
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePacked hybrid silica nanoparticles as sorbents with thermo-switchable surface chemistry and pore size for fast extraction of environmental pollutants†
Sushilkumar A. Jadhav ,
Roberto Nisticò‡
,
Giuliana Magnacca and
Dominique Scalarone*
,
Roberto Nisticò‡
,
Giuliana Magnacca and
Dominique Scalarone*
Department of Chemistry and NIS Research Centre, University of Torino, 10125 Torino, Italy. E-mail: dominique.scalarone@unito.it; Fax: +39 011 6707855; Tel: +39 011 6707546
First published on 3rd January 2018
Abstract
Thermoresponsive poly(N-isopropylacrylamide)-grafted silica nanoparticles (SiNPs) have been synthesized and fully characterized by ATR-FTIR, TGA, HRTEM, BET and DLS analysis. Hybrid solid phase extraction (SPE) beds with tuneable pore size and switchable surface chemistry were prepared by packing the polymer-grafted nanoparticles inside SPE cartridges. The cartridges were tested by checking the thermo-regulated elution of model compounds, namely methylene blue, caffeine and amoxicillin. Extraction of the analytes and regeneration of the interaction sites on the sorbent surface was carried out entirely in water solution by changing the external temperature below and above the lower critical solution temperature (LCST) of the polymer. The results demonstrate that the elution of model compounds depends on the temperature-regulated size of the inter-particle voids and on the change of surface properties of the PNIPAM-grafted nanoparticles from hydrophilic to hydrophobic.
1. Introduction
Environmental pollution is one of the biggest problems at a global level. Toxic pollutants released by industries, from farms, hospitals and household waste have harmful effects on human and animal health and it is essential to remove them effectively from the environment. The concentration of such pollutants is often very low. Therefore, there is the need to separate and concentrate these substances in order to make them detectable with analytical instruments. For this purpose, solid phase extraction (SPE) is a versatile technique used for rapid and selective separation of pollutant molecules. SPE can be used for many purposes such as the isolation and detection of pollutants in wastewater and food, isolation of pharmaceuticals from clinical samples, trace enrichment and fractionation of hydrocarbons or other organic compounds from complex matrices.1,2 Various materials are used as sorbents in SPE, which includes silica or functionalized silica,3 carbon or carbon nanotubes,4 magnetic nanomaterials,5 etc. Silica based materials, due to their high stability, chemical resistance and ease of surface functionalization, are among the most commonly used sorbent materials for the preparation of SPE beds.6,7 Especially, SPE systems based on silica particles grafted with smart polymers, such as thermoresponsive polymers, can be very versatile and stable filtration and separation systems because of their tunable porosity, resulting from particle packing and polymer properties. Packed columns and membranes based on silica particles grafted with thermoresponsive polymers have proven their usefulness in high resolution bio-separation,8 high-speed separation of biomolecules and peptides9 and in separation of steroids in aqueous chromatography.10,11 Improved performance by the thermoresponsive polymer-grafted silica particles in above mentioned applications was observed in comparison with the frequently used conventional bare silica particles. This is due to the modification of surface properties of the silica upon polymer grafting. However, the use of thermoresponsive polymer-grafted silica particles as sorbents in the preparation of SPE columns has not been much exploited yet.Grafting of thermoresponsive polymers on pre-formed structures made of bare or functionalized silica particles inside chromatographic columns,10 or the preparation of separation columns from thermoresponsive polymer-grafted silica particles are reported.12–15 Instead, SPE beds or columns made from thermoresponsive particles have not been investigated with much interest even if they can be ideal sorbent materials with switchable surface properties which can be controlled by an external stimulus (e.g. temperature). The testing of hybrid thermoresponsive materials as SPE sorbents will extend their potential applications beyond their use in chromatographic columns. As these hybrid materials have improved properties, they may serve as more efficient and smart sorbents. Especially, porous thermoresponsive SPE sorbents casted into portable devices, such as cartridges, can be the most convenient filtration or separation tools.
As an important step forward towards the development of smart SPE filtration devices, in the present work we report synthesis, characterization and testing of hybrid SPE cartridges based on poly(N-isopropylacrylamide) (PNIPAM)-grafted silica nanoparticles (SiNPs). Our approach involves first the synthesis and characterization of thermoresponsive PNIPAM-grafted SiNPs as sorbents for SPE, optimization of the polymer grafting procedure and characterization of the polymer-grafted nanoparticles, followed by the preparation and preliminary testing of thermoresponsive mesoporous SPE cartridges for thermo-regulated molecular transport of selected model organic compounds. Three model organic compounds, namely methylene blue (MB), caffeine (CF) and amoxicillin (AMX), from three different classes of pollutants, were chosen for the testing of the cartridges. MB is toxic and carcinogenic and it can cause some harmful health effects on acute exposure.16 It is released mainly by the textile dying industry and it is one of the major causes of colored wastewaters.17 CF is a major emerging pollutant and in certain areas of earth its levels appear to be sufficiently high to approach threshold toxicity values,18 while AMX is a pharmaceutical pollutant from the antibiotics category.19 In addition, the choice of these compounds for testing the thermoresponsive cartridges was also based on their different chemical properties as MB is positively charged, CF is neutral and AMX is amphoteric. This makes MB, CF and AMX good choices to study the role of PNIPAM in the retention mechanism. Moreover, it allows differentiating sorbent-molecule ionic interactions from the effect of PNIPAM chains on the sorbent porosity and hydrophilic–hydrophobic surface properties.
A remarkable advantage of the proposed sorbent material is the possibility to operate in mild conditions, that is without the use of organic solvents and the regeneration of the interaction sites by little temperature changes. To the best of our knowledge this is the first attempt to use PNIPAM-functionalized SiNPs to prepare thermoresponsive SPE cartridges.
2. Experimental
2.1 Materials
Two types of solid commercial silica nanoparticles named AOX50 and FK320 (Sipernat) by Evonik were used in this work. N-Isopropylacrylamide (NIPAM), azobisisobutyronitrile (AIBN), 3-(methacryloxypropyl)-trimethoxysilane (MPS), 1,4-dioxane, methylene blue (MB), caffeine (CF) and amoxicillin (AMX) were purchased from Sigma-Aldrich, Italy. All the solvents used for the synthesis were of high purity and used as received. 1,4-Dioxane was kept on molecular sieves overnight before using for polymerization reaction.2.2 Instruments and methods
Fourier-transform infrared (FTIR) spectra were recorded on a Perkin Elmer Spectrum 100 in the attenuated total reflectance (ATR) mode with a diamond crystal, using 16 scans per spectrum and a resolution of 4 cm−1 in the spectral range of 4000–600 cm−1. DTGS was used as detector.Thermogravimetric analyses (TGA) were carried out on a TA Q500 model from TA Instruments by heating samples in alumina pans at a rate of 10 °C min−1 from 25 to 600 °C in a nitrogen flow and from 600 to 800 °C in air. Change of the gas at 600 °C was used to remove completely the carbonaceous residues from pyrolysis reactions and measure the exact amount of organic component.
Gas-volumetric analysis, specific surface area (SSA), pore volume and size were measured by N2 adsorption–desorption isotherms at 77 K using an ASAP 2020 (Micromeritics) gas-volumetric analyzer. SSA was calculated using the Brunauer–Emmett–Teller (BET) method; average pore size and volume were calculated on the adsorption branch of the isotherms according to the Barrett–Joyner–Halenda (BJH) method (Kruk–Jaroniec–Sayari equations). Samples were outgassed at RT overnight before analyses.
High Resolution Transmission Electron Microscopy (HRTEM) images were obtained with a JEOL 2010 instrument (300 kV) equipped with a LaB6 filament. For specimen preparation powdery samples were supported onto holed carbon coated copper grids by dry deposition.
Dynamic Light Scattering (DLS) measurements were carried out by using Malvern ZS 90 Zetasizer instrument. App. 0.1% suspensions of nanoparticles were prepared in deionized water and these suspensions were sonicated for 20 minutes before the analysis.
UV-Vis analyses were performed using a Lambda 25 spectrophotometer (Perkin Elmer). For quantification of the probe molecules used for testing of the temperature-regulated elution of model organic compounds, calibration curves were constructed by plotting absorbance vs. concentration between 10−5 and 10−8 M (ε = 111 M−1, R2 = 0.997) for methylene blue, between 10−4 and 10−9 M (ε = 185.1 M−1, R2 = 0.995) for caffeine and between 10−2 and 10−6 M (ε = 115.8 M−1, R2 = 0.998) for amoxicillin.
2.3 Synthesis of PNIPAM-grafted silica nanoparticles
2.4 Preparation and testing of porous SPE cartridges
The SPE cartridges were prepared by compressing bare as well as polymer-grafted hybrid nanoparticles between two polyethylene frits in an empty SPE syringe. Stable compact discs were formed by packed hybrid silica nanoparticles which showed good mechanical stability. The solutions of known concentration of three model molecules, namely methylene blue, caffeine and amoxicillin, were allowed to pass across the cartridges at 20 and 40 °C. These two temperatures are below and above the LCST of PNIPAM which is around 32 °C. The elution was accelerated by connecting the other end of the cartridge to a peristaltic pump. The flow rate was adjusted to 3 ml per minute. The eluents (3 ml) were collected and filtered through 0.45 μm syringe filters before analysis. Concentrations of the molecules were monitored by UV-Vis spectroscopy.3. Results and discussion
3.1 Synthesis of PNIPAM-grafted nanoparticles
Two types of commercial bare silica particles, namely AOX50, which are less hydrophilic fumed or pyrogenic SiNPs, and FK320, which are hydrophilic precipitated SiNPs, were used in this work. The characterization data of the bare particles are reported in Table 1. In addition, the aggregation kinetics of both types of SiNPs were studied by monitoring the change in hydrodynamic diameter of the stable clusters and the change in the polydispersity index (PI) of with time (ESI, Fig. S1 and S2†). Due to very strong particle–particle interactions the bare silica nanoparticles showed fast aggregation. Especially, FK320 SiNPs showed very fast aggregation tendency due to very small size and hydrophilic properties. These properties were modified upon polymer grafting. The two-step synthetic procedure used for the grafting of PNIPAM on the nanoparticles is depicted in Fig. 1. First the particles were functionalized with polymerizable acryloxy groups by grafting of MPS by the alkoxysilane grafting protocol.20–23 The reaction was carried out in cyclohexane with propyl amine as the mild base catalyst.24 In the second step of the reaction radical, polymerization of NIPAM in the presence of MPS-grafted nanoparticles was carried out in 1,4-dioxane by using AIBN as the initiator.25,26 The particles to NIPAM monomer ratio was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.5, while the monomer to initiator ratio was 50:1. The grafting of PNIPAM on the nanoparticles was confirmed by ATR-FTIR, TGA, N2 adsorption and DLS analysis. The results of these analyses are summarized in Table 1.
:0.5, while the monomer to initiator ratio was 50:1. The grafting of PNIPAM on the nanoparticles was confirmed by ATR-FTIR, TGA, N2 adsorption and DLS analysis. The results of these analyses are summarized in Table 1.
| Sample | Diameterb (nm) | SSAc (m2 g−1) | HD (nm) | Zp (mV) | pH | Graftingd (%) | Grafting density (mol m−2) |
|---|---|---|---|---|---|---|---|
| a SSA = specific surface area, HD = average hydrodynamic diameter of stable clusters, Zp = zeta potential.b Diameter values as measured by TEM.c SSA calculated using the Brunauer–Emmett–Teller (BET) method.d Grafting w/w% as determined by TGA. | |||||||
| AOX50 | 60–80 | 52 | 450 ± 17 | −34.6 ± 3.1 | 7.06 | — | — |
| AOX50–PNIPAM | 60–80 | 37 | 301 ± 10 | −22.3 ± 2.0 | 7.10 | 2.5 | 0.385 |
| FK320 | 20–30 | 180 | 190 ± 11 | −27.0 ± 3.7 | 7.10 | — | — |
| FK320–PNIPAM | 20–30 | 110 | 255 ± 13 | −17.7 ± 1.9 | 7.12 | 10.0 | 0.180 |
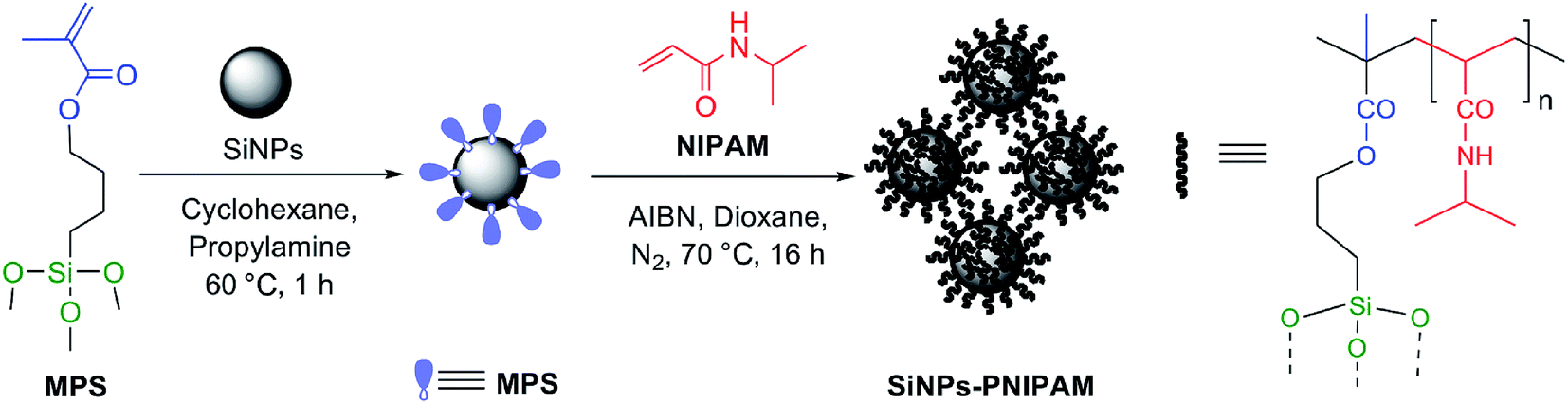 | ||
| Fig. 1 Synthesis protocol for PNIPAM-grafted SiNPs. | ||
3.2 Characterization of PNIPAM-grafted nanoparticles
The ATR-FTIR spectra of the starting bare silica nanoparticles, MPS-grafted silica particles and PNIPAM-grafted AOX50 and FK320 nanoparticles are shown in Fig. 2 and 3 respectively. The spectra of the bare and grafted silica particles are characterized by the intense absorption band arising from the asymmetric vibration of Si–O at 1070 cm−1.27 The spectra of MPS-functionalized particles show two peaks at 1720 and 1640 cm−1 due to carbonyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CC stretching vibrations, respectively.28 The presence of these two peaks confirmed the grafting of MPS on the surface of SiNPs. The spectra of PNIPAM-grafted silica nanoparticles (Fig. 2 spectrum C and Fig. 3 spectrum C) show the characteristic peaks of PNIPAM. Twin peaks of C–H bending coming from the isopropyl group were observed at 1365–1368 and 1385 cm−1, accompanied by corresponding C–H stretching modes below 3000 cm−1. Additional characteristic peaks of the polymer at 1459 and at approximately 2970, 1536–1537, 1645–1648 cm−1 are attributed to –CH3 bending and stretching, C–N stretching and CO stretching of the amide group, respectively.29–31 The carbonyl stretching peak at 1720 cm−1 from the acryloxy group of MPS is also present in the PNIPAM-grafted nanoparticles, which confirms the anchoring of the polymer on the surface of SiNPs. At high wavenumbers, the stretching signal of OH groups present on the FK320 surface can be observed above 3000 cm−1, witnessing the higher hydrophilic character of this silica with respect to AOX50, not presenting analogous behavior.
O and CC stretching vibrations, respectively.28 The presence of these two peaks confirmed the grafting of MPS on the surface of SiNPs. The spectra of PNIPAM-grafted silica nanoparticles (Fig. 2 spectrum C and Fig. 3 spectrum C) show the characteristic peaks of PNIPAM. Twin peaks of C–H bending coming from the isopropyl group were observed at 1365–1368 and 1385 cm−1, accompanied by corresponding C–H stretching modes below 3000 cm−1. Additional characteristic peaks of the polymer at 1459 and at approximately 2970, 1536–1537, 1645–1648 cm−1 are attributed to –CH3 bending and stretching, C–N stretching and CO stretching of the amide group, respectively.29–31 The carbonyl stretching peak at 1720 cm−1 from the acryloxy group of MPS is also present in the PNIPAM-grafted nanoparticles, which confirms the anchoring of the polymer on the surface of SiNPs. At high wavenumbers, the stretching signal of OH groups present on the FK320 surface can be observed above 3000 cm−1, witnessing the higher hydrophilic character of this silica with respect to AOX50, not presenting analogous behavior.
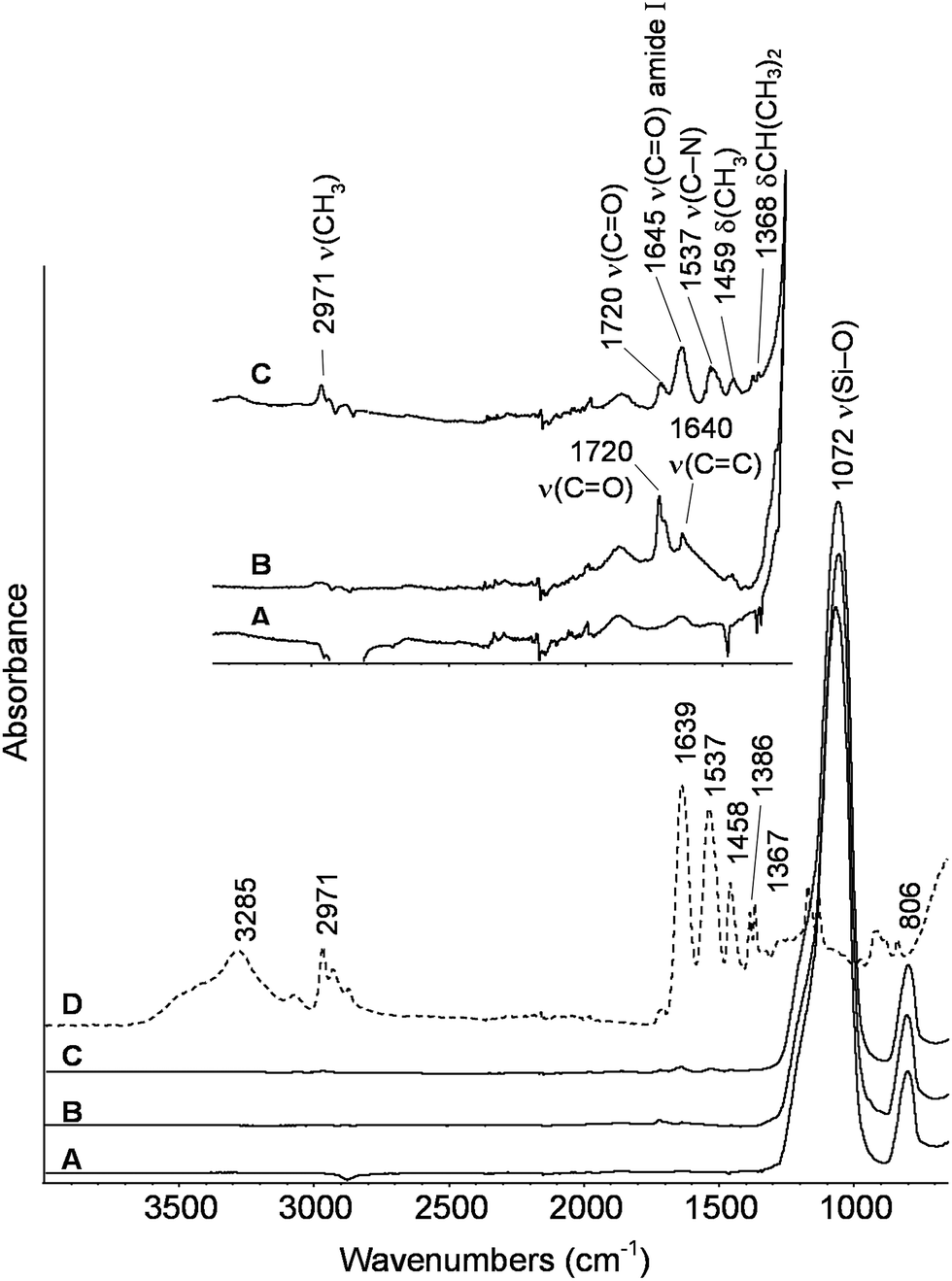 | ||
| Fig. 2 ATR-FTIR spectra of (A) bare (B) MPS-grafted (C) PNIPAM-grafted AOX50 silica nanoparticles and (D) PNIPAM reference. | ||
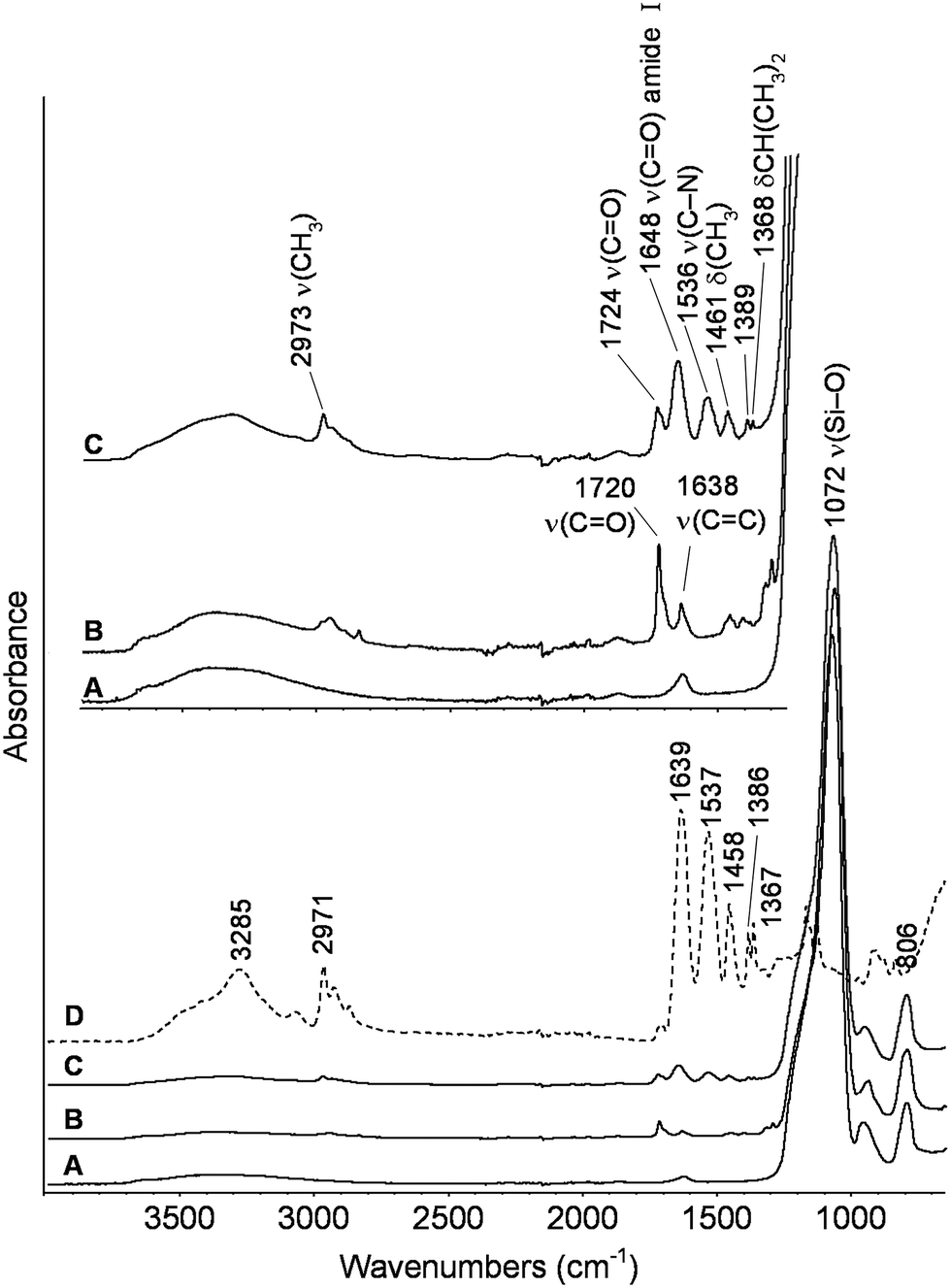 | ||
| Fig. 3 ATR-FTIR spectra of (A) bare (B) MPS-grafted (C) PNIPAM-grafted FK320 silica nanoparticles and (D) PNIPAM reference. | ||
The TGA curves of both types of bare, MPS-grafted nanoparticles and PNIPAM-grafted SiNPs are shown in Fig. 4. SiNPs showed good thermal stability upon programmed heating up to 800 °C. Only weight loss due to adsorbed water was observed for both samples. The TGA curve of FK320 silica show a higher weight loss due to adsorbed water with respect to AOX50 SiNPs. The weight losses for AOX50–MPS and FK320–MPS nanoparticles were 1.5 and 8.7% respectively. Instead the percentage of grafted polymer, calculated by subtracting the weight loss due to MPS grafting from the weight loss of the PNIPAM-grafted AOX50 and FK320 nanoparticles, was 2.5 and 10 wt% respectively (refer to Table 1). The higher amount of polymer grafting on FK320 SiNPs was obtained due to their smaller size, high surface area and more hydrophilic character coming from the large number of surface silanol groups (Si–OH). Higher numbers of surface silanol groups provide higher affinity to grafting molecules resulting in higher polymer grafting. The results of the ATR-FTIR and thermal analysis confirmed the grafting of the polymer on the nanoparticles.
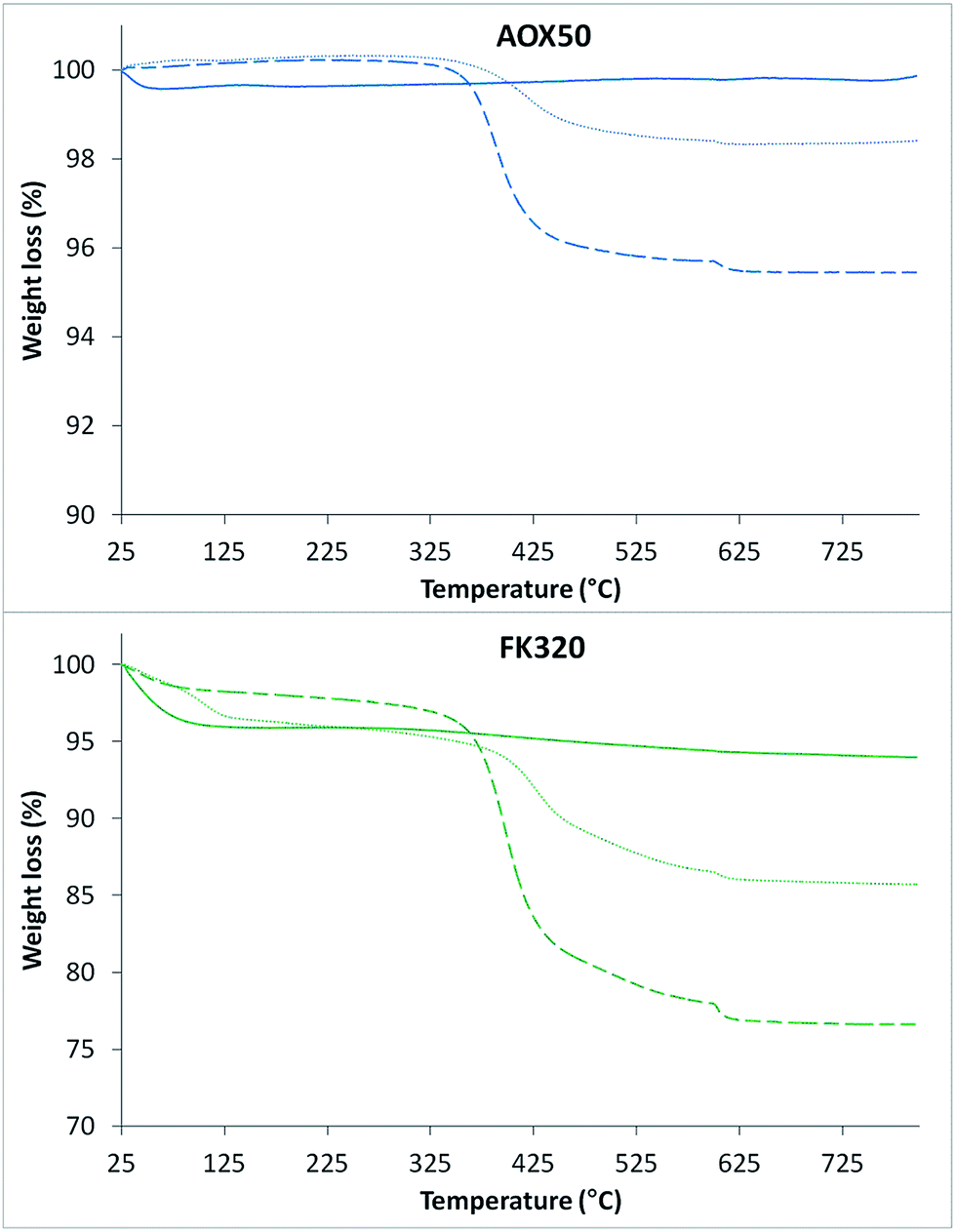 | ||
| Fig. 4 TGA curves of bare (solid line), MPS-grafted (dotted line) and PNIPAM-grafted (dashed line) silica nanoparticles. | ||
PNIPAM in aqueous solution shows a typical coil-to-globule transition above its lower critical solution temperature (LCST) which is around 32 °C. Upon heating, the water soluble hydrated extended chains of the polymer collapse to form compact insoluble and relatively hydrophobic globules.32,33 This behavior of the polymer can also be monitored on the surface of the PNIPAM-grafted nanoparticles in their aqueous suspensions. The bare silica particles (either AOX50 or FK320) tend to aggregate very fast due to particle–particle interactions. The hydrodynamic diameter (HD) of the stable cluster of particles at a given time of analysis is reported in Table 1. Upon polymer grafting the HD of AOX50 silica nanoparticles is decreased as the grafting of polymer has resulted in stabilization of smaller particles by minimizing the particle–particle interactions. This observation is also supported by zeta potential (Zp) values. Instead, in the case of FK320 nanoparticles the HD value of PNIPAM-grafted nanoparticles was found to be increased because the quantity of grafted polymer is much higher (10 wt%) than for AOX50 (2.5 wt%) nanoparticles. As shown in Fig. 5 the HD of the PNIPAM-grafted AOX50 and FK320 SiNPs in water suspensions have changed with increase in temperature and sudden decrease of the hydrodynamic diameter was observed at the LCST of the polymer.34–37 This transition was observed at 32 °C for AOX50–PNIPAM nanoparticles and at 31.6 °C for FK320–PNIPAM nanoparticles.
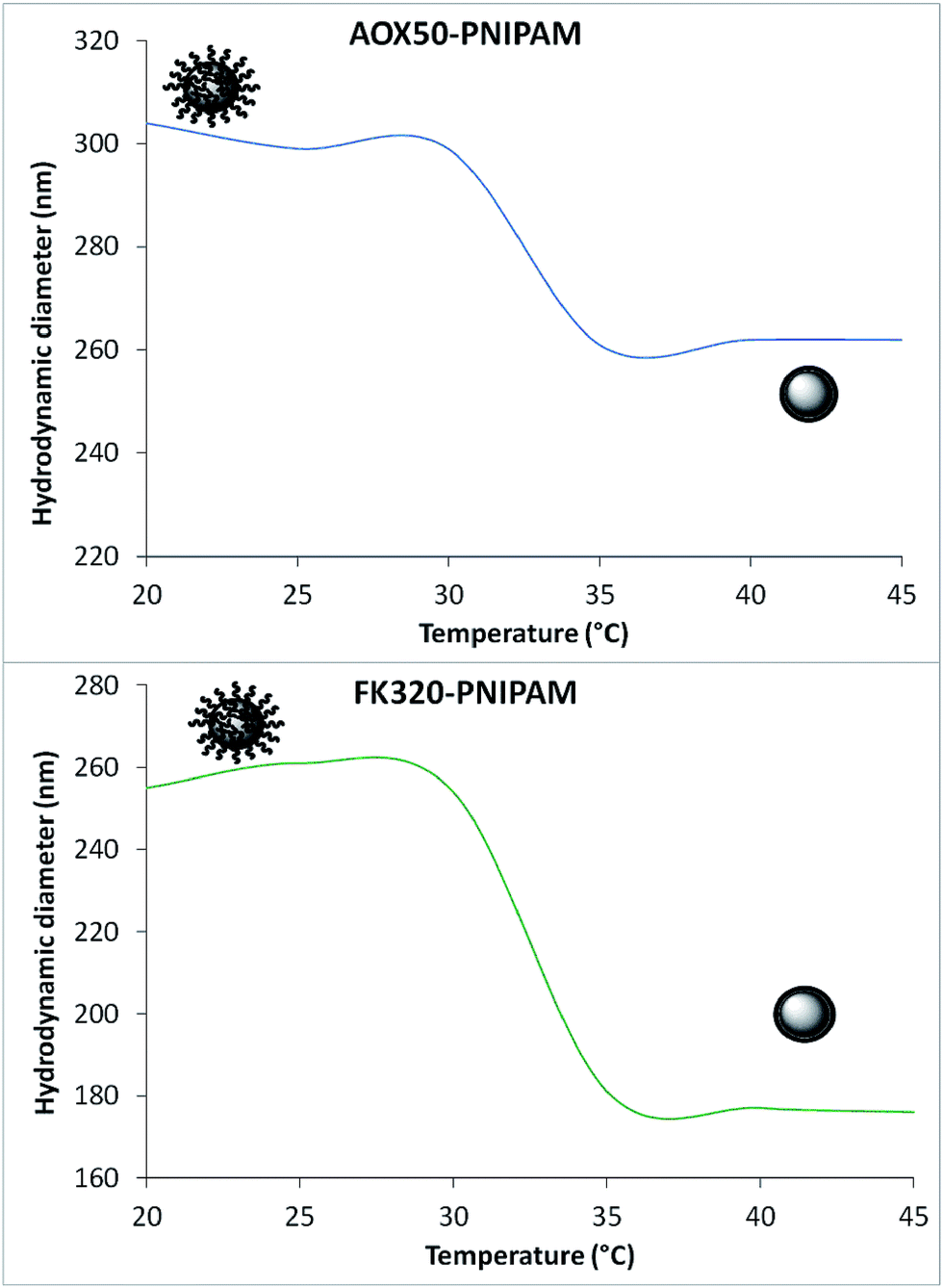 | ||
| Fig. 5 Average hydrodynamic diameter of PNIPAM-grafted SiNPs at different temperatures. | ||
HRTEM images of the bare AOX50 (Fig. 6A) and FK320 (Fig. 6C) SiNPs show that the particle powders are made up of nanoparticles of 20–80 nm in diameter. The TEM analysis of AOX50–PNIPAM (Fig. 6B) and FK320–PNIPAM (Fig. 6D) confirm that the starting nanoparticles remained stable upon polymer grafting. In the case of both types of SiNPs, the spherical or quasi-spherical shape was maintained after the two step polymerization grafting procedure explained in the experimental part. This observation confirm the suitability of the PNIPAM grafting procedure adapted for the surface modification of SiNPs. It is worth pointing out that no crosslinkers were used during the polymer grafting procedure thus avoiding formation of bigger agglomerates resulting from inter-particle cross linking.
 | ||
| Fig. 6 HRTEM images of the bare AOX50 (A), PNIPAM-grafted AOX50 (B), bare FK320 (C) and PNIPAM-grafted FK320 nanoparticles (D). | ||
The PNIPAM-grafted nanoparticles were further characterized by nitrogen adsorption–desorption analysis. The porosimetry data not only confirmed the polymer grafting on SiNPs, but also confirmed the modified surface properties of the particles (Table 1, Fig. 7). The specific surface areas (SSA) of the bare and PNIPAM-grafted AOX50 SiNPs are 52 and 37 m2 g−1 respectively. Although these values are very low for SPE sorbents, it was observed that upon polymer grafting the particle packing changes. PNIPAM grafting resulted in less compact packing of the particles together thus forming inter-particle voids. In fact, while bare AOX50 silica nanoparticles form a non-porous material, which is a characteristic property of pyrogenic or fumed silica, PNIPAM-grafted AOX50 gives a packing with mesopores of big size and probably macropores too (Fig. 7A), which are not detected by nitrogen-adsorption measurements. Instead, as expected for precipitated silica particles, FK320 SiNPs gave a better packing with a specific surface area of 180 m2 g−1 and a mixed porosity. The pore size distribution determined using the Barrett–Joyner–Halenda (BJH) method and reported in Fig. 7B shows a maximum at 30 nm, but also a notable amount of larger pores. Both surface area and pore diameter decreased after PNIPAM grafting as the polymer occupies considerable space between the inter-particle voids. In general, the size of the inter-particle voids is much bigger than the size of PNIPAM chains, so the polymer contribution in terms of size selectivity in actual performance of the material as porous sorbent may be small and probably limited to large molecular analytes. However, the switching of the sorbent surface properties from hydrophilic to hydrophobic is also an important parameter affecting the extraction efficiency and the recovery of the stationary phase. PNIPAM dehydration and hydration induced by changing the temperature around the coil-to-globule transition temperature significantly affects the surface-analytes interactions in addition to the steric hindrance due to conformational changes. This was confirmed by the testing of the SPE cartridges reported in the following section.
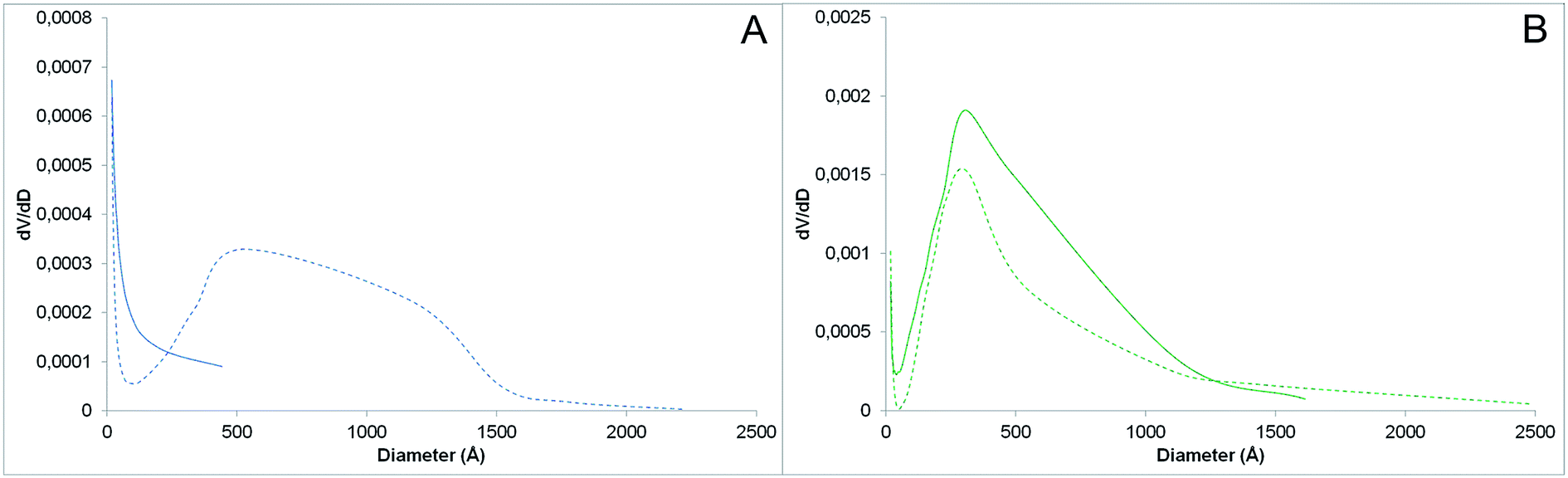 | ||
| Fig. 7 Pores size distribution of (A) bare (solid line) and AOX50–PNIPAM (dashed line), (B) bare (solid line) and FK320–PNIPAM nanoparticles (dashed line). | ||
3.3 Cartridge preparation and testing
The various components and preparation steps followed to prepare the SPE cartridges are shown in Fig. 8. Cartridges with porous discs of 1 cm in diameter × 0.5 cm in height were prepared from bare and PNIPAM-grafted nanoparticles. Adsorption membranes with porous voids, sometimes termed as nanovalves, in which molecular transport is controlled by temperature, utilize polymers whose conformation is responsive to the temperature changes. Thermoresponsive polymers, especially PNIPAM, have been most extensively used to create such nanovalves inside chromatographic columns or films.38 As stated before, below LCST, PNIPAM exists in the extended conformation that collapses above the LCST, thus providing a steric change that can be utilized to regulate the voids formed by interparticle spaces in the porous bed. Three model compounds namely methylene blue (indicator or dye), caffeine (found in food and beverages) and amoxicillin (pharmaceuticals) from three different classes of emerging organic pollutants (EOPs) were chosen to test the performances of the SPE cartridges. The structures of these compounds are shown in Fig. 9. The SPE bed consisting of porous interparticle voids showed increased diffusion rates at elevated temperature due to polymer brushes collapsing onto the surface of the nanoparticles. This diffusion is also assisted by switching of the surface properties of the particles from hydrophilic to hydrophobic as the compact globules of PNIPAM formed after the transition of the polymer are hydrophobic in nature and form a stable hydrophobic monolayer on the surface of the nanoparticles. The commonest commercial silica phases currently available include mostly alkyl-bonded silica (e.g. octadecyl- and octyl-bonded silica) for reverse-phase SPE, cyanopropyl- or diol-bonded silica for normal-phase SPE and silica grafted with organic functional groups, such as quaternary ammonium salts, sulfonic and carboxylic groups, for ion-exchange SPE. The surface properties and reactivity/affinity towards analytes of these materials remain the same during their use and the elution protocols involve the use of organic solvents. However, in the present study the use of thermoresponsive polymer-grafted silica particles has proved the advantage that the surface properties and to certain extent the charges on the particles can be thermally switched to trigger the adsorption or desorption process of the molecules of interest across the SPE bed, without the need to use organic solvents.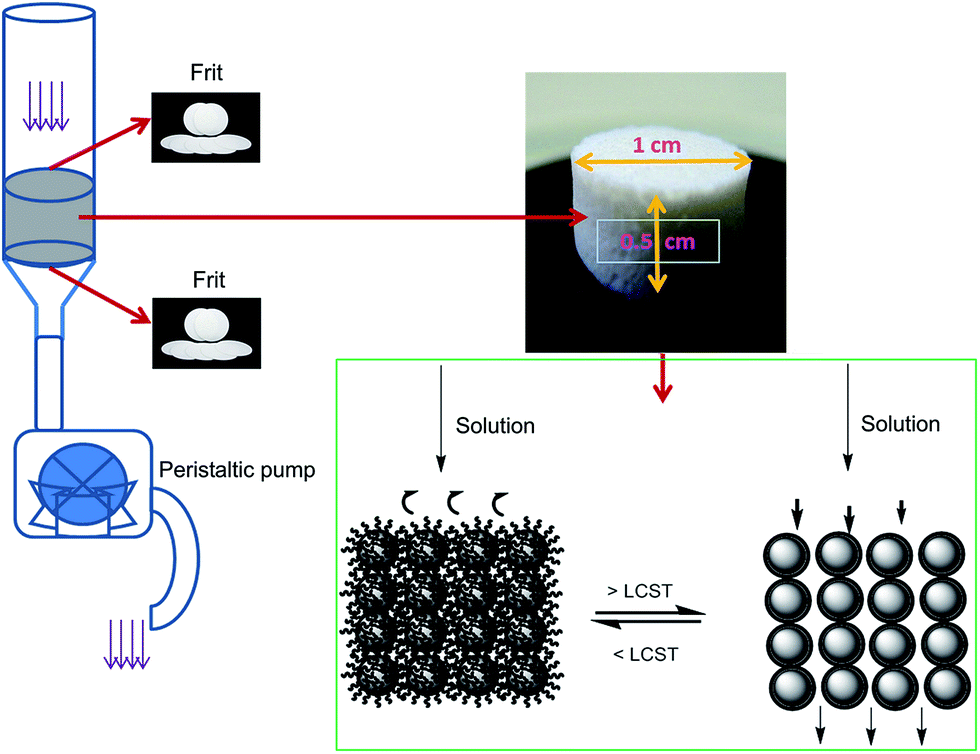 | ||
| Fig. 8 Pictures showing SPE cartridge preparation and its various components. Porous SPE disc formed and its shape and dimensions with mechanism of action. | ||
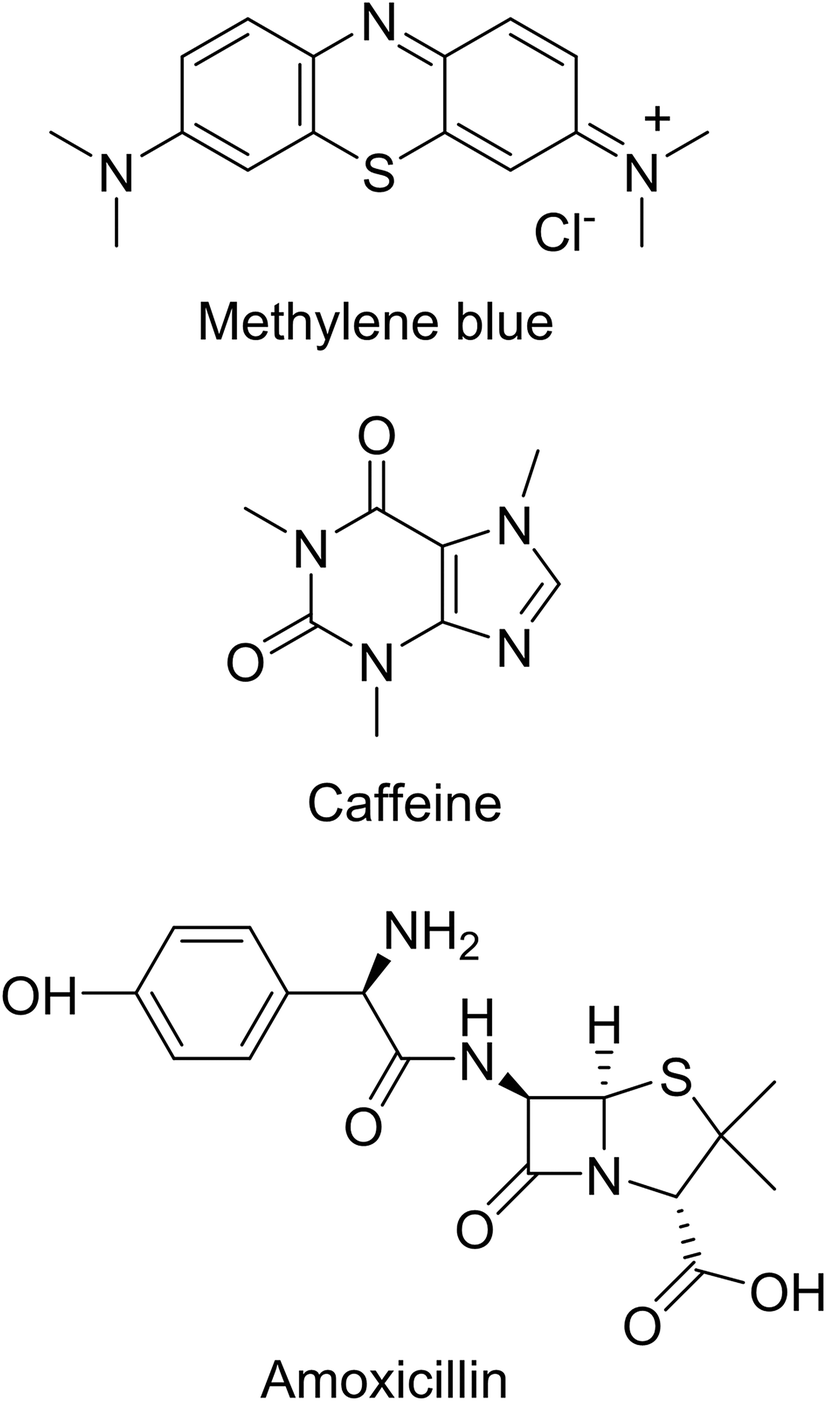 | ||
| Fig. 9 Structures of methylene blue (MB), caffeine (CF) and amoxicillin (AMX). | ||
The results obtained for the elution of the three model compounds in the form of concentration changes are summarized in Table 2. Temperature-induced modulation of the molecular transport across the SPE column was clearly evident in the case of methylene blue and amoxicillin solutions. Significant difference in the extraction efficiency of the thermoresponsive sorbent below and above the LCST of the polymer was observed, the main results obtained are shown in Fig. 10. It shows that AOX50–PNIPAM and FK320–PNIPAM nanoparticles almost completely or completely block the diffusion of methylene blue and amoxicillin at temperatures below the LCST of PNIPAM. On the contrary, unexpected odd results are shown by caffeine with both functionalized or non-functionalized SiNPs. Possible explanations concerning the molecular transport control of the prepared hybrid silica SPE cartridges towards the model compounds are summarized below considering two different behaviors, size and chemistry of the selected pollutants.
| Sample | Conc. g L−1 MB solution | Conc. g L−1 at 20 °C | Conc. g L−1 40 °C |
|---|---|---|---|
| AOX50 silica NPs | 3.16 × 10−4 | n.d. | n.d. |
| AOX50–PNIPAM | 3.16 × 10−4 | 3.75 × 10−5 | 2.93 × 10−4 |
| FK320 silica NPs | 3.16 × 10−4 | n.d. | n.d. |
| FK320–PNIPAM | 3.16 × 10−4 | n.d. | 4.88 × 10−5 |
| Sample | Conc. g L−1 CF solution | Conc. g L−1 at 20 °C | Conc. g L−1 40 °C |
|---|---|---|---|
| AOX50 silica NPs | 4.00 × 10−4 | 4.00 × 10−4 | 4.00 × 10−4 |
| AOX50–PNIPAM | 4.00 × 10−4 | 3.25 × 10−4 | 3.99 × 10−4 |
| FK320 silica NPs | 4.00 × 10−4 | 4.00 × 10−4 | 4.00 × 10−4 |
| FK320–PNIPAM | 4.00 × 10−4 | 3.69 × 10−4 | 4.00 × 10−4 |
| Sample | Conc. g L−1 AMX solution | Conc. g L−1 at 20 °C | Conc. g L−1 40 °C |
|---|---|---|---|
| AOX50 silica NPs | 4.2 × 10−2 | n.d. | 1.02 × 10−3 |
| AOX50–PNIPAM | 4.2 × 10−2 | n.d. | 3.17 × 10−2 |
| FK320 silica NPs | 4.2 × 10−2 | n.d. | n.d. |
| FK320–PNIPAM | 4.2 × 10−2 | n.d. | 2.98 × 10−2 |
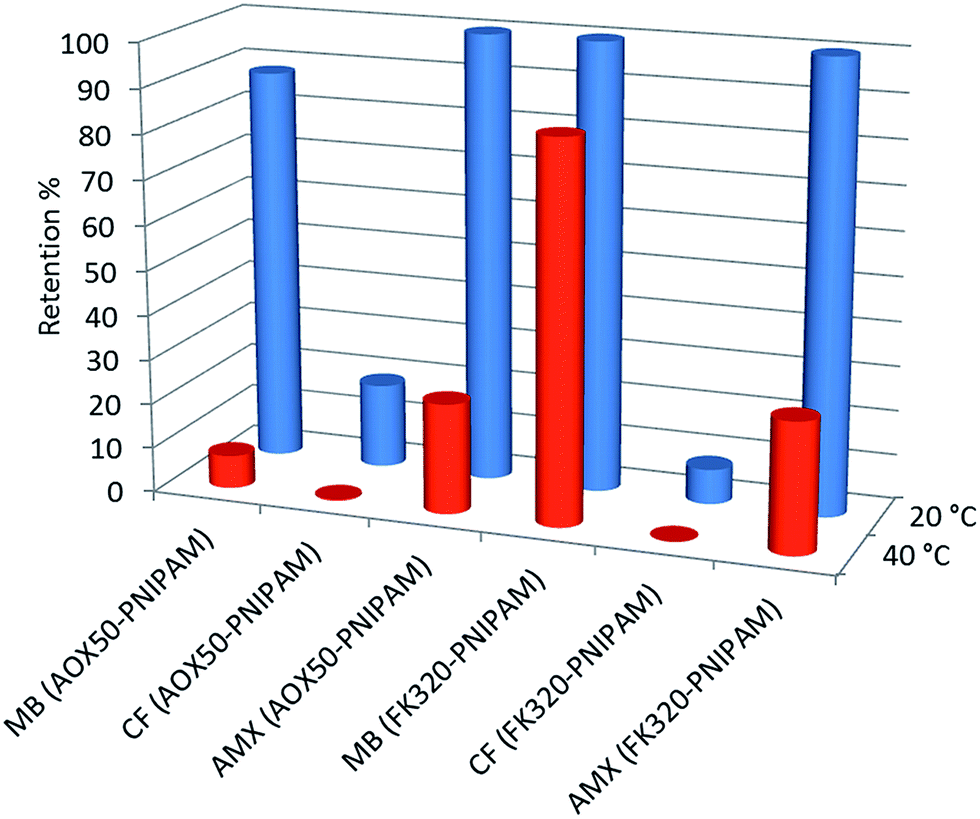 | ||
| Fig. 10 Retention of methylene blue (MB), caffeine (CF) and amoxicillin (AMX) on AOX50–PNIPAM and FK320–PNIPAM at 20 °C (blue) and 40 °C (red). | ||
MB is positively charged and is strongly adsorbed by bare SiNPs since they present Si–O− groups above pH 4 which act as adsorption sites for MB. For AOX50–PNIPAM and FK320–PNIPAM, retention of MB at 20 °C is almost complete and it is therefore mainly due to electrostatic interactions, nevertheless the effect of the thermoresponsivity of the sorbent material can be observed as grafting of PNIPAM decreases the zeta potential value of the nanoparticles and this trend results even more evident at T > LCST when PNIPAM chains collapse on the silica surface. For AOX50–PNIPAM, by increasing the temperature most of MB is released and recovery at 40 °C is 93% because collapsed polymer chains mask the silica surface charges. For FK320–PNIPAM, recovery at 40 °C is only 15% as the surface area of the silica sorbent is much higher and the collapsed PNIPAM chains cannot efficiently shield the negative surface charges.
CF, which is a small and non-charged molecule, does not interact with bare SiNPs and adsorption of CF on the unmodified cartridges is null, independently from the temperature. At 20 °C AOX50–PNIPAM and FK320–PNIPAM retain only 19% and 8% respectively of CF because the extended and hydrated polymer chains partially block the pore volume, but the small size of CF molecules allows the passage through the brushes of immobilized PNIPAM molecules. At 40 °C PNIPAM chains collapse on the surface of the SiNPs allowing free diffusion of caffeine molecules.
AMX is amphoteric, due to the presence of a carboxylic group and a primary amine group. Therefore, in a solution it could exist in anionic, cationic, or zwitterionic form. If the molecule exists in either cationic or zwitterionic form, the attractive electrostatic interaction with silica is involved in the retention mechanism and causes a stronger retention of the compound. AMX has pKa1 = 2.4 and pKa2 = 7.4, thus at pH > 2.4 it mainly exists in the zwitterionic or cationic form. Also, the phenol group in the structure of amoxicillin increases its polarity and probably provides a way to form hydrogen bonds with silica. Moreover, the size of AMX molecule is the largest examined in this study allowing some consideration concerning the size selective effect of the cartridge. These factors provide an explanation for the retention of AMX. Rising the temperature at 40 °C PNIPAM chains dehydrate and collapse making the silica surface more hydrophobic, thus breaking hydrophilic–hydrophilic interactions and allowing a recovery of AMX equal to 75% for AOX50–PNIPAM and 71% for FK320–PNIPAM.39,40 When the temperature decreases to 20 °C the PNIPAM molecules exhibit the coil conformation and AMX molecules are not allowed to pass through the cartridge.
4. Conclusions
In this work, PNIPAM-grafted solid SiNPs were synthesized, characterized and tested as new advanced materials for SPE beds. The SPE cartridges from PNIPAM-grafted SiNPs were prepared and tested for their performance in thermosensitive elution for the selective filtration of model compounds. Although preliminary, the presented results show that the transport of the selected molecules across the SPE bed can be regulated by changing the temperature above or below the LCST of PNIPAM grafted on silica particles. This allows controlling the inter-particle voids and switching the surface properties of the silica nanoparticles by virtue of the transition of PNIPAM chains from hydrophilic/extended to hydrophobic/collapsed. This work constitutes an important step towards the development of hybrid thermoresponsive SPE beds casted into portable devices. Additionally this work has the more general scope of presenting a simplified synthetic protocol for the preparation of thermoresponsive polymer-grafted particles that can be applied to prepare thermoresponsive nanoparticles of various oxide materials as components for hybrid sorbents and nanovalves with thermoresponsive surface properties.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Marie Sklodowska-Curie Research and Innovation Staff Exchange project funded by the European Commission H2020-MSCA-RISE-2014 [research project Mat4treaT, project number: 645551]; Compagnia di San Paolo and University of Torino [Project Torino_call2014_L2_126 through “Bando per il finanziamento di progetti di ricerca di Ateneo – anno 2014”, Project acronym: Microbusters]. Dr S. A. Jadhav thanks MIUR Italy for financial support. Moreover, authors thank Prof. A. Bianco Prevot and Dr D. Fabbri for assistance in the preparation of the thermoresponsive SPE cartridges and for useful discussion.References
- M. C. Hennion and V. Pinchon, Environ. Sci. Technol., 1994, 28, 576–583 CrossRef PubMed.
- A. Andrade-Eiroa, M. Canle, V. Leroy-Cancellieri and V. Cerdà, TrAC, Trends Anal. Chem., 2016, 80, 655–667 CrossRef CAS.
- J. S. Fritz, Analytical Solid-Phase Extraction, Wiley, New York, 1999 Search PubMed.
- K. Pyrzynska, Sep. Purif. Rev., 2008, 37, 372–389 CrossRef CAS.
- D. Huang, C. Deng and X. Zhang, Anal. Methods, 2014, 6, 7130–7141 RSC.
- X. Ma, M. Zhao, F. Zhao, H. Guo, J. Crittenden, Y. Zhu and Y. Chen, J. Sol-Gel Sci. Technol., 2016, 80, 87–95 CrossRef CAS.
- L. Vidal, O. Robin, J. Pashintsev, J. P. Mikkola and M. L. Reikkola, J. Chromatogr. A, 2013, 1285, 7–14 CrossRef CAS PubMed.
- K. Nagase, J. Kobayashi, A. Kikuchi, Y. Akiyama, H. Kanazawa and T. Okano, RSC Adv., 2015, 5, 66155–66167 RSC.
- K. Nagase, J. Kobayashi, A. Kikuchi, Y. Akiyama, H. Kanazawa and T. Okano, Biomacromolecules, 2014, 15, 1204–1215 CrossRef CAS PubMed.
- F. Roohi, Y. Fatoglu and M. M. Titirici, Anal. Methods, 2009, 1, 52–58 RSC.
- N. Li, L. Qi, Y. Shen, Y. Li and Y. Chen, ACS Appl. Mater. Interfaces, 2013, 5, 12441–12448 CAS.
- I. Tan, Z. Zarafshani, J. F. Lutz and M. M. Titirici, ACS Appl. Mater. Interfaces, 2009, 1, 1869–1872 CAS.
- F. Roohi, M. Antonietti and M. M. Titirici, J. Chromatogr. A, 2008, 1203, 160–167 CrossRef CAS PubMed.
- F. Roohi and M. Titirici, New J. Chem., 2008, 32, 1409–1414 RSC.
- S. A. Jadhav, I. Miletto, V. Brunella, G. Berlier and D. Scalarone, Polym. Adv. Technol., 2015, 26, 1070–1075 CrossRef CAS.
- G. Muthuraman, T. T. Teng, C. P. Leh and I. Norli, J. Hazard. Mater., 2009, 163, 363–369 CrossRef CAS PubMed.
- Y. Duan, Q. Wen, Y. Chen, T. Duan and Y. Zhou, Appl. Surf. Sci., 2014, 320, 746–755 CrossRef CAS.
- R. L. Seiler, S. D. Zaugg, J. M. Thomas and D. L. Howcroft, Groundwater, 1999, 37, 405–410 CrossRef CAS.
- M. K. Liu, Y. Y. Liu, D. D. Bao, G. Zhu, G. H. Yang, J. F. Geng and H. T. Li, Sci. Rep., 2017, 7, 43717 CrossRef PubMed.
- S. A. Jadhav, D. Scalarone, V. Brunella, E. Ugazio, S. Sapino and G. Berlier, eXPRESS Polym. Lett., 2017, 11, 96–105 CrossRef.
- S. A. Jadhav, V. Brunella, I. Miletto, G. Berlier and D. Scalarone, J. Appl. Polym. Sci., 2016, 133, 1–8 CrossRef.
- S. A. Jadhav, V. Brunella and D. Scalarone, Part. Part. Syst. Charact., 2015, 32, 417–428 CrossRef CAS.
- S. de Monredon, A. Pottier, J. Maquet, F. Babonneau and C. Sanchez, New J. Chem., 2006, 30, 797–802 RSC.
- I. D. Sideridou and M. M. Karabela, Dent. Mater., 2009, 25, 1315–1324 CrossRef CAS PubMed.
- F. M. Winnik, A. Adronov and H. Kitano, Can. J. Chem., 1995, 73, 2030–2040 CrossRef CAS.
- F. Eeckman, A. J. Moës and K. Amighi, Eur. Polym. J., 2004, 40, 873–881 CrossRef CAS.
- C. O. Metin, L. W. Lake, C. R. Miranda and Q. P. Nguyen, J. Nanopart. Res., 2001, 13, 839–850 CrossRef.
- P. S. Chinthamanipeta, S. Kobukata, H. Nakata and D. A. Shipp, Polymer, 2008, 49, 5636–5642 CrossRef CAS.
- J. Chen, M. Liu, C. Chen, H. Gong and C. Gao, ACS Appl. Mater. Interfaces, 2011, 3, 3215–3223 CAS.
- Y. Yang, X. Yan, Y. Cui, Q. He, D. Li, A. Wang, J. Fei and J. Li, J. Mater. Chem., 2008, 18, 5731–5737 RSC.
- J. H. Park, Y. H. Lee and S. G. Oh, Macromol. Chem. Phys., 2007, 208, 2419–2427 CrossRef CAS.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- C. Hofmann and M. Schönhoff, Colloid Polym. Sci., 2009, 287, 1369–1376 CAS.
- H. Zou and H. Schlaad, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1260–1267 CrossRef CAS.
- H. Wang, W. Luo and J. Chen, J. Mater. Sci., 2012, 47, 5918–5925 CrossRef CAS.
- S. Won, D. J. Phillips, M. Walker and M. I. Gibson, J. Mater. Chem. B, 2004, 4, 5673–5682 RSC.
- C. Walldal and S. Wall, Colloid Polym. Sci., 2000, 278, 936–945 CAS.
- Q. Fu, G. V. Rama Rao, T. L. Ward, Y. Lu and G. P. Lopez, Langmuir, 2007, 23, 170–174 CrossRef CAS PubMed.
- M. Jovanovic, T. Rakic, B. Jancic, A. Malenovic, D. Ivanovic and M. Medenica, J. Sep. Sci., 2012, 35, 1424–1431 CrossRef CAS PubMed.
- M. Godejohann, J. Chromatogr. A, 2007, 1156, 87–93 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra11869d |
| ‡ Present address: Polytechnic of Torino, Department of Applied Science and Technology DISAT, C.so Duca degli Abruzzi 24, 10129 Torino, Italy. |
| This journal is © The Royal Society of Chemistry 2018 |