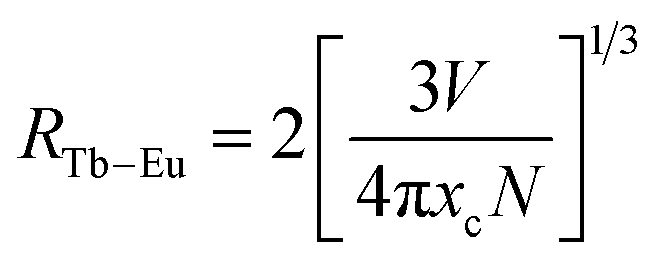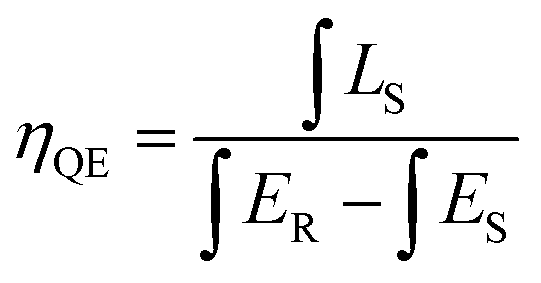DOI:
10.1039/C7RA12260H
(Paper)
RSC Adv., 2018,
8, 3530-3535
Tunable emission from green to red in the GdSr2AlO5:Tb3+,Eu3+ phosphor via efficient energy transfer
Received
9th November 2017
, Accepted 26th December 2017
First published on 17th January 2018
Abstract
Herein, a series of GdSr2AlO5:Tb3+,Eu3+ phosphors were successfully synthesized through a high temperature solid-state reaction, and their crystal structures as well as photoluminescence properties were investigated in detail. Compared to the intense emission of 5D0 → 7F1 or 5D0 → 7F2 transition of Eu3+, another strong emission corresponding to 5D0 → 7F4 was observed. Concentration quenching is not obvious in Tb3+ or Eu3+-doped GdSr2AlO5 because structure isolation and energy transfer (ET) of Gd3+ → Eu3+ and Gd3+ → Tb3+ were found. Moreover, the energy transfer process from Tb3+ to Eu3+ was verified by the overlap of luminescence spectra and the variation of lifetime. Energy transfer mechanism was determined to be a dipole–dipole interaction, and ET efficiency as well as quantum efficiency were also obtained. Moreover, the emission color of GdSr2AlO5:Tb3+,Eu3+ can be tuned from green to red by altering the ratio of Tb3+/Eu3+. These results indicate that the GdSr2AlO5:Tb3+,Eu3+ phosphor is a promising single-component white light-emitting phosphor.
1. Introduction
White light-emitting diodes (w-LEDs) with superior advantages, such as high energy efficiency, good physical and chemical stability, long lifetime, and environment friendliness, have been drawing attention and are regarded as the next generation of illumination sources.1–4 The most available material for light devices involves Ce3+-doped yttrium aluminum garnet (YAG:Ce3+) with blue-LED chips, which has been commercialized.5,6 Because of the scarcity of red component, it encounters a low color rendering index (Ra < 80) and a high color temperature (CCT > 7000 K).7,8 To overcome these drawbacks, the combination of a UV LED chip with red (Ca4(PO4)O:Eu2+),9 green (Na2Y2B2O7:Ce3+,Tb3+),10 and blue (NaxCa1−xAl2−xSi2+xO8:Eu2+)11 to achieve white-light with high Ra and appropriate CCT is another approach. However, the mixture of different phosphors brings about an inevitable problem of fluorescence re-absorption, resulting in the loss of luminous efficiency.12,13 Therefore, it is obligatory to develop a single-phase white light-emitting phosphor.
Rare earth ion-doped aluminate phosphors, such as LaSr2AlO5:Ce3+/Eu2+,14,15 (La,Gd)Sr2AlO5:Ce,16 and (La,Gd)Sr2(Al,B)O5:Ce,17 have been studied extensively due to their cheap raw materials and good chemical and physical stability. The structural properties of this aluminate-based phosphor and the effect on the luminescence properties have been investigated. However, the luminescence of Ce3+ and Eu2+ belong to the f–d transition.18 Upon reviewing the literature, it is observed that it is difficult to find other rare-earth ions with f–f transition for doping into the GdSr2AlO5 phosphor. As is well known, co-doping of different rare ions as a sensitizer and activator plays a significant role in obtaining emission via energy transfer processes.19
Tb3+ and Eu3+ with characteristic green20,21 and red22,23 emissions are widely used in luminescent materials, respectively. Compared to most Eu3+-doped phosphors with the dominant emission corresponding to 5D0 → 7F1 or 5D0 → 7F2, the Eu3+-doped GdSr2AlO5 phosphor shows an intensive emission related to 5D0 → 7F4. It is clear that Tb3+ functions as a good sensitizer to improve the luminescence efficiency of Eu3+ ions in Sr2MgSi2O7,24 Na3La(PO4)2,25 CaYAlO4,26 and Y2O3 (ref. 27) phosphors. In this study, a single-phased GdSr2AlO5:Tb3+,Eu3+ phosphor was synthesized by a conventional solid-state method. The efficient energy transfer from Tb3+ to Eu3+ was systematically investigated with steady state fluorescence, lifetime measurement, and the energy transfer efficiency. Moreover, the color of the total emission can be tuned by altering the ratio of Tb3+/Eu3+ in the GdSr2AlO5 host.
2. Experimental method
A series of phosphors with the composition of GdSr2AlO5:Tb3+, GdSr2AlO5:Eu3+, and GdSr2AlO5:Tb3+,Eu3+ were prepared by a conventional high temperature solid-state method under a reductive atmosphere (5% H2 + 95% N2) at 1500 °C. SrCO3 (A. R.), Al2O3 (A. R.), Gd2O3 (A. R.), Tb4O7 (99.99%), and Eu2O3 (99.99%) were used as raw materials. They were stoichiometrically weighed and mixed by grinding in an agate mortar for 30 min. After this, they were transferred into ceramic crucibles and calcined in a high-temperature tubular furnace at 1500 °C for 4 h at a heating rate of 3 °C min−1. Finally, the samples were cooled down to room temperature in the furnace and ground into powders for subsequent use.
Bruker D8 (voltage 40 kV and current 40 mA) over the 2θ range from 10° to 70° with CuKα radiation (λ = 1.54178 Å) was used to obtain the XRD patterns of GdSr2AlO5:Tb3+,Eu3+ samples. Hitachi F-7000 spectrophotometers with a 150 W xenon lamp light source were used to obtain the excitation and emission spectra. The morphology of the prepared phosphor was examined by a field-emission scanning electron microscope (Hitachi S-4800). The decay curves were obtained using the Hamamatsu Quantaurus-Tau C11367 fluorescence spectrophotometer. The chromaticity data were calculated by the CIE1931 software. Photoluminescence absolute quantum efficiency (QE) was determined via Hamamatsu Quantaurus-QY C11347 using an integrated sphere. All the measurements were conducted at room temperature.
3. Results and discussion
3.1 XRD analysis
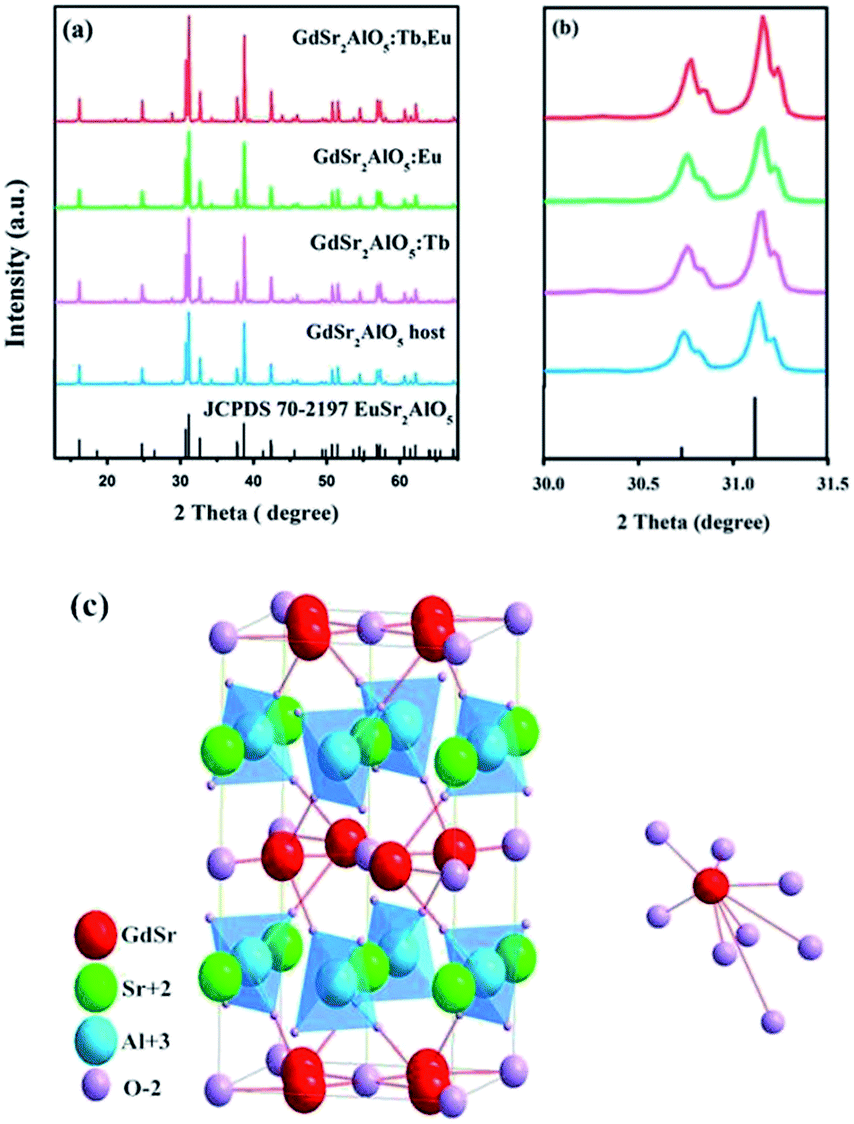
Fig. 1(a) shows the XRD patterns of the as-prepared GdSr2AlO5 host and Tb3+, Eu3+-doped phosphor as well as the standard patterns of EuSr2AlO5 (JCPDS 70-2197). The patterns agreed with the crystallographic structure of the JCPDS card 70-2197, and impurity peaks were not found. This result illustrates that the obtained samples are in single phase, and doping of Tb3+ and/or Eu3+ ions at low concentrations does not change the crystal structure. In addition, it can be observed from Fig. 1(b) that the diffraction peak positions of the samples are shifted slightly to a higher angle as compared to those of the standard patterns of EuSr2AlO5 (JCPDS 70-2197); this can be due to the replacement of large-size Eu3+ sites by the smaller-size trivalent ions. Hence, RE3+ ions were effectively doped into the lattice; this could be confirmed by the slight change in the diffraction peaks.
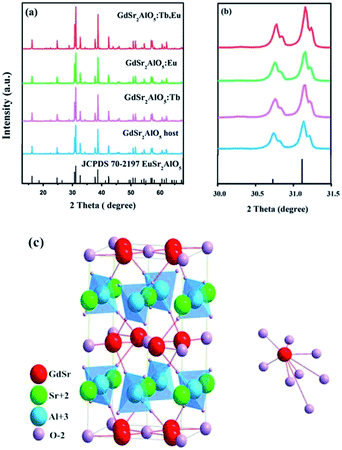 |
| | Fig. 1 (a) Representative XRD patterns of the samples. (b) Magnified XRD patterns between 30° and 31.5°. (c) Crystal structure of the GdSr2AlO5 and the coordination environment of GdO8. | |
Fig. 1(c) displays a partial structure of GdSr2AlO5. As reported previously, the crystal structure of GdSr2AlO5 is isostructural with the standard file of EuSr2AlO5 (JCPDS no. 70-2197, tetragonal, space group I4/mcm).28 Gd atoms will occupy the half 8h sites, and the other half is occupied by the Sr atoms. The cell also contains 4a sites, which are fully occupied by the Sr atoms, and the Al atoms will occupy the 4b sites. The 4c sites and 16l sites are occupied by O, denoted as O1 and O2, respectively. The GdO8 polyhedron consists of two O1 atoms and six O2 atoms, where the latter are shared by four AlO4 tetrahedral on the adjacent layers, forming an isolated structure. Fortunately, isolation in space weakens the interaction between the paired ions. To some extent, the concentrating quenching effect would be avoided.29
3.2 SEM with energy dispersive X-ray analysis
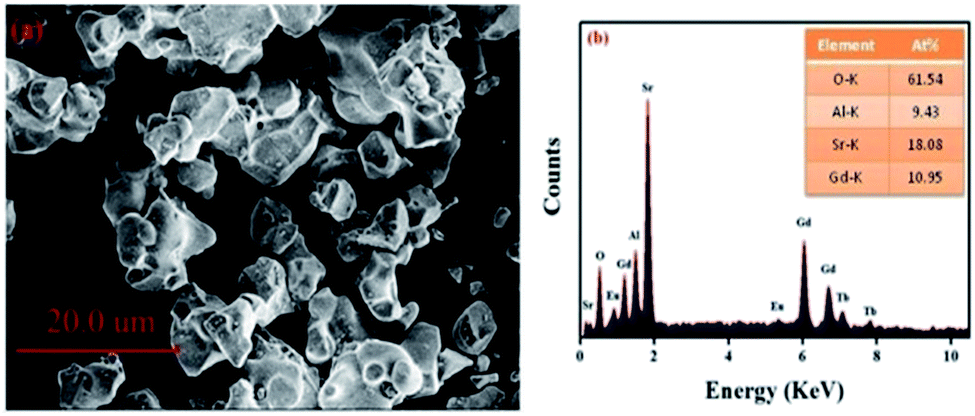
Fig. 2(a) displays the morphology of the GdSr2AlO5:Tb3+,Eu3+ sample. The surface image shows irregular particles with a size range of 5–10 μm. Moreover, as shown in Fig. 2(b), EDS is used to analyze the chemical composition of the GdSr2AlO5:Tb3+,Eu3+ phosphor, and the result confirms the presence of Gd, Sr, Al, O, Tb, and Eu elements in the phosphor. Compared with the abovementioned XRD results, the EDS results demonstrate that Tb/Eu ions have been successfully incorporated into the GdSr2AlO5 lattice.
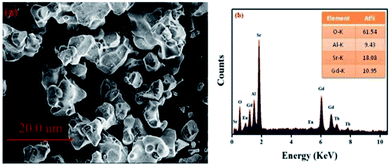 |
| | Fig. 2 SEM image (a) and EDS spectrum (b) of GdSr2AlO5:2%Tb3+,2%Eu3+. | |
3.3 Luminescence properties
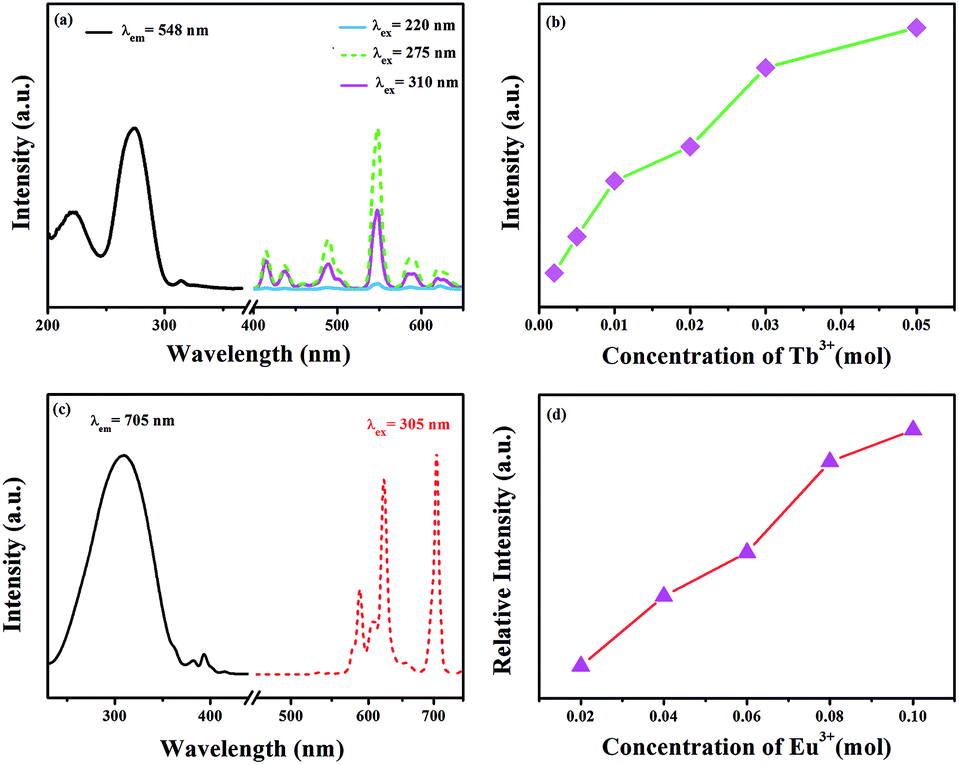
The excitation spectrum of GdSr2AlO5:2%Tb3+ is shown in Fig. 3(a). The excitation spectrum monitored at 548 nm exhibits a broad band and several weak peaks. The broad band from 200 nm to 250 nm centered at 220 nm is ascribed to the f–d transitions of the Tb3+ ions, whereas the excitation peaks at 275 and 310 nm originate from the transitions of 8S7/2 → 6I7/2 and 8S7/2 → 6P7/2 of the Gd3+ ions, respectively.30,31 The appearance of strong excitation sharp bands of Gd3+ ions confirms that there is an efficient energy transfer from Gd3+ to Tb3+. The emission spectra of the phosphor under 220, 275, and 310 nm excitation in the wavelength range from 400–650 nm are also depicted in Fig. 3(a). There are two groups of lines in the emission spectrum: one group in the region from 400 to 480 nm is derived from the 5D3 → 7FJ (J = 5, 4, and 3) transitions of Tb3+. The second groups of lines in the wavelength range from 480 to 650 nm originate from the 5D4 → 7FJ (J = 6, 5, 4, and 3) transitions. Among these emissions, the 5D4 → 7F5 transition with a maximum at about 548 nm exhibits the strongest emission intensity. The emission intensity increases with the increasing concentration of Tb3+, as presented in Fig. 3(b). Concentration quenching does not occur in GdSr2AlO5:Tb3+.
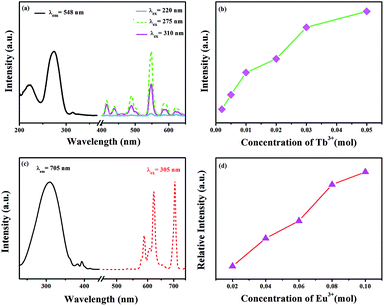 |
| | Fig. 3 (a) Excitation and emission spectra of the GdSr2AlO5:2%Tb3+ phosphor. (b) Concentration-dependent luminescence intensity of GdSr2AlO5:xTb3+. (c) Excitation and emission spectra of the GdSr2AlO5:2%Eu3+ phosphor and (d) concentration-dependent luminescence intensity of GdSr2AlO5:yEu3+. | |
Fig. 3(c) and (d) present the excitation and emission spectra as well as concentration-dependent luminescence intensity of GdSr2AlO5:yEu3+. The excitation spectrum of the Eu3+-activated sample monitoring the emission wavelength at 705 nm contains a broad band in the wavelength range from 220 to 350 nm, corresponding to the charge transfer transition of Eu3+ (O2− → Eu3+) and several characteristic narrow peaks at 383 nm and 393 nm. They are attributed to 7F0 → 5G2 and 7F0 → 5L6 electronic transitions of the Eu3+ ions. In this band, there should be absorption of transitions from 8S7/2 to 6I7/2 level of Gd3+, which is too weak and is overlapped with the strong CTB of the O2− → Eu3+.32,33 The emission spectrum consists of multiple band emissions at 587, 607, 622, 657, and 705 nm, which are assigned to the 5D0 → 7FJ (J = 0, 1, 2, 3, and 4) transitions of Eu3+ ions,34 respectively. Among these emission peaks, the two most intense lines are the emissions located at 622 and 705 nm. Obviously, the magnetic dipole transition of 5D0 → 7F1 is stronger than the 5D0 → 7F2 electronic transition; this indicates that Eu3+ ions in the GdSr2AlO5 host occupy a site with a non-inversion symmetry; this is consistent with its structure. Compared to other Eu3+ phosphors, GdSr2AlO5:Eu3+ provides more red components to improve Ra. Similarly, there is no evident concentration quenching with the change in the Eu3+ concentration.
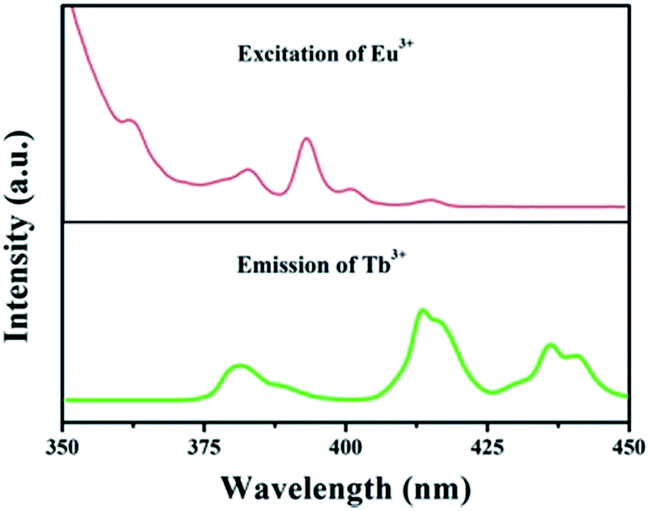
The foundation of energy transfer is the spectral overlapping Tb3+ emission and the Eu3+ excitation. As is clearly shown in Fig. 4, there are some overlaps between the emission of 5D3 → 7F6 (Tb3+) and the excitation of 7F0 → 5G2,3,4,5, 5L8 (Eu3+). Moreover, the excitation of 7F0 → 5D3, 5L6 (Eu3+) overlaps with the emission of 5D3 → 7F5 (Tb3+).35
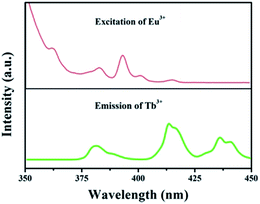 |
| | Fig. 4 Spectral overlapping of the PL spectrum of GdSr2AlO5:Tb3+ and the PLE spectrum of GdSr2AlO5:Eu3+. | |
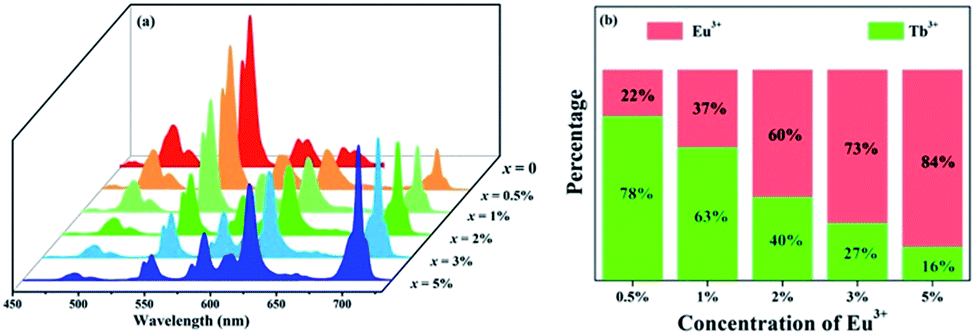
To further investigate the energy transfer process, a series of samples of GdSr2AlO5:2%Tb3+,x%Eu3+ (x = 0, 0.5, 1, 2, 3, and 5) were prepared whose emission spectra under the excitation at 275 nm are presented in Fig. 5(a). The characteristic sharp emission peaks of Tb3+ and Eu3+ are observed. The tendency in the variation of Tb3+ and Eu3+ differs significantly. The emission intensity of Tb3+ at 548 nm decreases remarkably with an increase in the concentration of Eu3+, whereas the emission intensity of Eu3+ increases monotonously. Fig. 5(b) depicts the contribution of different ions in the corresponding emission spectra. The G/R (green to red) ratio continuously decreases notably. Based on the prerequisite of the concentration of Tb3+ being a constant value, explanation for this phenomenon should be the evidence of energy transfer from Tb3+ to Eu3+.
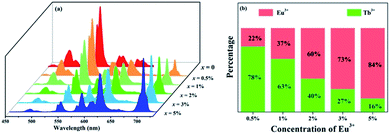 |
| | Fig. 5 (a) Emission spectra of GdSr2AlO5:2%Tb3+,x%Eu3+ (x = 0, 0.5, 1, 2, 3, and 5) under 275 nm excitation. (b) Variation tendency of the green emission of Tb3+ and the red emission of Eu3+. | |
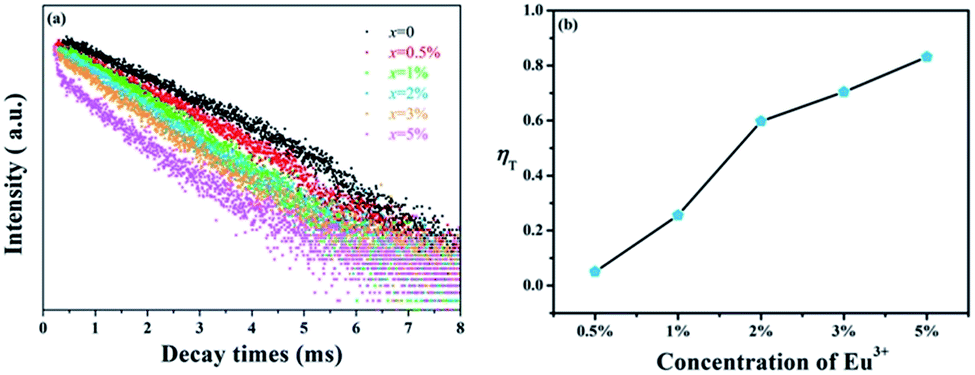
In spite of steady-state spectra, kinetic spectra are necessary to better analyze the energy transfer mechanism of Tb3+ → Eu3+ in GdSr2AlO5. Fig. 6(a) shows the fluorescence decay curves of Tb3+ emissions, which are obtained under 275 nm excitation and detected at 548 nm. The luminescence curves can be fitted well with the double-exponential expression:36,37
| |
I(t) = I0 + A1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−t/τ1) + A2exp(−t/τ2) exp(−t/τ1) + A2exp(−t/τ2)
| (1) |
where
I and
I0 represent the luminescence intensity at time
t and 0,
A1 and
A2 are equal to constants,
t is the time, and
τ1 and
τ2 are the rapid and slow lifetimes for exponential components, respectively. As a function of these parameters, the average luminescence lifetime (
τ) was determined by the following equation:
| | |
τ = (A1τ12 + A2τ22)/(A1τ1 + A2τ2)
| (2) |
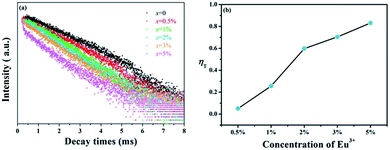 |
| | Fig. 6 (a) Decay curves of Tb3+ in GdSr2AlO5:2%Tb3+,x%Eu3+ (x = 0, 0.5, 1, 2, 3, and 5). (b) Energy transfer efficiencies (ηT) in GdSr2AlO5:2%Tb3+,x%Eu3+ as a function of different Eu3+ concentrations. | |
The values of τ are calculated to decrease when the Eu3+ concentration increases; this indicates that energy transfer occurs from Tb3+ to Eu3+, as expected. Furthermore, the energy transfer efficiency is calculated by the expression as follows:38
where
ηT represents the energy transfer efficiency, and
Is and
Iso represent the luminescence intensity of Tb
3+ in the presence and absence of Eu
3+, respectively. The results of
ηT are shown in
Fig. 6(b). It can be found that the efficiencies from Tb
3+ to Eu
3+ increase with the increasing Eu
3+ concentration, which can reach the maximum value of 83%. Therefore, the energy transfer from Tb
3+ to Eu
3+ ions is efficient to improve Eu
3+ luminescence.
3.4 Energy transfer mechanism
The type of energy transfer should depend on either the exchange interaction or the multipolar interaction. It relies on the critical distance between the Tb3+ (sensitizer) and Eu3+ (activator), and the critical distance is expressed by the following formula:39| |
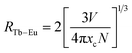 | (4) |
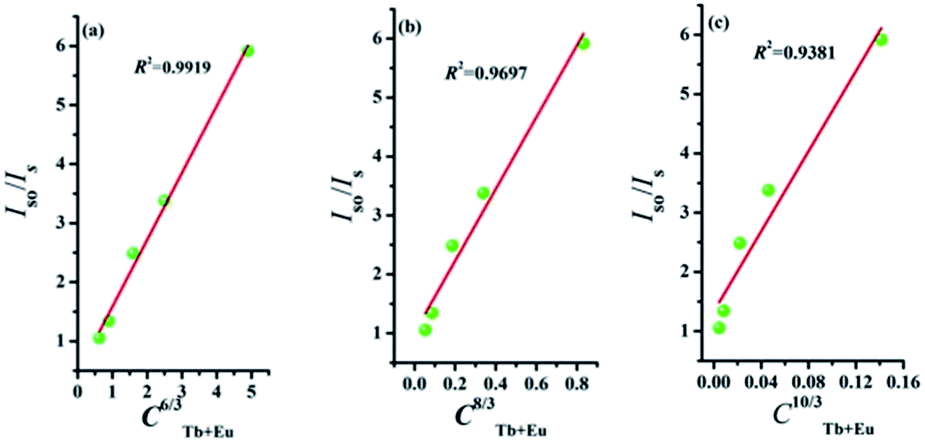
where V represents the cell volume, N is equal to the number of cations in the unit cell, and xc is the total concentration of Tb3+ and Eu3+. In this case, N = 4 and V = 498.64 Å3. The corresponding distance is calculated to be more than 5 Å, indicating that Tb3+ → Eu3+ energy transfer would tend to be a multipolar interaction. On account of energy transfer mechanism for multipolar interaction, the following expression can be given:40,41where Iso and Is are the luminescence intensity of the sensitizer without and with activator. C represents the doping concentration of Tb3+ and Eu3+ ion. The values for n = 6, 8, and 10 correspond to dipole–dipole, dipole–quadrupole, and quadrupole–quadrupole interactions. Results are illustrated in Fig. 7. The best linear fit was obtained when n = 6, which confirmed energy transfer through dipole–dipole mechanism.
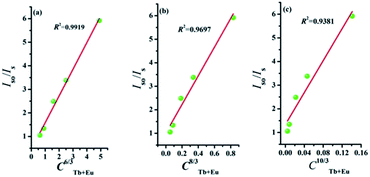 |
| | Fig. 7 Dependence of Iso/Is of Tb3+ on (a) C6/3, (b) C8/3, and (c) C10/3. | |
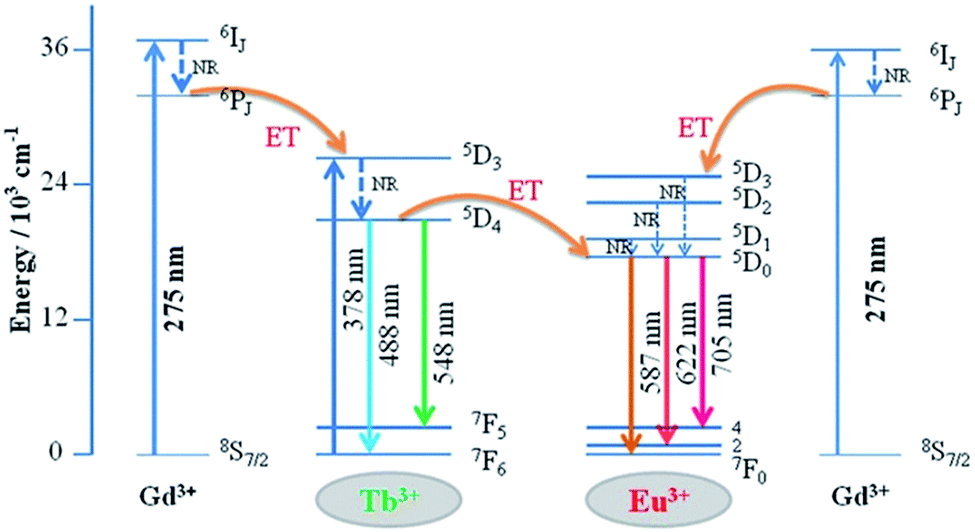
The detailed Gd3+ → Tb3+, Gd3+ → Eu3+, and Tb3+ → Eu3+ energy transfer processes are shown in Fig. 8. The electrons were excited from the 8S7/2 ground state to the 6IJ excited state of Gd3+ under 275 nm excitation. Because the energy levels 5D3 of Tb3+ and 5D1 state of Eu3+ are lower than the 6IJ state of Gd3+ in the diagram, the energy transfer processes from Gd3+ → Tb3+ and Gd3+ → Eu3+ occur simultaneously. In addition, the electrons absorbing energy were excited from the 7F6 ground state to the 5D3 excited state of Tb3+. The energy transfer process occurs between Tb3+ and Eu3+ activators since the 5D3 state of Tb3+ lies higher than the 5D1 state of Eu3+. Then, the electrons from the excited state of 5D3 (Tb3+) and 5D1 (Eu3+) relaxed to 5D4 and 5D0, respectively. Finally, green to red emission is yielded by electrons returning to their respective ground states.
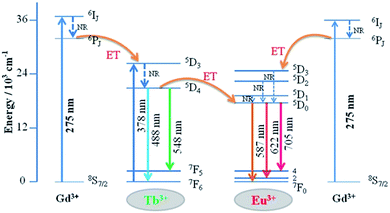 |
| | Fig. 8 Schematic of energy transfer from Tb3+ to Eu3+ in GdSr2AlO5. | |
3.5 CIE chromaticity coordinate and QE
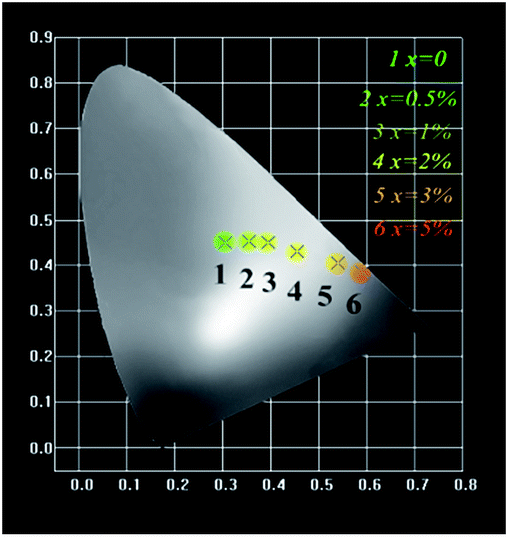
The Commission Internationale de L'Eclairage (CIE 1931) chromaticity coordinates of GdSr2AlO5:2%Tb3+,x%Eu3+ phosphors under the excitation at 275 nm are presented in Fig. 9. The CIE coordinates shifted from (0.320, 0.466) to (0.574, 0.403) for the samples x = 0 to x = 5% correspond to green, yellow-green, and red. It is clear that the coordinate Y changes linearly with the X, the expression of which can be shown as below:
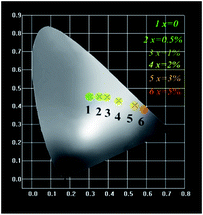 |
| | Fig. 9 CIE chromaticity diagram for GdSr2AlO5:2%Tb3+,x%Eu3+ (x = 0, 0.5, 1, 2, 3, and 5) samples. | |
The equation proves that the range of chromaticity coordinates can be obtained by adjusting the ratio of Tb3+ and Eu3+ mathematically. The as-prepared phosphor emits bright tunable visible light, implying its potential application in multicolor displays, and meets the requirement for application in n-UV LEDs.
For photo-luminescence application, the importance of quantum efficiency (QE) should be considered. According to the method described by De Mello et al., QE can be calculated by the following equation:42
| |
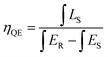 | (7) |
where
LS is the emission of the sample,
ES equals to the spectrum of the sample excited by the light, and
ER represents the spectrum of the excitation light without the sample. The results are listed in
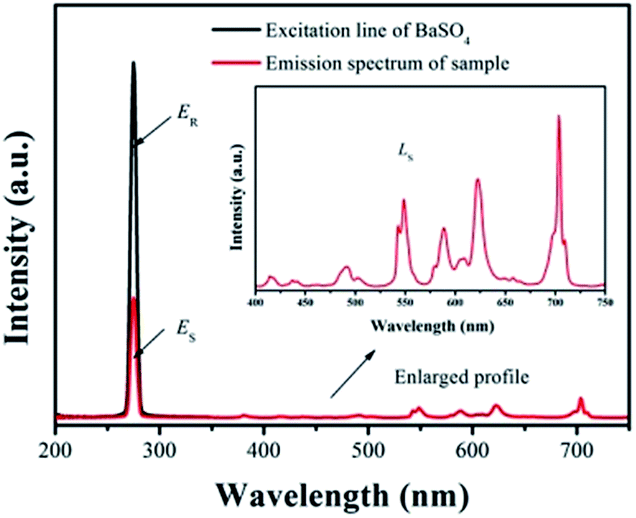
Fig. 10. Under 275 nm sexcitation, the calculated QE of GdSr
2AlO
5:2%Tb
3+,2%Eu
3+ is 78.5%, which demonstrates a high QE.
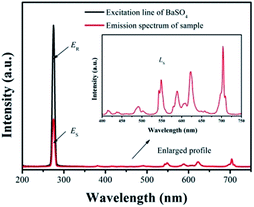 |
| | Fig. 10 Excitation line of BaSO4 and emission spectrum of the sample. Inset shows the magnification of the emission spectrum. | |
4. Conclusions
The GdSr2AlO5:Tb3+,Eu3+ phosphors were prepared by a high temperature solid-state reaction method, and their luminescence properties were investigated in detail. Due to REO8 structure isolation, damping of the concentration quenching would occur. Energy transfer from Gd3+ → Eu3+ and Gd3+ → Tb3+ was found in the excitation spectrum. Moreover, the luminescence properties and the transient spectra confirmed the efficiency energy transfer from Tb3+ to Eu3+. The mechanism it demonstrated is dipole–dipole interaction. GdSr2AlO5:Tb3+,Eu3+ phosphors were able to provide multi-color emission from green, yellow, orange, and finally to deep red by the different ratios of Tb3+/Eu3+. Additionally, the QE can reach about 78.5%. All the results indicate that the series of GdSr2AlO5:Tb3+,Eu3+ phosphors can be a candidate for color tunable luminescence materials used for n-UV LEDs.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The present work was supported by the National Natural Science Foundation of China (Grant No. 21671070, and 21571067), the Project for Construction of High-level University in Guangdong Province, the Teamwork Projects funded by the Guangdong Natural Science Foundation (Grant No. S2013030012842), the Guangzhou Science & Technology Project (No. 201704030086, 201605030005), and the Spectral Funds for the Cultivation of Guangdong College Student Scientific and Technological Innovation (“Climbing Program” Special Funds No. pdjh2017a0075).
References
- W. Li, J. Wang, H. Zhang, Y. Liu, B. Lei, J. Zhuang, J. Cui, M. Peng and Y. Zhu, RSC Adv., 2016, 6, 33076–33082 RSC.
- N. C. George, K. A. Denault and R. Seshadri, Annu. Rev. Mater. Res., 2013, 43, 481–501 CrossRef CAS.
- C. C. Lin and R. S. Liu, J. Phys. Chem. Lett., 2011, 2, 1268–1277 CrossRef CAS PubMed.
- M. Shang, C. Li and J. Lin, Chem. Soc. Rev., 2014, 43, 1372–1386 RSC.
- A. A. Setlur, Electrochem. Soc. Interface, 2009, 18, 32–36 CAS.
- D. J. Robbins, J. Electrochem. Soc., 1979, 126, 1550–1555 CrossRef CAS.
- S. Nakamura, MRS Bull., 2009, 34, 101–107 CrossRef CAS.
- S. Nakamura, Proc. SPIE, 1997, 3002, 26–35 CrossRef CAS.
- D. Deng, H. Yu, Y. Lin, Y. Hua, G. Jia and S. Zhao, J. Mater. Chem. C, 2013, 1, 3194–3199 RSC.
- D. Wen, H. Yang, G. Yang, J. Shi, M. Wu and Q. Su, J. Solid State Chem., 2014, 213, 65–71 CrossRef CAS.
- G. Lee, J. Y. Han, W. B. Im, S. H. Cheong and D. Y. Jeon, Inorg. Chem., 2012, 51, 10688–10694 CrossRef CAS PubMed.
- T. Nishida, T. Ban and N. Kobayashi, Appl. Phys. Lett., 2003, 82, 3817–3819 CrossRef CAS.
- Y. Liu, X. Zhang, Z. Hao, X. Wang and J. Zhang, Chem. Commun., 2011, 47, 10677–10679 RSC.
- W. B. Im, N. N. Fellows, S. P. DenBaars, R. Seshadri and Y. Kim, Chem. Mater., 2009, 21, 2957–2966 CrossRef CAS.
- C. R. García, J. Oliva and L. A. Díaz-Torres, Photochem. Photobiol., 2015, 91, 505–509 CrossRef PubMed.
- Z. Xia and Z. Yu, J. Mater. Chem. C, 2015, 44, 11629–11634 Search PubMed.
- J. Hyun Kim and K. Youl Jung, J. Lumin., 2012, 132, 1376–1381 CrossRef.
- G. Li, Y. Tian, Y. Zhao and J. Lin, Chem. Soc. Rev., 2015, 44, 8688–8713 RSC.
- K. Li, M. Shang, H. Lian and J. Lin, J. Mater. Chem. C, 2016, 4, 5507–5530 RSC.
- X. Yang, B. Zhang, T. Xu, L. Wang, J. Shen and Q. Zhang, J. Mater. Sci.: Mater. Electron., 2016, 27, 9448–9453 CrossRef CAS.
- C. Zhang, H. Liang, S. Zhang, C. Liu, D. Hou, L. Zhou, G. Zhang and J. Shi, J. Phys. Chem. C, 2012, 116, 15932–15937 CAS.
- Y. Meng, W. Zhao, L. Wang, Y. Zhou, M. He and Y. Zhu, J. Mater. Sci.: Mater. Electron., 2017, 28, 4984–4989 CrossRef CAS.
- X. Zhou and X. Wang, J. Lumin., 2014, 29, 143–146 CrossRef CAS PubMed.
- K. Pavani, J. Suresh Kumar and L. Rama Moorthy, J. Alloys Compd., 2014, 586, 722–729 CrossRef CAS.
- D. Wang, Y. Wang and J. He, Mater. Res. Bull., 2012, 47, 142–145 CrossRef CAS.
- D. Geng, G. Li, M. Shang, C. Peng, Y. Zhang, Z. Cheng and J. Lin, Dalton Trans., 2012, 41, 378–386 Search PubMed.
- M. Back, M. Boffelli, A. Massari, R. Marin, F. Enrichi and P. Riello, J. Nanopart. Res., 2013, 15, 1753–1767 CrossRef.
- L. S. D. Glasser and F. P. Glasser, Acta Crystallogr., 1965, 18, 453–455 CrossRef CAS.
- D. Wen, J. Feng, J. Li, J. Shi, M. Wu and Q. Su, J. Mater. Chem. C, 2015, 3, 2107–2114 RSC.
- H. Guan, G. Liu, J. Wang, X. Dong and W. Yu, Dalton Trans., 2014, 43, 181–188 Search PubMed.
- M. M. Yawalkar, G. D. Zade, V. Singh and S. J. Dhoble, J. Mater. Sci.: Mater. Electron., 2017, 28, 180–189 CrossRef CAS.
- J. Singh and J. Manam, J. Mater. Sci., 2016, 51, 2886–2901 CrossRef CAS.
- X. X. Li, Y. H. Wang and Z. Chen, J. Alloys Compd., 2008, 453, 392–394 CrossRef CAS.
- G. K. Behrh, R. Gautier, C. Latouche, S. Jobic and H. Serier-Brault, Inorg. Chem., 2016, 55, 9144–9146 CrossRef CAS PubMed.
- J. Li, Z. Zhang, X. Li, Y. Xu, Y. Ai, J. Shi and M. Wu, J. Mater. Chem. C, 2017, 5, 6294–6299 RSC.
- M. Yu, J. Lin and J. Fang, Chem. Mater., 2005, 17, 1783–1791 CrossRef CAS.
- X. Liu, C. Li, Z. Quan, Z. Cheng and J. Lin, J. Phys. Chem. C, 2007, 111, 16601–16607 CAS.
- M. Xu, L. Wang, D. Jia and H. Zhao, J. Am. Ceram. Soc., 2015, 98, 1536–1541 CrossRef CAS.
- S. P. Lee, C. H. Huang and T. Chen, J. Phys. Chem. C, 2014, 2, 8925–8931 CAS.
- D. L. Dexter and J. H. Schulman, J. Chem. Phys., 1954, 22, 1063–1070 CrossRef CAS.
- W. Yang, L. Luo, T. Chen and N. Wang, Chem. Mater., 2005, 17, 3883–3888 CrossRef CAS.
- L. O. P. Lsson and A. P. Monkman, Adv. Mater., 2002, 14, 757–758 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  * and
Bingfu Lei
* and
Bingfu Lei