
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIsolation and enantiostability of the B-chiral bis(salicylato)borate anions [BR(Sal)2] and [BS(Sal)2]†
Lawrence W.-Y. Wong,
Alex S.-F. Au Yeung,
Gemma S.-S. Tam,
Jack W.-H. Kan,
Herman H.-Y. Sung,
Fu Kit Sheong,
Zhenyang Lin and
Ian D. Williams*
and
Ian D. Williams*
Department of Chemistry, Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong, China. E-mail: chwill@ust.hk
First published on 4th January 2018
Abstract
The chiral spiroborate anions [BS(Sal)2] and [BR(Sal)2], (R and S subscripts indicate boron stereochemistry) have been isolated as 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 quininium and 1:2 sparteinium salts, [HQuin][BS(Sal)2] and [H2Spa][BR(Sal)2]2 respectively, by either cation metathesis or a simple one-pot synthesis involving reaction of boric and salicylic acids with the alkaloid base. Circular dichroism (CD) spectroscopy shows that the B-based chirality is stable in polar aprotic media, such as DMF or DMSO, though labile in protic solutions. Enantiopure salts with achiral counter-cations such as [NBu4][BR(Sal)2] may then be prepared by exchange, so these B-chiral anions may have use in metathesis-based resolutions. Due to a site disorder the anion in [H2Spa][BR(Sal)2]2 is limited to 70% ee, however an enantiopure analogue [H2Spa][BR(5-Cl-Sal)2]2 is readily formed using 5-chlorosalicylic acid. This also indicates a wide family of stable enantiopure B-chiral anions may be isolated by this approach.
:1 quininium and 1:2 sparteinium salts, [HQuin][BS(Sal)2] and [H2Spa][BR(Sal)2]2 respectively, by either cation metathesis or a simple one-pot synthesis involving reaction of boric and salicylic acids with the alkaloid base. Circular dichroism (CD) spectroscopy shows that the B-based chirality is stable in polar aprotic media, such as DMF or DMSO, though labile in protic solutions. Enantiopure salts with achiral counter-cations such as [NBu4][BR(Sal)2] may then be prepared by exchange, so these B-chiral anions may have use in metathesis-based resolutions. Due to a site disorder the anion in [H2Spa][BR(Sal)2]2 is limited to 70% ee, however an enantiopure analogue [H2Spa][BR(5-Cl-Sal)2]2 is readily formed using 5-chlorosalicylic acid. This also indicates a wide family of stable enantiopure B-chiral anions may be isolated by this approach.
Introduction/background
Spiroborate anions are readily prepared by direct reaction of boric acid, chelating oxyacids, such as diols, catechols, α-hydroxy acids and salicylic acids, together with a suitable base.1,2 Their salts may have a wide variety of properties and potential applications.2 As analogues of commonly used BF4− and BPh4− anions they may serve as crystallizing agents,3 or in the case of chiral anions like [B(Binol)2]−,4 or related aryl diborate anions,5 as resolving agents via diastereomeric salt formation.6 This method of optical resolution has found numerous important applications in the field of organic and pharmaceutical chemistry.7 Recently we explored the utility of bis(mandelato)-borate [BMan2] anions as resolving agents and found they offered highly efficient resolution of a chemically diverse range of racemic cations.8In this regard it was noted that the anions had permanent C-based chiral centres through use of either R-mandelic or S-mandelic acid as the starting oxyacid chelate. However the chelation of two asymmetric ligands to tetrahedral B also creates a stereochemical centre at boron. Chiral borate anions based on such asymmetric bis-chelation was discussed by Lacour in a review of anion design for resolution.9 The potential for chirality in such ions was first reported in 1925, with the partial resolution of chiral spiroborate anions derived from 3-nitro- and 4-chloro-catechol by brucine or strychnine.10 These were found to be configurationally labile. Racemization processes for chiral spiroborates were more recently examined by NMR, first by Hosseini et al. for a chiral boracryptand11 and subsequently by Nelson et al.12 for two chiral aryl spiroborates. This latter study derived a ΔG‡ of 85 kJ mol−1 for the activation energy for racemization for chiral bis(3-isopropylcatecholato)borate [BCat*2]− in an acidic medium (16 mM benzoic acid), implying rapid scrambling at room temperature (t1/2 ca. 2 min).
In the diastereomeric [BMan2]− system, DFT calculations indicated that anions with both B-based chiralities are similar in energy, whilst NMR spectra confirmed both co-exist in solution.8 However the two anions are markedly different in shape and in the solid state the precipitation of twisted [BS(R-Man)2] or [BR(S-Man)2] is always observed in crystallized salts, rather than ‘V-shaped’ [BR(R-Man)2] or [BS(S-Man)2].8,13 Circular dichroism and NMR spectra can be used to track the stability of the [BMan2] anions and suggested that in line with previous findings a slow equilibration of bis(mandelato)borate anions occurs in protic solvents such as methanol. However this was not the case in aprotic solvents, such as dimethylformamide (DMF), in which the anions were found to be configurationally stable.8
Given this finding we determined to reinvestigate whether B-chiral spiroborates arising from bis-ligation of two asymmetric chelates could first be fully resolved and then similarly shown to exhibit B-based enantiostability in the absence of protic sources. The most obvious choice, with possibility for extending to a wider class, is the use of salicylic acid to form the bis(salicylato)borate anion [BSal2] (Fig. 1).
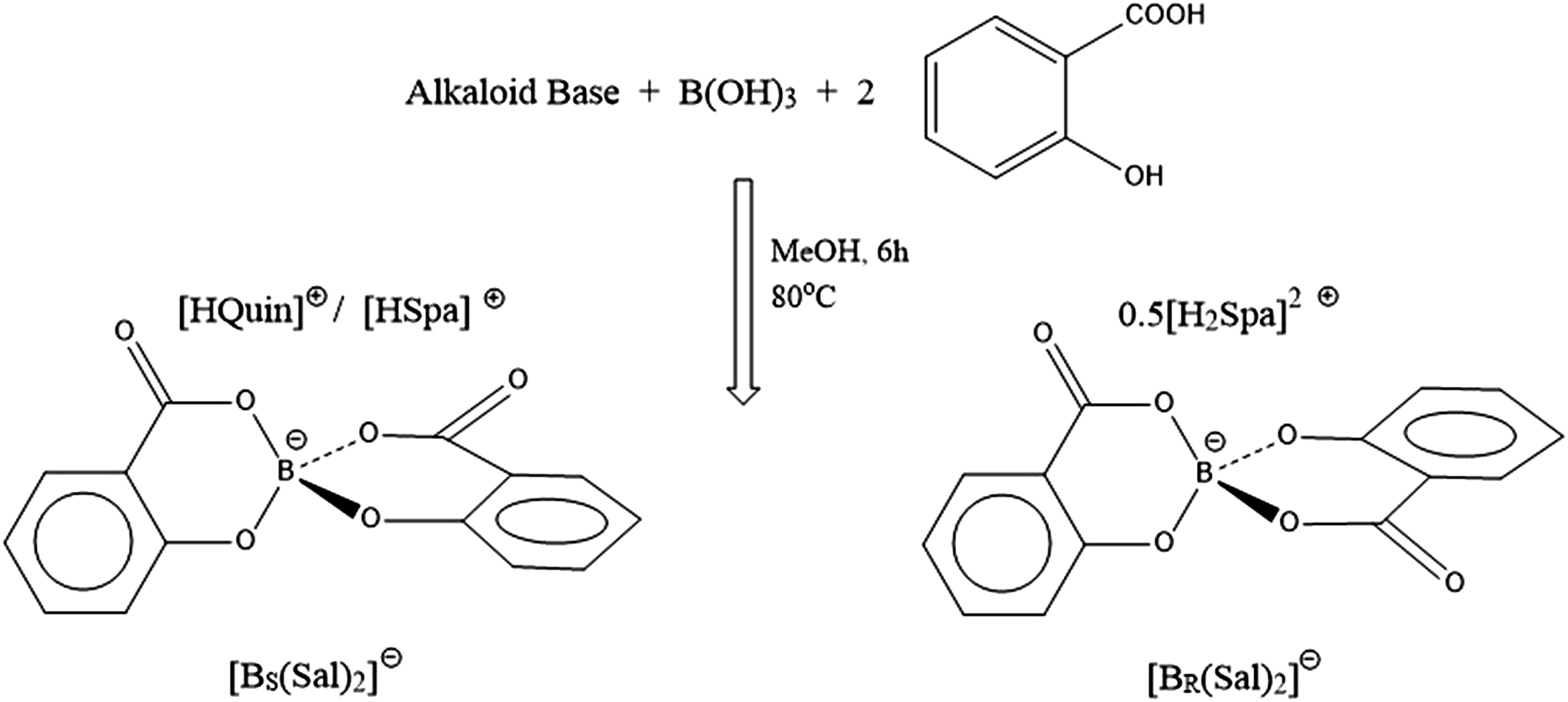 | ||
| Fig. 1 Preparation of chiral bis(salicylato)borate ions [BR(Sal)2] and [BS(Sal)2]. The subscripted BR and BS notations indicate the Cahn–Ingold–Prelog stereochemistry at the chiral B centre. | ||
Herein we report the successful solid-state isolation of both the [BS(R-Man)2] and [BS(Sal)2] enantiomers, their X-ray structures, solution circular dichroism (CD) spectra. Furthermore their enantiostability in aprotic solution, as well as their ability to participate in metathesis crystallizations, is demonstrated.
Results/discussion
Isolation of [BS(Sal)2] salts
Quinine (abbreviated here as Quin) is a chiral alkaloid base isolated from the bark of cinchona sp. with much classical use as a resolving cation and continues to show application for example in the recent reports of resolution of malates and mandelates.14 It has two basic sites, a tertiary quinuclidine amine and an aromatic quinoline nitrogen. The former pKa is much higher and so typically mono-cations are formed. In the case of bis(salicylato)borate single crystals of a well-resolved salt [HQuin][BS(Sal)2] 1 may be obtained from a simple metathesis crystallization between an aqueous solution of sodium bis(salicylato)borate Na[BSal2] and methanolic quinine free base. Alternatively heating quinine, boric acid and 2 equivalents of salicylic acid in methanol to reflux, or using solvothermal conditions for several hours can result in formation of 1 upon cooling (Fig. 1). Single crystal structure determination of 1 readily showed that resolution of the [BSal2] ions present in solution was not only possible, but due to dynamic nature of equilibrium between the two anions also in high yield and purity. The experimental details of the crystal structure are given in Table 1 and key geometric features of the [BSal2] anions in Table 2.| Compound | 1 | 2 | 2-RT |
| Abbreviated name | [HQuin][BS(Sal)2] | [HSpa][BS(Sal)2] | [HSpa][BS(Sal)2] |
| CSD number | 1581657 | 1581658 | 1581659 |
| Empirical formula | C34H33BN2O8 | C29H35BN2O6 | C29H35BN2O6 |
| Formula weight | 608.43 | 518.40 | 518.40 |
| Temperature/K | 100(2) | 100(2) | 293(2) |
| Crystal system | Triclinic | Monoclinic | Orthorhombic |
| Space group | P1 | P21 | P212121 |
| a/Å | 7.9518(3) | 11.5542(4) | 11.70439(17) |
| b/Å | 9.5337(5) | 18.7836(5) | 11.96490(18) |
| c/Å | 10.5702(3) | 11.9810(4) | 18.9233(2) |
| α/° | 89.020(3) | 90 | 90 |
| β/° | 76.831(3) | 95.040(3) | 90 |
| γ/° | 65.860(4) | 90 | 90 |
| Volume/Å3 | 709.41(6) | 2590.18(14) | 2650.06(6) |
| Z, Z′ | 1, 1 | 4, 2 | 4, 1 |
| ρcalc g cm−3 | 1.424 | 1.329 | 1.299 |
| Radiation, μ/mm−1 | Cu Kα (λ = 1.54184) 0.831 | Cu Kα (λ = 1.54184) 0.748 | Cu Kα (λ = 1.54184) 0.731 |
| F(000) | 320 | 1104 | 1104.0 |
| Crystal size/mm3 | 0.25 × 0.2 × 0.2 | 0.1 × 0.1 × 0.1 | 0.1 × 0.1 × 0.1 |
| 2Θ maximum/° | 135 | 135 | 135 |
| Index ranges | −9 ≤ h ≤ 9 | −13 ≤ h ≤ 13 | −13 ≤ h ≤ 10 |
| −10 ≤ k ≤ 11 | −22 ≤ k ≤ 22 | −13 ≤ k ≤ 14 | |
| −12 ≤ l ≤ 12 | −14 ≤ l ≤ 14 | −22 ≤ l ≤ 19 | |
| Independent reflections | 4775 | 14696 |
4763 |
| [R(int) = 0.0175, Rsigma = 0.0192] | [Rint = 0.0445, Rsigma = 0.0286] | [Rint = 0.0157, Rsigma = 0.0151] | |
| Data/restr./params. | 4775/3/415 | 14696/1/694 |
4763/213/482 |
| Goodness-of-fit F2 | 1.013 | 1.012 | 1.014 |
| Final R indexes [I ≥ 2σ(I)] | R1 = 0.0252, wR2 = 0.0661 | R1 = 0.0305, wR2 = 0.0811 | R1 = 0.0285, wR2 = 0.0751 |
| Final R indexes [all data] | R1 = 0.0252, wR2 = 0.0664 | R1 = 0.0342, wR2 = 0.0826 | R1 = 0.0310, wR2 = 0.0770 |
| Diff. peak/hole eÅ−3 | 0.17/−0.18 | 0.17/−0.13 | 0.11/−0.10 |
| Flack parameter | 0.07(8) | −0.01(8) | 0.00(6) |
| Compound | 3 | 4 | 5 |
| Abbreviated name | [H2Spa][BR(Sal)2]2 | [NBu4][BR(Sal)2] | [H2Spa][BR(5-Cl-Sal)2]2H2O |
| CSD number | 1581660 | 1581661 | 1581667 |
| Empirical formula | C43H44B2N2O12 | C30H44BNO6 | C43H42B2Cl4N2O13 |
| Formula weight | 802.42 | 525.47 | 958.20 |
| Temperature/K | 100(2) | 100(2) | 100(2) |
| Crystal system | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21 | P21 | P21 |
| a/Å | 12.29380(10) | 9.49961(15) | 12.1841(4) |
| b/Å | 19.4089(2) | 13.42524(18) | 10.7394(3) |
| c/Å | 16.2958(2) | 12.0837(2) | 16.5166(7) |
| α/° | 90 | 90 | 90 |
| β/° | 90.4770(10) | 110.1708(19) | 98.350(3) |
| γ/° | 90 | 90 | 90 |
| Volume/Å3 | 3888.19(7) | 1446.58(4) | 2138.29(13) |
| Z, Z′ | 4, 2 | 2, 1 | 2, 1 |
| ρcalc g cm−3 | 1.371 | 1.206 | 1.488 |
| Radiation, μ/mm−1 | Cu Kα (λ = 1.54184) 0.820 | Cu Kα (λ = 1.54184) 0.659 | Cu Kα (λ = 1.54184) 3.108 |
| F(000) | 1688.0 | 568.0 | 992.0 |
| Crystal size/mm3 | 0.40 × 0.20 × 0.20 | 0.03 × 0.02 × 0.02 | 0.35 × 0.03 × 0.02 |
| 2Θ maximum/° | 135 | 135 | 135 |
| Index ranges | −14 ≤ h ≤ 14 | −11 ≤ h ≤ 11 | −14 ≤ h ≤ 14 |
| −23 ≤ k ≤ 23 | −16 ≤ k ≤ 16 | −12 ≤ k ≤ 12 | |
| −13 ≤ l ≤ 19 | −14 ≤ l ≤ 14 | −19 ≤ l ≤ 19 | |
| Independent reflections | 13854 |
5215 | 13467 |
| [Rint = 0.0242, Rsigma = 0.0368] | [Rint = 0.0393, Rsigma = 0.0330] | [Rint = ‘twin’, Rsigma = 0.0285] | |
| Data/restr./params. | 13858/3/1159 |
5215/1/347 | 13467/2/594 |
| Goodness-of-fit F2 | 1.018 | 1.036 | 1.022 |
| Final R indexes [I ≥ 2σ(I)] | R1 = 0.0490, wR2 = 0.1269 | R1 = 0.0281, wR2 = 0.0640 | R1 = 0.0520, wR2 = 0.1422 |
| Final R indexes [all data] | R1 = 0.0501, wR2 = 0.1278 | R1 = 0.0309, wR2 = 0.0652 | R1 = 0.0564, wR2 = 0.1445 |
| Diff. peak/hole eÅ−3 | 0.33/−0.22 | 0.13/−0.15 | 0.45/−0.35 |
| Flack parameter | −0.20(10) | −0.03(7) | 0.019(12) |
| 1 | 2 | 3 | 4 | 5 | |
|---|---|---|---|---|---|
| [HQuin][BS(Sal)2] | [HSpa][BS(Sal)2] | [H2Spa][BR(Sal)2]2 | [NBu4][BR(Sal2)] | [H2Spa] [BR(5-Cl-Sal)2]2 | |
| a Geometries concur with other [BSal2] structures found in the CSD. (see ref. 18). | |||||
| Distances (Å) | |||||
| B–O(11) phenolate | 1.508, 1.468 | 1.503, 1.464, 1.493, 1.468 | 1.503, 1.473, 1.506, 1.395, 1.506, 1.475, 1.445, 1.503, Ave. 1.476 | 1.490, 1.486 | 1.505, 1.482, 1.483, 1.511 |
| B–O(12) carboxylate | 1.451, 1.443 | 1.443, 1.467, 1.449, 1.459 | 1.444, 1.449, 1.434, 1.487, 1.437, 1.434, 1.437, 1.437, Ave. 1.445 | 1.446, 1.450 | 1.431, 1.453, 1.438, 1.447 |
|
|||||
| Angles (°) | |||||
| O(11)–B–O(12) | 111.7, 114.1 | 112.0, 112.7, 112.8, 113.4 | 111.5, 114.9, 116.0, 111.0, 114.6, 111.5, 114.6, 111.7, Ave. 113.2 | 112.7, 112.8 | 114.0, 111.1, 113.9, 111.5 |
| O(11)–B–O(11)′ | 106.6 | 107.4, 106.5 | 107.0, 106.0, 106.2, 106.7, Ave. 106.5 | 106.9 | 107.0, 106.9 |
| O(12)–B–O(12)′ | 107.7 | 108.8, 109.3 | 107.7, 105.3, 107.8, 108.1, Ave. 107.2 | 107.2 | 108.4, 108.8 |
| O(11)–B–O(12)′ | 107.6, 109.3 | 107.5, 108.1, 106.6, 108.3 | 109.2, 106.5, 105.4, 113.0, 109.7, 107.1, 106.5, 109.2, Ave. 108.3 | 108.4, 108.9 | 107.8, 108.5, 108.2, 107.6 |
| Torsion angle: C(10)–O(11)–B–O(11)′ | 80.7, 80.9 | 159.0, 80.5, 141.8, 96.1 | −89.1, −84.4, −85.9, −95.9, −100.2, −85.5, −104.1, −84.9 | −99.0, −91.6 | −81.4, −97.0, −100.8, −89.6 |
| Dihedral: chelate ring hinge angle | +31.4, +33.2 | −33.9 + 37.9, −22.5, +23.0 | +29.7, +31.3, +29.3, +29.7, +33.6, +16.2, +12.5, +34.2 | +19.3, +23.5 | +34.0, +23.7, +28.7, +21.9 |
The X-ray structure of 1 (Fig. 2) shows an intermolecular H-bond between N1–H of the quininium ion to a keto oxygen O(10) of the [BS(Sal)2] anion with N⋯O 2.800 Å. The [HQuin] alcohol forms a O(1)–H⋯π hydrogen bond to aromatic ring of the [BSal2] anion with the shortest contact O(1)⋯C(33) 3.214 Å. The crystal system is triclinic, space group P1 with one molecular formula per unit cell. The packing may be regarded as reasonably efficient with the π-systems of the quinoline and one salicylate ligand arm aligning in parallel.
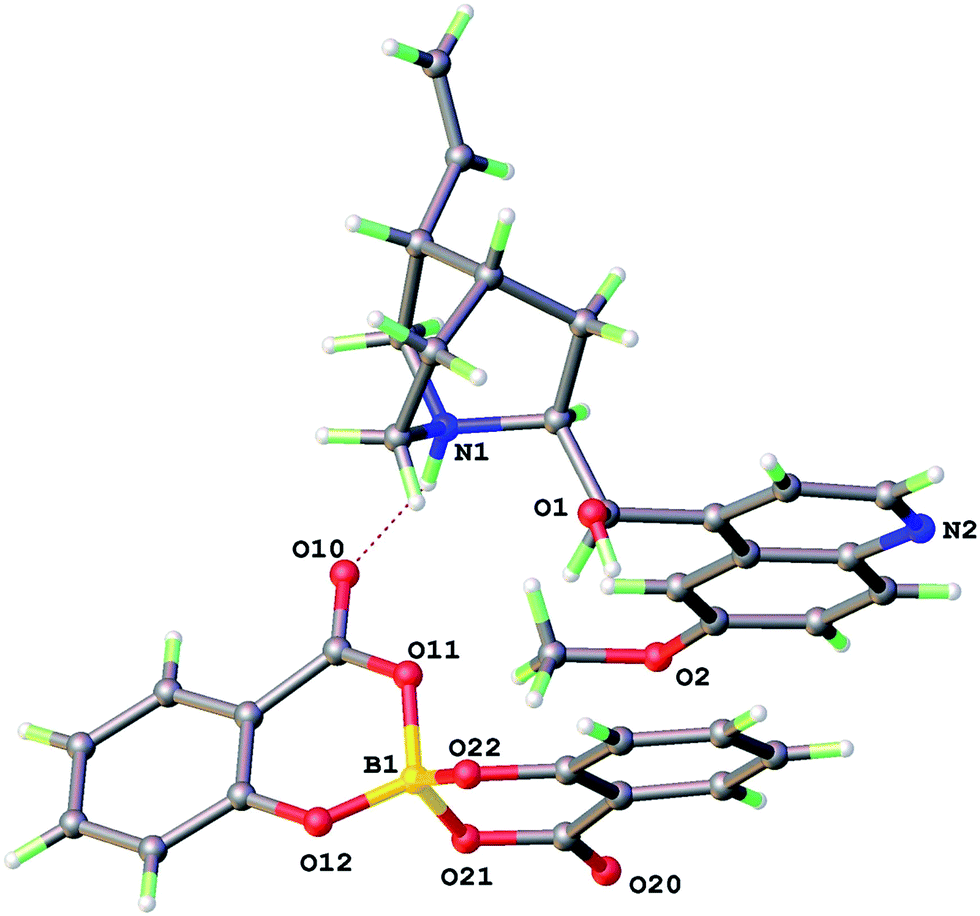 | ||
| Fig. 2 X-ray structure of ion pair in 1 [HQuin][BS(Sal)2]. | ||
As might be expected the geometry of the [BSal2] anion (Table 2) is that of a distorted tetrahedron. The B–O bonds are shorter to phenolic oxygen (1.443(2) and 1.446(2) Å) but longer to the carboxylate oxygens O(12) and O(22) (average 1.49 Å). The external hydrogen bond to keto O(10) has the effect of modifying the carboxylate resonance forms and the B–O(11) 1.502(4) is significantly longer than B–O(21) 1.469(4) Å. In general the stronger B–O bonds are to ligand O that are from the stronger conjugate base. This was also seen previously in the bis(mandelato)borate [BMan2] anions which have roughly planar 5-membered chelate rings.8,13 The ring expansion in [BSal2] to six-membered chelates also comes with a greater propensity for ring non-planarity. This is manifested in a considerable deviation of the boron atom from the ring plane of around 0.4 Å.
The non-planarity can also be characterized by the dihedral angle ϕ formed by the BO2 plane with the planar salicylate. The salicylate can be bent towards the carboxylate (ϕ defined as +ve) or the phenolate O (ϕ defined as −ve) of the distal ligand with ϕ angles typically between ±20 to 30°. Density functional calculations (DFT) at the B3-LYP/6-31G* level indicate that such solid-state distortions are energetically preferable to planar 6-membered rings. However the potential surface is shallow <0.5 kcal mol−1, until it starts to rise sharply above dihedral hinge angles ϕ in excess of ±35° (Fig. 3). The consequence of this is that [B(Sal)2] anions are conformationally semi-rigid with a considerable degree of shape flexibility which may assist efficient packing and crystallizations.
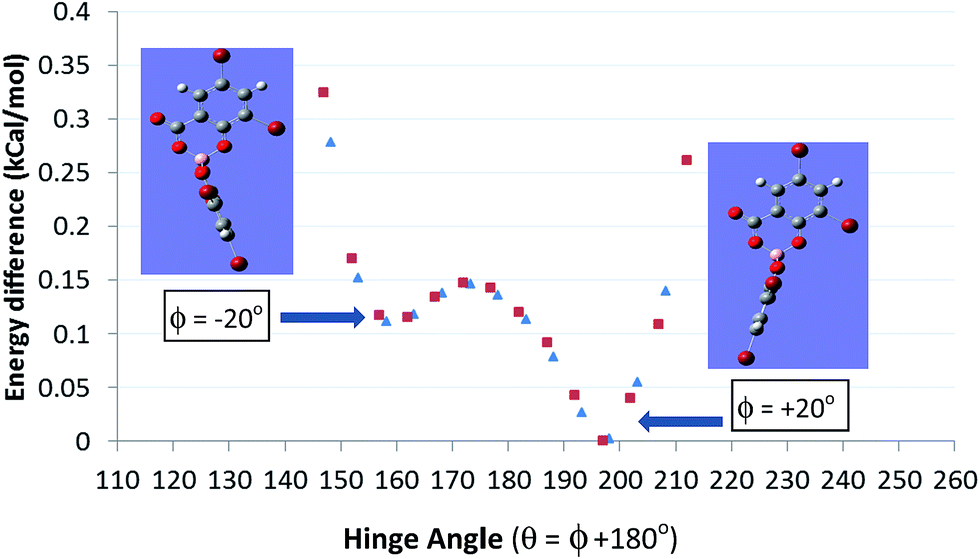 | ||
| Fig. 3 Energy profile (kcal mol−1) for chelate ring hinge angle in [BSal*2] ions. | ||
Once isolated, our main interest in the [BS(Sal)2] anions was to see whether the boron based chirality could be stable in solution. A solution of [HQuin][BS(Sal)2] 1 was prepared and circular dichroism spectroscopy was carried out. Strong features due to the chiral spiroborate anion were observed, however the CD spectrum of quininium hydrochloride indicated the region above 300 nm was partly overlapped by features arising from the cation as well. Nevertheless, studies of the stability of the CD spectral signals indicated that the [BS(Sal)2] anion was enantiomerically stable in aprotic polar solvents, such as dimethylformamide (DMF). This encouraged its further isolation with a counter-ion which would be CD-silent in the region of interest.
Screening for suitable salt formation with a number of chiral cations, including others derived from alkaloid bases, was carried out. In the case of (−)-sparteine, a dibasic alkaloid frequently used as a chiral auxiliary in organo-lithiation14 and coordination catalysis,15 an analogous salt 2, [HSpa][BS(Sal)2] was prepared in good yield after a short reaction. A single crystal structure of 2 confirmed this was a 1:1 salt involving the mono-protonated form of the base (Fig. 4). The crystal structure was found to be monoclinic with space group P21 (Table 1). However, a check of the powder X-ray diffraction (pXRD) pattern of the bulk material indicated no clear correspondence of peak positions to those simulated from the single crystal structure, although unit cells of further specimens from the sample were in agreement with the original specimen. Further inspection of the structure for 2 showed there were two molecular ion pairs per asymmetric unit (Z′ = 2) which appeared to be related by pseudo-symmetry. Accordingly the single crystal was re-inspected at room temperature (RT) and found to undergo a phase transition to a higher symmetry orthorhombic form with Z′ = 1 and space group P212121. Details of 2-RT are given for comparison in Table 1. The cell undergoes a remarkable change of β-angle from 95.1 to 90.0°. There is a slightly larger cell volume but anisotropy in the thermal expansion, so that diffraction peaks move to both lower and higher Bragg angles. The simulated pXRD pattern from the RT form 2-RT was then found in excellent agreement with the experimental pattern from the bulk solid indicating phase purity.
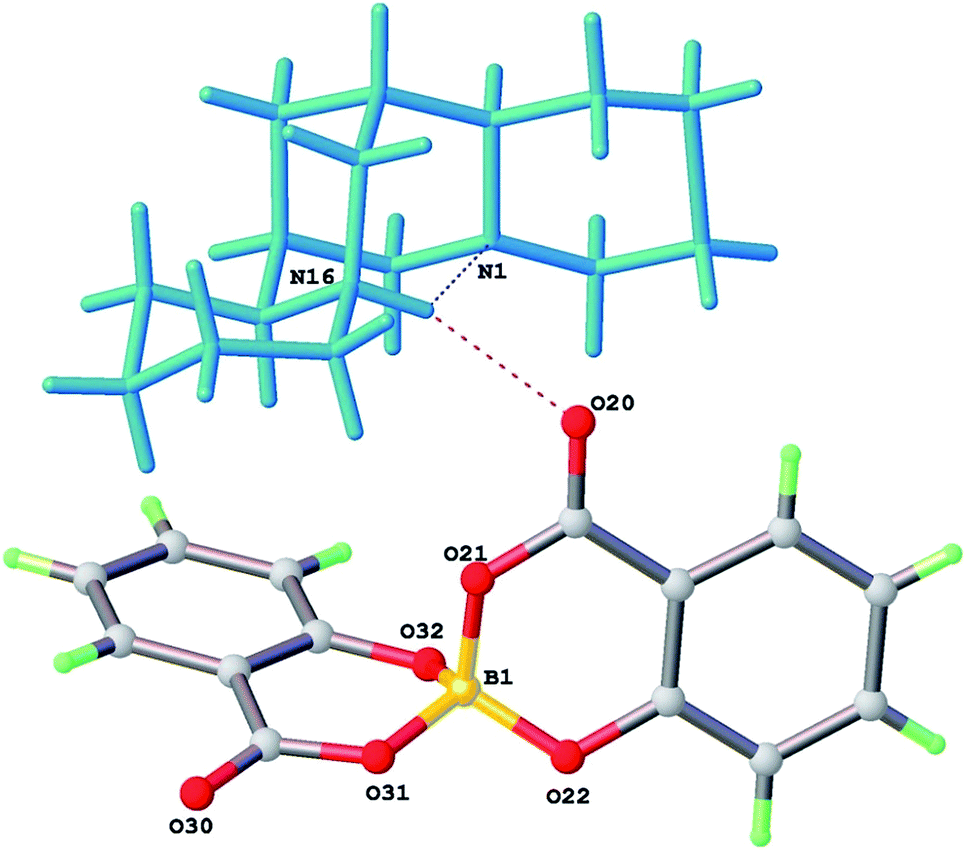 | ||
| Fig. 4 Ion pair from 2 [HSpa][BS(Sal)2] showing bifurcated H-bond from H(16). | ||
The stereochemistry of both crystallographically independent bis(salicylato)borate anions in 2 are the same as 1. Once again the BS stereochemistry is exclusively found. As is the case for 1 residual electron density peaks and holes from the structural refinement are below 0.2 eÅ−3, consistent with no appreciable disorder of the [Bs(Sal)2] anions. The two [HSpa]+ cations in 2 have essentially identical geometry in the low temperature structure and an intramolecular N(16)H⋯N(1) hydrogen bond, which is positionally ordered.
The nitrogen atoms N(1) at the ring A/B junction and N(16) at the C/D ring junction have axial and equatorial lone pairs – protonation of the equatorial position is favoured, so [HSpa]+ has N(16) as the protonated site (Fig. 3). The N(16)H proton also forms a weak H-bond bifurcation to O(20) of a neighbouring [BS(Sal)2] anion with N⋯O separation of 3.09 Å an ion pair from 2 is shown in Fig. 4.
Unlike the case for the quininium salt 1, in 2 the CD spectrum between 270-350 nm is mostly unaffected by contributions from the monoprotonated sparteinium cation [HSpa]+ which is an aliphatic compound with no unsaturated bonds. The CD spectrum for 2 is shown in Fig. 5. The [BS(Sal)2] anion shows a pronounced negative Cotton effect. The molar ellipticity at the negative trough at 316 nm is –θ = −226 deg M−1 cm−1. The CD spectrum has a cross-over at the absorption λmax of 304 nm and a slightly weaker positive peak at 293 nm with +θ = +176 deg M−1 cm−1. The CD spectra of 2 were measured under various conditions to establish [BS(Sal)2] enantiostability in aprotic media. In fact DMF solutions of both 1 and 2 give unchanged CD signals over prolonged periods (>1 week). Further tests to explore the addition of water show that moderate amounts (e.g. 4DMF:1H2O) can be tolerated indefinitely, but in pure water the CD signal of 2 is effectively lost over 4 h.
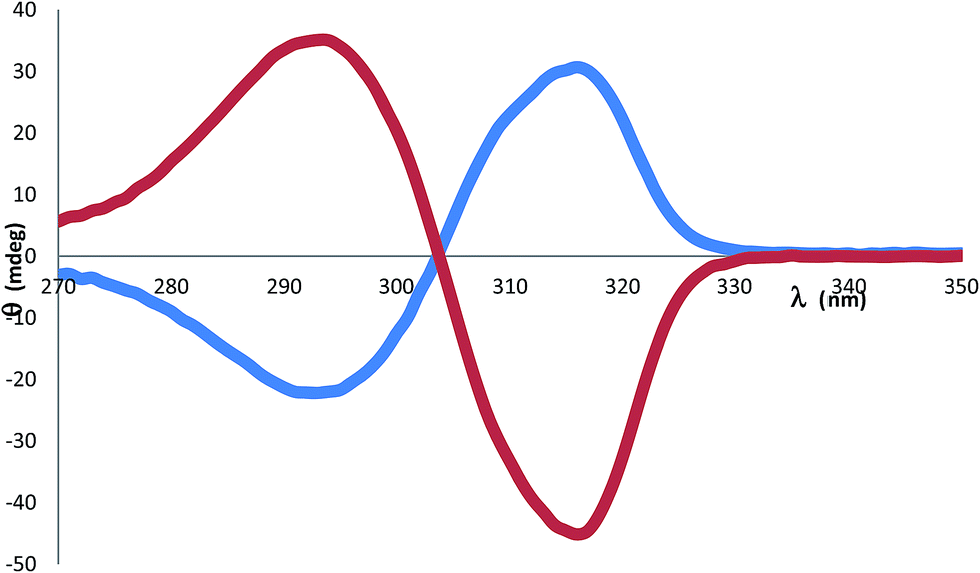 | ||
| Fig. 5 Circular dichroism spectra for 2 [HSpa][BS(Sal)2] 0.2 mM (red) and 3 [H2Spa][BR(Sal)2]2 0.1 mM (blue) in dimethylformamide, indicating 70% ee for 3. | ||
Isolation of [BR(Sal)2] salts
The successful isolation of chiral [BS(Sal)2] salts and the finding that these could be enantiostable in aprotic media led us to also seek salts of the mirror image [BR(Sal)2] anions. Quinine is unavailable as its mirror image form – quinidine being a diastereomer rather than an enantiomer, whilst (+)-sparteine, a natural product also known as pachycarpine, is commercially available, but expensive.Fortunately our studies with (−)-sparteine indicated that if longer crystallization times were used, a different phase 3 was then produced rather than 2. This phase was also favoured if (−)-sparteine sulphate rather than sparteine free base was employed. Since (−)-sparteine is dibasic and known with both mono and diprotonated forms,16 it was reasoned that phase 3 might contain the dication. A single crystal specimen was selected and the structure analysis (Table 1) indicated that 3 was indeed a 1:2 salt and best formulated as [H2Spa][BR(Sal)2]2. The diprotonated form of sparteinium has a major shape change17 (Fig. 6) and in the 1:2 salt preferentially selects for BR configurations at the two crystallographically independent [BSal2] sites. However structure refinement was not perfectly smooth and residual peaks of about 0.5 eÅ−3 could be found at the anion sites consistent with a minor disorder [BS(Sal)2], such that the enantio-excess was estimated to be about 80% ee. This was subsequently borne out by the resulting CD spectrum of 3, which gave an inverted CD spectrum to 2, but with signals of approximately 70% ee for the bulk material (Fig. 5). The packing diagram for 3 looking along the a-axis is shown in Fig. 7 and can help explain why BR/BS disorder may occur in this salt. As was the case for 2, the asymmetric unit of 3 has Z′ = 2 at low temperature, this means there are two independent [H2Spa] and four [BSal2] ions. The two cations are coloured dark and pale green and the four anions labelled as B1–B4. Each of these make self-stacks along the a-axis direction. In order to prepare a salt with enantiopure [BR(Sal)2] anions we decided to carry out a metathesis crystallization from the impure salt 3. By exchanging the cation it was hoped the diastereomeric structural disorder found in 3 could be removed.
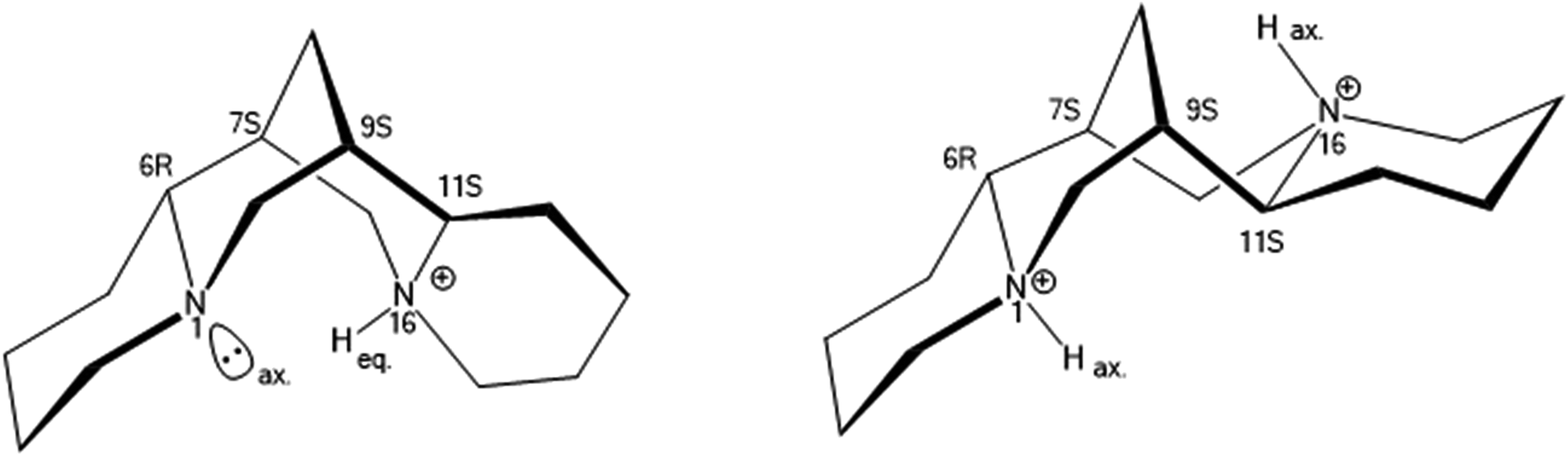 | ||
| Fig. 6 Spatial relationship of mono and diprotonated sparteinium cations, [HSpa]+ and [H2Spa]2+ showing shape change. | ||
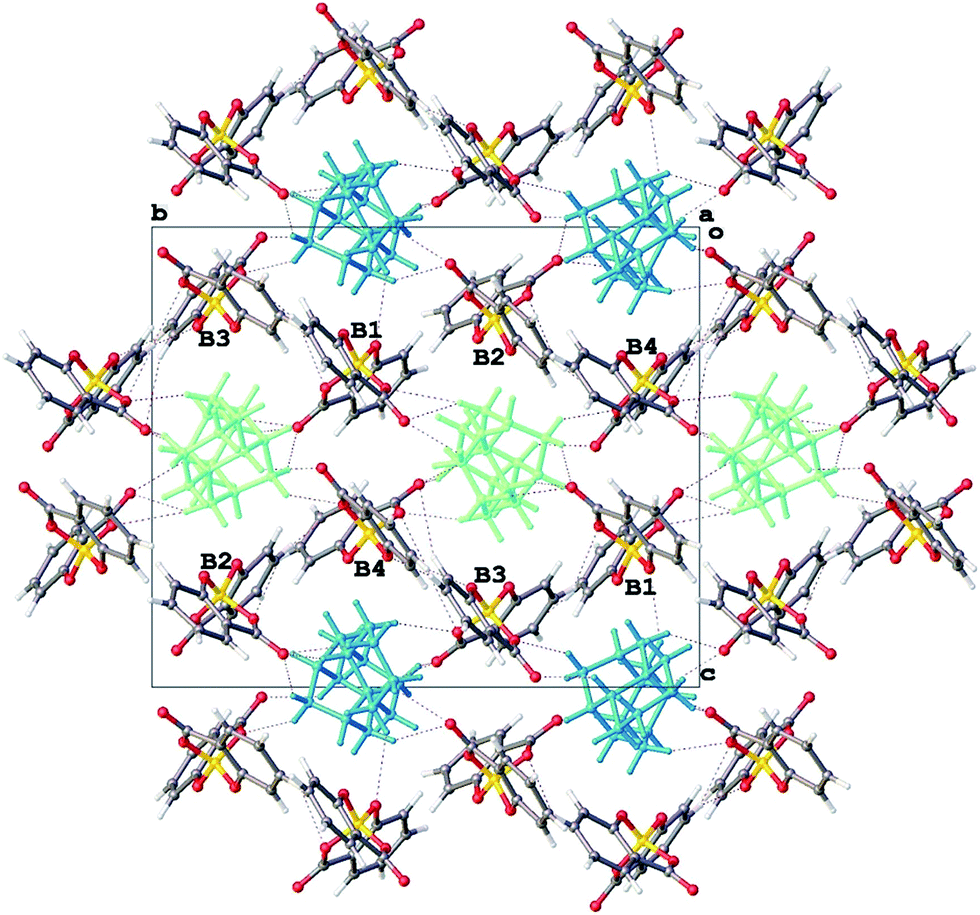 | ||
| Fig. 7 Packing diagram for 3, [H2Spa][B(Sal)2]2 along [100]. | ||
A dry methanolic solution of 3 was prepared and excess 1 M methanolic tetra-n-butylammonium hydroxide was added with stirring. A crystalline precipitate formed within 15 min and was filtered off and characterized. A subsequent crystal structure analysis (Table 1) showed the product was 4 [NBu4][BR(Sal)2] (Fig. 8).
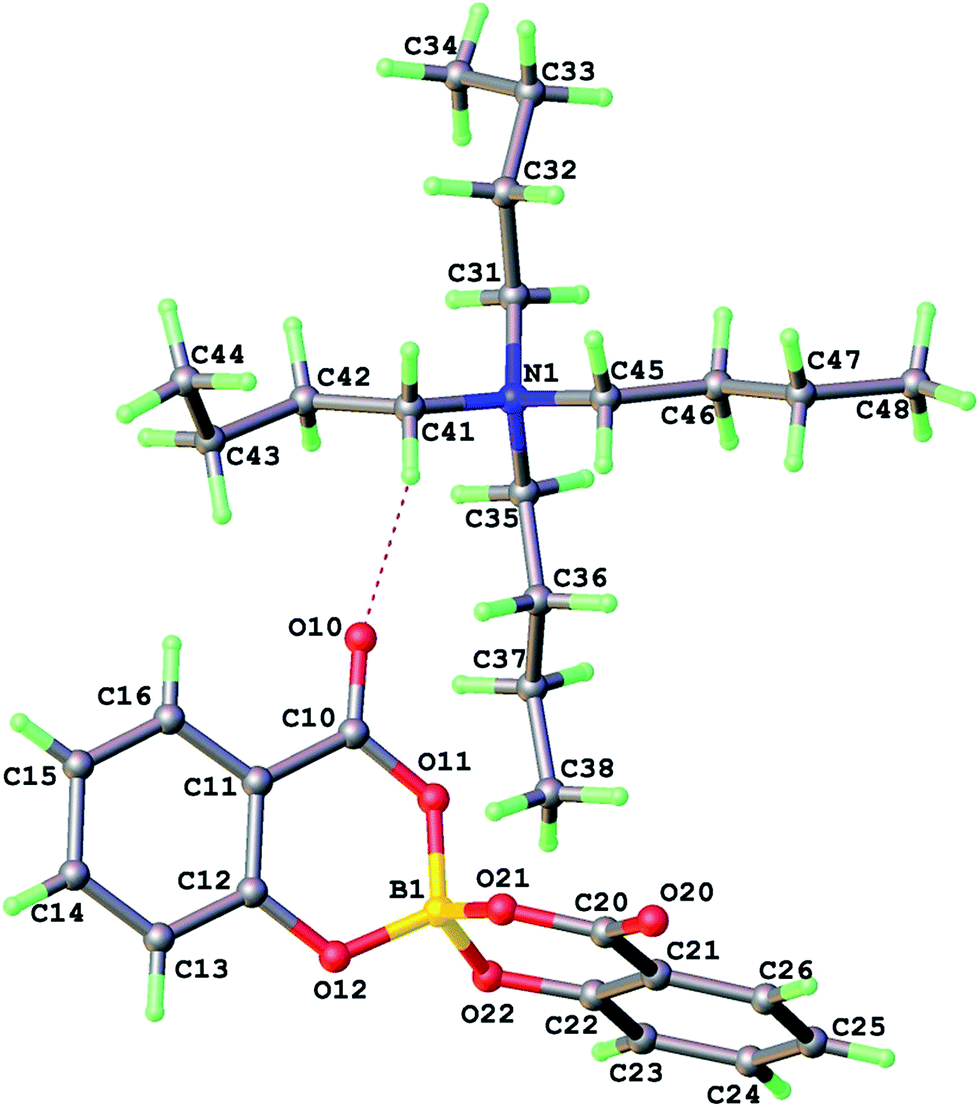 | ||
| Fig. 8 Crystal structure of the salt [NBu4][BR(Sal)2] 4, showing the ion pair of the asymmetric unit. | ||
Structural refinement for 4 is well-behaved and no ghost peaks indicating anion disorder are found – residual peak/hole are +0.13/−0.15 eÅ−3. Furthermore the Flack parameter −0.03(7) refines to an acceptable value close to zero with small standard uncertainty implying racemic twinning in this crystal specimen of 4 is negligible. p-XRD pattern of bulk powder indicates the crystalline peaks belong to a mixture of enantiopure 4 and a racemic phase that can be formed from direct reaction of [NBu4]OH with boric and salicylic acids. The CD spectrum of this powder mixture showed the overall enantiopurity of the material was unchanged, based on the molar values established for [BS(Sal)2] in 2.
The importance of this material's isolation was that metathesis crystallization of chiral bis(salicylato)borate anions is possible with retention of configuration. Since the [NBu4]+ counterion is a simple aliphatic one and unlike quininium or sparteinium ions has no protic site, this is a more enantio-robust salt, so may be more suitable for use in further resolutions. One final point worth mentioning for 4 is that its' bis(mandelato)borate analogue [NBu4][BR(S-Man)2] is a low melting solid and differential scanning calorimetry (DSC) indicates it melts just below 30 °C.13 In the case of [NBu4][BSal2] 4 we have found a higher melting point at 115 °C, underscoring the idea that the bis(salicylato)borate ion is more rigid. Further engineering of ionic liquids by varying counter cation and salicylate substituents could be of some interest, given the demonstration of enantiostability of the [BSal2] anions in aprotic media.
In principle therefore either hand of [BSal2] could be prepared from suitable reaction with sparteine base, albeit a simpler one-step procedure for [BR(Sal)2] is still being sought. Indeed the disorder present in the structure of 3 [H2Spa][BR(Sal)2]2 was rather surprising, but can be seen as a consequence of similar non-bonded contacts being formed for the two salicylate orientations. In principle appropriate ring substitution should break the approximate degeneracy of packing and yield cleaner disorder-free salts.
To prove this and show that chiral spiroborates derived from salicylic acids can be readily extended we carried out reaction of sparteine, boric and 5-chlorosalicylic acid and make an analogous salt to 3. The product was formed in excellent yield and purity and a crystal structure revealed 5 to be the monohydrate of the 1:2 salt [H2Spa][BR(5-Cl-Sal)2]2·H2O. The two independent anion sites in the crystal are both consistent with enantiopure [BR(5-Cl-Sal)2] configurations. They are shown together as an overlay in Fig. 9 which emphasizes their very similar geometry. The dihedral ‘hinge’ angles of the chelate rings are +34.0 and +23.9 for B(1) anion and +28.8 and +22.2° for the B(2) anion (Table 2), positive values implying the hinge is bent towards the distal carboxy O rather than phenoxy O.
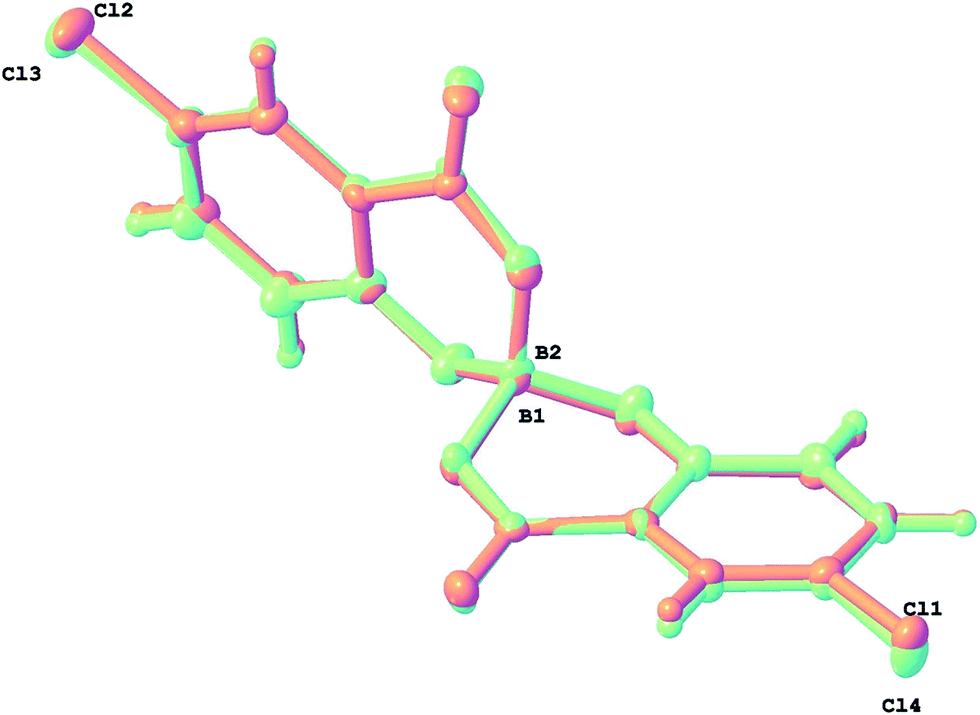 | ||
| Fig. 9 Overlay of the two independent [BR(5-Cl-Sal)2] anions in 5 - all chelate rings are ‘hinged’ towards the carboxy O of the other chelating ligand. | ||
The electronic perturbation of the salicylate anion by introduction of the 5-chloro substituent has a subtle effect on the CD spectrum, which was measured after dehydration of the solid. There is a distinct red shift of all the CD spectral features, which can be attributed to the corresponding shift in the UV-vis absorption spectrum (λmax = 317 nm). Assuming the isolated salt 5 is enantiopure, the molar ellipticity values at the Cotton effect peak +θ(330 nm) = +252 deg M−1 cm−1 and trough −θ(305 nm) = −164 deg M−1 cm−1 for [BR(5-Cl-Sal)2]. (CD spectrum shown in Fig. S10, ESI data†). Table 3 lists these for comparison with the CD parameters for the [BS(Sal)2] anion, which was discussed earlier. The inverted BR chirality in the case of 5 leads to a positive Cotton effect.
| Anion | [BS(Sal)2] | [BR(5-Cl-Sal)2] |
|---|---|---|
| a (See ESI data for CD spectra showing racemization or enantiostability of [BR(Sal)2] ions in different media and CD spectrum of [BR(5-Cl-Sal)2]). | ||
| UV-vis absorption | ||
| λmax (nm) | 304 | 317 |
| εmax (M−1 cm−1) | 9725 | 8210 |
|
||
| Circular dichroism | ||
| λ(+θ) (nm) | 293 | 330 |
| +θ (deg M−1 cm−1) | +176 | +252 |
| +Δε (M−1 cm−1) | +53.2 | +76.4 |
| λ(−θ) (nm) | 316 | 305 |
| −θ (deg M−1 cm−1) | −226 | −164 |
| −Δε (M−1 cm−1) | −68.5 | −49.7 |
| Cotton effect | Negative | Positive |
Successful isolation of 5 and the enantiopurity and enantiostability of such spiroborate compounds implies that a much wider family of bis(salicylato)borate anions [B(Sal*)2] with substituted salicylates may be prepared and isolated as either their BR or BS salts using appropriate resolving cations. These may then have their own prospective use in either metathesis based resolutions, or even in chiral ionic liquid formulations.19
Experimental
Materials and methods
All chemicals were reagent grade (99%+) from reputable commercial sources (Sigma-Aldrich or Acros). Quinine was used as its free base, sparteine either as free base or as diprotonated sulphate salt pentahydrate. Tetrabutylammonium hydroxide (TBAH) was a 1 M methanolic solution. Elemental combustion analyses were carried out by Medac Ltd., Surrey, U.K.Synthetic procedures
Salts 2, 3 and 5 were prepared by a facile one-pot procedure involving direct reaction of the resolving alkaloid base with boric and salicylic acids in appropriate molar ratio. Salt 1 is more reliably prepared by a prior isolation of anhydrous Na[BSal2] from boric flux20 reaction of NaOH, boric acid and salicylic acid in 1:1:2 ratio at 110 °C for 1 day. Salt 4 is prepared from a cation metathesis exchange from 3.
:1) were mixed in a Teflon cup, and heated at 80 °C for 12 hours. The resulting white sample was dried and the crystal was filtered and washed with small amount of ethanol to give white block with 76% yield. Combustion analysis: calculated for C43H44B2N2O12 (found) % C = 64.36 (62.35); % H = 5.53 (4.58); % N = 3.49 (3.02).Method A. 5-Chlorosalicylic acid 263 mg, (2 mmol), 31 mg (0.5 mmol) boric acid, 28 mg (0.5 mmol) KOH, were mixed with (−)-sparteine sulphate (0.25 mmol, 106 mg) and EtOH mixed in glass vial, sealed and heated at 80 °C for 4 h. After cooling to ambient temperature, colourless crystals were filtered and washed by minimal amounts of water and acetone to give 74% yield of 5 as its monohydrate.
Method B. Sodium bis(5-chloro-salicylato)borate (1 mmol, Fw 379.09, 0.379 g) in 3 mL of ethanol were layered with (−)-sparteinium sulphate pentahydrate (1 mmol, Fw 422.54, 0.422 g) in 3 mL water. The mixture was layered in a crystallization tube for 3 days. The resulting colorless crystal was filtered and washed by minimal amount of ethanol (0.601 g, Fw 958.23, 63% yield).
Synthesis of racemic Na[B(Sal)2] and Na[B(5-Cl-Sal)2] salts
Salicylic acid (20 mmol, Fw 138.12, 2.76 g)/or 5-chlorosalicylic acid (20 mmol, Fw 172.57, 3.45 g), boric acid (10 mmol, Fw 61.8, 0.620 g), sodium hydroxide (10 mmol, Fw 40.0, 0.40 g), in 30 mL ethanol were heated at reflux in a 100 mL RB-flask for 4 hours. The solutions were concentrated to around 10 mL and diethyl ether (20 mL) added until precipitation was complete. The white solid samples were filtered and washed with minimal amount of cold ethanol (76% and 72% yield respectively), which were used for metathesis crystallizations without further purification.Circular dichroism (CD) spectroscopy
Combined UV-vis absorption and circular dichroism spectra were run in dimethylformamide (DMF) solution on a Chirascan™ (Applied Photophysics) spectrophotometer between 270–370 nm with 0.2 nm steps. Typical concentration ranges were 0.1–0.01 mmol. Spectral features (Cotton effect) for chiral [B(Sal)2] anions were found around 300 nm in which range marked interference from quininium, but not sparteinium cations was found. Spectral stability was measured at ambient temperature in pure water, pure DMF or mixed H2O/DMF solutions for periods up to 1 week (Fig. S3–S9†).Crystallography
Single crystal structures for 1–5 (Table 1) were carried out on a Rigaku OD Supernova diffractometer operating with Cu-Kα radiation at 100 K unless otherwise noted. Structure solution and refinement were carried out with the Olex2 software package21 or SHELXTL22 configured to run within that GUI platform. All structures solved and refined in Sohncke (chiral) space groups. Twin laws were applied to structures of 2 and 5 substantially improving their refinement and final discrepancy indices. In the case of 5 this results in difficulty assigning an appropriate Rint for merging data. These were rotational not enantiomorphic twins, since these phases involve sparteinium ions of fixed chirality. Phase 3 showed positional disorder for the two independent anion sites with a minor component of [BS(Sal)2] anion juxtaposed at the predominantly [BR(Sal)2] sites with approximate occupancy factors of at the B(1) and at the B(2) site. All non-hydrogen atoms were refined with anisotropic thermal parameters excepting these minor component atoms which were isotropic and with bond length restraints applied. Protonation states of alkaloid cations were clearly established by identification of sensibly located electron density peaks for H associated with N heteroatoms. The Flack parameters of all three structures are zero or slightly negative with s.u.'s below 0.1, supporting the expected absolute chirality for quininium and (−)-sparteinium ions and establishing the handedness of the [BSal2] ions in these structures.Powder X-ray diffraction measurements for bulk samples of solids 1–5 were made in the range 2Θ 5–40° with step size of 0.02° on a PANalytical Xpert Pro powder diffractometer fitted with Xcelerator detector strip. These were in good agreement with the patterns simulated from single crystal structure determinations (see ESI data†) with the exception of compound 2. This resulted from a low temperature structural phase transition which transformed the cell from orthorhombic to monoclinic symmetry with a change of β > 5°. An ambient temperature structure determination gave a modified simulated p-XRD pattern which then agreed with that measured experimentally.
Computation
DFT calculations on idealized [BSal2] anions were conducted using Gaussian 09 program23 with 6-31G* basis set employing a Becke 3-parameter Lee–Yang–Parr functional (B3LYP).24 Starting geometries were obtained from crystal structure and energies optimized with constraint applied to salicylate hinge angles, which was varied in 5° steps. These indicated coplanar rings were disfavoured by 0.25 kcal mol−1 compared to hinged minima.Conclusions
Salts of both enantiomeric hands of the B-chiral bis(salicylato)borate [BSal2] anions may be been prepared by facile one-pot or metathesis procedures. The 1:1 quininium salt [HQuin][BS(Sal)2] and 1:2 sparteinium salt [SpaH2][BR(Sal)2]2 were formed in high yield. Single crystal structure determinations indicate high enantiopurity in the former and about 70% ee in the latter, due to disorder at one of four independent anion sites. By using 5-chloromandelic acid the analogous salt [H2Spa][BR(5-Cl-Sal)2]2 can be isolated in enantiomerically pure form. Circular dichroism (CD) measurements indicate a strong characteristic spectrum is found for the [BSal2] anion. In pure water the resolved [BSal2] ions rapidly lose their enantiopurity over time and are fully racemic after about 2 h. However in aprotic DMF solution the CD signals remain completely unchanged after 1 week and even remain unperturbed by addition of up to 25% water. The B-chiral anions can be also be metathesized into salts such as [NBu4][BR(Sal)2], implying that they may have prospects for use in both crystallizations and resolutions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for support of this work from the Research Grants Council of Hong Kong (grant 605511). We also thank Ms Carmen Tse for assistance with the DFT calculations.References
- C. M. Vogels and S. A. Westcott, Chem. Soc. Rev., 2011, 40, 1446–1458 RSC.
- D. M. Schubert, Struct. Bonding, 2003, 105, 1–40 CAS.
- W. Clegg, A. J. Scott, F. J. Lawlor, N. C. Norman, T. B. Marder, C. Y. Dai and P. Nguyen, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1998, 54, 1875–1880 Search PubMed; W. Clegg, M. R. J. Elsegood, A. J. Scott, T. B. Marder, C. Y. Dai, N. C. Norman, N. L. Pickett and E. G. Robins, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1999, 55, 733–739 Search PubMed.
- M. Periasamy, N. S. Kumar, S. Sivakumar, V. D. Rao, C. R. Ramanathan and L. Venkatraman, J. Org. Chem., 2001, 66, 3828–3833 CrossRef CAS PubMed; M. Periasamy, L. Venkatraman, S. Sivakumar, N. Sampathkumar and C. R. Ramanathan, J. Org. Chem., 1999, 63, 7643–7645 CrossRef; M. Periasamy, C. R. Ramanathan and N. S. Kumar, Tetrahedron: Asymmetry, 1999, 10, 2307–2310 CrossRef.
- Y. Loewer, C. Weiss, A. T. Biju, R. Frohlich and F. Glorius, J. Org. Chem., 2011, 76, 2324–2327 CrossRef CAS PubMed.
- E. L. Eliel, S. H. Wilen and L. N. Mander, Stereochemistry of Organic Compounds, Wiley, New York, 1994 Search PubMed.
- R. Siedlecka, Tetrahedron, 2013, 69, 6331–6363 CrossRef CAS; F. Faigl, E. Fogassy, M. Nogradi, E. Palovics and J. Schindler, Tetrahedron: Asymmetry, 2008, 19, 519–536 CrossRef.
- L. W.-Y. Wong, J. W.-H. Kan, T. Nguyen, H. H.-Y. Sung, D. Li, A. S.-F. Au Yeung, R. Sharma, Z. Lin and I. D. Williams, Chem. Commun., 2015, 51, 15760–15763 RSC.
- J. Lacour and V. Hebbe-Viton, Chem. Soc. Rev., 2003, 32, 373–382 RSC.
- J. Böeseken and J. A. Mijis, Recl. Trav. Chim. Pays-Bas, 1925, 44, 758 CrossRef.
- E. Graf, R. Graff, M. W. Hosseini, C. Huguenard and F. Taulelle, Chem. Commun., 1997, 1459–1460 RSC.
- S. Green, A. Nelson, S. Warriner and B. Whittaker, J. Chem. Soc., Perkin Trans. 1, 2000, 4403–4408 RSC.
- L. W.-Y. Wong, J. W.-H. Kan, H. H.-Y. Sung and I. D. Williams, Acta Crystallogr., Sect. C: Struct. Chem., 2017, 73, 625–631 CAS.
- N. B. Bathori, A. Jacobs, L. R. Nassimbeni and B. K. Sebogisi, S. Afr. J. Chem., 2014, 67, 160–166 Search PubMed; N. B. Bathori, L. R. Nassimbeni and C. L. Oliver, Chem. Commun., 2011, 47, 2670–2672 RSC.
- C. Strohmann, K. Strohfeldt and D. Schildbach, J. Am. Chem. Soc., 2003, 125, 13672–13673 CrossRef CAS PubMed.
- A. G. De Crisci, V. T. Annibale, G. K. Hamer, A. J. Lough and U. Fekl, Dalton Trans., 2010, 39, 2888–2902 RSC.
- B. Wiberg and W. F. Bailey, J. Mol. Struct., 2000, 556, 239 CrossRef.
- A. Downard, M. Nieuwenhuyzen, K. R. Seddon, J. A. van den Berg, M. A. Schmidt, J. F. S. Vaughan and U. Welz-Biermann, Cryst. Growth Des., 2002, 2, 111–119 Search PubMed; H. X. Liu, Q. Liu, H. M. Guo, K. Q. Ye, J. Z. Xiao, X. S. Tai and G. Zeng, Asian J. Chem., 2015, 25, 10601 Search PubMed; Y. Yang, S. Pan, X. Hou, J. Guo, F. Li, J. Han, J. Guo and D. Jia, Inorg. Chim. Acta, 2011, 365, 20–24 CrossRef CAS.
- M. Taher, F. U. Shah, A. Filippov, P. de Baets, S. Glavatskih and O. N. Antzukin, RSC Adv., 2014, 4, 30617–30623 RSC.
- M. M. Wu, T. S.-C. Law, H. H.-Y. Sung, J. W. Cai and I. D. Williams, Chem. Commun., 2005, 1827–1829 RSC; H. H.-Y. Sung, M. M. Wu and I. D. Williams, Inorg. Chem. Commun., 2000, 3, 401–404 CrossRef CAS; I. D. Williams, M. M. Wu, H. H.-Y. Sung, X. X. Zhang and J. H. Yu, Chem. Commun., 1998, 2463–2464 RSC.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS; L. J. Bourhis, O. V. Dolomanov, R. J. Gildea, J. A. K. Howard and H. Puschmann, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 59–75 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; C. Lee, W. Yang and G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1581657–1581661 and 1581667. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra11997f |
| This journal is © The Royal Society of Chemistry 2018 |