
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Vortex-assisted low density solvent and surfactant based dispersive liquid–liquid microextraction for sensitive spectrophotometric determination of cobalt
Patiwat Chaiyamate,
Ketsarin Seebunrueng and
Supalax Srijaranai *
*
Materials Chemistry Research Center, Department of Chemistry, Center of Excellence for Innovation in Chemistry, Faculty of Science, Khon Kaen University, Khon Kaen 40002, Thailand. E-mail: supalax@kku.ac.th; Tel: +66 43 009700 ext. 42175
First published on 14th February 2018
Abstract
This study describes the development of vortex-assisted low density solvent and surfactant based dispersive liquid–liquid microextraction (VALS-DLLME) for Co(II) prior to its spectrophotometric detection. The method consisted of the complexation of Co(II) with pyrocatechol violet (PV) followed by the preconcentration of the Co(II)–PV complex using VALS-DLLME and then an absorption measurement at 600 nm. The optimum conditions for complex formation were a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 mole ratio of Co(II) and PV at pH 7.5, while the conditions for VALS-DLLME were 300 μL 1-dodecanol as extraction solvent, and 300 μL acetonitrile as dispersive solvent under a vortex for 20 s with the addition of cationic surfactant (0.02 mmol L−1 CTAB). Under the optimum conditions, good linearity was in the range of 0.1–10 mg L−1, the enrichment factor (EF) was 13.5 and the low limit of detection (LOD) was 0.04 mg L−1. The method was applied to the analysis of Co(II) in water, green leaf vegetable and vitamin B12 samples. The proposed method provided good recoveries in the range of 86–104%, which were comparable to those obtained from flame atomic absorption spectrophotometry.
:3 mole ratio of Co(II) and PV at pH 7.5, while the conditions for VALS-DLLME were 300 μL 1-dodecanol as extraction solvent, and 300 μL acetonitrile as dispersive solvent under a vortex for 20 s with the addition of cationic surfactant (0.02 mmol L−1 CTAB). Under the optimum conditions, good linearity was in the range of 0.1–10 mg L−1, the enrichment factor (EF) was 13.5 and the low limit of detection (LOD) was 0.04 mg L−1. The method was applied to the analysis of Co(II) in water, green leaf vegetable and vitamin B12 samples. The proposed method provided good recoveries in the range of 86–104%, which were comparable to those obtained from flame atomic absorption spectrophotometry.
Introduction
Cobalt is an essential element in the human body since it plays an important role in many vital processes including blood formation, synthesis of hormones, hemoglobin, neurotransmitters and other compounds, such as bile acids and DNA.1 Moreover, cobalt influences the functionality of vitamins such as vitamin C (ascorbic acid) and vitamin B12.2 Generally, cobalt is supplied to humans through food and drink. The determination or monitoring of cobalt in body fluids is important for controlling nutritional deficiencies and also the prevention of toxicity from the exposure to a high amount of cobalt. Deficiency of cobalt leads to several diseases such as pernicious anemia. On the other hand, toxicities to humans from the intake of a large amount of cobalt are vasodilation, flushing and cardiomyopathy.3,4 Thus, the development of analytical methods for trace cobalt determination is still required.The sensitive and selective method for the determination of metal ions including cobalt is atomic spectrometry; both atomic absorption spectrometry (AAS) and inductive couple plasma optical emission spectrometry (ICP-OES) have been widely used. However, these techniques are sophisticated and expensive. An alternative technique for detection of metal ions is molecular spectrometry. Visible spectrometry has been accepted as a simple and cost effective instrumental method for chromophore analytes. The unique property of metal complexes enables the determination of metal ions by visible spectrometry. Pre-derivatization of the metal ions by complexation is, therefore, necessitated before their spectrometric determination. Complexation provides not only selectivity from appropriate ligands for metal ions but also sensitivity from high molar absorptivity of the obtained complexes. There are a number of ligands used for the determination of cobalt by spectrometry such as ninhydrin,5 hydroxytriazene6 and 5-(2-benzothiazolylazo)-8-hydroxyquinolene.2 However, the use of visible spectrometry is still limited by its low sensitivity, especially for trace analysis. To increase the detection sensitivity, preconcentration is recommended.
Dispersive liquid–liquid microextraction (DLLME) has become the most popular technique in liquid phase microextraction since its introduction in 2006 by Assadi and co-workers7 due to many advantages such as simplicity, high enrichment factors and rapidness. DLLME is based on ternary solvent system including an aqueous solution, a water immiscible solvent (extraction solvent) and a water miscible solvent (dispersive solvent).8,9 A mixture of an extraction solvent and a dispersive solvent is injected rapidly into an aqueous solution. Then, a stable emulsion is formed containing fine droplets of the extraction solvent dispersed in the aqueous solution resulting in a large increase in contact area between the two phases. The analytes are easily transferred into the extraction phase, thus enriched. After extraction, the emulsion is separated into two phases using centrifugation and the extraction phase is subjected for analysis. DLLME has been successfully applied for various analytes including organic compounds such as pesticides10–12 biogenic amine13 and metal ions.14,15 However, there are some limitations in DLLME including (i) the use of hazardous chlorinated solvents having density higher than water, (ii) the use of large amounts of dispersive solvents, resulting in a decrease in partition coefficients of analytes and (iii) requirement of extra time for the centrifugation step.16–18 To overcome these drawbacks and to achieve green extraction, various methods based on DLLME have been proposed such as using other extraction solvent groups that are more environmentally friendly than conventional DLLME,19–21 using external forces (ultrasound and vortex) in the dispersion process22–24 and omitting the centrifugation step to reduce time consumption.16,25,26
To our knowledge, the reports on DLLME as a preconcentration technique for the spectrometric analysis of cobalt are mostly based on the analysis by flame atomic absorption spectrometry (FAAS).27–30 Only three articles have been reported on the use of DLLME coupled with ultraviolet-visible (UV-vis) spectrophotometric detection of cobalt.26–28 For instance, 1-(2-pyridylazo)-2-napthol (PAN)31,32 and newly synthesized cinnamoyl pyrones33 were used as ligands for the complexation of cobalt prior to DLLME and UV-vis detection.
This research was aimed at the development of a selective and sensitive spectrometric method for the determination of cobalt. Cobalt was first derivatized via complexation with pyrocatechol violet (PV). After that, the cobalt complex was enriched by the modified DLLME, named vortex-assisted low density solvent and surfactant based DLLME (VALS-DLLME).
Experimental
Chemicals and reagents
All chemicals used are of at least analytical reagent grade. Cobalt chloride hexahydrate (CoCl2·6H2O), metal ions studied for interferences, pyrocatechol violet and nitric acid were obtained from Carlo-Erba, France. 1-Undecanol, 1-dodecanol, dodecyltrimethyl ammonium bromide (DTAB), trimethyltetradecyl ammonium bromide (TTAB), and cethyltrimethyl ammonium bromide (CTAB) were purchased from Sigma-Aldrich (USA). 1-Octanol was obtained from Panreac (Spain). Methanol (HPLC grade) and acetonitrile (HPLC grade) were obtained from Merck (Germany). Acetone was purchased from Q-rac (Malaysia). Sodium sulphate anhydrous was purchased from Fluka (Japan). Sodium chloride, sodium hydrogen phosphate and sodium dihydrogen phosphate were purchased from Ajax Finechem (Australia). Aluminium chloride was obtained from Ajax Finechem Pty Ltd (New Zealand). All solutions were prepared using deionized water with the resistivity of 18.2 MΩ cm from RiOs™ Type I Simplicity 185 (Millipore Waters, USA). Stock solution of standard Co(II) 100 mg L−1 (or 1.695 mmol L−1) was prepared and used throughout.Instruments
Absorbance measurements and spectra recording were performed on a spectrophotometer (Agilent Technologies Cary 60 UV-Visible Spectroscopy System, Germany). A 1 cm micro quartz cell was used throughout the experiments. An analytical balance (Scaltech, Germany) was used. A pH meter (Model 251, Denver Instrument, USA) was used for pH measurement. Vortex (Scientific Industries, INC., USA) was used for mixing.Quantitative determination of Co(II) was also performed on a flame atomic absorption spectrophotometer (Perkin Elmer Instrument AA Analyst 100, England).
Sample preparation
:1 HNO3, heated to near dryness and subsequently digested by 8 mL of 1:1 HCl. The residue was dissolved by adding an appropriate amount of water to 50 mL and filtered through Whatman (no. 42) filter paper before analysis.Preconcentration procedure and determination
Standard solution of Co(II) or the sample solution 5 mL was placed in a 10 mL volumetric flask. Aliquot 2.30 mL of 0.26 mmol L−1 PV, 50 μL of 1 mol L−1 phosphate buffer (pH 7.5), 200 μL of 1 mmol L−1 CTAB and 0.2 g Na2SO4 were added. Then, a mixture of 300 μL of 1-dodecanol (extraction solvent) and 300 μL of acetonitrile (dispersive solvent) was rapidly injected into the solution. After that water was added to make a final volume of 10 mL. The solution was then mixed using a vortex mixer at 3200 rpm for 40 s. The Co(II)–PV complex was extracted into the fine droplet of the extraction solvent. After leaving the solution to stand, the extraction phase at the top of the solution was collected and diluted with 600 μL of methanol and detected by a spectrophotometer at 600 nm.Results and discussion
Optimization for the formation of Co(II)–PV complex
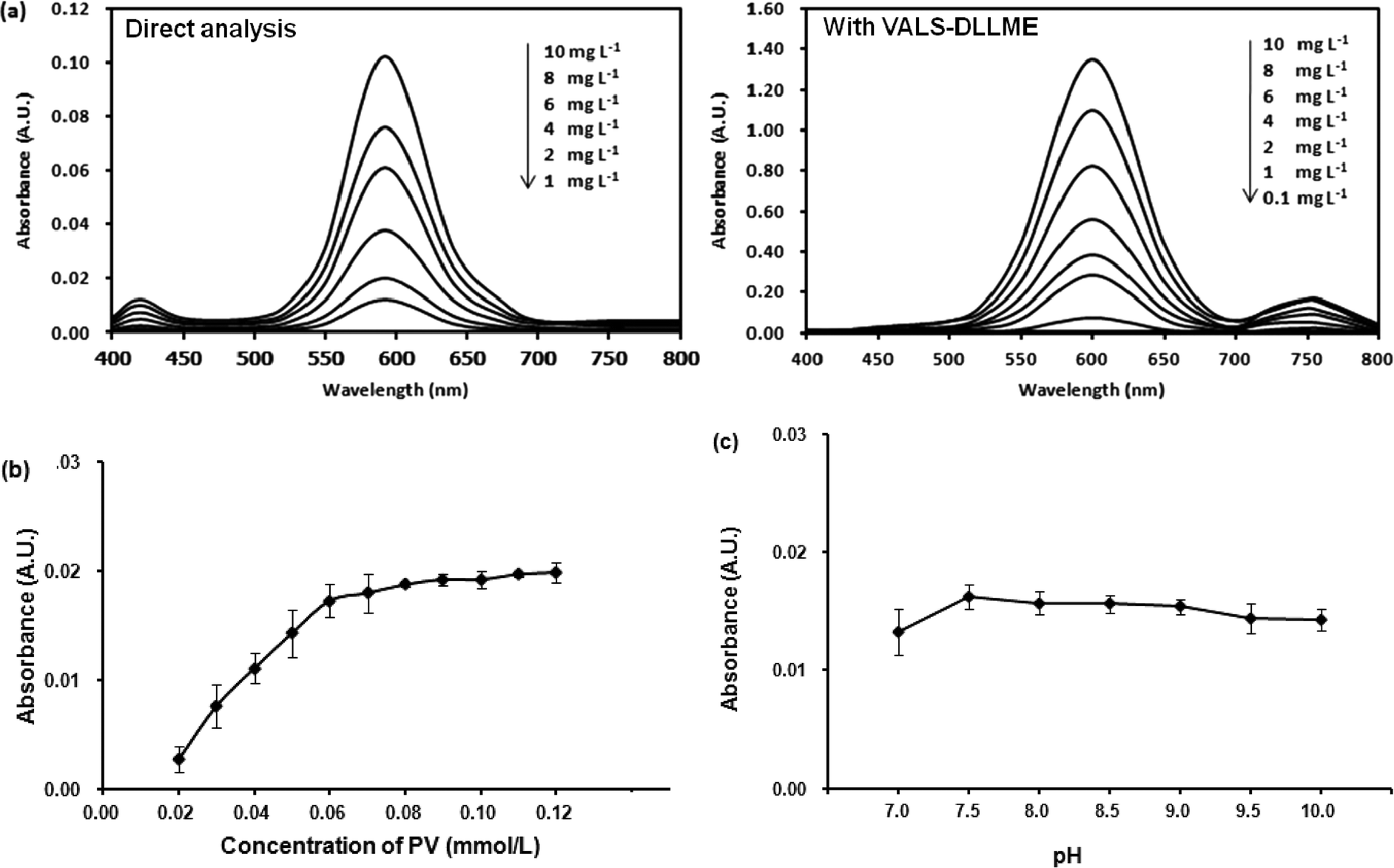 | ||
| Fig. 1 (a) Absorption spectra of Co(II)–PV complex at different concentrations of Co(II) obtained from direct analysis (left) and with VALS-DLLME (right). Conditions of complexation: molar ratio (Co(II):PV) of 1:3, phosphate buffer pH 7.5. Condition of VALS-DLLME: 300 μL 1-dodecanol, 0.02 mmol L−1 CTAB, 300 μL acetonitrile, 0.2 g Na2SO4, vortex at 3200 rpm for 40 s. (b) Effect of the PV concentration. Conditions: 1.00 mg L−1 Co(II) and phosphate buffer pH 7.5. (c) Effect of pH. Conditions: 1.00 mg L−1 Co(II) and 0.06 mmol L−1 PV in phosphate buffer. | ||
:3.5 implying 1:3 mole ratio of M:L, which is in good agreement with an earlier report.35Moreover, the stability of the Co(II)–PV complex was studied by leaving the complex solution for 1 hour before absorption measurement every 10 min. The results indicated that the Co(II)–PV complex was stable within 1 hour of the studied time.
Optimization of the VALS-DLLME
The preliminary study for extraction of Co(II)–PV complex by DLLME was unsuccessful as the obtained Co(II)–PV complex is anionic. Thus, it is necessary to neutralize the negative charge of Co(II)–PV complex by an addition of cationic surfactants into the solution which led to the successful extraction of the complex from the solution. However, after VALS-DLLME, the greenish blue solution of the complex was obtained, showing that there was a bathochromic shift of the maximum absorption wavelength from 590 nm to 600 nm (Fig. 1(a); with VALS-DLLME). In addition, the dominant bathochromic shift was observed for PV,36 by which the maximum absorption wavelength shifted from 445 nm to 720 nm. To obtain the highest extraction efficiency, parameters affecting the efficiency of VALS-DLLME were studied and optimized using 1.00 mg L−1 Co(II) and 0.059 mmol L−1 PV throughout the subsequent studies.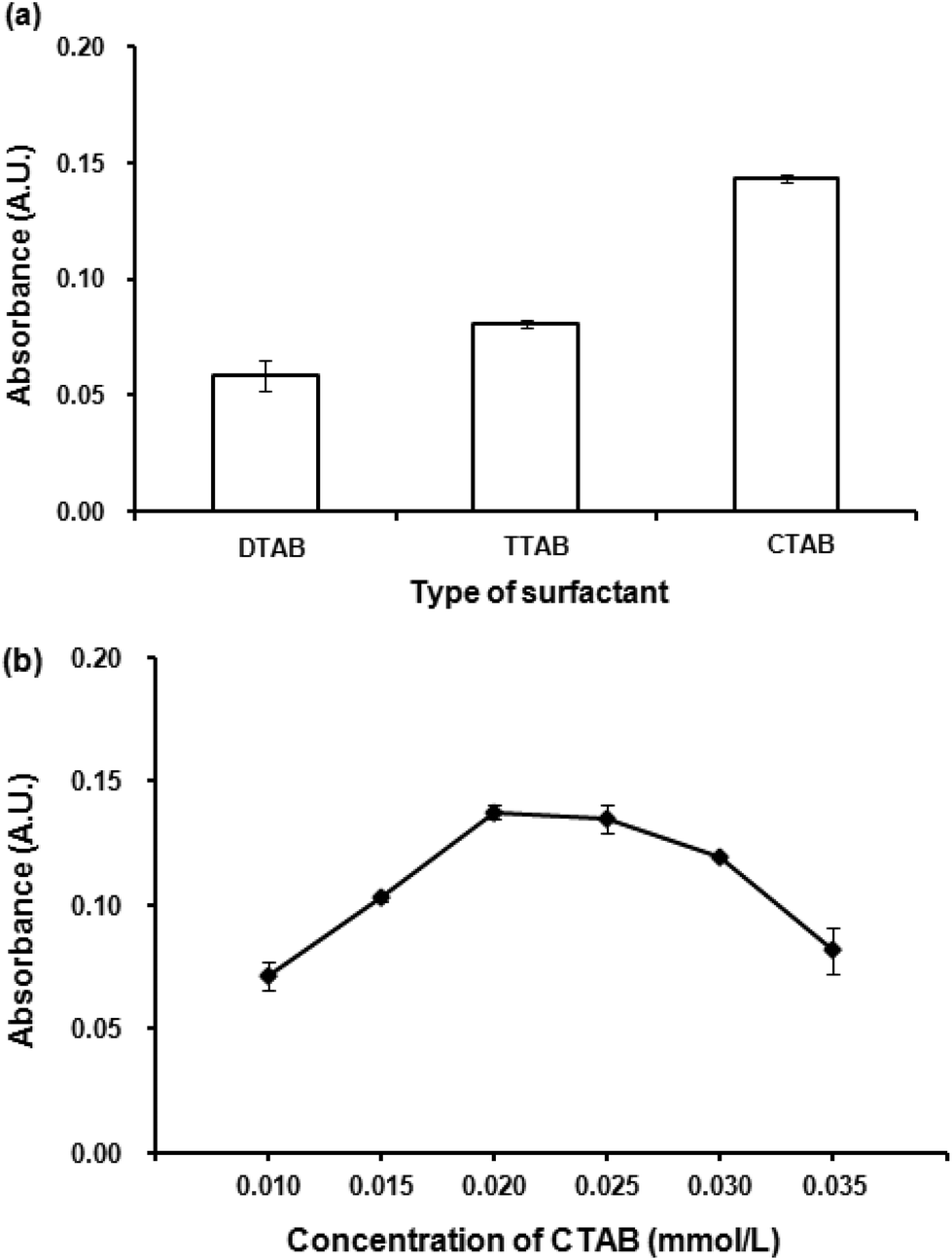 | ||
| Fig. 2 (a) Effect of surfactant. Conditions for VALS-DLLME: 300 μL 1-dodecanol, 0.02 mmol L−1 surfactant, vortex at 3200 rpm for 40 s. (b) Effect of CTAB concentration. Conditions: as described in (a) except CTAB concentrations were varied. | ||
Different concentrations of CTAB in the range of 0.010 to 0.035 mmol L−1 were investigated. Fig. 2(b) shows the dependence of absorbance upon the concentrations of CTAB. The absorbance increased with an increasing concentration of CTAB from 0.010 mmol L−1 to 0.020 mmol L−1. This can be explained by insufficient balancing of the anionic complex with low concentration of the positively charged CTAB. In addition, it was observed that the aqueous solution was still of the greenish blue color when the concentration of CTAB was less than 0.020 mmol L−1. The highest absorbance was obtained at 0.020 mmol L−1 CTAB and then slightly decreased. This is probably attributable to an excess of surfactant, thus increasing the solubility of the complex in an aqueous phase.38,39 The concentration of CTAB at 0.020 mmol L−1, which provided the highest absorbance was therefore selected for further experiments.
Kow = 3.15), 1-undecanol (logKow = 4.72) and 1-dodecanol (logKow = 5.13) were investigated. It is clearly seen (Fig. 3(a)) that the most hydrophobic solvent, 1-dodecanol, was the most efficient to extract the Co(II)–PV complex as it gave the highest absorbance. In addition, it was observed that 1-dodecanol provided good phase separation compared to the others. Consequently, 1-dodecanol was chosen for further studies.
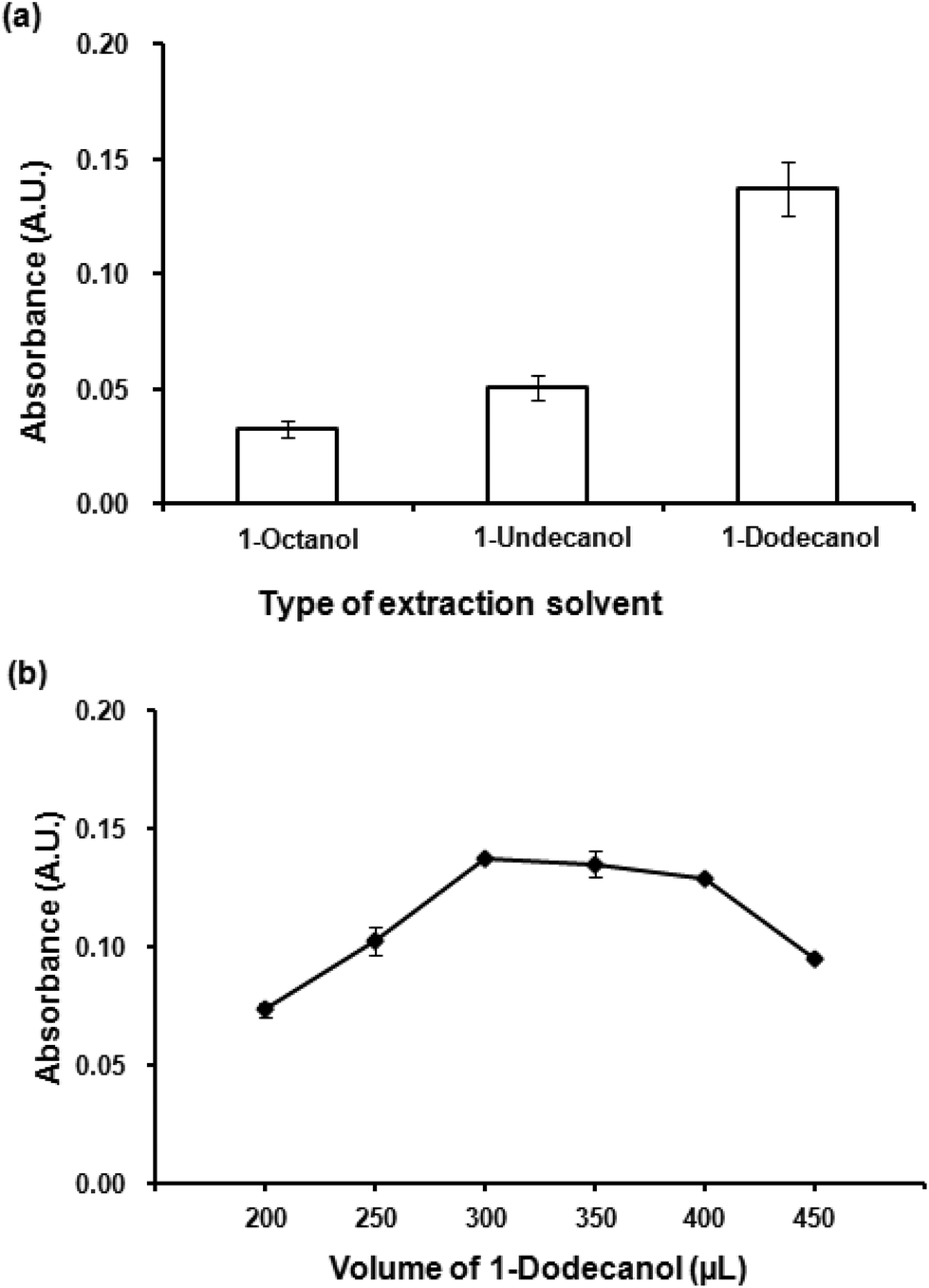 | ||
| Fig. 3 (a) Effect of extraction solvent. Conditions for VALS-DLLME: 300 μL 1-dodecanol, 0.02 mmol L−1 CTAB, vortex at 3200 rpm for 40 s. (b) Effect of extraction solvent volume. Conditions: as described in (a) except volumes of 1-dodecanol were varied. | ||
To optimize the volume of the extraction solvent, different volumes of 1-dodecanol from 200 μL to 450 μL were investigated. Fig. 3(b) depicts the effect of extraction solvent on extraction efficiency (as absorbance), volumes less than 300 μL was insufficient to extract all of the Co(II)–PV complex, while the volumes larger than 400 μL showed the dilution effect. The volume of 300 μL that produced the highest absorbance was chosen for the subsequent experiments.
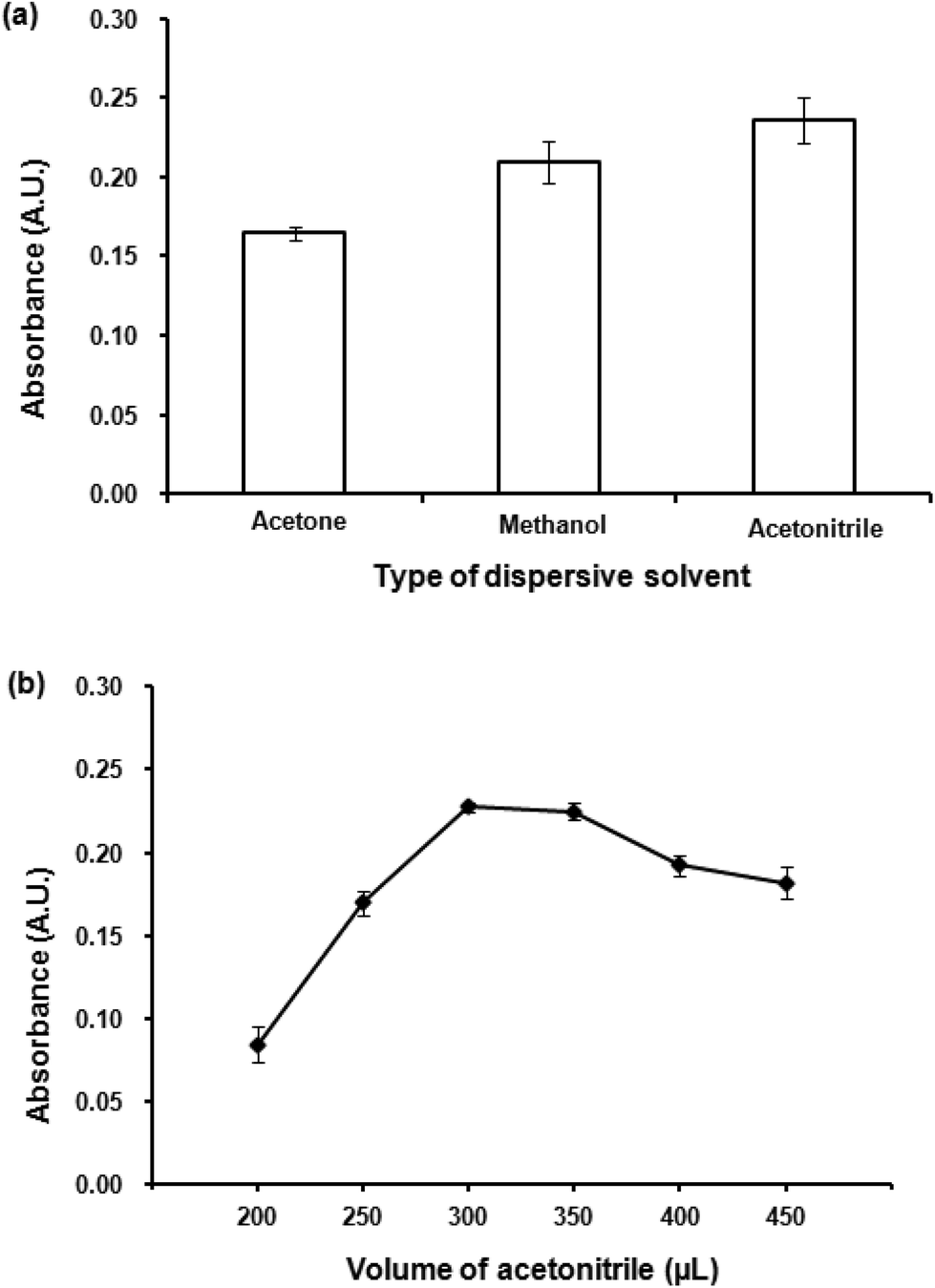 | ||
| Fig. 4 (a) Effect of dispersive solvent. Conditions: 300 μL 1-dodecanol as extraction solvent, 300 μL dispersive solvent, 0.02 mmol L−1 CTAB, vortex time at 3200 rpm for 40 s. (b) Effect of dispersive solvent volume. Conditions: as described in (a) except volumes of acetonitrile were varied. | ||
Different volumes acetonitrile (200–450 μL) were studied. Fig. 4(b) shows the dependence of absorbance upon the volume of acetonitrile. Low volume of acetonitrile provided insufficient contact area between the extraction solvent and aqueous solution preventing a good formation of the cloudy solution. The highest absorbance was obtained at 300 μL, after that an increase in acetonitrile volume resulted in a decrease in absorbance. Large volumes of the dispersive solvent increase the solubility of the analytes in the aqueous phase, leading to a decrease in extraction efficiency resulted from a reduction in distribution coefficient.42 Therefore, 300 μL of acetonitrile was selected as the optimum volume.
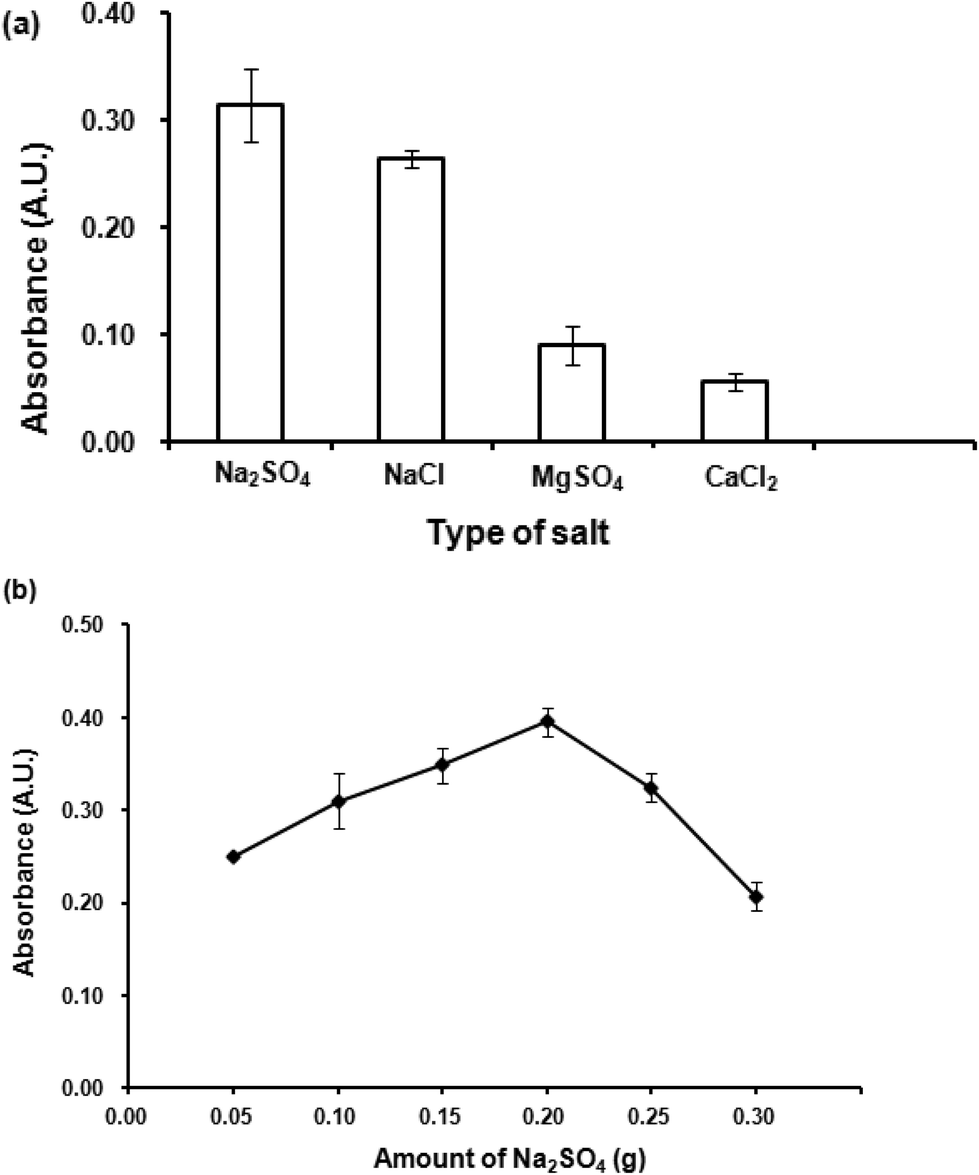 | ||
| Fig. 5 (a) Effect of salt addition. Conditions: 300 μL 1-dodecanol as extraction solvent, 300 μL acetonitrile as dispersive solvent, 0.02 mmol L−1 CTAB, 0.1 g (1.0% w/v) salt, vortex at 3200 rpm for 40 s. (b) Effect of amount of salt. Conditions: as described in (a) except amounts of Na2SO4 were varied. | ||
Effect of Na2SO4 amount was then studied by varying amounts of Na2SO4 from 0.05 to 0.3 g. The results (Fig. 5(b)) indicated that 0.2 g Na2SO4 gave the highest absorbance. Beyond this point, it was found that higher amounts of Na2SO4 could not improve the extraction efficiency. A high amount of salt increases viscosity of the aqueous phase, thus obstructing the migration of the analytes into the organic extraction phase. Therefore, 0.2 g Na2SO4 was selected for further studies.
Quantitative analysis and method validation
The analytical performances and validation of the proposed method were then investigated including linear range, limit of detection (LOD), limit of quantification (LOQ), precisions (intra-day and inter-day) and interference study. The LOD and LOQ were calculated as Co(II) concentration giving a signal equal to 3 SD and 10 SD, respectively, where SD is the standard deviation obtained from the measurement of ten blank samples. As summarized in Table 1, good linearity was observed in the range of 0.1–10.0 mg L−1 with R2 = 0.9995. LOD and LOQ were 0.04 mg L−1 and 0.13 mg L−1, respectively. High precision was obtained with the RSDs of less than 2.10%. The efficiency of the developed DLLME was evaluated in terms of enrichment factor (EF) as the slope ratio of two calibration curves for analyte with and without the preconcentration procedure (direct analysis). The proposed method provided high EF of 13.5.| Co(II)–PV complex | Linearity (mg L−1) | Linear equation | R2 | LOD (mg L−1) | LOQ (mg L−1) | EF | Intra-day (n = 6), %RSD | Inter-day (n = 6 × 6 days), %RSD |
|---|---|---|---|---|---|---|---|---|
| Direct analysis | 1.0–10.0 | Y = 0.0101X + 0.0003 | 0.9996 | 0.45 | 1.50 | — | 1.86 | 1.98 |
| With VALS-DLLME | 0.1–10.0 | Y = 0.1350X + 0.1459 | 0.9995 | 0.04 | 0.13 | 13.5 | 2.10 | 2.35 |
The selectivity of the proposed spectrophotometric method was determined by adding different amounts of potential interfering species into 1 mg L−1 standard Co(II) before the analysis. The tolerance limit was taken the concentration of the interfering species giving an error of absorbance lower than ±5%. The obtained results are summarized in Table 2. It can be classified into two groups i.e. the ions which increased the absorbance (positive bias) including Mg(II), Mn(II), Ni(II) and Fe(II) and the negative bias (decreased the absorbance) such as Cu(II), Cd(II) and Zn(II), Sn(II), Al(III), Fe(III) and Sb(III). The studied ions at various concentrations affected the detection of Co(II), notifying that the tolerance limits for studied metal ions including Ni(II), Fe(II), Cu(II), Cd(II) and Zn(II) were higher than 10 mg L−1, while those for Mg(II), Mn(II), Sn(II), Al(III), Fe(III) and Sb(III) were higher than 30 mg L−1.
| Foreign metal ion | Tolerance limit (mg L−1) |
|---|---|
| Cu(II) | 10.1 |
| Fe(II) | 10.2 |
| Zn(II) | 11.1 |
| Ni(II) | 12.4 |
| Cd(II) | 13.2 |
| Mg(II) | 31.0 |
| Mn(II) | 31.2 |
| Al(III) | 32.0 |
| Fe(III) | 34.1 |
| Sn(II) | 35.2 |
| Sb(III) | 35.4 |
Application to real samples
The proposed method was applied to determine Co(II) residue in water samples (tap water, ground water, surface water, agricultural water and wastewater), green leaf vegetables (Chinese cabbage, mint, spinach, cabbage and kale) and five vitamin B12 tablets (sample no. I, II, III, IV and V). Co(II) was not detected in any water or green leaf vegetable samples. However, Co(II) was detected in some vitamin B12 in the range of 0.036–0.729 mg g−1. These results were in accordance with those obtained from FAAS (Tables 3–5).| Sample | Spiked (mg L−1) | VALS-DLLME | FAAS | ||||
|---|---|---|---|---|---|---|---|
| Found (mg L−1) | Recovery (%) | RSD (%) | Found (mg L−1) | Recovery (%) | RSD (%) | ||
| a ND: not detected. | |||||||
| Tap water | 0.0 | NDa | — | — | ND | — | — |
| 0.5 | 0.497 | 99.4 | 0.50 | 0.492 | 98.5 | 1.23 | |
| 1.0 | 0.978 | 97.8 | 2.82 | 0.987 | 98.7 | 1.28 | |
| 3.0 | 2.912 | 97.1 | 2.77 | 3.041 | 101.4 | 1.97 | |
| Ground water | 0.0 | ND | — | — | ND | — | — |
| 0.5 | 0.483 | 96.6 | 0.78 | 0.489 | 97.8 | 2.78 | |
| 1.0 | 0.963 | 96.3 | 1.28 | 0.991 | 99.1 | 3.20 | |
| 3.0 | 3.098 | 103.3 | 3.18 | 3.016 | 100.5 | 1.21 | |
| Surface water | 0.0 | ND | — | — | ND | — | — |
| 0.5 | 0.472 | 94.4 | 1.40 | 0.479 | 95.8 | 1.90 | |
| 1.0 | 0.973 | 97.3 | 3.41 | 0.982 | 98.2 | 2.07 | |
| 3.0 | 2.892 | 96.4 | 1.18 | 3.104 | 103.5 | 3.42 | |
| Agricultural water | 0.0 | ND | — | — | ND | — | — |
| 0.5 | 0.481 | 96.2 | 2.63 | 0.487 | 97.3 | 1.63 | |
| 1.0 | 0.982 | 98.2 | 1.42 | 0.987 | 98.7 | 2.57 | |
| 3.0 | 2.875 | 95.8 | 2.79 | 3.006 | 100.2 | 0.71 | |
| Wastewater | 0.0 | ND | — | — | ND | — | — |
| 0.5 | 0.462 | 92.4 | 1.63 | 0.467 | 93.5 | 0.51 | |
| 1.0 | 0.971 | 97.1 | 3.20 | 0.985 | 98.5 | 3.30 | |
| 3.0 | 2.975 | 99.2 | 3.78 | 3.011 | 100.4 | 3.78 | |
| Sample | Spiked (mg g−1) | VALS-DLLME | FAAS | ||||
|---|---|---|---|---|---|---|---|
| Found (mg g−1) | Recovery (%) | RSD (%) | Found (mg g−1) | Recovery (%) | RSD (%) | ||
| a ND: not detected. | |||||||
| Mint | 0.00 | NDa | — | — | ND | — | — |
| 0.25 | 0.246 | 98.4 | 0.78 | 0.249 | 99.7 | 1.21 | |
| 0.50 | 0.495 | 99.0 | 1.67 | 0.496 | 99.2 | 2.65 | |
| 1.50 | 1.507 | 100.5 | 1.87 | 1.512 | 100.8 | 2.13 | |
| Spinach | 0.00 | ND | — | — | ND | — | — |
| 0.25 | 0.226 | 90.2 | 0.98 | 0.228 | 91.3 | 0.59 | |
| 0.50 | 0.491 | 98.2 | 2.13 | 0.507 | 101.3 | 1.24 | |
| 1.50 | 1.503 | 100.2 | 2.43 | 1.521 | 101.4 | 2.57 | |
| Kale | 0.00 | ND | — | — | ND | — | — |
| 0.25 | 0.236 | 94.2 | 2.53 | 0.248 | 99.1 | 0.76 | |
| 0.50 | 0.489 | 97.9 | 2.36 | 0.491 | 98.1 | 1.43 | |
| 1.50 | 1.554 | 103.6 | 1.89 | 1.523 | 101.5 | 0.98 | |
| Cabbage | 0.00 | ND | — | — | ND | — | — |
| 0.25 | 0.242 | 96.8 | 1.09 | 0.246 | 98.4 | 1.32 | |
| 0.50 | 0.496 | 99.1 | 2.34 | 0.506 | 101.2 | 2.36 | |
| 1.50 | 1.456 | 97.1 | 1.54 | 1.556 | 103.7 | 2.41 | |
| Chinese cabbage | 0.00 | ND | — | — | ND | — | — |
| 0.25 | 0.241 | 96.4 | 1.24 | 0.244 | 97.6 | 0.97 | |
| 0.50 | 0.493 | 98.6 | 2.32 | 0.503 | 100.6 | 1.23 | |
| 1.50 | 1.498 | 99.9 | 1.14 | 1.515 | 101.1 | 1.09 | |
| Sample | Spiked (mg g−1) | VALS-DLLME | FAAS | ||||
|---|---|---|---|---|---|---|---|
| Found (mg g−1) | Recovery (%) | RSD (%) | Found (mg g−1) | Recovery (%) | RSD (%) | ||
| No. I (1 tablet = 0.1509 g) | 0.000 | 0.729 | — | — | 0.762 | — | — |
| 0.165 | 0.868 | 94.1 | 1.24 | 0.888 | 94.0 | 2.49 | |
| 0.331 | 1.054 | 99.6 | 2.43 | 1.067 | 101.8 | 1.57 | |
| 0.994 | 1.736 | 101.1 | 1.09 | 1.749 | 102.3 | 0.98 | |
| No. II (1 tablet = 0.1374 g) | 0.000 | 0.699 | — | — | 0.706 | — | — |
| 0.182 | 0.859 | 86.0 | 1.54 | 0.888 | 99.0 | 1.26 | |
| 0.364 | 1.077 | 102.7 | 0.98 | 1.084 | 103.0 | 2.37 | |
| 1.092 | 1.776 | 99.7 | 2.32 | 1.849 | 104.3 | 1.57 | |
| No. III (1 tablet = 0.1270 g) | 0.000 | 0.701 | — | — | 0.702 | — | — |
| 0.197 | 0.882 | 92.0 | 1.43 | 0.898 | 98.0 | 1.43 | |
| 0.394 | 1.087 | 99.5 | 1.57 | 1.118 | 104.5 | 1.89 | |
| 1.181 | 1.882 | 100.7 | 0.69 | 1.953 | 105.7 | 0.94 | |
| No. IV (1 tablet = 0.3056 g) | 0.000 | 0.111 | — | — | 0.115 | — | — |
| 0.081 | 0.187 | 92.0 | 2.12 | 0.193 | 93.1 | 2.31 | |
| 0.164 | 0.272 | 97.0 | 3.45 | 0.275 | 99.0 | 1.54 | |
| 0.491 | 0.605 | 100.7 | 1.34 | 0.612 | 101.0 | 0.92 | |
| No. V (1 tablet = 0.3924 g) | 0.000 | 0.036 | — | — | 0.038 | — | — |
| 0.064 | 0.097 | 96.0 | 1.57 | 0.092 | 84.0 | 1.27 | |
| 0.127 | 0.163 | 98.5 | 2.58 | 0.168 | 102.4 | 0.93 | |
| 0.382 | 0.415 | 99.3 | 2.63 | 0.426 | 101.3 | 1.38 | |
The accuracy of the proposed method was also investigated as recovery by spiking known concentrations at three levels of Co(II) into sample solutions before acid digestion and ashing for vitamin B12 and vegetable sample, respectively. The obtained solutions were then subjected to VALS-DLLME and spectrophotometric analysis. All experiments were performed in triplicate. The recoveries of water, vegetable and vitamin B12 samples (Tables 3–5) were obtained in the range of 92.4–103.3%, 90.2–103.6% and 86.0–102.7% with RSD less than 3.78, 2.53 and 3.45%, respectively. In addition, the accuracy of the proposed method was studied by comparing the results with those obtained from FAAS. The results indicated an insignificant difference (p = 0.05) between the proposed method and FAAS.
Conclusions
A simple and sensitive spectrophotometric method has been successfully developed for the determination of Co(II) in water, green leaf vegetable and vitamin B12 samples. The method is based on the complexation of Co(II) with pyrocatechol violet (PV) and the preconcentration of Co(II)–PV complex by vortex-assisted low density solvent and surfactant based dispersive liquid–liquid microextraction (VALS-DLLME) before measurement of the absorbance at visible wavelength. The proposed method provided high precision, low LOD and high accuracy. Moreover, the method employs visible spectrophotometer, an unsophisticated instrument, providing an economical alternative to FAAS for the determination of Co(II) in real samples.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Project for the Promotion of Science and Mathematics talented Teachers (PSMT) from the Institute for the Promotion of teaching Science and Technology (IPST), Thailand and the Post-doctoral Program from Research Affairs and Graduate School, Khon Kaen University (58335).References
- F. R. Adolfo, P. C. Nascimento, D. Bohrer, L. M. Carvalho, C. Viana, A. Guarda, A. N. Colim and P. Mattiazzi, Talanta, 2016, 147, 241 CrossRef CAS PubMed.
- A. S. Amin, Arabian J. Chem., 2014, 7, 715 CrossRef CAS.
- J. W. Zhang, X. J. Ke, Y. K. Wang, X. Du, J. J. Ma and J. C. Li, J. Chin. Chem. Soc., 2011, 58, 911 CrossRef CAS.
- M. Hoshino, M. Matsushita, M. Samma, M. Asano, T. Yamaguchi and Y. Fujita, Chem. Pharm. Bull., 2011, 59, 721 CrossRef CAS PubMed.
- K. Mahwood, F. H. Wattoo, M. H. S. Wattoo, M. Imran, M. J. Asad, S. A. Tirmizi and A. Wadood, Saudi J. Biol. Sci., 2012, 19, 247 CrossRef PubMed.
- O. Ombaka and J. M. Gichumbi, Afr. J. Pure Appl. Chem., 2011, 5, 494 CAS.
- M. Rezaee, Y. Assadi and M. R. M. Hosseini, J. Chromatogr. A, 2006, 1116, 1 CrossRef CAS PubMed.
- A. Ranji, M. G. Ravandi and M. A. Farajzadeh, Anal. Sci., 2008, 24, 623 CrossRef CAS PubMed.
- H. A. Mashayekhi, M. Rezaee, S. S. Garmaroudi, N. Montazeri and S. J. Ahmadi, Anal. Sci., 2011, 27, 865 CrossRef CAS PubMed.
- Q. Zhou, L. Pang, G. Xie, J. Xiao and H. Bai, Anal. Sci., 2009, 25, 73 CrossRef CAS PubMed.
- S. Boonchiangma, W. Ngeontae and S. Srijaranai, Talanta, 2012, 88, 209 CrossRef CAS PubMed.
- K. Seebunrueng, Y. Santaladchaiyakit and S. Srijaranai, Chemosphere, 2014, 103, 51 CrossRef CAS PubMed.
- J. Donthuan, S. Yunchalard and S. Srijaranai, J. Sep. Sci., 2014, 37, 3164 CrossRef CAS PubMed.
- C. B. Ojeda and F. S. Rojas, Sample Perp., 2014, 2, 13 Search PubMed.
- A. Asghari, M. Ghazaghi, M. Rajabi, M. Behzad and M. Ghaedi, J. Serb. Chem. Soc., 2014, 79, 63 CrossRef CAS.
- K. Seebunrueng, Y. Santaladchaiyakit and S. Srijaranai, Talanta, 2015, 132, 769 CrossRef CAS PubMed.
- B. Majidi and F. Shemirani, Talanta, 2012, 93, 245 CrossRef CAS PubMed.
- J. Regueiro, M. Llompart, C. Garcia-Jares, J. C. Garcia-Monteagudo and R. Cela, J. Chromatogr. A, 2008, 1190, 27 CrossRef CAS PubMed.
- L. Kocurova, I. S. Balogh, J. Sandrejova and V. Andruch, Microchem. J., 2012, 102, 11 CrossRef CAS.
- L. Ranjbar, Y. Yamini, A. Saleh, S. Seidi and M. Faraji, Microchim. Acta, 2012, 177, 119 CrossRef CAS.
- E. Ragheb, P. Hashemi, K. Alizadeh and M. R. Ganjali, Anal. Sci., 2015, 31, 119 CrossRef CAS PubMed.
- D. K. Acar and D. Kara, Water, Air, Soil Pollut., 2014, 1864, 1 Search PubMed.
- S. Yang, X. Fang, L. Duan, S. Yang, Z. Lei and X. Wen, Spectrochim. Acta. Part A: Mol. Biomol. Spectrosc., 2015, 148, 72 CrossRef CAS PubMed.
- J. Lu, Z. P. Wu, W. J. Che, Y. P. Xian, X. D. Guo, J. X. Lv and H. Li, Anal. Sci., 2016, 32, 407 CrossRef CAS PubMed.
- L. Guo, S. H. Chia and H. K. Lee, Anal. Chem., 2016, 88, 2548 CrossRef CAS PubMed.
- Y. Xia, M. Cheng, F. Guo, X. Wang and J. Cheng, Anal. Chim. Acta, 2012, 724, 47 CrossRef CAS PubMed.
- M. Ramezani and S. Rahmani, J. Iran Chem. Soc., 2010, 3, 279 Search PubMed.
- Y. Wang, X. Ke, J. Zhang, X. Du, J. Ma and J. Li, Bull. Chem. Soc. Ethiop., 2012, 26, 9 CrossRef CAS.
- F. S. Rojas, C. B. Ojeda and J. M. C. Pavon, European Scientific Journal, 2013, 19, 1857 Search PubMed.
- S. Bahare and B. Bahare, Iran. J. Anal. Chem., 2014, 1, 115 Search PubMed.
- M. Gharehbaghi, F. Shemirani and M. Baghdadi, Int. J. Environ. Anal. Chem., 2008, 88, 513 CrossRef CAS.
- R. Khani and F. Shemirani, Food Anal. Meth., 2013, 6, 386 CrossRef.
- L. Ele kova, I. S. Balogh, J. Imrich and V. Andruch, J. Anal. Chem., 2015, 70, 298 CrossRef.
- M. R. Moghadam, S. M. P. Jahromi and A. Darehkordi, Food Chem., 2016, 192, 424 CrossRef PubMed.
- I. Narin, M. Soylak, L. Elci and M. Dogan, Talanta, 2000, 52, 1041 CrossRef CAS PubMed.
- I. M. Steinberg, A. Lobnik and O. S. Wolfbeis, Sens. Actuators, B, 2003, 90, 230 CrossRef CAS.
- B. Buke, U. Divrikli, M. Soylak and L. Elci, J. Hazard. Mater., 2009, 163, 1298 CrossRef CAS PubMed.
- S. M. Yousefi and F. Shemirani, J. Hazard. Mater., 2013, 254–255, 134 CrossRef CAS PubMed.
- M. Ezoddin, K. Abdi and N. Esmaeili, Microchem. J., 2016, 129, 200 CrossRef CAS.
- M. S. El-Shahawi and H. M. Al-Saidi, Trends Anal. Chem., 2013, 44, 12 CrossRef CAS.
- A. Zgoła-Grzeskowiak and T. Grzeskowiak, Trends Anal. Chem., 2011, 30, 1382 CrossRef.
- P. Soisungnoen, R. Burakham and S. Srijaranai, Talanta, 2012, 98, 62 CrossRef CAS PubMed.
- T. Asadollahi, S. Dadfarnia and A. M. H. Shabani, Talanta, 2010, 82, 208 CrossRef CAS PubMed.
- S. Jafarvand and F. Shemirani, Microchim. Acta, 2011, 173, 353 CrossRef CAS.
- G. Leng, H. Yin, S. Li, Y. Chen and D. Dan, Talanta, 2012, 99, 631 CrossRef CAS PubMed.
- E. Yiantzi, E. Psillakis, K. Tyrovola and N. Alogerakis, Talanta, 2010, 80, 2057 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |