
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChiral protic imidazolium salts with a (−)-menthol fragment in the cation: synthesis, properties and use in the Diels–Alder reaction†
Ewa
Janus
 *a,
Marcin
Gano
a,
Joanna
Feder-Kubis
b and
Jacek
Sośnicki
c
*a,
Marcin
Gano
a,
Joanna
Feder-Kubis
b and
Jacek
Sośnicki
c
aWest Pomeranian University of Technology Szczecin, Faculty of Chemical Technology and Engineering, Institute of Chemical Organic Technology, Pułaski Str. 10, 70-322 Szczecin, Poland. E-mail: ejanus@zut.edu.pl; Tel: +48 91 4494584
bWrocław University of Science and Technology, Faculty of Chemistry, Wybrzeże Wyspiańskiego 27, 50-370 Wrocław, Poland
cWest Pomeranian University of Technology Szczecin, Faculty of Chemical Technology and Engineering, Department of Organic and Physical Chemistry, Al. Piastów 42, 71-065 Szczecin, Poland
First published on 13th March 2018
Abstract
New chiral protic imidazolium salts containing a (1R,2S,5R)-(−)-menthol substituent in the cation and four different anions (chloride, hexafluorophosphate, trifluoromethanesulfonate and bis(trifluoromethylsulfonyl)imide) were efficiently prepared and extensively characterized. Detailed NMR analysis was performed, and a comparison of the chemical shifts of the protons and carbons of the imidazolium cation as a function of the combined anion was discussed. The specific rotation, solubility in commonly used solvents, thermal properties including phase transition temperatures, and thermal stability were also determined. Three of the synthesized tertiary salts (Cl, PF6 or OTf anion) were crystalline solids; 1-H-3-[(1R,2S,5R)-(−)-menthoxymethyl]-imidazolium bis(trifluoromethylsulfonyl)imide, (−)[H-Ment-Im][NTf2] was a liquid at room temperature. The chiral protic salts were used in a Diels–Alder reaction as a test reaction, and the results were compared with those from aprotic chiral ionic liquids having the same chiral substituent in the cation (1R,2S,5R)-(−)-menthol with a bis(trifluoromethylsulfonyl)imide anion. Both protic and aprotic chiral salts, used in a Diels–Alder reaction, were pure (−)-enantiomers, which was determined by NMR with Δ-TRISPHAT tetrabutylammonium salt as a chiral shift reagent. Protic salts offered distinctly higher endo/exo ratios than aprotic ones, but an enantiomeric excess was not obtained. The stereoselectivity reached the same high level even after the fourth recycle of (−)[H-Ment-Im][NTf2] in the reaction of ethyl-vinyl ketone with cyclopentadiene at temperature of −35 °C.
Introduction
Ionic liquids (ILs) are salts with melting points below 100 °C. They are very attractive materials distinguished by many factors including a liquid state across a range of temperatures with extremely low volatility and the ability to tune viscosity, solvation, polarity, polarizability, and proton-donating/accepting properties. Therefore, ionic liquids are extensively used in organic synthesis where they act as alternative solvents and/or catalysts for new opportunities to conduct both uncatalyzed and catalyzed processes.1 The chiral version of ionic liquids with chirality in a cation or anion provides simple access to chiral solvents. Due to their ionic and highly organized nature, they are attractive in asymmetric synthesis.2–4 Chiral ionic liquids have been synthesized based on different raw materials mainly of natural origin.5–9 However, chiral cations in ionic liquids are less common than chiral anions because the synthesis of chiral cations typically requires several steps.There are only a few reports of protic ionic liquids with chirality in a cation and a proton located on the nitrogen atom.10–15 In contrast, protic ionic liquids (PILs) are particularly important ILs family. They are a hydrogen bond donor,16,17 and their catalytic utility is accompanied by hydrogen bonding interactions and Brønsted acidity18,19 that are crucial in many organic reactions. Many articles and reviews imply the potential of protic ionic liquids for catalysis and organic synthesis,20,21 as well as electrochemistry,22,23 chromatography,24 biomass conversion,25 biocatalysis,26 and protein stabilization. They also offer a simple synthesis via a combination of amines with Brønsted acids and the possibility for acidity alterations.18,27
Considering this, the investigation of PILs shown here evaluates chiral cations and is of great interest because it offers progress in the context of developing catalysis and organic synthesis using chiral protic salts as catalysts and solvents. Therefore, we focus here on the synthesis and properties of chiral protic imidazolium salts with chirality in the cation. Three optically active centers in the molecules are associated with the presence of natural raw material: (1R,2S,5R)-(−)-menthol, which belong to the monocyclic monoterpenes. We also studied the utility of synthesized protic chiral salts as organocatalysts in the representative Diels–Alder reaction and compared the results with those obtained from aprotic analogs composed of the same chiral (1R,2S,5R)-(−)-menthoxymethyl fragment in the cation.
The Diels–Alder reaction was one of the most noteworthy because it has great practical applications in the synthesis of cyclic compounds, which are very often impossible or difficult to obtain through other approaches. Often, it is only one of the many stages of multistage synthesis including the synthesis of natural products and biologically active substances in which it is crucial to obtain an optically pure enantiomer. Therefore, the Diels–Alder reaction is a powerful tool in chemists' hands and an attractive approach to testing different ionic liquids.
Experimental
All materials to the synthesis of chiral protic and aprotic salts, their purification methods and general preparation methods of substrates and aprotic chiral salts as well as analytical methods used to their characterization were included to ESI.†Materials used in Diels–Alder reaction
Dicyclopentadiene, methyl acrylate (99%), pent-1-en-3-on (ethyl-vinyl ketone) (97%), were provided from Aldrich. Methyl acrylate prior to use was sequentially washed with aqueous NaOH and distilled water. Then it was dried with CaCl2 and fractionally distilled under reduced pressure. It was stored under nitrogen at 0 °C in dark. Ethyl-vinyl ketone was dried with MgSO4 and distilled.Synthesis of protic imidazolium salts
The 1-H-3-[(1R,2S,5R)-(−)-menthoxymethyl]imidazolium chloride, (−)[H-Ment-Im][Cl] was synthesized following a modified procedure described elsewhere.28 The 1-(1R,2S,5R)-(−)-menthoxymethylimidazole (70.80 g; 0.3 mol) was introduced into a glass reactor with a cooling water system mechanical stirring, reflux condenser, and thermometer. A hydrochloric acid solution 37% (29.60 g; 0.3 mol) was added dropwise to the chiral substrate under vigorous stirring using a dropping funnel (approx. 30–45 min.). The process was conducted under isothermal conditions at 15–20 °C because the acid–base reaction was slightly exothermic. Stirring was continued for 3 hours at room temperature after addition of the acid to the 1-(1R,2S,5R)-(−)-menthoxymethylimidazole.Afterward, 1,2-dichloroethane (DCE) was added (approx. 300 g), and the mixture was distilled under normal pressure until the water–DCE hetero-azeotropic boiling point was reached (73 °C). Subsequently, the residual DCE was finally evaporated under reduced pressure to yield a white solid chloride. The product was crystallized from a mixture of hexane/acetone and dried in a vacuum drier (0.3 mmHg). The 1-H-3-[(1R,2S,5R)-(−)-menthoxymethyl]imidazolium chloride was prepared with 93.0% yield (76.4 g; 0.28 mol). In the same way the 1-H-3-[(±)-menthoxymethyl]imidazolium chloride was prepared with 90% yield from (±)-menthoxymethylimidazole as a starting material.
1H NMR (CDCl3, 400 MHz, 25 °C) δ [ppm]: 0.49 (d, J = 7.0 Hz; 3H; H9 or H10); 0.81–0.99 (m, 9H with two doublets inside at 0.86 (d, J = 7.2 Hz) and 0.92 (d, J = 6.4 Hz); Ha-4, H7, H9 or H10, Ha-6 and Ha-3); 1.21–1.28 (m, 1H; H2); 1.36–1.47 (m, 1H; H5); 1.59–1.67 (m, 2H; Hb-3 and Hb-4); 1.96 (sept d, J = 7.1 Hz and J = 2.6, 1H; H8); 2.08–2.11 (m, 1H; Hb-6); 3.35 (td, J = 10.6 Hz and J = 4.3 Hz, 1H; H1); 5.65 and 5.89 (d, J = 10,6 Hz, 2H, AB system, H11); 7.33 (t, J = 1.6 Hz, 1H; H12), 7.43 (t, J = 1.6 Hz, 1H; H13); 10.0 (s, 1H; H14). 13C NMR (CDCl3, 100 MHz; 25 °C) δ [ppm]: 15.67 (C9 or C10); 21.05 (C7); 22.25 (C9 or C10); 22.91 (C3); 25.53 (C8); 31.31 (C5); 34.14 (C4); 40.30 (C6); 47.87 (C2); 76.64 (C11); 79.58 (C1); 119.95 (C13); 120.32 (C12); 135.83 (C14). HRMS (ESI+): m/z (%) calcd for C14H25N2O: 237.1967, found: 237.1958. Elemental analysis calc. (%) for C14H25N2OCl (272.81): C 61.635, H 9.24, N 10.27, found: C 61.54, H 9.37, N 10.30.
General procedure for metathesis reaction
The metathesis reaction was conducted in aqueous solution. A saturated aqueous solution of the respective alkali salt NaOTf, KPF6, or LiNTf2 (0.120 mol) was added to an aqueous solution of prepared 1-H-3-[(1R,2S,5R)-(−)-menthoxymethyl]imidazolium chloride (27.28 g, 0.100 mol). The reaction mixture was stirred at room temperature for 24 h to produce trifluoromethanesulfonate and hexafluorophosphate salts, while bis(trifluoromethanesulfonyl)imide was obtained by stirring for two days. The crude product was then separated—the prepared salt was precipitated from the reaction mixture as a solid and filtered. Liquid product was phase-separated. Each protic salt was washed with distilled water thrice to eliminate the chloride formed during the metathesis reaction. Each salt was then dissolved in acetone and stirred for 30 minutes to precipitate any halide salt residues. These were filtered with a 0.2 μm filter. To complete the inorganic salt precipitation, the filtrate was placed at −5 °C overnight. Another filtration was performed for precipitation the halide salt, and the final filtrate was evaporated. Finally, tests with AgNO3 were carried out to determine whether any chloride remained in the resulting protic salts. The products were dried for 12 h at 60 °C in a vacuum (0.3 mmHg). Afterwards, the solid salts were crystallized.1-H-3-[(1R,2S,5R)-(−)-Menthoxymethyl]imidazolium trifluoromethanesulfonate, (−)[H-Ment-Im][OTf]
Yield: 95.5%. 1H NMR (CDCl3, 400 MHz, 25 °C) δ [ppm] = 0.50 (d, J = 7.0 Hz, 3H; H9 or H10); 0.83–1.00 (m with two doublets inside at 0.87 (d, J = 7.2 Hz) and 0.92 (d, J = 6.4 Hz); 9H; Ha-4, H7, H9 or H10, Ha-6 and Ha-3); 1.23–1.29 (m, 1H, H2); 1.35–1.42 (m, 1H; H5); 1.61–1.67 (m, 2H, Hb-3 and Hb-4); 1.91–2.00 (m, 2H, Hb-6 and H8); 3.27 (td, J = 10.6 Hz and J = 4.3 Hz, 1H; H1); 5.57 and 5.67 (d, J = 10,6 Hz, 2H, AB system, H11); 7.36 (t, J = 1.7 Hz, 1H; H12); 7.50 (t, J = 1.7 Hz, 1H; H13); 9.12 (s, 1H, H14). 13C NMR (CDCl3, 100 MHz; 25 °C) δ [ppm] = 15.37 (C9 or C10); 20.90 (C7); 22.01 (C9 or C10); 22.78 (C3); 25.43 (C8); 31.20 (C5); 33.97 (C4); 40.06 (C6); 47.70 (C2); 76.90 (C11); 79.96 (C1); 120.20 (C13); 120.88 (C12); 135.31 (C14). Elemental analysis calc. (%) for C15H25F3N2O4S (386.43): C 46.62, H 6.52, N 7.25, found: C 46.75, H 6.66, N 7.16.1-H-3-[(1R,2S,5R)-(−)-Menthoxymethyl]imidazolium bis(trifluoromethanesulfonyl)imide, (−)[H-Ment-Im][NTf2]
Yield: 93.7%. 1H NMR (CDCl3, 600 MHz, 25 °C) δ [ppm] = 0.51 (d, J = 6.6 Hz, 3H, H9 or H10); 0.80–0.97 (m with two doublets inside at 0.87 (d, J = 7.0 Hz) and 0.91 (d, J = 6.8 Hz); 9H; Ha-4, H7, H9 or H10, Ha-6 and Ha-3); 1.20–1.27 (m, 1H; H2); 1.38 (m, 1H; H5); 1.62–1.67 (m, 2H; Hb-3 and Hb-4); 1.95–1.97 (m, 2H, Hb-6 and H8); 3.25 (td, J = 10.6 Hz, J = 4.2 Hz, 1H; H1); 5.57 and 5.62 (d, J = 10.6 Hz and J = 10.8 Hz, 2H, AB system, H11); 7.45 (s, 1H, H12); 7.50 (s, 1H, H13); 8.75 (s, 1H, H14); 9–13 (br s, 1H, H15). 13C NMR (CDCl3, 150.2 MHz, 25 °C) δ [ppm] = 15.4 (C9 or C10); 20.96 (C7); 22.05 (C9 or C10); 22.9 (C3); 25.6 (C8); 31.3 (C5); 34.1 (C4); 40.2 (C6); 47.8 (C2); 76.6 (C11); 80.4 (C1); 120.75 (C13); 121.04 (C12); 134.49 (C14); anion: 116.57–122.95 (quartet, JC–F = 320 Hz). 15N HNMR (CDCl3) δ [ppm] = −208.59 (N–H); −189.88. Elemental analysis calc. (%) for C16H25F6N3O5S2 (517.51): C 37.13, H 4.87, N 8.12, found: C 37.29, H 4.98, N 8.01.1-H-3-[(±)-menthoxymethyl]imidazolium bis(trifluoromethanesulfonyl)imide, (±)[H-Ment-Im][NTf2] was also synthesized from 1-H-3-[(±)-menthoxymethyl]imidazolium chloride with 92% yield.
1-H-3-[(1R,2S,5R)-(−)-Menthoxymethyl]imidazolium hexafluorophosphate, (−)[H-Ment-Im][PF6]
Yield: 96.9%. 1H NMR (CDCl3, 400 MHz; 25 °C) δ [ppm] = 0.50 (d, J = 7.0 Hz, 3H; H9 or H10); 0.77–0.98 (m with two doublets inside at 0.86 (d, J = 7.2 Hz) and 0.91 (d, J = 7.0 Hz); 9H; Ha-4, H7, H9 or H10, Ha-6 and Ha-3); 1.23–1.29 (m, 1H, H2); 1.40 (m, 1H, H5); 1.60–1.67 (m, 2H; Hb-3 and Hb-4); 1.92–1.99 (m, 2H; Hb-6 and H8); 3.27 (td, J = 10.6 Hz, J = 4.3 Hz, 1H, H1); 5.55 and 5.61 (d, J = 10.8 Hz, 2H, AB system, H11); 7.37 (t, J = 1.7 Hz, 1H; H12); 7.46 (t, J = 1.7 Hz, 1H; H13); 8.69 (s, 1H; H14); ∼12–16 (br s, 1H, H16). 13C NMR (CDCl3, 100 MHz; 25 °C) δ [ppm] = 15.4 (C9 or C10); 20.9 (C7); 22.0 (C9 or C10); 22.76 (C3); 25.4 (C8); 31.1 (C5); 34.0 (C4); 40.02 (C6); 47.7 (C2); 77.2 (C11); 79.95 (C1); 120.35 (C13); 121.0 (C12); 134.8 (C14). 31P NMR (CDCl3, 25 °C) δ [ppm] = −12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 596 ÷ −15529 (sextet, J = 713 Hz). Elemental analysis calc. (%) for C14H25F6N2OP (382.325): C 43.98, H 6.59, N 7.33, found: C 43.83, H 6.73, N 7.37.
596 ÷ −15529 (sextet, J = 713 Hz). Elemental analysis calc. (%) for C14H25F6N2OP (382.325): C 43.98, H 6.59, N 7.33, found: C 43.83, H 6.73, N 7.37.
Determination of enantiomeric purity of prepared chiral protic and aprotic salts
To a solution of 3–4 mg of chiral salt in 0.5 mL of CDCl3, were added 1–3 equivalents of Δ-TRISPHAT tetrabutylammonium salt. The registered 1H NMR spectra were presented in ESI.†Diels–Alder reaction general procedure
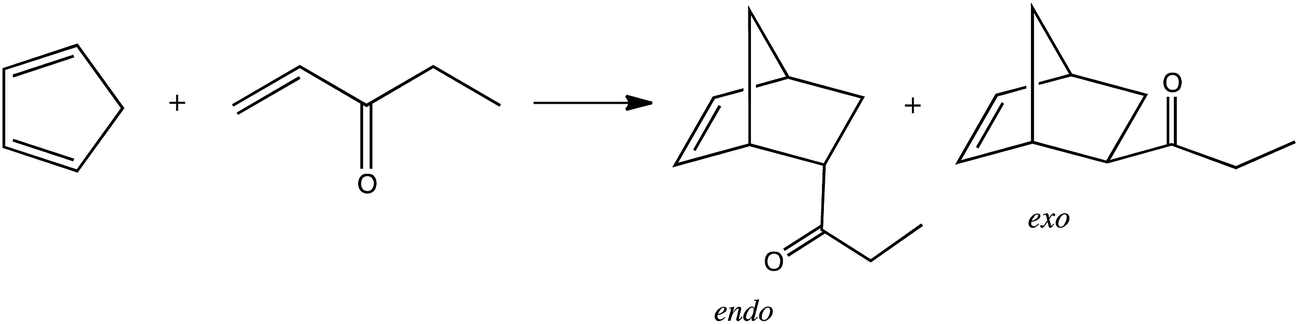
The amount of 1 mmol of chiral ionic liquids was placed in a 4 mL sealed vial followed by 2 mmol of freshly cracked cyclopentadiene. After mixing it, 1 mmol of dienophile was added. The reaction was performed at −35 °C or 25 °C. The reaction was monitored by collecting samples, extracting with hexane, and analyzing the extract with GC using the internal standard method. The yield of the product and the stereoselectivity were determined via GC analysis with a Rxi-17 column. The chiral column RT-BetaDEXsa was also used to check for the enantiomeric excess. Calculations of ee% were based on the surface area of the respective peaks. A representative chromatogram is presented in the ESI.†When the reaction was conducted in chloroform, 0.2 mmol of protic chiral salt was first dissolved in 0.5 mL of chloroform. Then, 2 mmol of freshly cracked cyclopentadiene was added followed by 1 mmol of methyl acrylate. The yield of product and endo/exo ratio were determined based on the 1H NMR spectrum registered for a reaction mixture. The proton integration data for the methyl ester group of the methyl acrylate and the products (2-endo-bicyclo[2.2.1]hept-5-ene-2-carboxylic acid methyl ester and 2-exo-bicyclo[2.2.1]hept-5-ene-2-carboxylic acid methyl ester) were used to calculate yield and endo:exo ratio. A chloroform was removed from taken samples (under vacuum), and residue was extracted with hexane. The hexane extract was analyzed by GC on a chiral column.
Gas chromatograph Model 8000 Top Series (ThermoQuest Italia S.p.A) was equipped with a flame ionization detector and a 30 m × 0.53 mm ID × 1.50 μm df Rxi®-17 column (Restek) or 60 m × 0.25 mm ID × 0.25 μm df Stabilwax® (Restek) column or 30 m × 0.32 mm ID × 0.25 mm df RT-BetaDEXsa® chiral column (Restek) for GC analysis. Hydrogen or helium were used as the carrier gas.
ATR-FTIR measurements
ATR-FTIR spectra were recorded on a Bruker Alpha Fourier Transform IR (FTIR) spectrometer equipped with a platinum ATR single reflection diamond-sampling module (Bruker Optics). The infrared spectra were collected as an average of 24 scans per sample between the wavenumber range of 4000–360 cm−1 at a resolution of 4 cm−1, controlled by Optics User Software (OPUS) version 7.5 (Bruker Optics). Air was used as reference background spectra. The ATR diamond surface was cleaned with acetone before each sample was scanned.Results and discussion
Synthesis, identification and properties of chiral protic imidazolium salts with (1R,2S,5R)-(−)-menthol substituent group in a cation
Four chiral protic imidazolium salts with different anionic species have been synthesized by introducing the chiral, commercially available pool, (1R,2S,5R)-(−)-menthol into imidazolium cation. The total synthesis route consisted of a few stages and is outlined in Scheme 1. | ||
| Scheme 1 Synthesis route of the chiral protic imidazolium salts. | ||
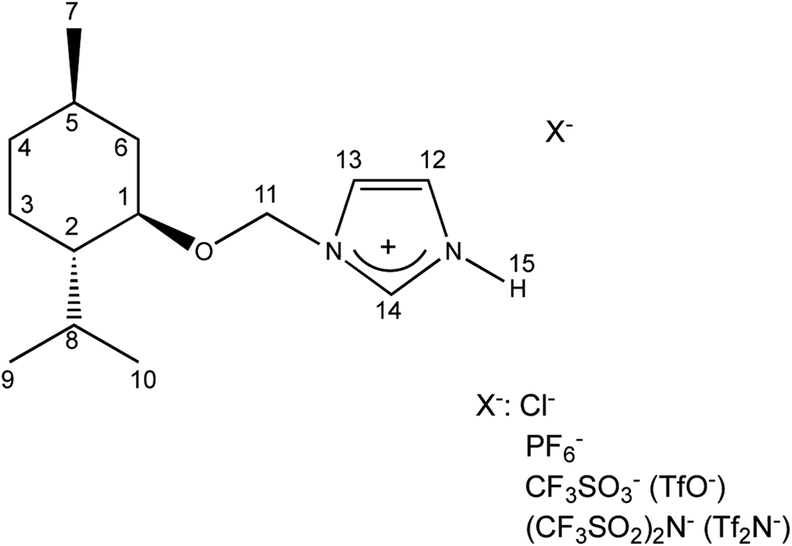
The (1R,2S,5R)-(−)-menthol was first converted to chloromethyl (1R,2S,5R)-(−)-menthyl ether, which was next purified by distillation and used to alkylate the imidazole in the presence of triethylamine to obtain 1-(1R,2S,5R)-(−)-menthoxymethylimidazole. The crystalline chiral amine was then protonated with hydrochloric acid, and the product was recrystallized from a hexane/acetone mixture. Successfully protonation was confirmed by HRMS analysis in high resolution positive ionization mode. The cation mass agreed with the mass calculated for the 1-H-3-[(1R,2S,5R)-(−)-menthoxymethyl]imidazolium cation.
The synthesized 1-H-3-[(1R,2S,5R)-(−)-menthoxymethyl]imidazolium chloride, (−)[H-Ment-Im][Cl] is an excellent reagents for obtaining protic salts via a simple exchange reaction. Therefore, the chloride anion can be changed to other anions such as OTf, NTf2, and PF6via a metathesis reaction with the appropriate salt in aqueous medium. The yield of each synthesis step was over 93% (Table 1). The surfactant content of the protic imidazolium salts were determined by a direct two-phase titration technique (EN ISO 2871-2:2010 standard) using water as the solvent for (−)[H-Ment-Im][Cl] and methanol for all other salts; the range was 99.1 to 99.7%. The remainder consists of water. However, the (−)[H-Ment-Im][NTf2] salt could not be estimated by the direct two-phase titration technique because the bis(trifluoromethanesulfonyl)imide anion in this salt is not interchangeable.
| Structure of protic imidazolium salts | Abbreviation, (−)[H-Ment-Im][X] | Yielda,b (%) | Surfactant contentc (%) |
|---|---|---|---|
| a Isolated yield after purification and drying. b Accuracy ± 0.1%. c Accuracy ± 0.1%. d Not applicable. | |||
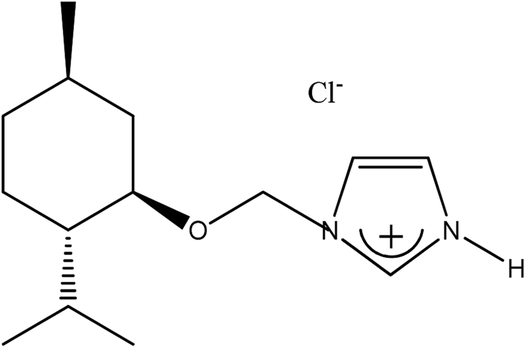
|
(−)[H-Ment-Im][Cl] | 93.0 | 99.7 |
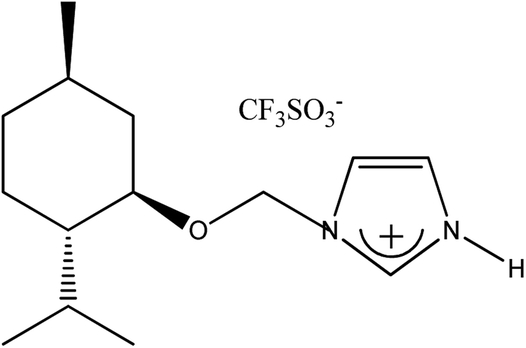
|
(−)[H-Ment-Im][OTf] | 95.5 | 99.1 |
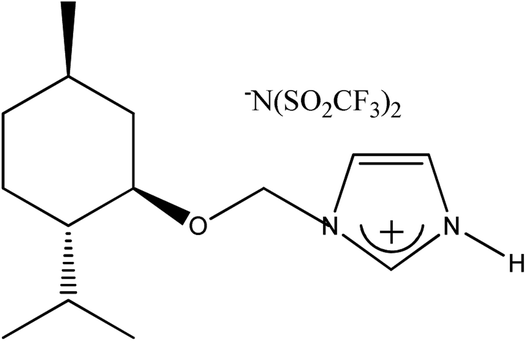
|
(−)[H-Ment-Im][NTf2] | 93.7 | —d |
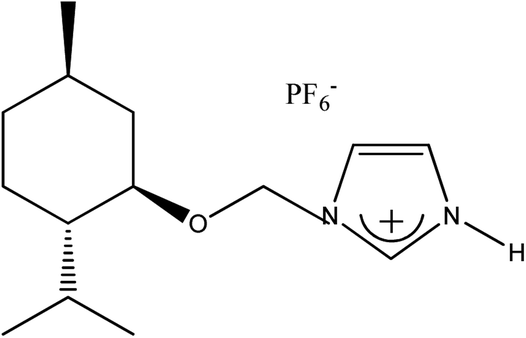
|
(−)[H-Ment-Im][PF6] | 96.9 | 99.6 |
The chiral protic salts were identified and characterized via NMR, elemental analysis, optical rotation, and thermogravimetric analysis. Salt with a NTf2 anion was studied with modulated differential scanning calorimetry (MDSC) in cooling and heating mode, and its refractive index as a function of temperature was determined. The properties of synthesized chiral protic salts are presented in Table 2. The 1H NMR, 13C NMR spectra, TG, DTG, and c-DTA curves registered from thermogravimetric and MDSC analysis are attached in the ESI.†
| Protic chiral salt | State at rt | Shape of crystals | Crystallization solvent | Phase transition temperature (°C) | Specific rotation [α]20D | T 5%d (°C) |
|---|---|---|---|---|---|---|
| a Determined as endothermic peak temperature in c-DTA curve registered from TG measurements. b Not applicable. c Determined as midpoint from MDSC measurements in a heating cycle. d Determined as midpoint from MDSC measurements in a second cooling cycle. | ||||||
| (−)[H-Ment-Im][Cl] | Solid | Irregular plates | Hexane–acetone | T m = 133.5a | −100.97, (c 1.040) | 155.6 |
| (−)[H-Ment-Im][OTf] | Solid | Irregular plates | Water–ethanol | T m = 107.5a | −82.3, (c 1.001) | 199.4 |
| (−)[H-Ment-Im][NTf2] | Liquid | —b | —b | T g(h) = −31.07c, Tg(2c) = −32.60d | −65.36, (c 1.149) | 222.0 |
| (−)[H-Ment-Im][PF6] | Solid | Needles | Water–ethanol | T m = 136.8a | −102.7, (c 0.96) | 200.9 |
Physical properties of chiral protic imidazolium salts
Of the four synthesized protic salts, three—(−)[H-Ment-Im][Cl], (−)[H-Ment-Im][OTf], (−)[H-Ment-Im][PF6] are solids. These solid salts have crystallization conditions elaborated. The 1-H-3-[(1R,2S,5R)-(−)-menthoxymethyl]imidazolium chloride easily crystallizes in the form of irregular plates from a hexane-acetone mixture. Salts with a PF6− and OTf− anion from a water–ethanol mixture in form of needles and irregular plates, respectively. Only 1-H-3-[(1R,2S,5R)-(−)-menthoxymethyl]imidazolium bis(trifluoromethylsulfonyl)imide is a liquid at room temperature.The melting points for the solids were determined using the c-DTA signal from TG analysis. They are 133.5 °C for (−)[H-Ment-Im][Cl], 107.5 °C for (−)[H-Ment-Im][OTf] and 136.8 °C for (−)[H-Ment-Im][PF6] (Table 2). Only the (−)[H-Ment-Im][NTf2] exhibited a glass transition temperature (Fig. 1). The reverse heat flow signal was registered by MDSC in subsequent cooling–heating–cooling cycles. However, no crystalline behavior was observed for this protic ionic liquid at this temperature range tested. During the heating cycle, the endothermal non-reversing signal accompanied the glass transition, and this may be attributed to enthalpic relaxation as the amorphous material approaches equilibrium. The glass transition temperature of (−)[H-Ment-Im][NTf2] was between −35.99 °C and −31.07 °C using a midpoint method. This temperature was about 20–30 °C higher than that determined for analogous aprotic chiral imidazolium salt with (1R,2S,5R)-(−)-menthoxymethyl group in the cation. This is compatible with general observations for aprotic and protic ionic liquids.
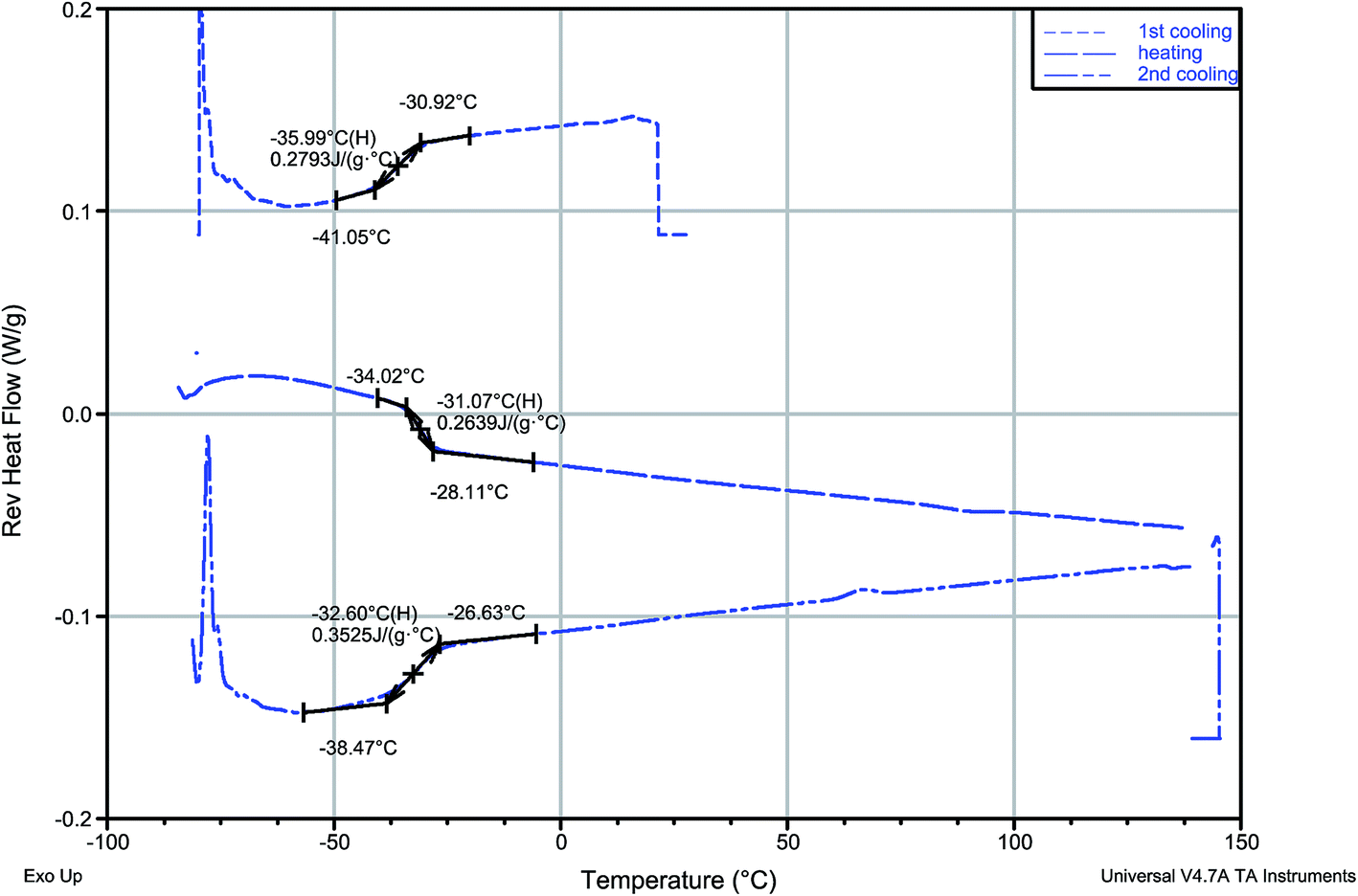 | ||
| Fig. 1 MDSC reversing heat flow signal registered in cooling–heating–cooling cycle of (−)[H-Ment-Im][NTf2]. | ||
The refractive index values for (−)[H-Ment-Im][NTf2] in the temperature range of 20–60 °C was also measured. Fig. 2 shows that the refractive index decreases with an increase in temperature. These values are similar to other reported protic ionic liquids.29
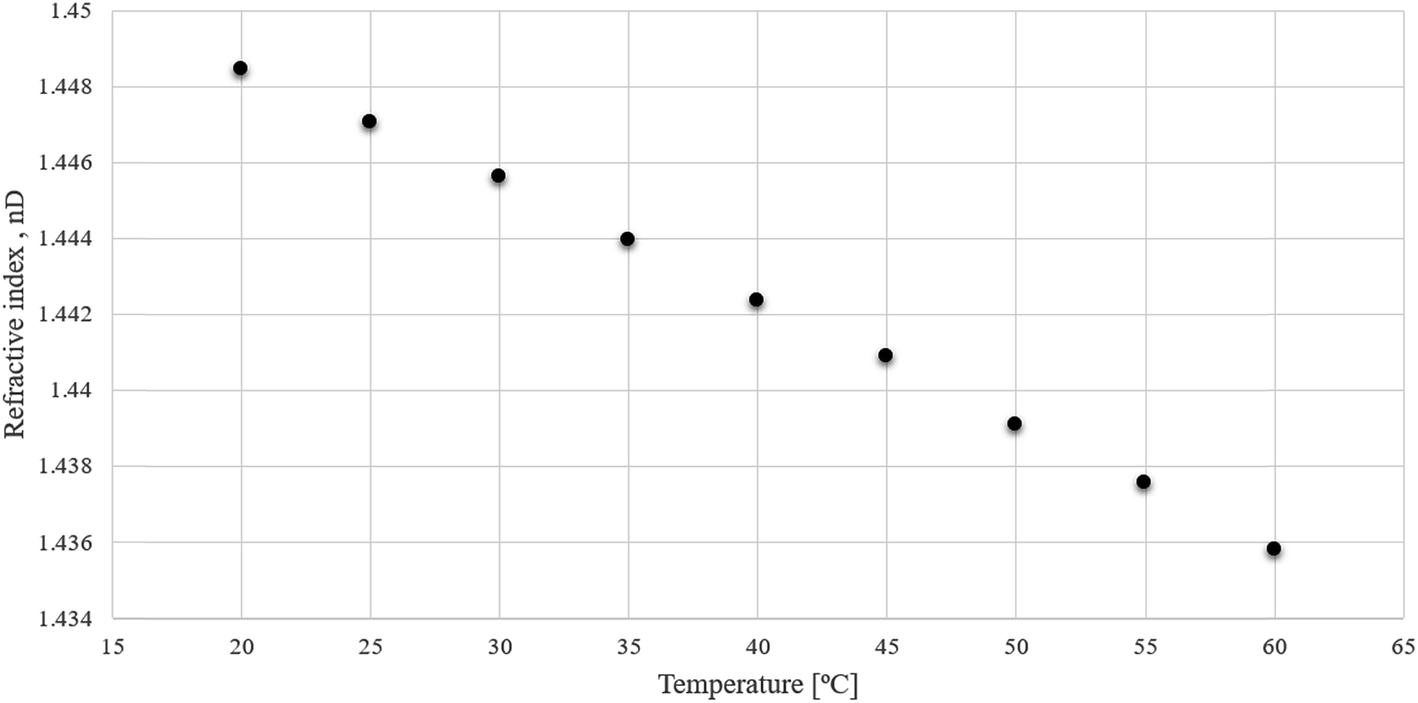 | ||
| Fig. 2 Refractive index (nD) as a function of temperature for (−)[H-Ment-Im][NTf2] ionic liquid. | ||
Temperatures corresponding to a 5% mass loss (T5%d), obtained for protic salts from thermogravimetric analysis are presented in Table 2. Of the synthesized protic salts, salt with a NTf2 anion is the most thermally stable with a T5%d = 222 °C. Salts containing PF6 and OTf anions have decomposition temperatures that are about 20 °C lower than (−)[H-Ment-Im][NTf2]. Salt with a chloride anion has the lowest decomposition temperature. The TG curves indicates two-stage decomposition of (−)[H-Ment-Im][OTf] and (−)[H-Ment-Im][NTf2]. The mass loss percentage in the first step suggests elimination of the menthyl fragment.
The solubility of chiral protic salts were studied in commonly used solvents (Table 4). All salts were soluble in alcohols, dimethylformamide, dimethylsulfoxide, chloroform, dichloromethane, toluene, and acetone but insoluble in hexane. Only (−)[H-Ment-Im][Cl] dissolves in water upon heating and is insoluble in ethyl acetate. Moreover, (−)[H-Ment-Im][NTf2] is the only salt that is soluble in diethyl ether.
|
|
||||||
|---|---|---|---|---|---|---|
| Compound abbreviation | Position | |||||
| 14 | 13 | 12 | 11 | 1 | ||
| (−)[Ment-Im] | δ H (ppm) | 7.59 | 7.08 | 7.05 | 5.32 | 3.12 |
| δ C (ppm) | 136.8 | 118.5 | 129.2 | 76.5 | 73.2 | |
| (−)[H-Ment-Im][Cl] | δ H (ppm) | 10.0 | 7.43 | 7.33 | 5.65 and 5.89 | 3.35 |
| δ C (ppm) | 135.83 | 119.95 | 120.32 | 76.64 | 79.58 | |
| (−)[H-Ment-Im][OTf] | δ H (ppm) | 9.12 | 7.50 | 7.36 | 5.57 and 5.67 | 3.27 |
| δ C (ppm) | 135.31 | 120.20 | 120.88 | 76.90 | 79.96 | |
| (−)[H-Ment-Im][NTf2] | δ H (ppm) | 8.75 | 7.50 | 7.45 | 5.57 and 5.62 | 3.25 |
| δ C (ppm) | 134.49 | 120.75 | 121.04 | 76.6 | 80.4 | |
| (−)[H-Ment-Im][PF6] | δ H (ppm) | 8.69 | 7.46 | 7.37 | 5.55 and 5.61 | 3.27 |
| δ C (ppm) | 134.8 | 120.35 | 121.0 | 77.2 | 79.95 | |
| Solvent | (−)[H-Ment-Im][Cl] | (−)[H-Ment-Im][NTf2] | (−)[H-Ment-Im][OTf] | (−)[H-Ment-Im][PF6] |
|---|---|---|---|---|
| a + Soluble at 25 °C. − Insoluble at 25 °C. b Heating is needed to dissolve. | ||||
| Hexane | − | − | − | − |
| Diethyl ether | − | + | − | − |
| Acetone | + | + | + | + |
| Ethyl acetate | − | + | + | + |
| Toluene | + | + | + | + |
| Chloroform | + | + | + | + |
| Dichloromethane | + | + | + | + |
| Water | +/−b | − | − | − |
| Methanol | + | + | + | + |
| Ethanol | + | + | + | + |
| 1-Propanol | + | + | + | + |
| DMF | + | + | + | + |
| DMSO | + | + | + | + |
NMR analysis of chiral protic salts
A comparison of the chemical shifts for protons and carbons in the protic imidazolium salts versus 1-(1R,2S,5R)-(−)-menthoxymethylimidazole indicates protonation of the imidazole ring. Moreover, the effect of the anion on the chemical shift is observed. The chemical shifts for the selected atoms are listed in Table 3. The signals of H-14, H-13, H-12, H-11, and H-1 protons in the imidazolium salts are shifted to lower field versus the starting material [Ment-Im]. This implies a reduction in electron density surrounding these atoms and a deshielding effect as a result of imidazole protonation. The most significant deshielding effect is observed at the H-14 position. Here, the signal is shifted to 1.10–2.41 ppm depending on the anion. However, protons H-13, H-12, H-11, and H-1 have a lower increase in chemical shift values—from 0.13 to 0.42 ppm. Changes in the chemical shift of another protons are minor.However, the extent of the deshielding effect for these protons is related to the counter ion that is ascribed the different electrostatic and steric interactions between the individual anions and the cationic head. The structure of the anion has only a minor effect on the changes in the chemical shift of H12, H13, H-11, and H-1 protons. For proton H-14, large anions with a greater steric hindrance such as OTf, PF6 and NTf2 have less deshielding effect than the chloride anion. In other words, H-14 is more shielded by large anions. This suggests that the steric effects are significant. This also demonstrates localization of the anion near the two nitrogen atoms of the imidazolium cation.
Notable differences are seen in the 13C NMR spectra of (1R,2S,5R)-(−)-menthoxymethylimidazole and protic salts (Table 3). Greater changes in the chemical shift are observed for the C-1 carbon located in menthyl fragment and slightly smaller for C-13 and C-11 carbons, connected with a quaternary nitrogen atom of the imidazolium ring. Carbons at these positions are more de-shielded in the imidazolium salts than in the starting menthoxymethylimidazole. The signal of the C-1 carbon is shifted from 73.2 in (1R,2S,5R)-(−)-menthoxymethylimidazole to about 80 ppm in 1-H-3-[(1R,2S,5R)-(−)-menthoxymethyl]imidazolium salts. As a result of protonation, there is an increase in the chemical shift values for C-13 and C-11 carbons. The changes are 1.45–2.25 ppm for C-13 and 0.1–0.4 ppm for C-11. The ordering of anions according to their increasing deshielding capacities for carbons C-1, C-11 and C-13 are as follows: Cl < OTf < PF6<NTf2. Signals of C-12 and C-14 carbons adjacent to the protonated nitrogen atom are shifted in the spectrum of 1-H-3-[(1R,2S,5R)-(−)-menthoxymethyl]imidazolium salts in the direction of the higher field. The signal of C-12 shifts from 129.2 ppm in (1R,2S,5R)-(−)-menthoxymethylimidazole to about 121 ppm in 1-H-3-[(1R,2S,5R)-(−)-menthoxymethyl]imidazolium salts. The chemical shift of the C-14 carbon decreased about 1–2.3 ppm. A greater shielding effect was seen for larger anions.
An inverse 2D heteronuclear correlation experiment was performed for (−)[H-Ment-Im][NTf2] as the representative ionic liquid. This shows imidazolium ring protonation data as a function of proton position. In the 1H(15N)HMBCAD spectrum (in ESI†), we see two signals of nitrogen atoms in the imidazolium cation (at −208.59 ppm and −189.88 ppm). Both signals give peak correlations with signals of H14, H13, and H12 protons of imidazolium ring. However, only the nitrogen atom at −208.59 ppm correlates with the H15 proton at 12.48 ppm. Moreover, the signal of H15 proton is a doublet because it couples with the adjacent nitrogen atom (N–H group).
The peak correlation at 5.59 ppm/−189.88 ppm corresponds to the H11 proton and nitrogen atom to which menthoxymethyl group is attached.
Enantiomeric purity of protic and aprotic chiral salts
Enantiomeric purity of the prepared chiral protic (Table 1) and aprotic (Fig. 3) salts was determined based on NMR experiments with Δ-TRISPHAT tetrabutylammonium salt. Many research articles reported that Lacour's Δ-TRISPHAT anion is an efficient NMR chiral shift reagent for chiral cations.30–33 Our first aim was to demonstrate that this reagent can be used for enantiomeric purity determination of synthesized chiral salts. Effect of Δ-TRISPHAT addition on chemical shift and expected split of signals of enantiomers was presented on the example of protic salt [H-Ment-Im][NTf2] (Fig. 4) and aprotic salt [C4-Ment-Im][NTf2] (Fig. 5). To observe the differentiation of shifts of proton signal from both enantiomers, racemic [H-Ment-Im][NTf2] and (+)[C4-Ment-Im][NTf2] were synthesized and used in these NMR experiments.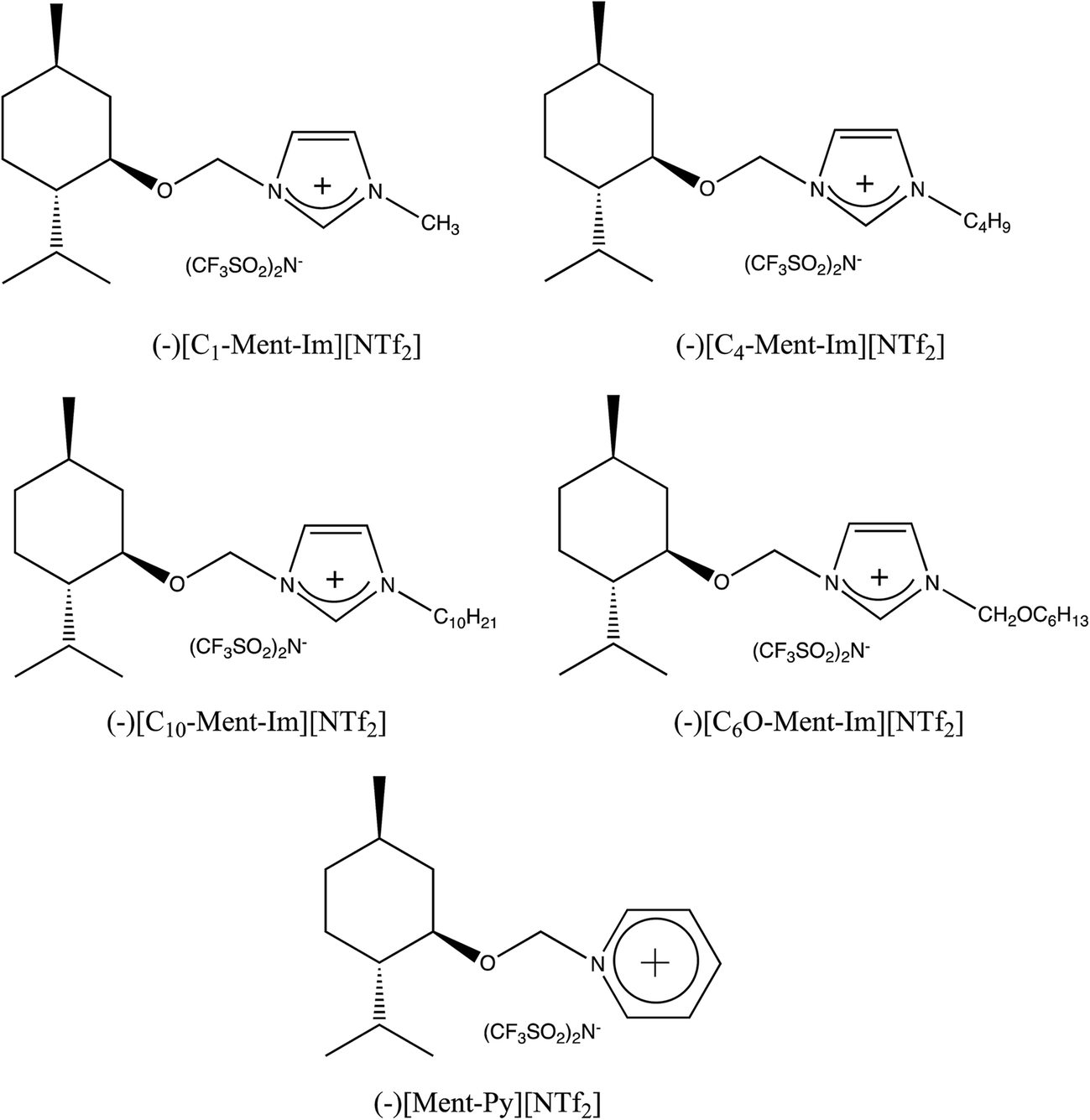 | ||
| Fig. 3 Structures and abbreviations of aprotic chiral ionic liquids. | ||
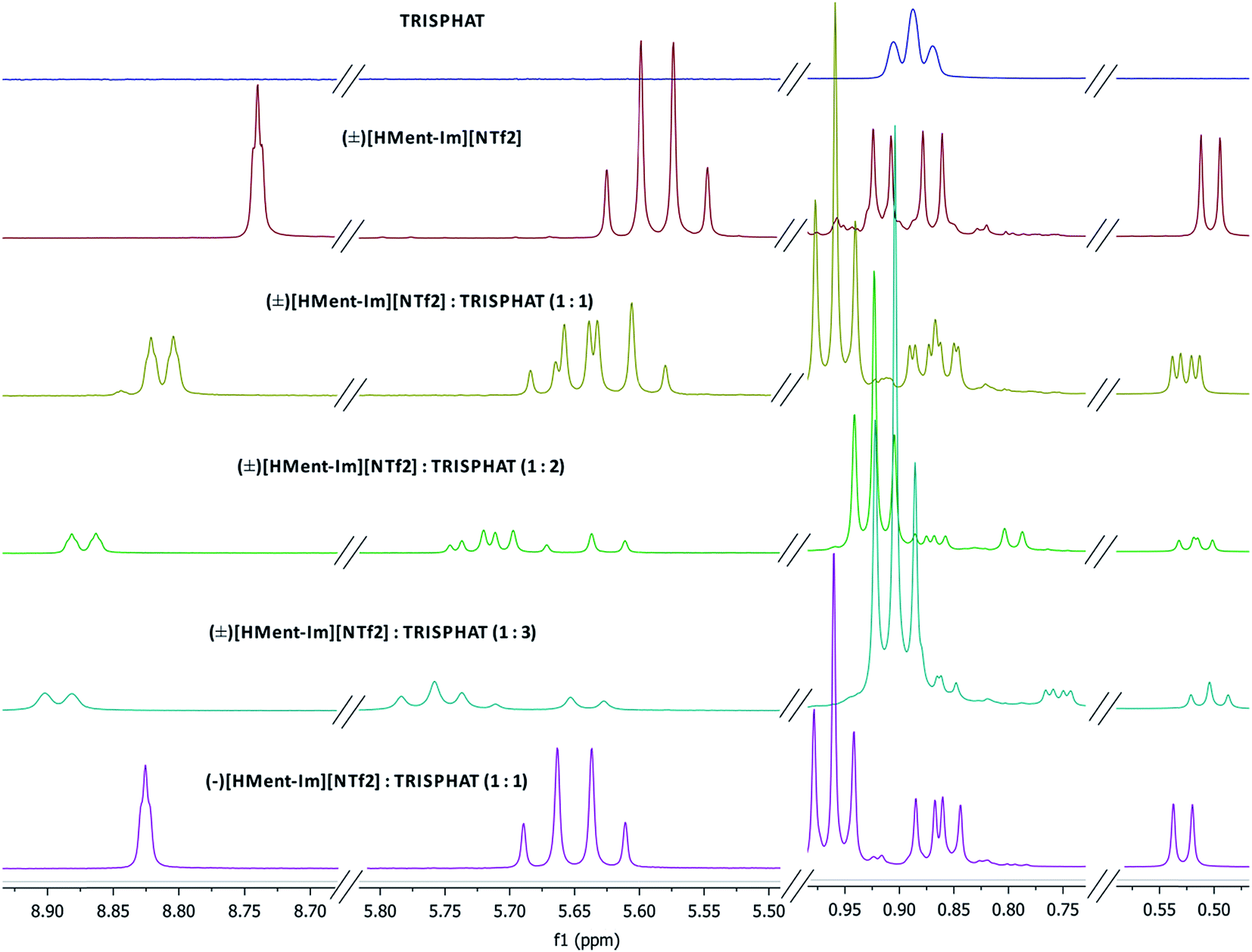 | ||
| Fig. 4 1H NMR spectra (400 MHz) of [n-Bu4N][Δ-TRISPHAT], (±)[H-Ment-Im][NTf2] with 1, 2 and 3 equivalents of [n-Bu4N][Δ-TRISPHAT], (−)[H-Ment-Im][NTf2] with 1 equivalent of [n-Bu4N][Δ-TRISPHAT]. | ||
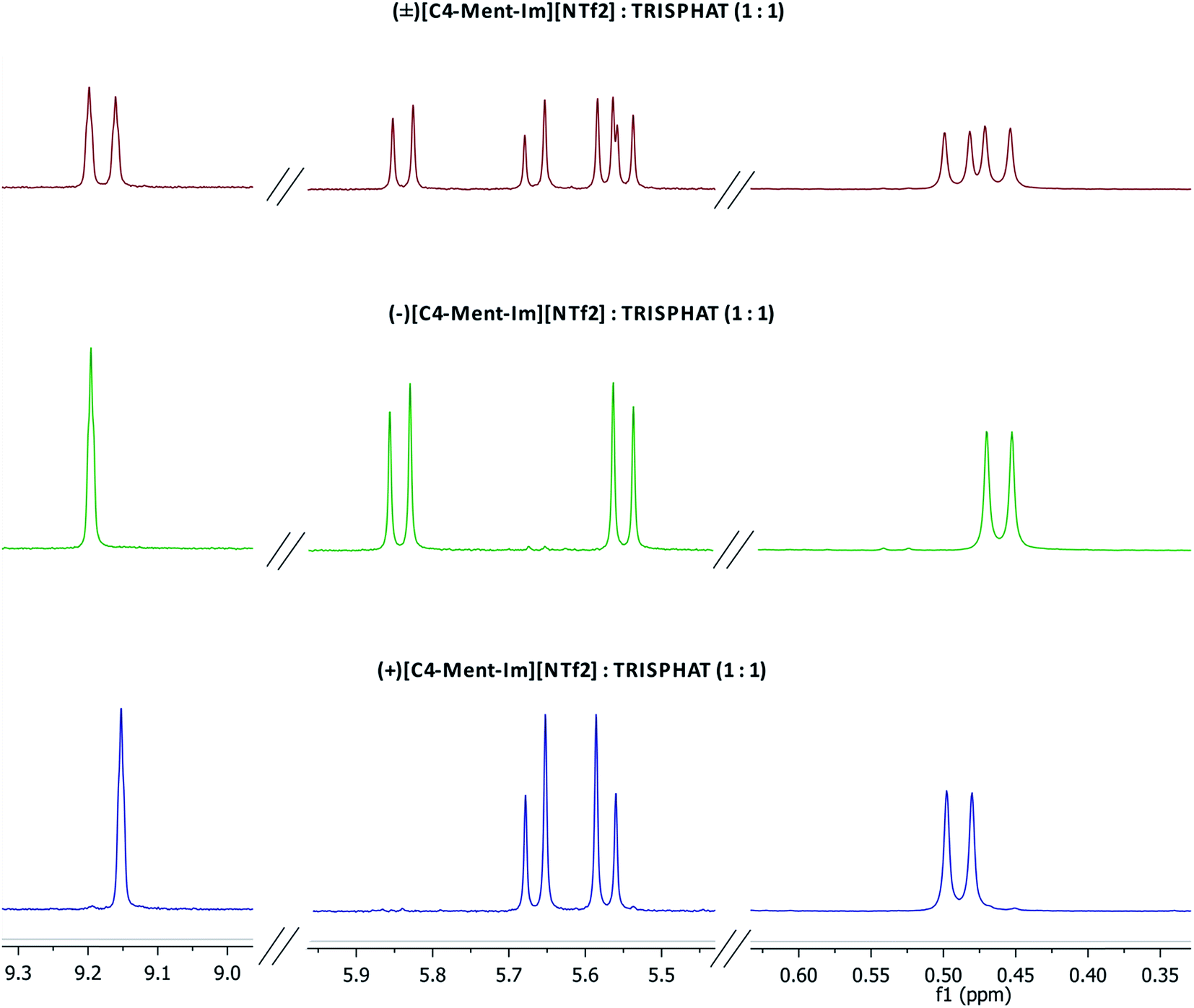 | ||
| Fig. 5 1H NMR spectra (400 MHz) of racemic mixture of (±)[C4-Ment-Im][NTf2] with 1 equivalent of [n-Bu4N][Δ-TRISPHAT] and each of the enantiomer with 1 equivalent of [n-Bu4N][Δ-TRISPHAT]. | ||
In Fig. 4 we can see, that already upon addition of 1 equivalent of TRISPHAT, signal of the H14 proton in [H-Ment-Im][NTf2] split and is shifted downfield from 8.740 ppm to 8.821 for (+)[H-Ment-Im][NTf2] and 8.804 ppm for (−)[H-Ment-Im][NTf2]. More equivalents of TRISPHAT is unfounded because showed broadening of this signal and lower differences in chemical shift for both enantiomers. Other signals of [H-Ment-Im][NTf2] were also shifted downfield and split, but differences in chemical shift were not sufficient and signals of both enantiomers were partly overlapped.
1H NMR spectra presented in Fig. 5 for both enantiomers of aprotic salt (+)[C4-Ment-Im][NTf2] and (−)[C4-Ment-Im][NTf2] and their racemic mixture with 1 equivalent of Δ-TRISPHAT showed also sufficient split of the signals of H14 proton with Δδ = 0.037 ppm. Signals of diastereotopic protons H11 and protons of methyl group of menthyl were split too and can be used to integrate the respective signals of each enantiomer and measure of enantiomeric purity based on proton integration.
Procedure with 1 equivalent of Δ-TRISPHAT were carried out for all salts derived from (−)-menthol and the registered 1H NMR spectra were included to the ESI.† It was determined that almost all chiral protic and aprotic salts were (−)-enantiomer of 100% ee, except of (−)[C10-Ment-Im][NTf2] purity, which was 83.5% ee.
Diels–Alder reaction in protic and aprotic chiral ionic liquids
Many articles have shown that the proton-donating properties of ionic liquids are crucial to increasing the stereoselectivity in Diels–Alder reactions. The hydrogen bond between the cation of the ionic liquid and the carbonyl oxygen of the dienophile influences the endo:exo ratio.34,35 The proton of C2 in the imidazolium ring and proton at nitrogen atom can be involved in hydrogen bond formation, but the later interacts more with the electron-withdrawing group of dienophiles.36 The role of the anion in these H-bonding interactions is also significant.37
Most recently, the “clamp” effect was shown as being crucial to explain some features of the Diels–Alder reaction in ionic liquids. This implies that the coordination of a dienophile by an ionic liquid cation is affected by the number and the nature of hydrogens available in the cation. Based on this theory, the dienophile is the most strongly “clamped” in protic imidazolium ionic liquids because the two acidic hydrogen atoms are directly bound to the imidazolium ring. This allows closer spatial approach between the cation and dienophile.38,39 Thus, the equilibrium geometries and electronic structures of the species in the presence of protic ionic liquid can be changed to affect reactivity and selectivity. Considering the above-discussed features of the imidazolium cation and previous explanations of its impact on the Diels–Alder reaction, we decided to study the ability to induce enantiomeric excess in the Diels–Alder reaction product by combining a proton at the nitrogen atom and the chiral substituent in the imidazolium ring. Until now, only aprotic chiral ionic liquids with chirality in an anion40 or cation8 were tested.
At first, we used the protic chiral ionic liquids in the Diels–Alder reaction between cyclopentadiene and methyl acrylate. For comparison, we also conducted this reaction in aprotic chiral ionic liquids with the same chiral (−)-menthoxymethyl substituent in the imidazolium or pyridinium cation and NTf2 anion. Their structures and abbreviations are presented in Fig. 3. Salts with imidazolium cation represent the biggest class of ILs with usually low melting point and viscosity.
The aprotic and protic chiral ionic liquids with NTf2 anion were liquids. They were used as a Diels–Alder reaction medium without addition of any other solvent. As was shown in Table 5 the reaction between cyclopentadiene and methyl acrylate had yields of 2-methoxycarbonyl-5-norbornene of 77–89% in chiral ionic liquids with NTf2 anion. Stereoselectivity endo/exo in the protic chiral ionic liquid (−)[H-Ment-Im][NTf2] was 4.9 and in aprotic chiral ILs between 3.7 and 4.1. However, no enantiomeric excess was achieved with both protic and aprotic chiral ILs. In addition, the effect of different anions of the chiral protic salts was compared in the reaction carried out in chloroform as a solvent, because two chiral protic salts were solids. There were slightly higher endo/exo ratios for the chiral protic salt containing the NTf2 anion (endo/exo 4.3) than in the ILs with another anions—3.7 and 4.1 with PF6− and TfO− respectively.
|
|
||
|---|---|---|
| Chiral protic salt | Yield (%) |
endo:exo ratio |
|
a Conditions of the reaction: 1:1 molar ratio of CIL:methyl acrylate; reaction temperature of 25 °C; endo/exo ratio was determined based on GC analysis.
b 0.5 mL of CHCl3, 0.2 mmol of CIL, 1 mmol of methyl acrylate, 2 mmol of cyclopentadiene. The yield and endo:exo ratio were determined based on 1H NMR analysis of reaction mixture and integration of protons of methyl ester group.
c 1-H-3-Methylimidazolium bis(trifluoromethylsulfonyl)imide.
|
||
| (−)[C1-Ment-Im][NTf2]a | 87 | 3.8 |
| (−)[C4-Ment-Im][NTf2]a | 88 | 3.7 |
| (−)[C10-Ment-Im][NTf2]a | 83 | 3.6 |
| (−)[C6O-Ment-Im][NTf2]a | 85 | 4.1 |
| (−)[Ment-Py][NTf2]a | 77 | 3.7 |
| (−)[H-Ment-Im][NTf2]a | 89 | 4.9 |
| (−)[H-Ment-Im][NTf2]b | 93 | 4.3 |
| (−)[H-Ment-Im][OTf]b | 97 | 4.1 |
| (−)[H-Ment-Im][PF6]b | 97 | 3.7 |
| [H-Me-Im][NTf2]a,c | 90 | 5.5 |
The higher stereoselectivity in the protic ionic liquid can be the effect of H-bonding between cation and carbonyl group of methyl acrylate. Hydrogen bond can stabilize the LUMO energy of the dienophile providing the endo product as the preferred isomer. The evidence for the H-bond interaction gave IR spectroscopy and comparison of carbonyl stretching band, νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O for pure methyl acrylate and its mixture with aprotic IL (−)[C4-Ment-Im][NTf2] (Fig. 6) and protic IL (−)[H-Ment-Im][NTf2] (Fig. 7). The νCO band for pure methyl acrylate is located at 1725.74 cm−1 which corresponds to non-H-bonded carbonyl. Adding protic IL to methyl acrylate we observed two peaks—one at 1721.82 cm−1 and second at 1712.84 cm−1 which provides an evidence on the H-bond presence. Furthermore, when aprotic ionic liquid was mixed with methyl acrylate, carbonyl stretching band was shifted only to 1724.38 cm−1 and can be rather attributed to non-H-bonded carbonyl group.
O for pure methyl acrylate and its mixture with aprotic IL (−)[C4-Ment-Im][NTf2] (Fig. 6) and protic IL (−)[H-Ment-Im][NTf2] (Fig. 7). The νCO band for pure methyl acrylate is located at 1725.74 cm−1 which corresponds to non-H-bonded carbonyl. Adding protic IL to methyl acrylate we observed two peaks—one at 1721.82 cm−1 and second at 1712.84 cm−1 which provides an evidence on the H-bond presence. Furthermore, when aprotic ionic liquid was mixed with methyl acrylate, carbonyl stretching band was shifted only to 1724.38 cm−1 and can be rather attributed to non-H-bonded carbonyl group.
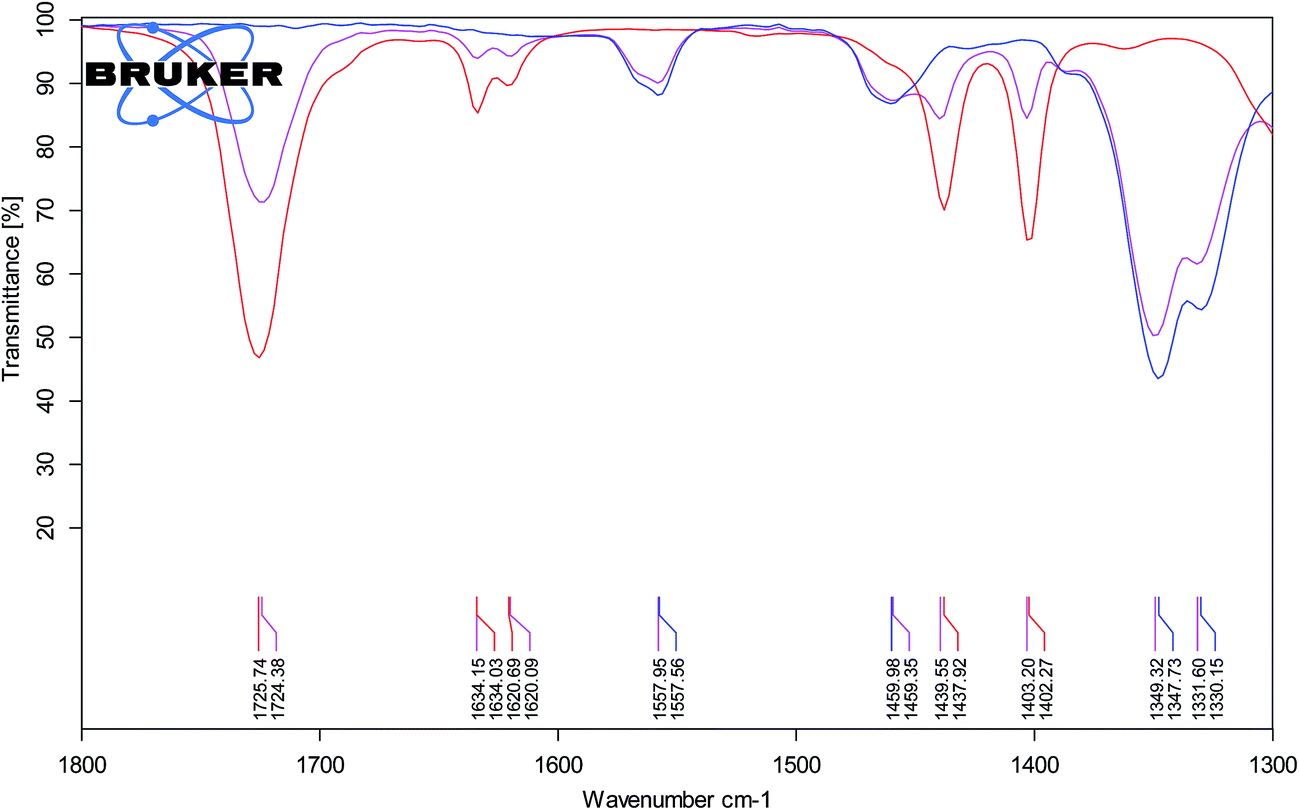 | ||
| Fig. 6 ATR-FTIR spectra of methyl acrylate (red line), (−)[C4-Ment-Im][NTf2] (blue line) and mixture of both (1:3, v/v) (pink line). | ||
 | ||
| Fig. 7 ATR-FTIR spectra of methyl acrylate (blue line), (−)[H-Ment-Im][NTf2] (green line) and mixture of both (1:3, v/v) (red line). | ||
Interestingly, it was found that the chiral protic IL allowed to achieve the same endo/exo stereoselectivity in the reaction between cyclopentadiene and methyl acrylate as previous reported by us in chiral pyrrolidinium ionic liquids with hydroxyethyl and (−)-menthol substituents in the cation.41 However, as was shown by a control experiment carried out in nonchiral aprotic bis(trifluoromethylsulfonyl)imide 1-H-3-methylimidazolium, (−)[H–Me-Im][NTf2], the (−)-menthol substituent in the protic imidazolium cation gave a little lower endo/exo ratio than methyl substituent.
Our results in aprotic chiral salts, were similar to the earlier reported for nonchiral aprotic imidazolium or pyridinium ionic liquids with NTf2 anion, which were in the range from 4.1 to 4.3, depending on the alkyl substituent in the cation.38
There is one report on the use the aprotic chiral ionic liquids, containing terpene group in the imidazolium cation and NTf2 anion.42 Authors obtained endo/exo stereoselectivity between 4.8 and 4.3 in the reaction of cyclopentadiene with acrylic acid, under similar conditions.
The second experiment was the reaction of cyclopentadiene with ethyl-vinyl ketone. The effect of the protic ionic liquid, (−)[H-Ment-Im][NTf2], on the increase in product rate formation is significant (Fig. 8) for this reaction. The reaction time needed to obtain a high yield (above 90%) was achieved in a two-fold shorter period of time in the protic ionic liquid than in the aprotic one. The differences in the reaction rate between imidazolium (−)[C1-Ment-Im][NTf2] and pyridinium cation (−)[Ment-Py][NTf2] was minimal. In addition, the endo/exo ratio was higher in the protic ionic liquid with a value of 9.5 at 25 °C. This endo/exo ratio was also higher than obtained in nonchiral protic ionic liquid (−)[H–Me-Im][NTf2] (endo/exo 8.4) (Table 6). Not able to facilitate enantiomeric excess we tried to conduct the reaction at lower temperature (Table 6). There are numerous data on enhanced selectivity of Diels–Alder reaction with lowering of reaction temperature.40,42 Our experiments showed that the endo/exo ratio almost doubled versus values at room temperature; enantioselectivity has not yet been achieved. The results suggest that the steric hindrance is too small to force the formation—albeit in slight excess—of one of the enantiomers of diastereomeric products.
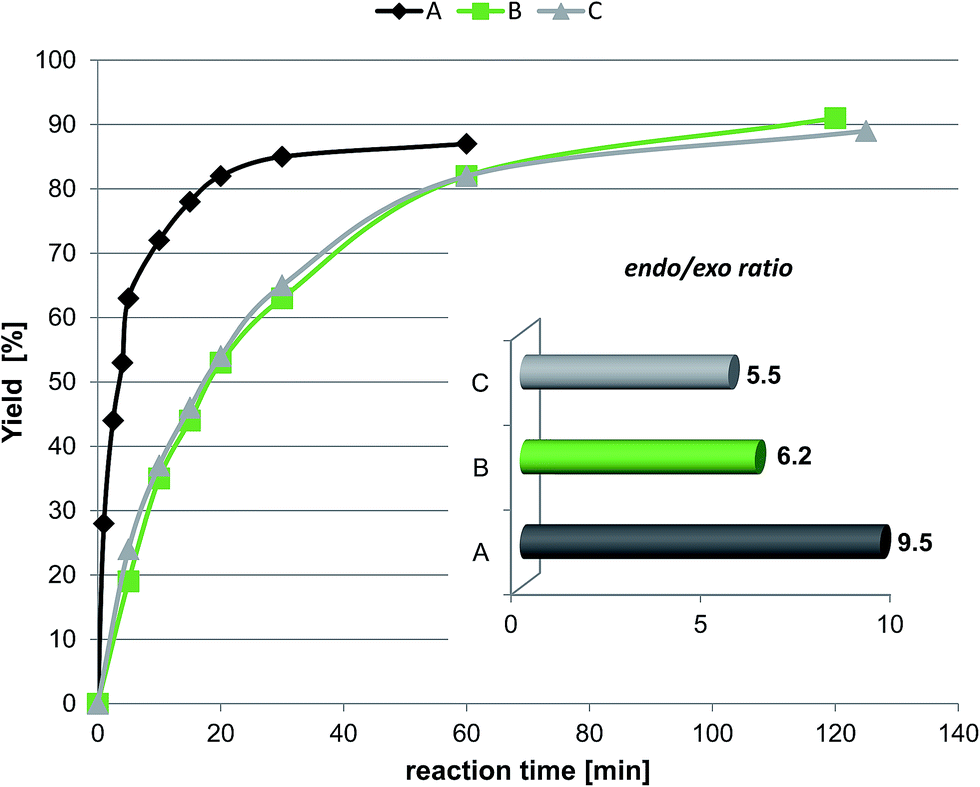 | ||
| Fig. 8 The effect of type of chiral protic imidazolium salt on the rate of product formation and stereoselectivity in the reaction of cyclopentadiene with methyl-vinyl ketone; A: (−)[H-Ment-Im][NTf2]; B: (−)[C1-Ment-Im][NTf2]; C: (−)[Ment-Py][NTf2]. Conditions of the reaction: 1.1 mmol of chiral ionic liquid; 1 mmol of methyl-vinyl ketone; reaction temperature of 25 °C; the endo/exo ratio was determined based on GC analysis. | ||
|
|
|||
|---|---|---|---|
| Entry | Chiral ionic liquid | Reaction temperature/time |
endo:exo ratioa |
| a Determined by GC. b The stereoselectivities are given in parentheses for the recycling experiments. | |||
| 1 | (−)[H-Ment-Im][NTf2] | 25 °C/1 h | 9.5 |
| 2 | (−)[H-Ment-Im][NTf2] | −35 °C/24 h | 18.7 (18.5; 19.1; 19.0; 18.9)b |
| 3 | (−)[C1-Ment-Im][NTf2] | 25 °C/2 h | 6.2 |
| 4 | (−)[C1-Ment-Im][NTf2] | −35 °C/24 h | 9.7 |
| 5 | (−)[Ment-Py][NTf2] | 25 °C/2 h | 5.5 |
| 6 | (−)[Ment-Py][NTf2] | −35 °C/24 h | 8.9 |
| 7 | [H-Me-Im][NTf2] | 25 °C/1 h | 8.4 |
The results definitely favor ionic liquids as solvents. They offer reusability, easier product separation, simple immobilization, and catalyst recycling. Therefore, we have undertaken experiments to recycle (−)[H-Ment-Im][NTf2]. The products were separated from the reaction mixture via hexane extraction. Stereoselectivity reached a similarly high value even on the 5th synthesis in series (Table 6, entry 2).
Conclusion
The preparation of new protic salts with a chiral (1R,2S,5R)-(−)-menthoxymethyl group attached to one nitrogen atom of the imidazolium cation was efficiently performed from imidazole and (1R,2S,5R)-(−)-menthol as the main starting materials. The yield was higher than 90% in each of the four synthesis stages. We studied the general properties such as specific rotation, thermal properties, and solubility in solvents for the chiral protic salts.Three of these salts (comprising of Cl−, CF3SO3−, or PF6− anions) were crystalline solids and salts with a (CF3SO2)2N− anion as a liquid at room temperature. The melting points for the solid salts (determined based on c-DTA signal from thermogravimetric analysis) were above 100 °C. The modulated DSC measurements indicated that the liquid salt, (−)[H-Ment-Im][NTf2], tends to supercool and transform into a glassy state; however, it could not be crystallized or melted. The very slow diffusion of ions can contribute to a lack of crystal formation. Thermal stability of the protic salts can be ordered in relation to the anion with an increasing trend as follows: Cl < OTf < PF6<NTf2. Comparison of NMR data indicated, the effect of the anion on the chemical shift of the protons and carbons of 1-H-3-[(1R,2S,5R)-(−)-menthoxymethyl]imidazolium cation. The strong effect of the anion was seen for the proton and carbon located between the two nitrogen atoms of the imidazolium cation. These are more shielded by the large anions and suggesting localization of the anion near two nitrogen atoms of the imidazolium cation.
Additionally, the presence of a proton on the nitrogen atom was confirmed by 1H(15N)HMBCAD.
Comparative studies on the use of chiral protic and aprotic salts with the same chiral substituent in the cation during Diels–Alder reactions indicates that the H-bonding catalysis results in a distinctly higher endo/exo ratios for protic rather than aprotic salts. The stereoselectivity reached the same high level even after the fourth round of recycling for (−)[H-Ment-Im][NTf2] in the reaction of ethyl-vinyl ketone with cyclopentadiene at −35 °C. However, no enantiomeric excess was obtained for either group.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financed by the National Science Centre of Poland (grant no. UMO-2012/05/B/ST5/01689).Notes and references
-
Ionic liquids in synthesis, ed. P. Wasserscheid and T. Welton, Wiley-VCH Verlag GmbH&Co. KGaA, Weinhem, Germany, 2nd edn, 2008, ISBN: 978-3-527-31239-9 Search PubMed
.
- K. Bica and P. Gaertner, Applications of Chiral Ionic Liquids, Eur. J. Org. Chem., 2008, 3235–3250, DOI:10.1002/ejoc.200701107
- T. Payagala and D. W. Armstrong, Chiral Ionic Liquids: A Compendium of Syntheses and Applications (2005–2012), Chirality, 2012, 24, 17–53, DOI:10.1002/chir.21975
- M. L. Patil and H. Sasai, Recent Developments on Chiral Ionic Liquids: Design, Synthesis and Applications, Chem. Rev., 2008, 8, 98–108, DOI:10.1002/tcr.20143
- F. Santamarta, M. Vilas, E. Tojo and Y. Fall, Synthesis and properties of novel chiral imidazolium-based ionic liquids derived from carvone, RSC Adv., 2016, 6, 31177–31180, 10.1039/c6ra00654j
- R. A. F. Matos and C. K. Z. Andrade, Synthesis of new chiral ionic liquids based on (−)-menthol and (−)-borneol, Tetrahedron Lett., 2008, 49, 1652–1655, DOI:10.1016/j.tetlet.2008.01.011
- M. Vasiloiu, S. Leder, P. Gaertner and K. Bica, Coordinating chiral ionic liquids, Org. Biomol. Chem., 2013, 11, 8092–8102, 10.1039/c3ob41635f
- K. Bica, G. Gmeiner, C. Reichel, B. Lendl and P. Gaertner, Microwave-assisted synthesis of camphor-derived chiral imidazolium ionic liquids and their application in diastereoselective Diels-Alder reaction, Synthesis, 2007, 1333–1338, DOI:10.1055/s-2007-966018
- A. Brown, V. Hogan, J. Perry and R. Manchanayakage, Chiral pool based synthesis of pyrrolidinium ionic liquids, Tetrahedron Lett., 2017, 58, 1061–1065, DOI:10.1016/j.tetlet.2017.01.102
- S. L. De Rooy, M. Li, D. K. Bwambok, B. El-Zahab, S. Challa and I. M. Warner, Ephedrinium-Based Protic Chiral Ionic Liquids for Enantiomeric Recognition, Chirality, 2011, 23, 54–62, DOI:10.1002/chir.20920.
- S. Cherukuvada and A. Nangia, Salts and ionic liquid of the antituberculosis drug S,S-Ethambutol, Cryst. Growth Des., 2013, 13, 1752–1760, DOI:10.1021/cg400071d
- M. B. Foreiter, H. Q. N. Gunaratne, P. Nockemann, K. R. Seddon and G. Srinivasan, Novel chiral ionic liquids: physicochemical properties and investigation of the internal rotameric behavior in the neat system, Phys. Chem. Chem. Phys., 2014, 16, 1208–1226, 10.1039/c3cp53472c
- O. J. Curnow, M. T. Holmes, L. C. Ratten, K. J. Walst and R. Yunis, A facile route to functionalized, protic and chiral ionic liquids based on the triaminocyclopropenium cation, RSC Adv., 2012, 2, 10794–10797, 10.1039/c2ra22078d
- D. K. Bwambok, S. K. Challa, M. Lowry and I. M. Warner, Amino acid-based fluorescent chiral ionic liquid for enantiomeric recognition, Anal. Chem., 2010, 82, 5028–5037, DOI:10.1021/ac9027774
- L. Zhang, L. He, C. B. Hong, S. Qin and G. H. Tao, Brønsted acidity of bio-protic ionic liquids: the acidic scale of [AA]X amino acid ionic liquids, Green Chem., 2015, 17, 5154–5163, 10.1039/c5gc01913c
- D. Paschek, B. Golub and R. Ludwig, Hydrogen bonding in a mixture of protic ionic liquids: a molecular dynamics simulation study, Phys. Chem. Chem. Phys., 2015, 17, 8431–8440, 10.1039/c4cp05432f
- D. Shang, X. Zhang, S. Zeng, K. Jiang, H. Gao, H. Dong, Q. Yang and S. Zhang, Protic ionic liquid [Bim][NTf2] with strong hydrogen bond donating ability for highly efficient ammonia absorption, Green Chem., 2017, 19, 937–945, 10.1039/c6gc03026b
- K. Matuszek, A. Chrobok, F. Coleman, K. R. Seddon and M. Swadźba-Kwaśny, Tailoring ionic liquid catalysts: structure, acidity and catalytic activity of protonic ionic liquids based on anionic clusters,
[(HSO4)(HSO4)x]− (x=0, 1, or 2), Green Chem., 2014, 16, 3463–3471, 10.1039/c4gc00415a
- S. Majumdar, J. De, A. Chakraborty and D. K. Maiti, General solvent-free highly selective N-tert-butyloxycarbonylation strategy using protic ionic liquid as an efficient catalyst, RSC Adv., 2014, 4(47), 24544–24550, 10.1039/c4ra02670e
- T. L. Greaves and C. J. Drummond, Protic ionic liquids, Chem. Rev., 2008, 108(1), 206–237, DOI:10.1021/cr068040u
- T. L. Greaves and C. J. Drummond, Protic ionic liquids: evolving structure-property relationships and expanding applications, Chem. Rev., 2015, 115(20), 11379–11448, DOI:10.1021/acs.chemrev.5b00158
- S. Menne, T. Vogl and A. Balducci, The synthesis and electrochemical characterization of bis(trifluorosulfonyl)imide-based protic ionic liquids,, Chem. Commun., 2015, 51, 3656, 10.1039/c4cc09665g
- L. Timperman, P. Skowron, A. Boisset, H. Galiano, D. Lemordant, E. Frąckowiak, F. Beguin and M. Anouti, Triethylammonium bis(trifluoromethylsulfonyl)amide protic ionic liquid as an electrolyte for electrical double-layer capacitors, Phys. Chem. Chem. Phys., 2012, 14, 8199–8207, 10.1039/c2cp40315c
- M. P. Collins, L. Zhou, S. E. Camp and N. D. Danielson, Isopropylammonium Formate as a Mobile Phase Modifier for Liquid Chromatography, J. Chromatogr. Sci., 2012, 50, 869–876, DOI:10.1093/chromsci/bms084
- E. C. Achinivu, R. M. Howard, G. Li, H. Gracz and W. A. Henderson, Lignin Extraction from Biomass with Protic Ionic Liquids, Green Chem., 2014, 16, 1114–1119, 10.1039/c3gc42306a
- M. J. Hernaiz, A. R. Alcantara, J. I. Garcia and J. V. Sinisterra, Applied Biotransformations in Green Solvents, Chem.–Eur. J., 2010, 16, 9422–9437, DOI:10.1002/chem.201000798
- J. A. McCune, P. He, M. Petkovic, F. Coleman, J. Estager, J. D. Holbrey, K. R. Seddon and M. Swadźba-Kwaśny, Brønsted acids in ionic liquids: how acidity depends on the liquid structure, Phys. Chem. Chem. Phys., 2014, 16, 23233–23243, 10.1039/c4cp03217a
- L. Timperman, P. Skowron, A. Boisset, H. Galiano, D. Lemordant, E. Frackowiak, F. Beguin and M. Anouti, Triethylammonium Bis(Tetrafluoromethylsulfonyl)Amide Protic Ionic Liquid as an Electrolyte for Electrical Double-Layer Capacitors, Phys. Chem. Chem. Phys., 2012, 14, 8199–8207, 10.1039/c2cp40315c
- Z. Ullah, M. A. Bustam, Z. Man, N. Muhammad and A. S. Khan, Synthesis, characterization and the effect of temperature on different physicochemical properties of protic ionic liquids, RSC Adv., 2015, 5, 71449–71461, 10.1039/c5ra07656k
- J. Lacour, J. J. Jodry, C. Ginglinger and S. Torche-Haldimann, Diastereoselective Ion Pairing of TRISPHAT Anions and Tris(4,49-dimethyl-2,29-bipyridine)iron(II), Angew. Chem., Int. Ed., 1998, 37(17), 2379–2380, DOI:10.1002/(SICI)1521-3773(19980918)37:17<2379::AID-ANIE2379>3.0.CO;2-C
- J. J. Jodry and J. Lacour, Efficient Resolution of a Dinuclear Triple Helicate by Asymmetric Extraction/Precipitation with TRISPHAT Anions as Resolving Agents, Chem.–Eur. J., 2000, 6(23), 4297–4304, DOI:10.1002/1521-3765(20001201)6:23<4297::aid-chem4297>3.0.co;2-#
- J. Lacour, C. Goujon-Ginglinger, S. Torche-Haldimann and J. J. Jodry, Efficient Enantioselective Extraction of Tris(diimine)ruthenium(II) Complexes by Chiral, Lipophilic TRISPHAT Anions, Angew. Chem., Int. Ed., 2000, 39(20), 3695–3697, DOI:10.1002/1521-3773(20001016)39:20<3695::AID-ANIE3695>3.0.CO;2-M
- J. J. Jodry and K. Mikami, New chiral imidazolium ionic liquids: 3D-network of hydrogen bonding, Tetrahedron Lett., 2004, 45, 4429–4431, DOI:10.1016/j.tetlet.2004.04.063
- T. Fisher, A. Sethi, T. Welton and J. Woolf, Diels-Alder reactions in room-temperature ionic liquids, Tetrahedron
Lett., 1999, 40, 793–796 CrossRef
- A. Aggarwal, N. L. Lancaster, A. R. Sethi and T. Welton, The role of hydrogen bonding in controlling the selectivity of Diels–Alder reactions in room-temperature ionic liquids, Green Chem., 2002, 4, 517–520, 10.1039/b206472c
- E. Janus, I. Goc-Maciejewska, M. Łożyński and J. Pernak, Diels-Alder reaction in protic ionic liquids, Tetrahedron Lett., 2006, 47, 4079–4083, DOI:10.1016/j.tetlet.2006.03.172
- K. Nobuoka, S. Kitaoka, M. Iio, T. Harran and Y. Ishikawa, Solute-solvent interactions in imidazole camphorsulfonate ionic liquids, Phys. Chem. Chem. Phys., 2007, 9, 5891–5896, 10.1039/b709407h
- R. Bini, C. Chiappe, V. L. Mestre, C. P. Pomelli and T. Welton, A theoretical study of the solvent effect on Diels-Alder reaction in room temperature ionic liquids using a supermolecular approach, Theor. Chem. Acc., 2009, 123, 347–352, DOI:10.1007/s00214-009-0525-0
- C. Chiappe, M. Malvaldi and C. S. Pomelli, The solvent effect on the Diels-Alder reaction in ionic liquids: multiparameter linear solvation energy relationships and theoretical analysis, Green Chem., 2010, 12, 1330–1339, 10.1039/c0gc00074d
- K. Nobuoka, S. Kitaoka, K. Kunimitsu, M. Iio, T. Harran, A. Wakisaka and Y. Ishikawa, Camphor ionic liquid: correlation between stereoselectivity and cation-anion interaction, J. Org. Chem., 2005, 70, 10106–10108, DOI:10.1021/jo051669x
- E. Janus and M. Gano, Chiral pyrrolidinium salts derived from menthol as precursor – synthesis and properties, Pol. J. Chem. Technol., 2017, 19(3), 92–98, DOI:10.1515/pjct-2017-0054
- K. Bica, G. Gmeiner, C. Reichel, B. Lendl and P. Gaertner, Microwave-assisted synthesis of camphor-derived chiral imidazolium ionic liquids and their application in diastereoselective Diels-Alder reaction, Synthesis, 2007, 9, 1333–1338, DOI:10.1055/s-2007-966018
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra12176h |
| This journal is © The Royal Society of Chemistry 2018 |