
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA high-activity cobalt-based MOF catalyst for [2 + 2 + 2] cycloaddition of diynes and alkynes: insights into alkyne affinity and selectivity control†
Fen Xu ,
Xiao-Ju Si,
Xiao-Ning Wang,
Hao-Dong Kou,
Di-Ming Chen,
Chun-Sen Liu* and
Miao Du*
,
Xiao-Ju Si,
Xiao-Ning Wang,
Hao-Dong Kou,
Di-Ming Chen,
Chun-Sen Liu* and
Miao Du*
College of Material and Chemical Engineering, Zhengzhou University of Light Industry, Zhengzhou 450002, China. E-mail: chunsenliu@zzuli.edu.cn; dumiao@zzuli.edu.cn
First published on 29th January 2018
Abstract
This work develops a highly efficient metal–organic framework (MOF) catalyst for the straightforward synthesis of functionalized benzenes via [2 + 2 + 2] cycloaddition. As efficient cooperative catalyst for such reactions, Co-MOF-1, shows great sustainability and efficiency. This new catalytic system can lead to the generation of a series of structurally diverse benzenes in good to excellent yields (up to 95%).
Cycloaddition is a promising strategy used to develop a new synthetic methodology with high atom economy, providing efficient routes to construct various chemical bonds resulting in molecular complexity.1 For example, the [2 + 2 + 2] cycloaddition of diynes and alkynes has been a powerful tool for the synthesis of functionalized benzenes in one step.2 A variety of transition metal catalysts, including Rh,3 Ru,4 Pd,5 Ir6 and Ni7 have shown their capacity for [2 + 2 + 2] annulations. However, in this connection, three crucial problems need to be resolved. (1) The competing dimerization or trimerization remains a challenging issue for [2 + 2 + 2] cycloaddition with high selectivity. (2) These unrecoverable homogeneous catalysts will take a major responsibility for energy waste and environmental pollution. (3) The sensitivity to air or moisture for the known catalysts may greatly reduce the reaction efficiency and make the operation complicated. As a result, the development of easily separable and recoverable heterogeneous catalysts is of great significance and interest in this respect.
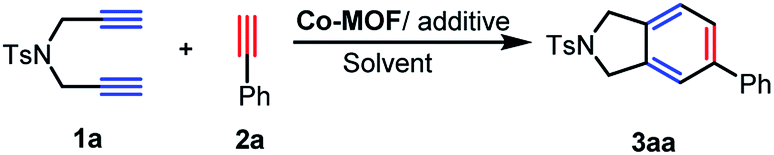
Metal–organic frameworks (MOFs) represent a rising class of porous crystalline materials with exceptional surface areas and uniformly dispersed metal ions.8 Such inherent properties make MOFs as advanced materials for gas storage and separation,9 drug delivery,10 heterogeneous catalysis,11 and so on. Recently, we have designed a series of MOFs materials with diverse pore shapes, sizes and compositions for targeted applications12 and also developed diversified metal-catalyzed cycloadditions involving alkynes.13 Thus, it will be anticipated to explore the potentials of MOFs as the new-type catalysts for cycloadditions, which may avoid the restrictions for the known examples. Notably, Co(I) or Co(II)/additive14 catalytic system has shown activity in [2 + 2 + 2] cycloaddition, though the dimerization or trimerization of terminal diynes usually occurs.15 On this account, a porous MOF [Co2(HCOO)2(CPT)2](NMF)5(H2O)2 (Co-MOF-1) with high affinity of acetylene was successfully constructed based on a bi-functional ligand 4-(4-carboxyphenyl)-1,2,4-triazole (HCPT).16 As a result, a variety of benzene derivatives can be obtained via Co-MOF catalyzed [2 + 2 + 2] cycloaddition in high yields with great tolerance of substrates and the crystalline defect of catalyst will account for the significant selectivity of the reactions (Scheme 1).
 | ||
| Scheme 1 Catalytic strategy for [2 + 2 + 2] cycloaddition in this work. | ||
The Co-MOF-1 was harvested by reaction of Co(NO3)2·6H2O and HCPT in NMF-water. Single-crystal X-ray diffraction analysis indicates that Co-MOF-1 crystallizes in the trigonal space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c.16 A mentionable structural feature is the presence of distorted octahedral cages with the inner diameter of 7.9 Å, constituted by twelve Co(II) and twelve CPT− for each cage (Fig. 1a and S2†). The octahedral cages are sequentially extended via sharing the trigonal windows. As a result, the 3D framework displays 1D hexagonal channels along a axis with the pore diameter of 5.4 Å (Fig. 1b, S3 and S4†) and the available void of 16
c.16 A mentionable structural feature is the presence of distorted octahedral cages with the inner diameter of 7.9 Å, constituted by twelve Co(II) and twelve CPT− for each cage (Fig. 1a and S2†). The octahedral cages are sequentially extended via sharing the trigonal windows. As a result, the 3D framework displays 1D hexagonal channels along a axis with the pore diameter of 5.4 Å (Fig. 1b, S3 and S4†) and the available void of 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 835 Å3, corresponding to 48.6% of the crystal volume (34647.6 Å3).
835 Å3, corresponding to 48.6% of the crystal volume (34647.6 Å3).
 | ||
| Fig. 1 (a) The serial octahedral cages in Co-MOF-1. (b) The 3D framework of Co-MOF-1 with 1D channels running along a axis. (c) N2 sorption isotherm at 77 K for Co-MOF-1′ (inset: pore size distribution plots). (d) C2H2 adsorption isotherms of Co-MOF-1′ at 273 and 298 K. | ||
The porosity of Co-MOF-1 was confirmed by N2 sorption at 77 K. The CH2Cl2-exchanged Co-MOF-1 was activated at 70 °C to afford the desolvated material Co-MOF-1′ (Fig. S5 and S6†). Notably, Co-MOF-1′ shows an initial type-I adsorption isotherm, as a typical microporous material and then, a type-IV adsorption with obvious hysteresis and a significant portion of the mesopore volume of 0.46 cm3 g−1 (Fig. 1c). This can be a consequence of the dissociation of Co(II) ions and formation of defect sites in lattice.17 The maximal N2 uptake is 307 cm3 g−1, corresponding to a BET surface area of 947 m2 g−1 and pore diameter of 5.4 Å. The pore size distribution is consistent with the observation from its crystal structure. The combination of high porosity and defect sites in Co-MOF-1′ promotes us to investigate its affinity toward acetylene, showing a high C2H2 adsorption capability (Fig. 1d) of 178 cm3 g−1 at 273 K and 1 bar and 120 cm3 g−1 at 298 K and 1 bar. The isosteric heat of sorption (Qst) for C2H2 molecule was calculated to evaluate the affinity between C2H2 and Co-MOF-1′ (Fig. S7 and S8†). The result shows a high Qst value of 36 kJ mol−1 at zero loading, revealing a preferred binding capacity for C2H2 compared with the reported MOFs without high density of active sites (Qst values: 13.3–32.0 kJ mol−1).18
Catalysis of [2 + 2 + 2] cycloaddition by Co-MOF-1 was initiated from a terminal NTs-diyne 1a and phenylacetylene 2a (Table 1), in the presence of 6 mol% 1,3-bis(diphenylphos phino)propane (dppp) and 10 mol% Zn powder in 1,2-dichloroethane (DCE). It is interesting that 3aa can be obtained directly in 40% yield after reaction at 80 °C for 24 h (entry 1). The yield was enhanced to 59% when Co-MOF-1′ was used and higher yield was achieved in the presence of 4 equiv. of phenylacetylene (entries 2 and 3). Subsequent tests indicated that reducing the temperature was greatly to the disadvantage of annulation (entries 5 and 6) and a control experiment illustrated that dppp was necessary for this transformation (entry 7). In addition, decreasing the loading of dppp also sharply cut down the yield of target product (entries 3 and 8). It was also found that the transformation was dramatically affected by the catalyst, and the combination of Co(NO3)2 and dppp was incompetent to produce 3aa in only 12% yield (entry 9). 3aa was not detected in the absence of Zn powder (entry 10). The air-stable and efficient Co-MOF-1′ catalyst can be separated and recycled that makes it much sustainable compared to known examples. Therefore, the optimal reaction conditions were determined as Co-MOF-1′ (10 mg), dppp (6 mol%) and Zn powder (10 mol%) in DCE (2.0 mL) at 80 °C for 24 h.
|
|||
|---|---|---|---|
| Entry | Catalyst | Temp. (°C) | Yieldb (%) |
| a Reaction conditions: diyne 1a (0.25 mmol), phenylacetylene 2a (2 equiv., 0.5 mmol), Co-MOF (10 mg), dppp (6 mol%), Zn powder (10 mol%) and DCE (2 mL) at 80 °C for 24 h.b Isolated yield.c 2a (4 equiv.).d Co-MOF-1′ (1 mg).e No dppp.f dppp (3 mol%).g Co(NO3)2 (5 mol%), dppp (6 mol%) and Zn powder (10 mol%).h In the absence of Zn powder, N.D (not detected). | |||
| 1 | Co-MOF-1 | 80 | 40 |
| 2 | Co-MOF-1′ | 80 | 59 |
| 3c | Co-MOF-1′ | 80 | 86 |
| 4d | Co-MOF-1′ | 80 | 28 |
| 5 | Co-MOF-1′ | 40 | Trace |
| 6 | Co-MOF-1′ | RT | Trace |
| 7e | Co-MOF-1′ | 80 | Trace |
| 8c,f | Co-MOF-1′ | 80 | 60 |
| 9g | Co(NO3)2 | 80 | 12 |
| 10h | Co-MOF-1′ | 80 | N.D |
The generality and functional-group tolerance was explored by different substituted alkynes. A broad range of phenylacetylenes with either electron-donating or -withdrawing groups, could react smoothly with NTs-diyne 1a and enable the formation of substituted benzenes 3aa–ad and 3af–ah in 86–95% yields (Scheme 2). Initially, 1a was treated with 2a, Co-MOF-1′ and dppp in DCE. This led to the formation of 3aa in 86% isolated yield. As illustrated, aromatic alkynes bearing Me– and F– groups served as the most proper substrates in this case and showed unconspicuous effect on the reaction efficiency (Scheme 2, 3ad and 3ag). In contrast, the ortho-substituted 1-ethynyl-2-methyl benzene showed extremely poor reactivity (Scheme 2, 3ae). Further, para- and meta-substitution was also tolerated, while a slight reduced yield was found for aromatic alkyne with meta-substitution (Scheme 2, 3ag and 3ah). Notably, the reaction of pharmaceutically important aromatic substituted alkynes, aiming at introducing fluorine and/or fluorinated building blocks into the aromatic ring, has remained a challenge thus far. Herein, 2g and 2h delicately decorated by fluorine were revealed as meaningful substrates as well, affording the corresponding products 3ag and 3ah in 93% and 86% yields. However, sterically hindered internal alkynes 2i and 2j were incompatible, forming dimer or trimer of diynes as side-products, where the bulky phenyl groups may prevent the contact of substrates with the active sites of catalyst.
 | ||
| Scheme 2 Formation of benzenes from diynes 1a–j and alkynes 2a–j. | ||
Subsequently, the scope of this reaction with respect to diynes was further extended. A variety of N-based diynes, ranging from N,N-di(prop-2-yn-1-yl)benzenesulfonamide 1c with electronically diverse groups on phenyl ring (1b–1d), N-benzyl-N-(prop-2-yn-1-yl)prop-2-yn-1-amine 1f, to N,N-di(prop-2-yn-1-yl) methanesulfonamide 1g, was used in this catalytic system. The corresponding products were obtained in 84–95% yields (Scheme 2, 3ba–ga), which are similar to those reactions with NTs-based diyne 1a. Notably, the electronic nature of phenyl ring for N-based diynes turned out to be significant for the cyclization. With Co-MOF-1′ and dppp in the presence of Zn powder, 3ba was achieved in a yield of 95%, whereas 3aa was produced in 86% yield. Significantly, cycloadduct 3da with F– substituent was obtained in high yield of 93%. In accordance with the observation that steric factor intensively influences the reactivity of alkyne substrates, internal diyne N,N-di(but-2-yn-1-yl)-4-methylbenzene sulfonamide 1h was failed to afford benzene scaffold 3ha. This work was also highlighted by substrate of ester tethered diyne, affording corresponding product 3ia in 87% yield. In addition to N-based or ester-tethered diyne, dipropargyl ether showing high reactivity for dimerization to arene byproduct,19 cyclized with 2a to afford the corresponding benzo[c]furan 3ja in moderate yield (Scheme 2).
The major advantage of heterogeneous catalysts is their reutilization, which is one of the most important indexes to evaluate the performance of catalyst effect and stability along the catalytic cycle. Recycling experiments were performed on Co-MOF-1′ for the annulation of 1b and 2a. The catalyst was recycled for five runs with no decrease of activity, giving 93–95% isolated yields (Fig. S9a†). After each case, the recovered Co-MOF-1′ catalyst was characterized by powder X-ray diffraction (PXRD) technique (Fig. S9b†). Notably, even after the fifth run, the PXRD pattern evidently confirmed that the catalyst remained excellent crystallinity and was not damaged in the catalytic process.
To further shed some light on the reaction mechanism, auxiliary experiment was designed and conducted to probe the active species of this transformation. As the presence of Co(II) ions in the initial reaction solution (25.5 mg L−1) was detected by ICP-MS, we suspected that such a reaction would begin with a sluggish release of Co(II) due to the crystal defect of catalyst, which was then reduced by Zn powder to give Co(I) species. The active Co(I) surrounded the surface of Co-MOF-1′ material to facilely capture the neighboring diyne A via ligand exchange, forming a 5-membered metallacyclopentadiene intermediate B through traditional oxidative addition. However, attempt to isolate the species B was failed. After that, B will either undergo alkyne insertion to form 7-membered azametallacycle C or [4 + 2] annulation to produce D, resulting in benzenes via reductive elimination to afford the final product E and also regenerate Co(I) for the next catalytic cycle (Fig. 2).
 | ||
| Fig. 2 Proposed mechanism for the transformation. | ||
To confirm the catalytic mechanism, two control experiments were further discussed. First, subjecting N,N-di(but-2-yn-1-yl)-4-methylbenzenesulfonamide 1h with 2a to this annulation failed to deliver the target product. In fact, 1h was largely retained after reaction, revealing that higher steric internal diyne will suppress the fast access to active Co(I) species on the surface of catalyst (eq. 1 in Fig. 3). Then, a compensatory experiment involving the coupling of terminal diyne 1a with alkyne 2j showed that the dimer of 1a was achieved instead of [2 + 2 + 2] annulation. This reveals that the internal alkyne will hardly get close to the active Co(I) due to the low affinity to Co-MOF-1′ (eq. 2 compared with eq. 3 and eq. 4 in Fig. 3). Thus, it can be concluded that the cycloaddition would proceed favorably near the catalyst surface with “spot-activated cobalt”, allowing only high-affinity alkynes to approach the metal center and thus high selectivity can be realized for cycloadditions. This strategy also provided the potentials for the development of template and precise catalysts.
 | ||
| Fig. 3 The auxiliary verification experiments for catalytic mechanism. | ||
Conclusions
In conclusion, a 3D Co(II)-based MOF was designed and prepared, showing serial window-sharing octahedral cages and 1D hexagonal channels. The activated Co-MOF-1′ has high affinity toward acetylene and serves as a heterogeneous and recoverable catalyst for [2 + 2 + 2] cycloaddition to afford highly substituted benzenes with remarkable efficiency, selectivity, and functional group tolerance. Notably, this transformation is also compatible with aromatic alkynes with fluorine substituent and thus provided products with potential pharmaceutical activity. Co-MOF-1′ with high alkyne affinity and “activated cobalt” can preclude the fast approach of sterically hindered substrates to the active sites, which may well account for the selectivity of achieved products. The results witnessed the great success of Co-MOF-1′ as catalyst for annulation and encouraged further developments of new MOFs catalysts for expedient synthetic transformations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the support from the National Natural Science Foundation of China (21471134, 21571158, and 21701148).Notes and references
- (a) S. Perreault and T. Rovis, Chem. Soc. Rev., 2009, 38, 3149 RSC; (b) H. Pellissier and H. Clavier, Chem. Rev., 2014, 114, 2775 CrossRef CAS PubMed; (c) D. M. Kuznetsov, O. A. Mukhina and A. G. Kutateladze, Angew. Chem., Int. Ed., 2016, 55, 6988 CrossRef CAS PubMed; (d) G. M. Torres, J. S. Quesnel, D. Bijou and B. A. Arndtsen, J. Am. Chem. Soc., 2016, 138, 7315 CrossRef CAS PubMed; (e) C. Ni and X. Tong, J. Am. Chem. Soc., 2016, 138, 7872 CrossRef CAS PubMed; (f) Y. Deng, L. A. Massey, Y. R. Núñez, H. Arman and M. P. Doyle, Angew. Chem., Int. Ed., 2017, 56, 12292 CrossRef CAS PubMed; (g) R. L. Sahani and R.-S. Liu, Angew. Chem., Int. Ed., 2017, 56, 12736 CrossRef CAS PubMed; (h) S. C. Schmid, I. A. Guzei and J. M. Schomaker, Angew. Chem., Int. Ed., 2017, 56, 12229 CrossRef CAS PubMed; (i) J.-L. Shih, S. Jansone-Popova, C. Huynh and J. A. May, Chem. Sci., 2017, 8, 7132 RSC; (j) L. Shen, K. Zhao, K. Doitomi, R. Ganguly, Y.-X. Li, Z.-L. Shen, H. Hirao and T.-P. Loh, J. Am. Chem. Soc., 2017, 139, 13570 CrossRef CAS PubMed.
- (a) M. R. Shaaban, R. El-Sayed and A. H. M. Elwahy, Tetrahedron, 2011, 67, 6095 CrossRef CAS; (b) S. Kotha, E. Brahmachary and K. Lahiri, Eur. J. Org. Chem., 2005, 4741 CrossRef CAS; (c) H. F. Jónsson, S. Evjen and A. Fiksdahl, Org. Lett., 2017, 19, 2202 CrossRef PubMed; (d) H. Ueda, K. Masutomi, Y. Shibata and K. Tanaka, Org. Lett., 2017, 19, 2913 CrossRef CAS PubMed; (e) D. Bhatt, H. Chowdhury and A. Goswami, Org. Lett., 2017, 19, 3350 CrossRef CAS PubMed; (f) D. Brenna, M. Villa, T. Gieshoff, F. Fischer, M. Hapke and A. J. Wangelin, Angew. Chem., Int. Ed., 2017, 56, 8451 CrossRef CAS PubMed.
- (a) R. Shintani, C. Takagi, T. Ito, M. Naito and K. Nozaki, Angew. Chem., Int. Ed., 2015, 54, 1616 CrossRef CAS PubMed; (b) R. P. Kaiser, F. Hessler, J. Mosinger, I. Císařová and M. Kotora, Chem.–Eur. J., 2015, 21, 13577 CrossRef CAS PubMed.
- (a) R. Karmakar, S. Y. Yun, J. Chen, Y. Xia and D. Lee, Angew. Chem., Int. Ed., 2015, 54, 6582 CrossRef CAS PubMed; (b) J. Jacquet, A. Auvinet, A. K. Mandadapu, M. Haddad, V. Ratovelomanana-Vidal and V. Micheleta, Adv. Synth. Catal., 2015, 357, 1387 CrossRef CAS; (c) H. Chowdhury and A. Goswami, Org. Biomol. Chem., 2017, 15, 5824 RSC.
- P. J. Parsons, D. R. Jones, A. C. Padgham, L. A. T. Allen, C. S. Penkett, R. A. Green and A. J. P. White, Chem.–Eur. J., 2016, 22, 3981 CrossRef CAS PubMed.
- G. Onodera, Y. Shimizu, J. Kimura, J. Kobayashi, Y. Ebihara, K. Kondo, K. Sakata and R. Takeuchi, J. Am. Chem. Soc., 2012, 134, 10515 CrossRef CAS PubMed.
- A. Thakur and J. Louie, Acc. Chem. Res., 2015, 48, 2354 CrossRef CAS PubMed.
- (a) P.-Z. Li, X.-J. Wang, S. Y. Tan, C. Yang, H. Chen, J. Liu, R. Zou and Y. Zhao, Angew. Chem., Int. Ed., 2015, 54, 12748 CrossRef CAS PubMed; (b) H. Deng, S. Grunder, K. E. Cordova, C. Valente, H. Furukawa, M. Hmadeh, F. Gándara, A. C. Whalley, Z. Liu, S. Asahina, H. Kazumori, M. O'Keeffe, O. Terasaki, J. F. Stoddart and O. M. Yaghi, Science, 2012, 336, 1018 CrossRef CAS PubMed; (c) J.-S. Qin, D.-Y. Du, W. Guan, X.-J. Bo, Y.-F. Li, L.-P. Guo, Z.-M. Su, Y.-Y. Wang, Y.-Q. Lan and H.-C. Zhou, J. Am. Chem. Soc., 2015, 137, 7169 CrossRef CAS PubMed; (d) A. Schoedel, M. Li, D. A. Schoedel, M. Li, D. Li, M. O'Keeffe and O. M. Yaghi, Chem. Rev., 2016, 116, 12466 CrossRef CAS PubMed; (e) Y. Long, L. Xiao, Q. Cao, X. Shi and Y. Wang, Chem. Commun., 2017, 53, 10831 RSC; (f) Y.-S. Wei, X.-P. Hu, Z. Han, X.-Y. Dong, S.-Q. Zang and T. C. W. Mak, J. Am. Chem. Soc., 2017, 139, 3505 CrossRef CAS PubMed.
- Y. He, W. Zhou, G. Qian and B. Chen, Chem. Soc. Rev., 2014, 43, 5657 RSC.
- H. Zheng, Y. Zhang, L. Liu, W. Wan, P. Guo, A. M. Nyström and X. Zou, J. Am. Chem. Soc., 2016, 138, 962 CrossRef CAS PubMed.
- (a) W.-Y. Gao, H. Wu, K. Leng, Y. Sun and S. Ma, Angew. Chem., Int. Ed., 2016, 55, 5472 CrossRef CAS PubMed; (b) J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C.-Y. Su, Chem. Soc. Rev., 2014, 43, 6011 RSC; (c) X.-L. Lv, K. Wang, B. Wang, J. Su, X. Zou, Y. Xie, J.-R. Li and H.-C. Zhou, J. Am. Chem. Soc., 2017, 139, 211 CrossRef CAS PubMed; (d) Y.-B. Huang, J. Liang, X.-S. Wang and R. Cao, Chem. Soc. Rev., 2017, 46, 126 RSC; (e) J. Liang, R.-P. Chen, X.-Y. Wang, T.-T. Liu, X.-S. Wang, Y.-B. Huang and R. Cao, Chem. Sci., 2017, 8, 1570 RSC.
- (a) M. Du, C.-P. Li, C.-S. Liu and S.-M. Fang, Coord. Chem. Rev., 2013, 257, 1282 CrossRef CAS; (b) M. Du, C.-P. Li, M. Chen, Z.-W. Ge, X. Wang, L. Wang and C.-S. Liu, J. Am. Chem. Soc., 2014, 136, 10906 CrossRef CAS PubMed; (c) M. Du, X. Wang, M. Chen, C.-P. Li, J.-Y. Tian, Z.-W. Wang and C.-S. Liu, Chem.–Eur. J., 2015, 21, 9713 CrossRef CAS PubMed; (d) D.-M. Chen, J.-Y. Tian, C.-S. Liu and M. Du, Chem. Commun., 2016, 52, 8413 RSC; (e) D.-M. Chen, J.-Y. Tian, M. Chen, C.-S. Liu and M. Du, ACS Appl. Mater. Interfaces, 2016, 8, 18043 CrossRef CAS PubMed.
- (a) F. Xu, C. Wang, D. Wang, X. Li and B. Wan, Chem.–Eur. J., 2013, 19, 2252 CrossRef CAS PubMed; (b) F. Xu, C. Wang, H. Wang, X. Li and B. Wan, Green Chem., 2015, 17, 799 RSC.
- (a) A. A. More and C. V. Ramana, J. Org. Chem., 2016, 81, 3400 CrossRef CAS PubMed; (b) Y. Sugiyama, R. Kato, T. Sakurada and S. Okamoto, J. Am. Chem. Soc., 2011, 133, 9712 CrossRef CAS PubMed.
- N. Saino, F. Amemiya, E. Tanabe, K. Kase and S. Okamoto, Org. Lett., 2006, 8, 1439 CrossRef CAS PubMed.
- D.-M. Chen, X.-H. Liu, J.-Y. Tian, J.-H. Zhang, C.-S. Liu and M. Du, Inorg. Chem., 2017, 56, 14767 CrossRef CAS PubMed.
- H. Wu, Y. S. Chua, V. Krungleviciute, M. Tyagi, P. Chen, T. Yildirim and W. Zhou, J. Am. Chem. Soc., 2013, 135, 10525 CrossRef CAS PubMed.
- (a) Y. He, Z. Zhang, S. Xiang, F. R. Fronczek, R. Krishna and B. Chen, Chem. Commun., 2012, 48, 6493 RSC; (b) S. Xiang, W. Zhou, J. M. Gallegos, Y. Liu and B. Chen, J. Am. Chem. Soc., 2009, 131, 12415 CrossRef CAS PubMed; (c) J. Pang, F. Jiang, M. Wu, C. Liu, K. Su, W. Lu, D. Yuan and M. Hong, Nat. Commun., 2015, 6, 7575 CrossRef PubMed; (d) Y. He, R. Krishna and B. Chen, Energy Environ. Sci., 2012, 5, 9107 RSC.
- R. Grigg, R. Scott and P. Stevenson, J. Chem. Soc., Perkin Trans. 1, 1988, 1357 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1491616–1491623. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra12136a |
| This journal is © The Royal Society of Chemistry 2018 |