. Salvadori, Chirality, 2009, 21, 519–524 CrossRef CAS PubMed.
- M. Birringer, D. Lington, S. Vertuani, S. Manfredini, D. Scharlau, M. Glei and M. Ristow, Free Radical Biol. Med., 2010, 49, 1315–1322 CrossRef CAS PubMed.
- F. D. Monache, M. Marta, M. M. Mac-Quhae and M. Nicoletti, Gazz. Chim. Ital., 1984, 114, 135–137 Search PubMed.
- K. Terashima, T. Shimamura, M. Tanabayashi, M. Aqil and J. A. Akinniyi, Heterocycles, 1997, 45, 1559–1566 CrossRef CAS.
- W. N. Setzer, T. J. Green, R. O. Lawton, D. M. Moriarity, R. B. Bates, S. Caldera and W. A. Haber, Planta Med., 1995, 61, 275–276 CrossRef CAS PubMed.
- A. Lavaud, P. Richomme, M. Litaudon, R. Andriantsitohaina and D. Guilet, J. Nat. Prod., 2013, 76, 2246–2252 CrossRef CAS PubMed.
- K. Alsabil, S. Suor-Cherer, A. Koeberle, G. Viault, A. Lavaud, V. Temml, B. Waltenberger, D. Schuster, M. Litaudon, S. Lorkowski, R. de Vaumas, J.-J. Helesbeux, D. Guilet, H. Stuppner, O. Werz, D. Seraphin and P. Richomme, Planta Med., 2016, 82, 1110–1116 CrossRef CAS PubMed.
- B. M. Maya, A. Abedini, S. C. Gangloff, A. Kabouche, Z. Kabouche and L. Voutquenne-Nazabadioko, Phytochem. Lett., 2017, 20, 252–258 CrossRef CAS.
- J. S. R. Teixeira, L. deMoreira, M. L. daGuedes and F. G. Cruz, J. Braz. Chem. Soc., 2006, 17, 812–815 CrossRef CAS.
- D. J. Maloney and S. M. Hecht, Org. Lett., 2005, 7, 4297–4300 CrossRef CAS PubMed.
- A. Lavaud, P. Richomme, J. Gatto, M.-C. Aumond, C. Poullain, M. Litaudon, R. Andriantsitohaina and D. Guilet, Phytochemistry, 2015, 109, 103–110 CrossRef CAS PubMed.
- Y.-C. Lin, J.-C. Chang, S.-Y. Cheng, C.-M. Wang, Y.-L. Jhan, I.-W. Lo, Y.-M. Hsu, C.-C. Liaw, C.-C. Hwang and C.-H. Chou, J. Agric. Food Chem., 2015, 63, 2472–2478 CrossRef CAS PubMed.
- D. H. S. Silva, F. C. Pereira, M. V. B. Zanoni and M. Yoshida, Phytochemistry, 2001, 57, 437–442 CrossRef CAS PubMed.
- P. C. Vieira, O. R. Gottlieb and H. E. Gottlieb, Phytochemistry, 1983, 22, 2281–2286 CrossRef CAS.
- D. H. S. Silva, Y. Zhang, L. A. Santos, V. S. Bolzani and M. G. Nair, J. Agric. Food Chem., 2007, 55, 2569–2574 CrossRef CAS PubMed.
- A. L. Pérez-Castorena, A. Arciniegas, M. T. Apan, J. L. Villaseñor and A. R. de Vivar, Planta Med., 2002, 68, 645–647 CrossRef PubMed.
- T. Kusumi, Y. Shibata, M. Ishitsuka, T. Kinoshita and H. Kakisawa, Chem. Lett., 1979, 277–278 CrossRef CAS.
- L. X. Dong, W. U. S. Hua, M. A. Y. Bao and W. U. D. Gang, Acta Bot. Yunnanica, 2001, 23, 515–520 Search PubMed.
- M. Bentinger, M. Tekle, K. Brismar, T. Chojnacki, E. Swiezewska and G. Dallner, J. Biol. Chem., 2008, 283, 14645–14653 CrossRef CAS PubMed.
- T. Kato, A. S. Kumanireng, I. Ichinose, Y. Kitahara, Y. Kakinuma and Y. Kato, Chem. Lett., 1975, 335–338 CrossRef CAS.
- M. Iwashima, J. Mori, X. Ting, T. Matsunaga, K. Hayashi, D. Shinoda, H. Saito, U. Sankawa and T. Hayashi, Biol. Pharm. Bull., 2005, 28, 374–377 CAS.
- E. Komai, T. Miyahara, J. Mori, N. Obi, H. Ochiai, H. Saito and T. Hayashi, Biol. Pharm. Bull., 2006, 29, 1980–1982 CAS.
- K. Hayashi, J. Mori, H. Saito and T. Hayashi, Biol. Pharm. Bull., 2006, 29, 1843–1847 CAS.
- J. T. Etse, A. I. Gray, P. G. Waterman, C. D. Bates, D. G. Watson and D. W. Thomas, J. Nat. Prod., 1988, 51, 314–318 CrossRef CAS.
- R. Geris and T. J. Simpson, Nat. Prod. Rep., 2009, 26, 1063–1094 RSC.
- Y. Matsuda and I. Abe, Nat. Prod. Rep., 2016, 33, 26–53 RSC.
- I. Ferreira, L. Barros and R. Abreu, Curr. Med. Chem., 2009, 16, 1543–1560 CrossRef CAS PubMed.
- G.-P. Hu, J. Yuan, L. Sun, Z.-G. She, J.-H. Wu, X.-J. Lan, X. Zhu, Y.-C. Lin and S.-P. Chen, Mar. Drugs, 2011, 9, 514–525 CrossRef PubMed.
- J. W. Blunt, B. R. Copp, W.-P. Hu, M. H. G. Munro, P. T. Northcote and M. R. Prinsep, Nat. Prod. Rep., 2009, 26, 170–244 RSC.
- V. Amico, Phytochemistry, 1995, 39, 1257–1279 CrossRef CAS.
- L. Jin, C. Quan, X. Hou and S. Fan, Mar. Drugs, 2016, 14, pii: E76 CrossRef PubMed.
- M. Menna, C. Imperatore, F. D'Aniello and A. Aiello, Mar. Drugs, 2013, 11, 1602–1643 CrossRef CAS PubMed.
- S. R. Yende, U. N. Harle and B. B. Chaugule, Pharmacogn. Rev., 2014, 8, 1–7 CrossRef PubMed.
- A. A. El Gamal, Saudi Pharm. J., 2010, 18, 1–25 CrossRef CAS PubMed.
- A. Jensen, J. Sci. Food Agric., 1969, 20, 622–626 CrossRef CAS PubMed.
- K. R. R. Rengasamy, M. G. Kulkarni, W. A. Stirk and J. van Staden, Biotechnol. Adv., 2014, 32, 1364–1381 CrossRef CAS PubMed.
- M. Gaysinski, A. Ortalo-Magné, O. P. Thomas and G. Culioli, Methods Mol. Biol., 2015, 1308, 207–223 Search PubMed.
- Y. Kakinuma, AIBS Bull., 1960, 10, 37 Search PubMed.
- K. H. Jang, B. H. Lee, B. W. Choi, H.-S. Lee and J. Shin, J. Nat. Prod., 2005, 68, 716–723 CrossRef CAS PubMed.
- J. I. Lee and Y. Seo, Chem. Pharm. Bull., 2011, 59, 757–761 CrossRef CAS PubMed.
- S.-C. Chung, K. H. Jang, J. Park, C.-H. Ahn, J. Shin and K.-B. Oh, Bioorg. Med. Chem. Lett., 2011, 21, 1958–1961 CrossRef CAS PubMed.
- S.-J. Heo, J. Jang, B.-R. Ye, M.-S. Kim, W.-J. Yoon, C. Oh, D.-H. Kang, J.-H. Lee, M.-C. Kang, Y.-J. Jeon, S.-M. Kang, D. Kim and K.-N. Kim, Food Chem. Toxicol., 2014, 67, 169–175 CrossRef CAS PubMed.
- B.-G. Park, W.-S. Shin, S. Oh, G.-M. Park, N. I. Kim and S. Lee, Bioorg. Med. Chem., 2017, 25, 4649–4655 CrossRef CAS PubMed.
- (a) S. K. Kim and J. A. Kim, US Pat., US2013338221 (A1), 2012; (b) J. H. Shin, T. Y. Kim, K. H. Chang, B. H. Lee and B. W. Choi, KR Pat., KR20060082214 (A), 2005.
- S.-J. Heo, K.-N. Kim, W.-J. Yoon, C. Oh, Y.-U. Choi, A. Affan, Y.-J. Lee, H.-S. Lee and D.-H. Kang, Food Chem. Toxicol., 2011, 49, 1998–2004 CrossRef CAS PubMed.
- J.-H. Lee, J.-Y. Ko, K. Samarakoon, J.-Y. Oh, S.-J. Heo, C.-Y. Kim, J.-W. Nah, M.-K. Jang, J.-S. Lee and Y.-J. Jeon, Food Chem. Toxicol., 2013, 62, 54–60 CrossRef CAS PubMed.
- W.-J. Yoon, S.-J. Heo, S.-C. Han, H.-J. Lee, G.-J. Kang, H.-K. Kang, J.-W. Hyun, Y.-S. Koh and E.-S. Yoo, Arch. Pharm. Res., 2012, 35, 1421–1430 CrossRef CAS PubMed.
- W.-J. Yoon, S.-J. Heo, S.-C. Han, H.-J. Lee, G.-J. Kang, E.-J. Yang, S.-S. Park, H.-K. Kang and E.-S. Yoo, Food Chem. Toxicol., 2012, 50, 3273–3279 CrossRef CAS PubMed.
- W.-J. Yoon, K.-N. Kim, S.-J. Heo, S.-C. Han, J. Kim, Y.-J. Ko, H.-K. Kang and E.-S. Yoo, Biochem. Biophys. Res. Commun., 2013, 434, 892–897 CrossRef CAS PubMed.
- I. P. S. Fernando, J.-W. Nah and Y.-J. Jeon, Environ. Toxicol. Pharmacol., 2016, 48, 22–30 CrossRef CAS PubMed.
- J.-A. Kim, B.-N. Ahn, C.-S. Kong and S.-K. Kim, Br. J. Dermatol., 2013, 168, 968–976 CrossRef CAS PubMed.
- M. Makoto Iwashima, N. Tako, T. Hayakawa, T. Matsunaga, J. Mori and H. Saito, Chem. Pharm. Bull., 2008, 56, 124–128 CrossRef PubMed.
- (a) J. W. Blunt, B. R. Copp, W.-P. Hu, M. H. G. Munro, P. T. Northcote and M. R. Prinsep, Nat. Prod. Rep., 2008, 25, 35–94 RSC; (b) Y. Seo, K. E. Park, Y. A. Kim, H.-J. Lee, J.-S. Yoo, J.-W. Ahn and B.-J. Lee, Chem. Pharm. Bull., 2006, 54, 1730–1733 CrossRef CAS PubMed.
- A. G. González, J. Darias and J. D. Martín, Tetrahedron Lett., 1971, 12, 2729–2732 CrossRef.
- O. M. M. Sabry, S. Andrews, K. L. McPhail, D. E. Goeger, A. Yokochi, K. T. LePageh, T. F. Murray and W. H. Gerwick, J. Nat. Prod., 2005, 68, 1022–1030 CrossRef CAS PubMed.
- F. Sanchez-Ferrando and A. San-Martin, J. Org. Chem., 1995, 60, 1475–1478 CrossRef CAS.
- C. Areche, A. San-Martín, J. Rovirosa, M. A. Muñoz, A. Hernández-Barragán, M. A. Bucio and P. Joseph-Nathan, J. Nat. Prod., 2010, 73, 79–82 CrossRef CAS PubMed.
- A. G. Gonzales, M. A. Alvarez, J. Darias and J. D. Martin, J. Chem. Soc., Perkin Trans. 1, 1973, 2637–2642 RSC.
- J. Rovirosa, M. Sepulveda, E. Quezada and A. San-Martin, Phytochemistry, 1992, 31, 2679–2681 CrossRef CAS.
- (a) W. H. Gerwick and W. Fenical, J. Org. Chem., 1981, 46, 22–27 CrossRef CAS; (b) A. R. Soares, B. A. P. da Gama, A. P. da Cunha, V. L. Teixeira and R. C. Pereira, Mar. Biotechnol., 2008, 10, 158–165 CrossRef CAS PubMed.
- A. R. Soares, J. L. Abrantes, T. M. Lopes Souza, C. F. Leite Fontes, R. C. Pereira, I. C. de Palmer Paixão Frugulhetti and V. L. Teixeira, Planta Med., 2007, 73, 1221–1224 CrossRef CAS PubMed.
- B. Gil, M. L. Ferrándiz, M. J. Sanz, M. C. Terencio, A. Ubeda, J. Rovirosa, A. San-Martin, M. J. Alcaraz and M. Payá, Life Sci., 1995, 57, PL25–PL30 CrossRef CAS PubMed.
- D. M. Pereira, J. Cheel, C. Areche, A. San-Martin, J. Rovirosa, L. R. Silva, P. Valentao and P. B. Andrade, Mar. Drugs, 2011, 9, 852–862 CrossRef CAS PubMed.
- C. Areche, A. San-Martín, J. Rovirosa and B. Sepúlveda, Nat. Prod. Commun., 2011, 6, 1073–1074 CAS.
- G. Mendes, A. R. Soares, L. Sigiliano, F. Machado, C. Kaiser, N. Romeiro, L. Gestinari, N. Santos and M. T. V. Romanos, Molecules, 2011, 16, 8437–8450 CrossRef CAS PubMed.
- C. Areche, J. Benites, A. Cornejo, L. M. Ruiz, O. García-Beltrán, M. J. Simirgiotis and B. Sepúlveda, Mar. Drugs, 2015, 13, 1726–1738 CrossRef CAS PubMed.
- R. Valls, L. Piovetti, B. Banaigst and A. Praud, Phytochemistry, 1993, 32, 961–966 CrossRef CAS.
- M. D. Guiry and G. M. Guiry, AlgaeBase, http://www.algaebase.org.
- C. Francisco, B. Banaigs, J. Teste and A. Cave, J. Org. Chem., 1986, 51, 1115–1120 CrossRef CAS.
- M. Fadli, J. M. Aracil, G. Jeanty, B. Banaigs, C. Francisco and S. Moreau, Tetrahedron Lett., 1991, 32, 2477–2480 CrossRef CAS.
- M. El Hattab, G. Genta-Jouve, N. Bouzidi, A. Ortalo-Magné, C. Hellio, J.-P. Maréchal, L. Piovetti, O. P. Thomas and G. Culioli, J. Nat. Prod., 2015, 78, 1663–1670 CrossRef CAS PubMed.
- (a) C. Francisco, B. Banaigs, M. Rakba, J. Teste and A. Cave, J. Org. Chem., 1986, 51, 2707–2711 CrossRef CAS; (b) C. Francisco, B. Banaigs, L. Codomier and A. Cave, Tetrahedron Lett., 1985, 26, 4919–4922 CrossRef CAS.
- M. Fadli, J. M. Aracil, G. Jeanty, B. Banaigs and C. Francisco, J. Nat. Prod., 1991, 54, 261–264 CrossRef CAS.
- V. Amico, F. Consulo, G. Oriente and M. Piattelli, J. Nat. Prod., 1984, 47, 947–952 CrossRef CAS.
- R. Valls, V. Mesguiche, L. Piovetti, M. Prost and G. Peiffer, Phytochemistry, 1996, 41, 1367–1371 CrossRef CAS.
- C. Vizetto-Duarte, L. Custódio, G. Acosta, J. H. G. Lago, T. R. Morais, C. Bruno de Sousa, K. N. Gangadhar, M. J. Rodrigues, H. Pereira, R. T. Lima, M. H. Vasconcelos, L. Barreira, A. P. Rauter, F. Albericio and J. Varela, PeerJ, 2016, 4, e1704 Search PubMed.
- G. Navarro, J. J. Fernández and M. Norte, J. Nat. Prod., 2004, 67, 495–499 CrossRef CAS PubMed.
- D. J. Faulkner, Nat. Prod. Rep., 1995, 223–269 RSC.
- E. M. Balboa, Y.-X. Li, B.-N. Ahn, S.-H. Eom, H. Domínguez, C. Jiménez and J. Rodríguez, J. Photochem. Photobiol., B, 2015, 148, 51–58 CrossRef CAS PubMed.
- C. Bruno de Sousa, K. N. Gangadhar, T. R. Morais, G. A. A. Conserva, C. Vizetto-Duarte, H. Pereira, M. D. Laurenti, L. Campino, D. Levy, M. Uemi, L. Barreira, L. Custódio, L. F. D. Passero, J. H. G. Lago and J. Varela, Exp. Parasitol., 2017, 174, 1–9 CrossRef CAS PubMed.
- A. Numata, S. Kanbara, C. Takahashi, R. Fujiki, M. Yoneda, E. Fujita and Y. Nabeshima, Chem. Pharm. Bull., 1991, 39, 2129–2131 CrossRef CAS PubMed.
- N. C. Dlova, WO Pat., WO2013072855 (A2), 2012.
- A. Numata, S. Kanbara, C. Takahashi, R. Fujiki, M. Yoneda, Y. Usami and E. Fujita, Phytochemistry, 1992, 31, 1209–1213 CrossRef CAS.
- T. Kikuchi, Y. Mori, T. Yokoi, S. Nakazawa, H. Koruda, Y. Masada, K. Kitamura and K. Kuriyama, Chem. Pharm. Bull., 1983, 31, 106–113 CrossRef CAS.
- J. D. Faulkner, Nat. Prod. Rep., 1999, 16, 155–198 RSC.
- D. Davyt, W. Enz, E. Manta, G. Navarro and M. Norte, Nat. Prod. Lett., 1997, 9, 305–312 CrossRef CAS.
- (a) R. Brkljača and S. Urban, Mar. Drugs, 2014, 13, 102–127 CrossRef PubMed; (b) J. Kimura, C. Horie, H. Marushima, Y. Matsumoto, C. Sanjoba and Y. Osada, Japanese Patent, JP 2012–502793, 2012.
- B. W. Choi, G. Ryu, S. H. Park, E. S. Kim, J. Shin, S. S. Roh, H. C. Shin and B. H. Lee, Phytother. Res., 2007, 21, 423–426 CrossRef CAS PubMed.
- S. Hur, H. Lee, Y. Kim, B.-H. Lee, J. Shin and T.-Y. Kim, Eur. J. Pharmacol., 2008, 582, 1–11 CrossRef CAS PubMed.
- S. H. Seong, M. Y. Ali, H.-R. Kim, H. A. Jung and J. S. Choi, Bioorg. Med. Chem., 2017, 25, 3964–3970 CrossRef CAS PubMed.
- W.-G. Gwon, E.-J. Joung, M.-S. Kwon, S.-J. Lim, T. Utsuki and H.-R. Kim, Int. Immunopharmacol., 2017, 42, 81–89 CrossRef CAS PubMed.
- E.-J. Yang, Y. M. Ham, K.-W. Yang, N. H. Lee and C.-G. Hyun, Sci. World J., 2013, 2013, 712303 Search PubMed.
- J.-A. Kim, B.-N. Ahn, C.-S. Kong and S.-K. Kim, Exp. Dermatol., 2012, 21, 630–631 CrossRef CAS PubMed.
- C. K. Tsang, A. Ina, T. Goto and Y. Kamei, Neuroscience, 2005, 132, 633–643 CrossRef CAS PubMed.
- P. M. D. J. Fernando, M. J. Piao, S. R. K. M. Hewage, H. K. Kang, E. S. Yoo, Y. S. Koh, M. H. Ko, C. S. Ko, S. H. Byeong, S. R. Mun, N. H. Lee and J. W. Hyun, Environ. Toxicol. Pharmacol., 2016, 43, 112–119 CrossRef CAS PubMed.
- P. Reddy and S. Urban, Phytochemistry, 2009, 70, 250–255 CrossRef CAS PubMed.
- S. Kim, M.-S. Lee, B. Lee, W.-G. Gwon, E.-J. Joung, N.-Y. Yoon and H.-R. Kim, BMC Complementary Altern. Med., 2014, 14, 231 CrossRef PubMed.
- H. Choi, H. Hwang, J. Chin, E. Kim, J. Lee, S.-J. Nam, B. C. Lee, B. J. Rho and H. Kang, J. Nat. Prod., 2011, 74, 90–94 CrossRef CAS PubMed.
- C. L. Céspedes, P. Torres, J. C. Marín, A. Arciniegas, R. d. V. Alfonso, A. L. Pérez-Castorena and E. Aranda, Phytochemistry, 2004, 65, 1963–1975 CrossRef PubMed.
- (a) J. E. Simon, M. Wang, K. Gbewonyo, M. M. Rafi, D. F. Acquaye and Y. Asianowa, US Pat., US2005003030 (A1), 2004; (b) J. E. Simon, Q. L. Wu and W. Deng, US Pat., US2012136047 (A1), 2010; (c) T. Y. Kim, J. H. Shin and B. H. Lee, US Pat., US2010168222 (A1), 2007.
- J. Mori, T. Hayashi, M. Iwashima, T. Matsunaga and H. Saito, Biol. Pharm. Bull., 2006, 29, 1197–1201 CAS.
- (a) J. W. Blunt, B. R. Copp, M. H. G. Munro, P. T. Northcote and M. R. Prinsep, Nat. Prod. Rep., 2010, 27, 165–237 RSC; (b) G. Culioli, A. Ortalo-Magné, R. Valls, C. Hellio, A. S. Clare and L. Piovetti, J. Nat. Prod., 2008, 71, 1121–1126 CrossRef CAS PubMed.
- Y. Seo, K. E. Park and T. J. Nam, Bull. Korean Chem. Soc., 2007, 28, 1831–1833 CrossRef CAS.
- J. W. Blunt, B. R. Copp, M. H. G. Munro, P. T. Northcote and M. R. Prinsep, Nat. Prod. Rep., 2011, 28, 196–268 RSC.
- S. H. Cho, J. Y. Cho, S. E. Kang, Y. K. Hong and D. H. Ahn, J. Environ. Biol., 2008, 29, 479–484 CAS.
- R. Mokrini, M. B. Mesaoud, M. Daoudi, C. Hellio, J.-P. Maréchal, M. El Hattab, A. Ortalo-Magné, L. Piovetti and G. Culioli, J. Nat. Prod., 2008, 71, 1806–1811 CrossRef CAS PubMed.
- H. Safafar, J. van Wagenen, P. Møller and C. Jacobsen, Mar. Drugs, 2015, 13, 7339–7356 CrossRef CAS PubMed.
- (a) D. A. Esquivel-Hernández, V. H. López, J. Rodríguez-Rodríguez, G. S. Alemán-Nava, S. P. Cuéllar-Bermúdez, M. Rostro-Alanis and R. Parra-Saldívar, Int. J. Mol. Sci., 2016, 17, pii: E658 CrossRef PubMed; (b) D. J. M. Gómez-Coronado, E. Ibañez, F. J. Rupérez and C. Barbas, J. Chromatogr. A, 2004, 1054, 227–233 Search PubMed.
- B. A. Ruggeri, R. J. Gray, T. R. Watkins and R. I. Tomlins, Appl. Environ. Microbiol., 1985, 50, 1404–1408 CAS.
- (a) Y. Yamamoto, A. Fujisawa, A. Hara and W. C. Dunlap, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 13144–13148 CrossRef CAS PubMed; (b) Y. Yamamoto, N. Maita, A. Fujisawa, J. Takashima, Y. Ishii and W. C. Dunlap, J. Nat. Prod., 1999, 62, 1685–1687 CrossRef CAS PubMed.
- W. C. Dunlap, A. Fujisawa, Y. Yamamoto, T. J. Moylan and B. D. Sidell, Comp. Biochem. Physiol., Part B: Biochem. Mol. Biol., 2002, 133, 299–305 CrossRef.
- P. Yang, D.-Q. Liu, T.-J. Liang, J. Li, H.-Y. Zhang, A.-H. Liu, Y.-W. Guo and S.-C. Mao, Bioorg. Med. Chem., 2015, 23, 38–45 CrossRef PubMed.
- R. A. Keyzers and M. T. Davies-Coleman, Chem. Soc. Rev., 2005, 34, 355–365 RSC.
- H. Zhang, Z. G. Khalil and R. J. Capon, Tetrahedron, 2011, 67, 2591–2595 CrossRef CAS.
- V. A. Stonik, T. N. Makarieva and A. S. Dmitrenok, J. Nat. Prod., 1992, 55, 1256–1260 CrossRef CAS.
- Y. Venkateswarlu and M. V. R. Reddy, J. Nat. Prod., 1994, 57, 1286–1289 CrossRef CAS.
- R. H. Cichewicz, V. A. Kenyon, S. Whitman, N. M. Morales, J. F. Arguello, T. R. Holman and P. Crews, J. Am. Chem. Soc., 2004, 126, 14910–14920 CrossRef CAS PubMed.
- J. C. Braekman, D. Daloze, G. Hulot, B. Tursch and J. P. Declercq, Bull. Soc. Chim. Belg., 1978, 87, 917 CrossRef CAS.
- J. Salva and D. J. Faulkner, J. Org. Chem., 1990, 55, 1941–1943 CrossRef CAS.
- M. Balbin-Oliveros, R. A. Edrada, P. Proksch, V. Wray, L. Witte and R. W. van Soest, J. Nat. Prod., 1998, 61, 948–952 CrossRef CAS PubMed.
- Y.-C. Shen, M.-C. Hung, C. V. S. Prakash and J.-J. Wang, J. Chin. Chem. Soc., 2000, 47, 567–570 CrossRef CAS.
- H. Liu, M. Namikoshi, K. Akano, H. Kobayashi, H. Nagai and X. Yao, J. Asian Nat. Prod. Res., 2005, 7, 661–670 CrossRef CAS PubMed.
- A. Noda, E. Sakai, H. Kato, F. Losung, R. E. P. Mangindaan, N. J. de Voogd, H. Yokosawa and S. Tsukamoto, Bioorg. Med. Chem. Lett., 2015, 25, 2650–2653 CrossRef CAS PubMed.
- J.-S. Lee, D. B. Abdjul, H. Yamazaki, O. Takahashi, R. Kirikoshi, K. Ukai and M. Namikoshi, Bioorg. Med. Chem. Lett., 2015, 25, 3900–3902 CrossRef CAS PubMed.
- K. A. Mohammed, R. C. Jadulco, T. S. Bugni, M. K. Harper, M. Sturdy and C. M. Ireland, J. Med. Chem., 2008, 51, 1402–1405 CrossRef CAS PubMed.
- A. Hoshino, H. Mitome, H. Miyaoka, A. Shintani, Y. Yamada and R. W. M. van Soest, J. Nat. Prod., 2003, 66, 1600–1605 CrossRef CAS PubMed.
- W. Yu, P. Hjerrild, J. Overgaard and T. B. Poulsen, Angew. Chem., Int. Ed., 2016, 55, 8294–8298 CrossRef CAS PubMed.
- S. K. Palanisamy, N. M. Rajendran and A. Marino, Nat. Prod. Bioprospect., 2017, 7, 1–111 CrossRef CAS PubMed.
- (a) J. W. Blunt, B. R. Copp, R. A. Keyzers, M. H. G. Munro and M. R. Prinsep, Nat. Prod. Rep., 2012, 29, 144–222 RSC; (b) J. W. Blunt, B. R. Copp, R. A. Keyzers, M. H. G. Munro and M. R. Prinsep, Nat. Prod. Rep., 2013, 30, 237–323 RSC.
- W.-C. Wei, P.-J. Sung, C.-Y. Duh, B.-W. Chen, J.-H. Sheu and N.-S. Yang, Mar. Drugs, 2013, 11, 4083–4126 CrossRef PubMed.
- B. F. Bowden and J. C. Coll, Aust. J. Chem., 1981, 34, 2677 CrossRef CAS.
- S.-Y. Cheng, S.-T. Lin, S.-K. Wang and C.-Y. Duh, Bull. Chem. Soc. Jpn., 2011, 84, 783–787 CrossRef CAS.
- J. E. Banfield, D. S. Black, D. J. Collins, B. P. M. Hyland, J. J. Lee and S. R. Pranowo, Aust. J. Chem., 1994, 47, 587 CrossRef CAS.
- C.-G. Zhao, M.-J. Yao, J.-W. Yang, Y.-L. Chai, X.-D. Sun and C.-S. Yuan, Nat. Prod. Res., 2014, 28, 169–173 CrossRef CAS PubMed.
- P.-S. Yang, M.-J. Cheng, C.-F. Peng, J.-J. Chen and I.-S. Chen, J. Nat. Prod., 2009, 72, 53–58 CrossRef CAS PubMed.
- B. Muckensturm, F. Diyani, J.-P. Reduron and M. Hildenbrand, Phytochemistry, 1997, 45, 549–550 CrossRef CAS.
- M. C. Gonzalez, A. Serrano, M. C. Zafra-Polo, D. Cortes and K. S. Rao, J. Nat. Prod., 1995, 58, 1278–1284 CrossRef CAS.
- M. Carmen González, M. A. Sentandreu, K. Sundar Rao, M. Carmen Zafra-Polo and D. Cortes, Phytochemistry, 1996, 43, 1361–1364 CrossRef.
- M. C. Zafra-Polo, M. C. González, J. R. Tormo, E. Estornell and D. Cortes, J. Nat. Prod., 1996, 59, 913–916 CrossRef CAS PubMed.
- N. B. Perry and L. M. Foster, J. Nat. Prod., 1995, 58, 1131–1135 CrossRef CAS.
- J. Luo, C. Zhang, H. Zhu, X. Jin, S. Cao, M. Jin, Z. Jiang, M. Zheng and G. Li, Nat. Prod. Res., 2015, 29, 698–702 CrossRef CAS PubMed.
- X.-R. Peng, J.-Q. Liu, L.-S. Wan, X.-N. Li, Y.-X. Yan and M.-H. Qiu, Org. Lett., 2014, 16, 5262–5265 CrossRef PubMed.
- M. Dou, L. Di, L.-L. Zhou, Y.-M. Yan, X.-L. Wang, F.-J. Zhou, Z.-L. Yang, R.-T. Li, F.-F. Hou and Y.-X. Cheng, Org. Lett., 2014, 16, 6064–6067 CrossRef CAS PubMed.
- J. W. Blunt, B. R. Copp, M. H. G. Munro, P. T. Northcote and M. R. Prinsep, Nat. Prod. Rep., 2004, 21, 1–49 RSC.
- F. Ishibashi, S. Sato, K. Sakai, S. Hirao and K. Kuwano, Biosci., Biotechnol., Biochem., 2013, 77, 1120–1122 CrossRef CAS PubMed.
- W. Fenical and O. McConnell, Experientia, 1975, 31, 1004–1005 CrossRef CAS.
- K. Aoki, M. Takahashi, M. Hashimoto, T. Okuno, K. Kurata and M. Suzuki, Biosci., Biotechnol., Biochem., 2002, 66, 1915–1924 CrossRef CAS PubMed.
- K. Aoki, M. Takahashi, M. Hashimoto, T. Okuno, K. Kurata and M. Suzuki, Biosci., Biotechnol., Biochem., 2014, 66, 1915–1924 CrossRef.
- K. Kurata, K. Taniguchi and M. Suzuki, Phytochemistry, 1996, 41, 749–752 CrossRef CAS.
- P. Djura, D. B. Stierle, B. Sullivan, D. J. Faulkner, E. V. Arnold and J. Clardy, J. Org. Chem., 1980, 45, 1435–1441 CrossRef CAS.
- A. Aiello, E. Fattorusso and M. Menna, Z. Naturforsch., B: J. Chem. Sci., 1993, 48, 209–212 CAS.
- R. E. Longley, O. J. McConnell, E. Essich and D. Harmody, J. Nat. Prod., 1993, 56, 915–920 CrossRef CAS.
- M. Nakamura, A. Suzuki, M. Nakatani, T. Fuchikami, M. Inoue and T. Katoh, Tetrahedron Lett., 2002, 43, 6929–6932 CrossRef CAS.
- K. Hagiwara, J. E. Garcia Hernandez, M. K. Harper, A. Carroll, C. A. Motti, J. Awaya, H.-Y. Nguyen and A. D. Wright, J. Nat. Prod., 2015, 78, 325–329 CrossRef CAS PubMed.
- M. Jaspars, P. A. Horton, L. H. Madrid and P. Crews, J. Nat. Prod., 1995, 58, 609–612 CrossRef CAS.
- G. Cimino, S. de Stefano and L. Minale, Tetrahedron, 1973, 29, 2565–2570 CrossRef CAS.
- A. Casapullo, L. Minale and F. Zollo, J. Nat. Prod., 1993, 56, 527–533 CrossRef CAS.
- E. Zubía, M. J. Ortega, J. Luis Carballo and J. Salvá, Tetrahedron, 1994, 50, 8153–8160 CrossRef.
- Y. Venkateswarlu, D. J. Faulkner, J. L. R. Steiner, E. Corcoran and J. Clardy, J. Org. Chem., 1991, 56, 6271–6274 CrossRef CAS.
- A. Rudi, Y. Benayahu and Y. Kashman, Org. Lett., 2004, 6, 4013–4016 CrossRef CAS PubMed.
- R. A. Davis, A. R. Carroll and R. J. Quinn, J. Nat. Prod., 1999, 62, 1405–1409 CrossRef CAS.
- A. Putz, G. M. König and H. Wägele, Nat. Prod. Rep., 2010, 27, 1386–1402 RSC.
- K. L. McPhail, M. T. Davies-Coleman and J. Starmer, J. Nat. Prod., 2001, 64, 1183–1190 CrossRef CAS.
- L.-F. Liang and Y.-W. Guo, Chem. Biodiversity, 2013, 10, 2161–2196 CAS.
- S.-Y. Cheng, K.-J. Huang, S.-K. Wang, Z.-H. Wen, P.-W. Chen and C.-Y. Duh, J. Nat. Prod., 2010, 73, 771–775 CrossRef CAS PubMed.
- J. C. Coll, N. Liyanage, G. J. Stokie, I. van Altena, J. N. E. Nemorin, S. Sternhell and R. Kazlauskas, Chem. Informationsdienst, 1978, 9, 157 Search PubMed.
- (a) M. V. Sobral, A. L. Xavier, T. C. Lima and D. P. de Sousa, Sci. World J., 2014, 2014, 953451 Search PubMed; (b) R. de Cássia da Silveira e Sá, L. N. Andrade and D. P. de Sousa, Molecules, 2013, 18, 1227–1254 CrossRef PubMed.
- G. D. Manners and L. Jurd, J. Chem. Soc., Perkin Trans. 1, 1977, 405 RSC.
- (a) H.-E. Högberg, R. H. Thomson and T. J. King, J. Chem. Soc., Perkin Trans. 1, 1976, 1696–1701 RSC; (b) D. M. Estrada, J. D. Martín and C. Pérez, J. Nat. Prod., 1987, 50, 735–737 CrossRef CAS.
- E. Dorta, J. Darias, A. San Martín and M. Cueto, J. Nat. Prod., 2002, 65, 329–333 CrossRef CAS.
- M. E. Wall, M. C. Wani, G. Manikumar, H. Taylor, T. J. Hughes, K. Gaetano, W. H. Gerwick, A. T. McPhail and D. R. McPhail, J. Nat. Prod., 1989, 52, 1092–1099 CrossRef CAS.
- M. Park, W. Fenical and M. E. Hay, Phytochemistry, 1992, 31, 4115–4118 CrossRef CAS.
- N. M. Targett and W. S. Keeran, J. Nat. Prod., 1984, 47, 556–557 CrossRef CAS.
- A. F. Benslimane, Y. F. Pouchus, J.-F. Verbist, J.-Y. Petit, E. N. Khettab, L. Welin and J. D. Brion, J. Clin. Pharmacol., 1992, 32, 37–40 CrossRef CAS PubMed.
- A. F. Benslimane, Y. F. Pouchus, J. Le Boterff, J. F. Verbist, C. Roussakis and F. Monniot, J. Nat. Prod., 1988, 51, 582–583 CrossRef CAS.
- A. Simon-Levert, A. Arrault, N. Bontemps-Subielos, C. Canal and B. Banaigs, J. Nat. Prod., 2005, 68, 1412–1415 CrossRef CAS PubMed.
- A. Sato, T. Shindo, N. Kasanuki and K. Hasegawa, J. Nat. Prod., 1989, 52, 975–981 CrossRef CAS.
- L. Garrido, E. Zubía, M. J. Ortega and J. Salvá, J. Nat. Prod., 2002, 65, 1328–1331 CrossRef CAS.
- A. F. Benslimane, Y. F. Pouchus, J. F. Verbist, J. Y. Petit, J. D. Brion and L. Welin, J. Clin. Pharmacol., 1995, 35, 298–301 CrossRef CAS PubMed.
- A. R. Carroll, B. F. Bowden and J. C. Coll, Aust. J. Chem., 1993, 46, 1079 CrossRef CAS.
- A. Simon-Levert, C. Menniti, L. Soulère, A.-M. Genevière, C. Barthomeuf, B. Banaigs and A. Witczak, Mar. Drugs, 2010, 8, 347–358 CrossRef CAS PubMed.
- S. J. Rochfort, R. Metzger, L. Hobbs and R. J. Capon, Aust. J. Chem., 1997, 28, 1217–1219 Search PubMed.
- S. Wang, X.-M. Li, F. Teuscher, D.-L. Li, A. Diesel, R. Ebel, P. Proksch and B.-G. Wang, J. Nat. Prod., 2006, 69, 1622–1625 CrossRef CAS PubMed.
- H. Schildknecht, F. Straub and V. Scheidel, Justus Liebigs Ann. Chem., 1976, 1976, 1295–1306 CrossRef.
- L. K. Ho, M. J. Don, H. C. Chen, S. F. Yeh and J. M. Chen, J. Nat. Prod., 1996, 59, 330–333 CrossRef CAS PubMed.
- L. C. Chang, D. Chávez, J. J. Gills, H. H. S. Fong, J. M. Pezzuto and A. D. Kinghorn, Tetrahedron Lett., 2000, 41, 7157–7162 CrossRef CAS.
- S. M. Kim, H. S. Park, D. Y. Jun, H. J. Woo, M. H. Woo, C. H. Yang and Y. H. Kim, Toxicol. Appl. Pharmacol., 2009, 241, 210–220 CrossRef CAS PubMed.
- D. Y. Jun, C. R. Han, M. S. Choi, M. A. Bae, M. H. Woo and Y. H. Kim, Phytother. Res., 2011, 25, 724–731 CrossRef CAS PubMed.
- T. P. Tran, H. G. Kim, J. H. Choi, M.-K. Na and H. G. Jeong, Phytomedicine, 2013, 20, 622–631 CrossRef CAS PubMed.
- (a) Y. D. Jahng, S. H. Lee, J. K. Son, Y. S. Kim and M. H. Woo, KR Pat., KR20090047718 (A), 2007; (b) C. T. Kim, KR Pat., KR20110051476 (A), 2009; (c) J. A. Kim, Y. R. Lee, K. J. Kim, X. Wang and J. S. Lee, KR Pat., KR20100112922 (A), 2009; (d) W. Kang and W. Zhang, CN Pat., CN102875517 (A), 2012.
- (a) N. Tanaka, M. Kudo, T. Taniguchi, T. Murakami, Y. Saiki and C. Chen, Chem. Pharm. Bull., 1978, 26, 1339–1342 CrossRef CAS; (b) S. E. Drewes, F. Khan, S. F. van Vuuren and A. M. Viljoen, Phytochemistry, 2005, 66, 1812–1816 CrossRef CAS PubMed.
- S. Banerjee, J. Jakupovic, F. Bohlmann, R. M. King and H. Robinson, Phytochemistry, 1985, 24, 2681–2683 CrossRef CAS.
- W. S. Bowers, T. Ohta, J. S. Cleere and P. A. Marsella, Science, 1976, 193, 542–547 CAS.
- A. R. Burnett and R. H. Thomson, J. Chem. Soc. C, 1968, 850–853 RSC.
- G. Schmeda-Hirschmann and F. Papastergiou, J. Nat. Sci., 2003, 58c, 495–501 Search PubMed.
- P.-Y. Zhuang, G.-J. Zhang, X.-J. Wang, Y. Zhang, S.-S. Yu, S.-G. Ma, Y.-B. Liu, J. Qu, Y. Li, S. Xu, H.-N. Lü, X. Chen, L. Li, Y.-K. Si and D. Zhang, Phytochemistry, 2013, 86, 176–183 CrossRef CAS PubMed.
- K. Morimura, C. Yamazaki, Y. Hattori, H. Makabe, T. Kamo and M. Hirota, Biosci., Biotechnol., Biochem., 2007, 71, 2837–2840 CrossRef CAS PubMed.
- P. Gebhardt, K. Dornberger, F. A. Gollmick, U. Gräfe, A. Härtl, H. Görls, B. Schlegel and C. Hertweck, Bioorg. Med. Chem. Lett., 2007, 17, 2558–2560 CrossRef CAS PubMed.
- K. Morimura, K. Hiramatsu, C. Yamazaki, Y. Hattori, H. Makabe and M. Hirota, Biosci., Biotechnol., Biochem., 2009, 73, 627–632 CrossRef CAS PubMed.
- M. Tanaka, F. Nara, Y. Yamasato, Y. Ono and T. Ogita, J. Antibiot., 1999, 52, 827–830 CrossRef CAS PubMed.
- L. Schmölz, M. Birringer, S. Lorkowski and M. Wallert, World J. Biol. Chem., 2016, 7, 14–43 CrossRef PubMed.
- Q. Jiang, Free Radical Biol. Med., 2014, 72, 76–90 CrossRef CAS PubMed.
- D. J. Mustacich, S. W. Leonard, N. K. Patel and M. G. Traber, Free Radical Biol. Med., 2010, 48, 73–81 CrossRef CAS PubMed.
- S. Ciffolilli, M. Wallert, D. Bartolini, V. Krauth, O. Werz, M. Piroddi, B. Sebastiani, P. Torquato, S. Lorkowski, M. Birringer and F. Galli, Free Radical Biol. Med., 2015, 89, 952–962 CrossRef CAS PubMed.
- Y. Zhao, M.-J. Lee, C. Cheung, J.-H. Ju, Y.-K. Chen, B. Liu, L.-Q. Hu and C. S. Yang, J. Agric. Food Chem., 2010, 58, 4844–4852 CrossRef CAS PubMed.
- (a) M. Schultz, M. Leist, M. Petrzika, B. Gassmann and R. Brigelius-Flohé, Am. J. Clin. Nutr., 1995, 62, 1527S–1534S CrossRef CAS PubMed; (b) S. Chiku, K. Hamamura and T. Nakamura, J. Lipid Res., 1984, 25, 40–48 CAS; (c) J. E. Swanson, R. N. Ben, G. W. Burton and R. S. Parker, J. Lipid Res., 1999, 40, 665–671 CAS.
- P. Grammas, L. Hamdheydari, E. J. Benaksas, S. Mou, Q. N. Pye, W. J. Wechter, R. A. Floyd, C. Stewart and K. Hensley, Biochem. Biophys. Res. Commun., 2004, 319, 1047–1052 CrossRef CAS PubMed.
- Q. Jiang, X. Yin, M. A. Lill, M. L. Danielson, H. Freiser and J. Huang, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 20464–20469 CrossRef CAS PubMed.
- M. Birringer, P. Pfluger, D. Kluth, N. Landes and R. Brigelius-Flohé, J. Nutr., 2002, 132, 3113–3118 CrossRef CAS PubMed.
- H. Freiser and Q. Jiang, J. Nutr., 2009, 139, 884–889 CrossRef CAS PubMed.
- S. A. Bardowell, X. Ding and R. S. Parker, J. Lipid Res., 2012, 53, 2667–2676 CrossRef CAS PubMed.
- S. A. Bardowell, F. Duan, D. Manor, J. E. Swanson and R. S. Parker, J. Biol. Chem., 2012, 287, 26077–26086 CrossRef CAS PubMed.
- H. Bruunsgaard, H. E. Poulsen, B. K. Pedersen, K. Nyyssönen, J. Kaikkonen and J. T. Salonen, J. Nutr., 2003, 133, 1170–1173 CrossRef CAS PubMed.
- S. Devaraj, R. Tang, B. Adams-Huet, A. Harris, T. Seenivasan, J. A. de Lemos and I. Jialal, Am. J. Clin. Nutr., 2007, 86, 1392–1398 CAS.
- E. J. Goetzl, Nature, 1980, 288, 183–185 CrossRef CAS PubMed.
- Z. Jiang, X. Yin and Q. Jiang, J. Immunol., 2011, 186, 1173–1179 CrossRef CAS PubMed.
- A. Koeberle, personal communication.
- M. Wallert, L. Schmölz, A. Koeberle, V. Krauth, M. Glei, F. Galli, O. Werz, M. Birringer and S. Lorkowski, Mol. Nutr. Food Res., 2015, 59, 1524–1534 CAS.
- L. Schmölz, M. Wallert, N. Rozzino, A. Cignarella, F. Galli, M. Glei, O. Werz, A. Koeberle, M. Birringer and S. Lorkowski, Mol. Nutr. Food Res., 2017, 61(12) DOI:10.1002/mnfr.201700562.
- Y. Jang, N.-Y. Park, A. L. Rostgaard-Hansen, J. Huang and Q. Jiang, Free Radical Biol. Med., 2016, 95, 190–199 CrossRef CAS PubMed.
- N. Grebenstein, M. Schumacher, L. Graeve and J. Frank, Mol. Nutr. Food Res., 2014, 58, 1052–1060 CAS.
- C. Panagabko, S. Morley, M. Hernandez, P. Cassolato, H. Gordon, R. Parsons, D. Manor and J. Atkinson, Biochemistry, 2003, 42, 6467–6474 CrossRef CAS PubMed.
- M. Wallert, personal communication.
- S.-J. Oh, E.-J. Joung, M.-S. Kwon, B. Lee, T. Utsuki, C.-W. Oh and H.-R. Kim, J. Med. Food, 2016, 19, 1023–1031 CrossRef CAS PubMed.
- (a) A. Aljada, R. Garg, H. Ghanim, P. Mohanty, W. Hamouda, E. Assian and P. Dandona, J. Clin. Endocrinol. Metab., 2001, 86, 3250–3256 CAS; (b) V. A. Dixit and P. V. Bharatam, Chem. Res. Toxicol., 2011, 24, 1113–1122 CrossRef CAS PubMed; (c) K. Kassahun, P. G. Pearson, W. Tang, I. McIntosh, K. Leung, C. Elmore, D. Dean, R. Wang, G. Doss and T. A. Baillie, Chem. Res. Toxicol., 2001, 14, 62–70 CrossRef CAS PubMed; (d) C. Funk, C. Ponelle, G. Scheuermann and M. Pantze, Mol. Pharmacol., 2001, 59, 627–635 CrossRef CAS PubMed.
- A. K. Smolarek and N. Suh, Nutrients, 2011, 3, 962–986 CrossRef CAS PubMed.
- (a) E. A. Klein, I. M. Thompson, C. M. Tangen, J. J. Crowley, M. S. Lucia, P. J. Goodman, L. M. Minasian, L. G. Ford, H. L. Parnes, J. M. Gaziano, D. D. Karp, M. M. Lieber, P. J. Walther, L. Klotz, J. K. Parsons, J. L. Chin, A. K. Darke, S. M. Lippman, G. E. Goodman, F. L. Meyskens and L. H. Baker, JAMA, J. Am. Med. Assoc., 2011, 306, 1549–1556 CrossRef CAS PubMed; (b) C. S. Yang, N. Suh and A.-N. T. Kong, Cancer Prev. Res., 2012, 5, 701–705 CrossRef CAS PubMed; (c) E. R. Miller, R. Pastor-Barriuso, D. Dalal, R. A. Riemersma, L. J. Appel and E. Guallar, Ann. Intern. Med., 2005, 142, 37–46 CrossRef CAS PubMed; (d) D. Q. Pham and R. Plakogiannis, Ann. Pharmacother., 2005, 39, 1870–1878 CrossRef CAS PubMed.
- J. Ju, S. C. Picinich, Z. Yang, Y. Zhao, N. Suh, A.-N. Kong and C. S. Yang, Carcinogenesis, 2010, 31, 533–542 CrossRef CAS PubMed.
- J. Neuzil, M. Tomasetti, Y. Zhao, L.-F. Dong, M. Birringer, X.-F. Wang, P. Low, K. Wu, B. A. Salvatore and S. J. Ralph, Mol. Pharmacol., 2007, 71, 1185–1199 CrossRef CAS PubMed.
- M. Birringer, J. H. EyTina, B. A. Salvatore and J. Neuzil, Br. J. Cancer, 2003, 88, 1948–1955 CrossRef CAS PubMed.
- (a) L.-F. Dong, G. Grant, H. Massa, R. Zobalova, E. Akporiaye and J. Neuzil, Int. J. Cancer, 2012, 131, 1052–1058 CrossRef CAS PubMed; (b) L.-F. Dong, R. Freeman, J. Liu, R. Zobalova, A. Marin-Hernandez, M. Stantic, J. Rohlena, K. Valis, S. Rodriguez-Enriquez, B. Butcher, J. Goodwin, U. T. Brunk, P. K. Witting, R. Moreno-Sanchez, I. E. Scheffler, S. J. Ralph and J. Neuzil, Clin. Cancer Res., 2009, 15, 1593–1600 CrossRef CAS PubMed.
- S. E. Campbell, W. L. Stone, S. Lee, S. Whaley, H. Yang, M. Qui, P. Goforth, D. Sherman, D. McHaffie and K. Krishnan, BMC Cancer, 2006, 6, 13 CrossRef PubMed.
- (a) R. Gysin, A. Azzi and T. Visarius, FASEB J., 2002, 16, 1952–1954 CrossRef CAS PubMed; (b) Q. Jiang, J. Wong and B. N. Ames, Ann. N. Y. Acad. Sci., 2004, 1031, 399–400 CrossRef CAS PubMed.
- (a) P. W. Sylvester, M. R. Akl, A. Malaviya, P. Parajuli, S. Ananthula, R. V. Tiwari and N. M. Ayoub, BioFactors, 2014, 40, 49–58 CrossRef CAS PubMed; (b) W. Xu, Y. Mi, P. He, S. He and L. Niu, Molecules, 2017, 22, pii: E1299 CrossRef PubMed.
- M. M. Kanchi, M. K. Shanmugam, G. Rane, G. Sethi and A. P. Kumar, Drug Discovery Today, 2017, 12, 1765–1781 CrossRef PubMed.
- N. Guthrie, A. Gapor, A. F. Chambers and K. K. Carroll, J. Nutr., 1997, 127, 544S–548S CAS.
- N. Guthrie, A. Gapor, A. F. Chambers and K. K. Carroll, Asia Pac. J. Clin. Nutr., 1997, 6, 41–45 CAS.
- O. A. Alawin, R. A. Ahmed, B. A. Ibrahim, K. P. Briski and P. W. Sylvester, J. Nutr. Biochem., 2016, 27, 266–277 CrossRef CAS PubMed.
- K. Nesaretnam, R. Stephen, R. Dils and P. Darbre, Lipids, 1998, 33, 461–469 CrossRef CAS PubMed.
- M. Wallert, S. Mosig, K. Rennert, H. Funke, M. Ristow, R. M. Pellegrino, G. Cruciani, F. Galli, S. Lorkowski and M. Birringer, Free Radical Biol. Med., 2014, 68, 43–51 CrossRef CAS PubMed.
- H. H. Sun, N. M. Ferrara, O. J. McConnell and W. Fenical, Tetrahedron Lett., 1980, 21, 3123–3126 CrossRef CAS.
- Q. Jiang, I. Elson-Schwab, C. Courtemanche and B. N. Ames, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 11494–11499 CrossRef CAS PubMed.
- M.-L. Yam, S. R. Abdul Hafid, H.-M. Cheng and K. Nesaretnam, Lipids, 2009, 44, 787–797 CrossRef CAS PubMed.
- R. Loganathan, K. R. Selvaduray, K. Nesaretnam and A. K. Radhakrishnan, Cell Proliferation, 2013, 46, 203–213 CrossRef CAS PubMed.
- F. Galli, A. M. Stabile, M. Betti, C. Conte, A. Pistilli, M. Rende, A. Floridi and A. Azzi, Arch. Biochem. Biophys., 2004, 423, 97–102 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra11819h |
| This journal is © The Royal Society of Chemistry 2018 |