
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel steroidal saponins with cytotoxic activities from the roots of Ophiopogon japonicus (L. f.) Ker-Gawl†
Yan Wu‡
,
Su-Xia Bi‡,
Zhen Huang,
Jin Qi* and
Bo-Yang Yu *
*
Jiangsu Key Laboratory of TCM Evaluation and Translational Research, China Pharmaceutical University, Nanjing 211198, People's Republic of China. E-mail: boyangyu59@163.com; yaoyuelingxing@163.com; Fax: +86-25-86185158; Tel: +86-25-86185157
First published on 11th January 2018
Abstract
Six new steroidal saponins (1–6) and one known steroidal saponin (7) were obtained from the roots of Ophiopogon japonicus (L. f.) Ker-Gawl. Their structures were determined by the detailed analysis of extensive nuclear magnetic resonance and mass spectroscopic data. The in vitro cytotoxic activities of these compounds against MDA-MB-435, HepG2 and A549 cell lines were also investigated.
Introduction
Ophiopogon japonicus (L.f.) Ker-Gawl is an evergreen perennial in the family Liliaceae and mainly distributed in the Southern of China, Japan, Vietnam and India. O. japonicas is widely artificially cultivated in Zhejiang and Sichuan Provinces of China.1 The tuber of O. japonicus, commonly known as Maidong, is a famous herb used to treat coughs, sore throats, constipation and insomnia for thousands of years in Traditional Chinese Medicine.2Pharmacological studies indicated that the tubers of O. japonicus exhibit various biological activities such as immunomodulation, anti-diabetes, cardiovascular protection, anti-oxidation and anti-cancer.3–7 In recent years, the significant pharmacological effects of the tubers of O. japonicus on cardiovascular disease and cancer have drawn much attention from medicinal researchers. Previous phytochemical investigations on the tubers of O. japonicas resulted in the isolation of homoisoflavonoids,8–13 organic acids,14–16 saccharides17,18 and steroidal saponins.19–23 However, the bioactive ingredients of this herb are still not fully elucidated. As part of our ongoing progress to search for bioactive constituents from traditional Chinese medicine, a 75% EtOH extract of the roots of O. japonicus was investigated which led to the isolation of six new steroidal saponins (1–6) and one known steroidal saponin (7) (Fig. 1). The in vitro cytotoxic activities of these compounds against MDA-MB-435, HepG2 and A549 cell lines were also investigated. In this paper, we describe the isolation, structural elucidation and cytotoxic activities evaluation of these steroidal saponins.
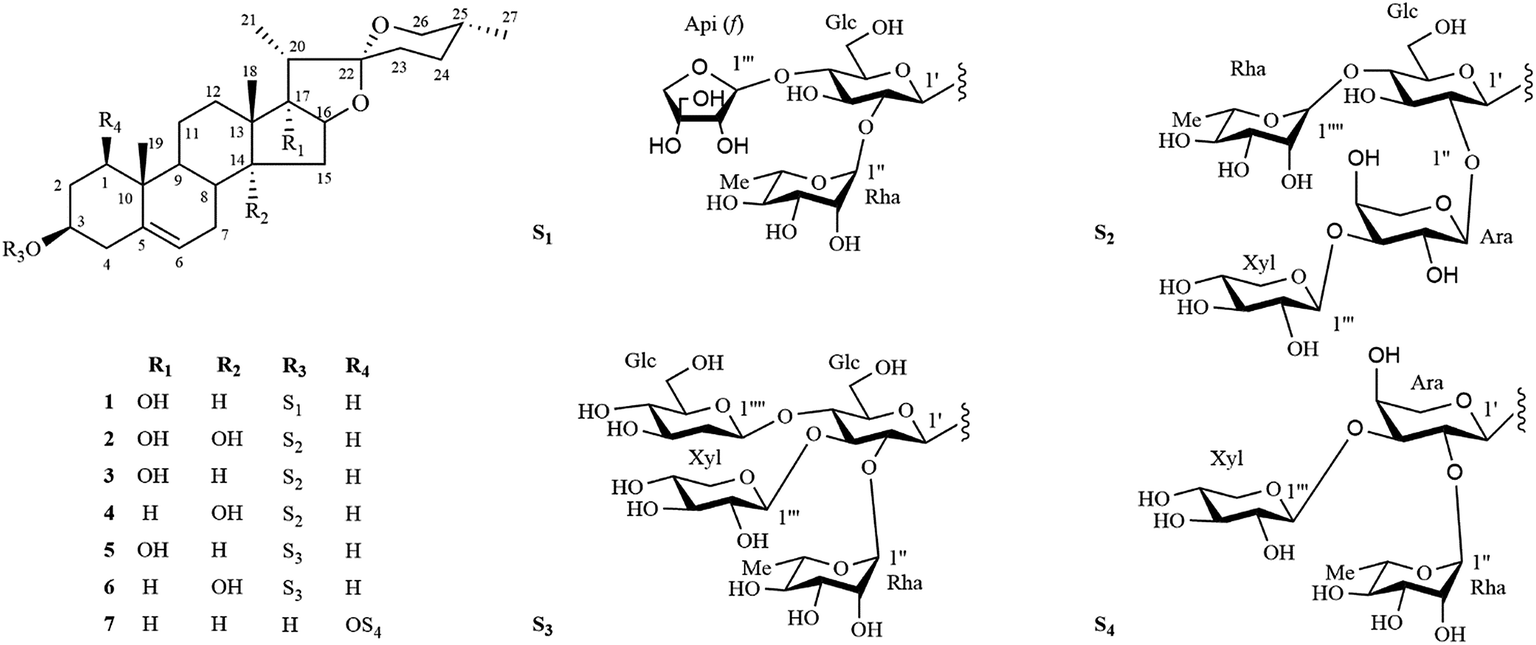 | ||
| Fig. 1 Chemical structures of compounds 1–7. | ||
Results and discussion
The phytochemical investigation of a 75% EtOH extract of the roots of Ophiopogon japonicus (L. f.) Ker-Gawl resulted in the isolation of seven steroidal saponins (1–7), including six new steroidal saponins (1–6) and one known steroidal saponin (7). The known compound (7) was identified as (25R)-ruscogenin-1-O-α-L-rhamnopyranosyl-(1 → 2)-[β-D-xylopyranosyl-(1 → 3)]-α-L-arabinopyranoside by comparison of its MS, 1H NMR and 13C NMR data with published data in the literature.24Compound 1 was isolated as an amorphous solid and gave a positive Liebermann–Burchard reaction. Its molecular formula was assigned to be C44H70O17 by the HRESI-QTOF-MS ion peak at m/z 893.4447 [M + Na]+ (calcd for C44H70O17Na, 893.4505). The 1H and 13C spectra of 1 (Tables 1–4) displayed characteristic signals for four steroidal methyl groups [δH 0.68 (3H, d, J = 4.5 Hz), 0.96 (3H, s), 1.09 (3H, s) and 1.24 (3H, d, J = 7.0 Hz)], an olefinic group [δC 141.2, 122.2; δH 5.31 (1H, d, J = 5.0 Hz)], three anomeric protons [δH 4.92 (1H, d, J = 7.5 Hz), 5.90 (1H, d, J = 3.0 Hz) and 6.24 (1H, br s)] and three anomeric carbon signals at δC 100.5, 111.6, 102.4. These spectra data together with the observation that three anomeric protons signals at δH 4.92, 5.90 and 6.24 giving correlations with three anomeric carbon signals at δC 100.5, 111.6, 102.4 in the HSQC spectrum, implied that 1 was a steroidal derivative containing three sugar units.
| Position | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| a NMR data were measured at 500 MHz for 1H and at 125 MHz for 13C in pyridine-d5. Assignments are based on HSQC and HMBC experiments. | ||||||
| 1 | 37.6 | 38.1 | 38.0 | 37.9 | 37.5 | 38.1 |
| 2 | 30.8 | 30.4 | 31.0 | 30.8 | 30.8 | 30.4 |
| 3 | 78.5 | 78.2 | 78.3 | 78.3 | 78.3 | 78.2 |
| 4 | 39.4 | 39.0 | 39.1 | 40.3 | 39.7 | 39.1 |
| 5 | 141.2 | 140.6 | 141.2 | 140.7 | 141.2 | 140.7 |
| 6 | 122.2 | 122.7 | 122.8 | 122.3 | 122.2 | 122.8 |
| 7 | 32.2 | 26.5 | 32.8 | 27.1 | 32.2 | 27.1 |
| 8 | 30.6 | 36.6 | 30.8 | 36.0 | 30.4 | 36.0 |
| 9 | 50.7 | 43.9 | 50.7 | 44.0 | 50.6 | 44.0 |
| 10 | 38.0 | 37.8 | 37.6 | 37.6 | 37.9 | 37.9 |
| 11 | 21.4 | 20.5 | 21.4 | 20.8 | 21.3 | 20.8 |
| 12 | 32.2 | 26.9 | 32.8 | 32.2 | 32.4 | 32.4 |
| 13 | 45.2 | 48.7 | 45.5 | 45.5 | 45.2 | 45.4 |
| 14 | 53.5 | 88.1 | 53.5 | 86.8 | 53.4 | 86.8 |
| 15 | 32.5 | 40.7 | 32.9 | 39.1 | 32.7 | 40.3 |
| 16 | 90.5 | 90.9 | 90.5 | 82.3 | 90.4 | 82.2 |
| 17 | 90.6 | 91.5 | 90.6 | 60.3 | 90.5 | 60.3 |
| 18 | 17.5 | 21.0 | 17.5 | 20.4 | 17.5 | 20.4 |
| 19 | 19.8 | 19.8 | 19.8 | 19.7 | 19.8 | 19.7 |
| 20 | 45.5 | 45.5 | 45.2 | 42.5 | 45.5 | 42.4 |
| 21 | 10.1 | 10.1 | 10.1 | 15.7 | 10.0 | 15.7 |
| 22 | 110.2 | 109.9 | 110.2 | 110.0 | 110.1 | 109.9 |
| 23 | 32.8 | 32.5 | 32.4 | 31.0 | 32.8 | 31.0 |
| 24 | 29.2 | 29.6 | 29.2 | 29.8 | 29.2 | 29.7 |
| 25 | 32.9 | 30.8 | 30.4 | 30.4 | 32.8 | 30.3 |
| 26 | 67.1 | 67.2 | 67.1 | 67.2 | 66.2 | 67.2 |
| 27 | 17.7 | 17.6 | 17.7 | 17.5 | 17.6 | 17.7 |
| Position | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| a NMR data were measured at 500 MHz for 1H and at 125 MHz for 13C in pyridine-d5. Assignments are based on HSQC and HMBC experiments. | ||||||
| 1′-Glc-1′ | 100.5 | 100.2 | 100.3 | 100.3 | 100.3 | 100.3 |
| 2′ | 78.0 | 79.3 | 79.3 | 79.3 | 76.3 | 76.3 |
| 3′ | 77.8 | 74.2 | 74.3 | 74.3 | 80.7 | 80.7 |
| 4′ | 79.8 | 79.4 | 79.4 | 79.4 | 80.7 | 80.7 |
| 5′ | 77.0 | 77.6 | 77.7 | 77.7 | 78.3 | 78.5 |
| 6′ | 61.7 | 61.4 | 61.4 | 61.4 | 62.4 | 62.4 |
| 2′′-Ara-1′′ | 103.6 | 103.9 | 103.9 | |||
| 2′′ | 73.5 | 73.6 | 73.6 | |||
| 3′′ | 75.8 | 75.9 | 75.9 | |||
| 4′′ | 71.2 | 71.3 | 71.3 | |||
| 5′′ | 66.4 | 66.5 | 66.5 | |||
| 2′′-Rha-1′′ | 102.4 | 102.5 | 102.5 | |||
| 2′′ | 72.9 | 72.8 | 72.8 | |||
| 3′′ | 73.2 | 72.8 | 72.8 | |||
| 4′′ | 74.6 | 73.8 | 73.9 | |||
| 5′′ | 69.9 | 70.3 | 70.3 | |||
| 6′′ | 19.0 | 19.0 | 19.0 | |||
| 3′′′-Api (f)-1′′′ | 111.6 | |||||
| 2′′′ | 78.1 | |||||
| 3′′′ | 80.5 | |||||
| 4′′′ | 75.4 | |||||
| 5′′′ | 65.2 | |||||
| 3′′′-Xyl-1′′′ | 103.8 | 103.7 | 103.7 | 102.9 | 102.9 | |
| 2′′′ | 74.2 | 74.3 | 74.3 | 73.8 | 73.1 | |
| 3′′′ | 77.6 | 77.0 | 77.0 | 78.3 | 78.9 | |
| 4′′′ | 71.1 | 71.1 | 71.1 | 70.9 | 70.9 | |
| 5′′′ | 65.8 | 65.9 | 65.9 | 64.8 | 66.2 | |
| 4′′′′-Rha-1′′′′ | 102.9 | 102.9 | 102.9 | |||
| 2′′′′ | 73.1 | 72.8 | 72.8 | |||
| 3′′′′ | 73.1 | 73.2 | 73.2 | |||
| 4′′′′ | 74.3 | 74.4 | 74.4 | |||
| 5′′′′ | 70.3 | 70.3 | 70.3 | |||
| 6′′′′ | 19.0 | 19.1 | 19.1 | |||
| 4′′-Glc-1′′′′ | 102.9 | 102.9 | ||||
| 2′′′′ | 74.3 | 74.3 | ||||
| 3′′′′ | 75.6 | 75.6 | ||||
| 4′′′′ | 71.1 | 71.1 | ||||
| 5′′′′ | 77.2 | 77.2 | ||||
| 6′′′′ | 61.8 | 62.4 | ||||
| Position | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| a NMR data were measured at 500 MHz for 1H and at 125 MHz for 13C in pyridine-d5. Assignments are based on HSQC and HMBC experiments. | ||||||
| 1 | 0.97 | 1.06 | 0.98 | 1.01 | 0.96 | 0.96 |
| 1.78 | 1.78 | 1.76 | 1.81 | 1.79 | 1.78 | |
| 2 | 1.87 m | 2.03 | 1.85 | 1.84 | 1.83 | 1.83 |
| 2.10 | 1.83 | 2.00 | 2.01 | 2.03 | 2.02 | |
| 3 | 3.96 m | 3.84 | 3.82 | 3.82 | 3.85 | 3.80 |
| 4 | 2.77 m | 2.76 | 2.72 | 2.73 | 2.71 | 2.75 |
| 6 | 5.31 d (5.0 Hz) | 5.40 br.s | 5.34 d (4.5 Hz) | 5.42 br.s | 5.35 d (6.0 Hz) | 5.42 d (5.0 Hz) |
| 7 | 1.54 | 1.87 | 1.56 | 1.86 | 2.23 | 2.46 |
| 2.26 m | 2.58 | 2.29 | 2.53 | 1.56 | 1.86 | |
| 8 | 1.61 m | 2.06 | 1.65 | 2.01 | 1.62 | 2.05 |
| 9 | 0.98 m | 1.79 | 0.97 | 1.82 | 0.98 | 1.78 |
| 11 | 1.60 | 1.12 | 1.60 | 1.57 | 1.62 | 1.57 |
| 12 | 1.54 | 1.35 | 1.53 | 1.46 | 1.49 | 1.73 |
| 1.74 | 1.73 | 1.74 | 1.72 | 1.45 | ||
| 14 | 2.01 m | — | 2.01 | — | 2.06 | — |
| 15 | 1.64 m | 1.87 | 1.65 | 1.90 | 1.68 | 1.88 |
| 2.57 | 2.35 | 2.33 | ||||
| 16 | 4.46 | 4.79 | 4.44 | 5.01 | 4.42 | 5.06 |
| 17 | — | — | — | 2.78 | — | 2.75 |
| 18 | 0.96 s | 1.11 | 0.97 s | 1.07 s | 0.96 s | 1.08 |
| 19 | 1.09 s | 1.12 | 1.09 s | 1.13 s | 1.09 s | 1.06 |
| 20 | 2.29 d (7.0 Hz) | 2.40 d (7.0 Hz) | 2.23 d (7.5 Hz) | 2.10 m | 2.27 d (7.5 Hz) | 2.06 m |
| 21 | 1.24 d (7.0 Hz) | 1.27 d (7.0 Hz) | 1.23 d (7.5 Hz) | 1.27 d (7.0 Hz) | 1.23 d (7.5 Hz) | 1.19 d (7.0 Hz) |
| 23 | 1.95 | 1.70 | 1.95 | 1.28 | 1.98 | 1.27 |
| 24 | 1.61 | 1.59 | 1.59 | 1.32 | 1.60 | 1.32 |
| 25 | 1.95 | 1.85 | 1.83 | 2.03 | 1.98 | 2.02 |
| 2.16 | 2.05 | 2.01 | 2.16 | |||
| 26 | 3.54 m | 3.51 | 3.55 | 3.52 | 3.55 | 3.47 |
| 27 | 0.68 d (4.5 Hz) | 0.69 d (4.0 Hz) | 0.70 d (4.5 Hz) | 0.68 d (5.0 Hz) | 0.68 d (6.5 Hz) | 0.68 d (6.0 Hz) |
| Position | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| a NMR data were measured at 500 MHz for 1H and at 125 MHz for 13C in pyridine-d5. Assignments are based on HSQC and HMBC experiments. | ||||||
| 1′-Glc-1′ | 4.92 d (7.5 Hz) | 4.93 d (7.5 Hz) | 4.91 d (7.5 Hz) | 4.91 d (7.0 Hz) | 4.88 d (7.5 Hz) | 4.89 d (8.0 Hz) |
| 2′ | 4.16 | 4.15 | 4.17 | 4.16 | 4.14 | 4.14 |
| 3′ | 4.74 | 4.30 | 4.30 | 4.30 | 4.68 | 4.64 |
| 4′ | 3.85 | 4.51 | 4.52 | 4.52 | 4.68 | 4.64 |
| 5′ | 3.74 | 3.73 | 3.73 | 3.73 | 3.73 | 3.72 |
| 6′ | 4.22 | 4.31 | 4.31 | 4.31 | 4.30 | 4.29 |
| 4.29 | 4.47 | 4.48 | 4.48 | 4.38 | 4.39 | |
| 2′′-Ara-1′′ | 5.46 d (5.5 Hz) | 5.46 d (5.5 Hz) | 5.46 d (6.0 Hz) | |||
| 2′′ | 4.20 | 4.20 | 4.20 | |||
| 3′′ | 4.13 | 4.13 | 4.13 | |||
| 4′′ | 4.12 | 4.13 | 4.13 | |||
| 5′′ | 3.67 | 3.67 | 3.67 | |||
| 4.52 | 4.52 | 4.52 | ||||
| 2′′-Rha-1′′ | 6.24 br.s | 6.23 br.s | 6.21 br.s | |||
| 2′′ | 4.77 | 4.83 | 4.83 | |||
| 3′′ | 4.60 | 4.54 | 4.52 | |||
| 4′′ | 4.31 | 4.31 | 4.19 | |||
| 5′′ | 4.91 | 4.81 | 4.81 | |||
| 6′′ | 1.75 d (6.5 Hz) | 1.73 d (6.5 Hz) | 1.74 d (7.0 Hz) | |||
| 3′′′-Api (f)-1′′′ | 5.90 d (3.0 Hz) | |||||
| 2′′′ | 4.74 | |||||
| 3′′′ | 4.18 | |||||
| 4′′′ | 4.30 | |||||
| 5′′′ | 4.13 | |||||
| 3′′′-Xyl-1′′′ | 5.48 d (5.5 Hz) | 5.49 d (5.5 Hz) | 5.48 d (6.0 Hz) | 5.51 d (6.0 Hz) | 5.50 d (6.0 Hz) | |
| 2′′′ | 4.12 | 4.13 | 4.13 | 4.12 | 4.10 | |
| 3′′′ | 4.13 | 4.15 | 4.15 | 4.28 | 4.27 | |
| 4′′′ | 4.13 | 4.13 | 4.13 | 4.16 | 4.12 | |
| 5′′′ | 3.67 | 3.67 | 3.67 | 3.59 | 3.65 | |
| 4.65 | 4.68 d (8.0 Hz) | 4.68 d (8.5 Hz) | 4.67 | 4.57 | ||
| 4′′′′-Rha-1′′′′ | 6.05 br.s | 6.07 br.s | 6.07 br.s | |||
| 2′′′′ | 4.80 | 4.83 | 4.83 | |||
| 3′′′′ | 4.52 | 4.56 | 4.56 | |||
| 4′′′′ | 4.31 | 4.32 | 4.32 | |||
| 5′′′′ | 4.86 | 4.83 | 4.83 | |||
| 6′′′′ | 1.74 d (6.5 Hz) | 1.74 d (6.5 Hz) | 1.74 d (6.0 Hz) | |||
| 4′′′′-Glc-1′′′′ | 5.42 d (8.0 Hz) | 5.42 d (8.0 Hz) | ||||
| 2′′′′ | 4.29 | 4.32 | ||||
| 4.11 | 4.02 | |||||
| 3′′′′ | 4.05 | 4.14 | ||||
| 4′′′′ | 4.20 | 4.19 | ||||
| 4.13 | 4.09 | |||||
| 5′′′′ | 3.83 | 3.80 | ||||
| 6′′′′ | 4.37 | 4.80 | ||||
The 13C-NMR chemical shifts at δC 32.8 (C-23), 29.2 (C-24), 32.9 (C-25) and 67.1 (C-26) as well as the characteristic absorptions of a 25(R) spiroketal unit at 981, 918, 892, 836 (intensity 918 < 892 cm−1) in the IR spectrum indicated that the C-25 configuration of the aglycone was R. The aglycone moiety of 1 was further deduced to be pennogenin by comparison its 1H and 13C NMR data to those reported in the literature.25–27 Complete assignments of each sugar unit were achieved by analyses of their chemical shifts and coupling constants obtained from extensive 1D and 2D NMR experiments, allowing the identification one β-glucopyranosyl (Glc), one α-rhamnopyranosyl (Rha) and one β-apiofuranosyl (Api) units in 1. Their absolute configurations were further determined to be D for Glc/Api and L for Rha by GC comparison of their corresponding trimethylsilylthiazolidine derivatives to the authentic samples prepared in the same procedure. In the HMBC spectrum (Fig. 2), the correlations between δH 6.24 (br s, Rha-1′′) and δC 78.0 (Glc-2′), between δH 5.90 (d, J = 3.0 Hz, Api-1′′′) and δC 79.8 (Glc-4′), between δH 4.92 (d, J = 7.5 Hz, Glc-1′) and δC 78.5 (Agly C-3), characterized that the sequence of the sugar chain was α-L-Rha-(1 → 2)-[β-D-Api-(1 → 4)]-β-D-Glc, and that the glycosidic site was at C-3. Therefore, the structure of 1 was established as pennogenin-3-O-α-L-rhamnopyranosyl-(1 → 2)-[β-D-apiofuranosyl-(1 → 4)]-β-D-glucopyranoside.
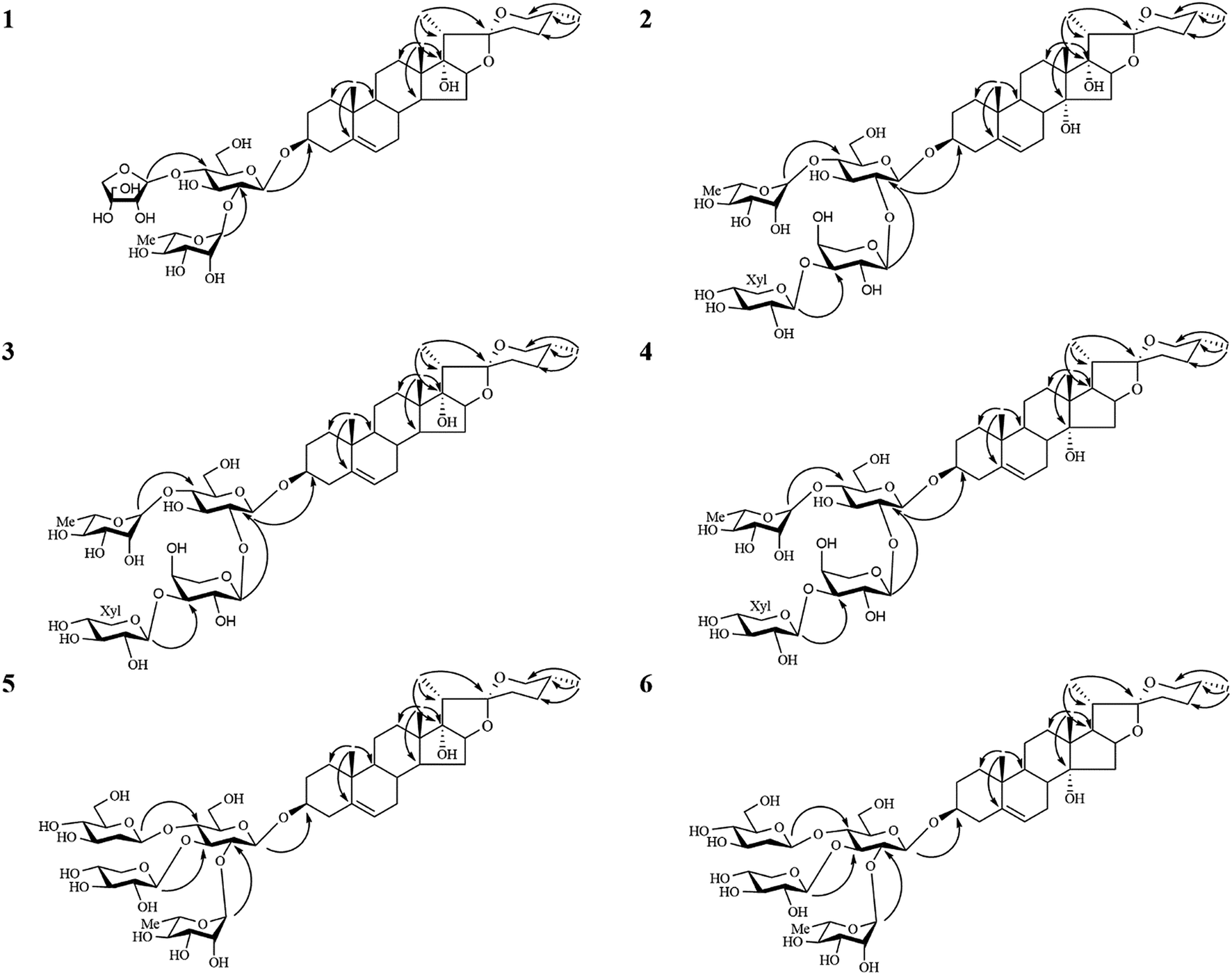 | ||
| Fig. 2 Key HMBC correlations of compounds 1–6. | ||
Compound 2 gave a sodium adduct ion at m/z 1041.4809 [M + Na]+ (calcd for C49H78O22Na, 1041.4877) in the HRESI-QTOF-MS, corresponding to the molecular formula C49H78O22. Comparison of the NMR data of 2 obtained from 1D and 2D NMR spectra (Tables 1–4) to those of lirigramoside A28 indicated that their chemical structures were closely similar. The major difference was that the chemical shifts at δC 32.8 (C-23), 29.2 (C-24), 32.9 (C-25) and 67.1 (C-26) in the 13C-NMR spectrum had lower field resonances than those of lirigramoside A, as well as an additional hydroxyl group in the aglycone moiety of 2. These observations suggested a (25R)-spirostanol aglycone moiety containing three hydroxyl groups for 2. The aglycone moiety of 2 was further identified as ophiogenin by comparison its spectroscopic data to those reported in the literature.29 The HMBC correlations (Fig. 2) between δH 6.05 (brs, Rha-1′′′′) and δC 79.4 (Glc-4′), between δH 5.48 (d, J = 5.5 Hz, Xyl-1′′′) and δC 75.8 (Ara-3′′), between δH 5.46 (d, J = 5.5 Hz, Ara-1′′) and δC 79.3 (Glc-2′), between δH 4.93 (d, J = 7.5 Hz, Glc-1′) and δC 78.2 (Agly C-3) indicated 2 contained the same sugar moiety of α-L-Rha-(1 → 4)-[β-D-Xyl-(1 → 3)-α-L-Ara(1 → 2)]-β-D-Glc at C-3 as lirigramoside A. Compound 2 was therefore assigned as ophiopogenin-3-O-α-L-rhamnopyranosyl-(1 → 4)-[β-D-xylopyranosyl-(1 → 3)-α-L-arabinopyranosyl-(1 → 2)]-β-D-glucopyranoside.
Compounds 3 and 4 possessed the same molecular formula C49H78O21 as deduced from their HRESI-QTOF-MS data (m/z 1025.4871 [M + Na]+, calcd for C49H78O21Na, 1025.4928). Detailed analysis of their 1H and 13C NMR data (Tables 1–4) suggested that they possessed the same sugar chain at C-3 as 2, but differed slightly in the aglycone moiety. The only difference was the absent of a hydroxyl group in the aglycone moiety of 3 and 4, respectively. The differences in chemical shifts of C-7 (Δδ + 6.3), C-8 (Δδ − 5.8), C-9 (Δδ + 6.8), C-12 (Δδ + 5.9), C-13 (Δδ − 3.2), C-14 (Δδ − 34.6) and C-15 (Δδ − 7.8) observed from comparative analysis of the 13C NMR data of 2 and 3, proved the absent hydroxyl group was attached at C-14 in 2, indicating a pennogenin25–27,30 aglycone moiety for 3. Similarly, the differences in chemical shifts of C-12 (Δδ + 5.3), C-13 (Δδ − 3.2), C-14 (Δδ − 1.3), C-16 (Δδ − 7.4), C-17 (Δδ − 31.2) and C-21 (Δδ + 5.6) in the 13C NMR spectra of 4, compared with those for 2 implied the absent hydroxyl group was at C-17 in 2, the aglycone moiety of 4 was therefore identified to be prazerigenin A.31 The structures of the aglycone moiety and the sequence of the sugar chains of 3 and 4 were further confirmed by the correlations in their HMBC spectrum (Fig. 2). Thus, the structures of 3 and 4 were determined to be pennogenin-3-O-α-L-rhamnopyranosy l-(1 → 4)-[β-D-xylopyranosyl-(1 → 3)-α-L-arabinopyranoseyl-(1 → 2)]-β-D-glucopyranoside and praze rigenin A-3-O-α-L-rhamnopyranosyl-(1 → 4)-[β-D-xylopyranosyl-(1 → 3)-α-L-arabinopyranosyl-(1 → 2)]-β-D-glucopyranoside, respectively.
The molecular formulas of compounds 5 and 6 were established as C50H80O22 from their HRESI-QTOF-MS ion peak at m/z 1055.4968 (calcd for C50H80O22Na, 1055.5033). A comprehensive analysis of the 1H and 13C NMR spectra (Tables 1–4) of 5 disclosed that these data were in good agreement with those of Cixi-ophiopogon B.27 The only difference was the absent of a hydroxyl group in the aglycone moiety of 5. The aglycone moiety of 5 was further assigned as pennogenin by comparison its 1H and 13C NMR data to those of 3. In the HMBC spectrum (Fig. 2), the correlations between δH 6.23 (brs, Rha-1′′) and δC 76.3 (Glc-2′), between δH 5.51 (d, J = 6.0 Hz, Xyl-1′′′) and δC 80.7 (Glc-3′), between δH 5.42 (d, J = 8.0 Hz, Glc-1′′′′) and δC 80.7 (Glc-4′), between δH 4.88 (d, J = 7.5 Hz, Glc-1′) and δC 78.3 (Agly C-3) suggested a α-L-Rha-(1 → 2)-[β-D-Xyl-(1 → 3)]-[β-D-Glc-(1 → 4)]-β-D-Glc sugar moiety at C-3 for 5, which was identical to that of Cixi-ophiopogon B. The structure of 5 was therefore established to be pennogenin-3-O-α-L-rhamnopyranosyl-(1 → 2)-[β-D-xylopyranosyl-(1 → 3)]-[β-D-glucopyranosyl-(1 → 4)]-β-D-glucopyranoside. The aglycone moiety of compound 6 was deduced to be prazerigenin A by detailed comparison its NMR data (Tables 1–4) to those of 4. Moreover, 6 was established to own the same sugar moiety at C-3 as 5 by the correlations between δH 6.21 (brs, Rha-1′′) and δC 76.3 (Glc-2′), between δH 5.50 (d, J = 6.0 Hz, Xyl-1′′′) and δC 80.7 (Glc-3′), between δH 5.42 (d, J = 8.0 Hz, Glc-1′′′′) and δC 80.7 (Glc-4′), between δH 4.89 (d, J = 8.0 Hz, Glc-1′) and δC 78.2 (Agly C-3) in the HMBC spectrum (Fig. 2). The structure of 6 was therefore established to be prazerigenin A-3-O-α-L-rhamnopyranosyl-(1 → 2)-[β-D-xylopyranosyl-(1 → 3)]-[β-D-glucopyranosyl-(1 → 4)]-β-D-glucopyranoside.
All the isolates were biologically evaluated for their in vitro cytotoxic activities against MDA-MB-435, HepG2 and A549 cell lines except for compounds 3 and 4 due to insufficient amount of compounds. According to the cytotoxicity data summarized in Table 5, compound 1 exhibited the best cytotoxicity against three tested cell lines with IC50 values ranging from 1.69 to 4.39 μM, compounds 5 and 7 showed moderate cytotoxicity with IC50 values ranging from 9.13 to 29.12 μM, whereas compounds 2 and 6 were nearly inactive (IC50 > 50 μM).
| Compounds | IC50 (μM) | ||
|---|---|---|---|
| HepG2 | MDA-MB-435 | A549 | |
| a No activity (IC50 > 50 μM).b Not measured due to insufficient amount of compounds. | |||
| 1 | 1.69 ± 0.18 | 1.90 ± 0.17 | 4.39 ± 0.37 |
| 2 | NAa | NAa | NAa |
| 3 | —b | —b | —b |
| 4 | —b | —b | —b |
| 5 | 21.18 ± 1.87 | 9.13 ± 1.43 | 21.27 ± 2.53 |
| 6 | NAa | NAa | NAa |
| 7 | NAa | 10.32 ± 2.37 | 29.12 ± 4.66 |
| 5-Fluorouracil | 87.3 ± 12.10 | 120.5 ± 15.53 | 256.8 ± 19.03 |
Experimental
General
IR data (KBr disks, in cm−1) were recorded on a Bruker Tensor 27 spectrometer (Bruker Corporation, Faellanden, Switzerland). Optical rotations were acquired using a JASCO P-1020 digital polarimeter (JASCO Corporation, Easton, MD, USA). NMR spectra were obtained on a Bruker Avance 500 NMR spectrometer (Bruker Corporation, Faellanden, Switzerland) using pyridine-d5 and tetramethylsilane as solvent and internal standard, respectively. Melting points were measured on a Buchi melting point B-545 apparatus (Buchi Instrument, Switzerland) without correction. ESI-MS and HRESI-Q-TOF-MS spectroscopic data were acquired on an Agilent 1100 Series MSD Trap mass spectrometer (Agilent Technologies, Santa Clara, CA, USA) and Agilent 6520 ESI-Q-TOF spectrometer (Agilent Technologies, Santa Clara, CA, USA), respectively. D101 macroporous resin (Shaanxi Lanshen Special Resin Co., Xian, China), silica gel (100–200 mesh and 200–300 mesh, Qingdao Marine Chemical Factory, Qingdao, China), and C18 reversed-phase silica gel (50 mesh, YMC, Tokyo, Japan) were used for column chromatography (CC). Preparative high-performance liquid chromatography (PHPLC) experiments were performed on an Agilent 1100 Series HPLC instrument (Agilent Technologies, Santa Clara, CA, USA) equipped with an evaporative light scattering detector and a diode array detector. Fractions obtained from CC were analyzed by TLC using silica gel GF254 (Qingdao Marine Chemical Factory, Qingdao, China) plates. GC analysis was measured with an Agilent 6890 gas chromatograph (Agilent Technologies, Santa Clara, CA, USA). Standard L-rhamnose, D-xylose, L-arabinose, D-glucose, D-apiofuranose, hexamethyldisilazane, L-cysteine methyl ester hydrochloride, and trimethylchlorosilane were purchased from Sigma-Aldrich Trading Co. Ltd. (Shanghai, China).Plant material
Dried tubers of O. japonicas were collected from Cixi city, Zhejiang Province, People's Republic of China, in May 2013 and authenticated by one of the authors (B. Y, Y.). A voucher specimen (no. 20130510) was deposited at the herbarium of Jiangsu Key Laboratory of TCM Evaluation and Translational Research, China Pharmaceutical University, Nanjing, People's Republic of China.Extraction and isolation
Dried roots of O. japonicas (15.5 kg) were powdered and extracted with 75% EtOH (1.5 h, 3 × 60 L), and then the solvent was removed by evaporation to afford a crude residue (825 g). The crude residue was suspended in H2O and then subjected to D101 macroporous resin CC, eluted successively with EtOH–H2O (0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100, 30:70, 70:30, 95:5, v/v) to afford four sub-fractions (Fr. A–D). After concentration under vacuum, Fr. B and Fr. C were suspended in H2O and extracted successively with ethyl acetate (10 L × 3) and n-butyl alcohol (10 L × 3). The n-butyl alcohol extract of Fr. B was subjected to silica gel CC eluted with a gradient of CHCl3–MeOH–H2O (90:10:1 to 10:90:5, v/v) to afford five sub-fractions (Fr. Ba–e). Fr. Ba was further purified using a Waters Xbridge OBD Prep C-18 column, eluted with CH3CN–H2O (38:62, v/v) to yield compounds 2 (23 mg), 5 (9 mg), 6 (11 mg) and 7 (35 mg). The n-butyl alcohol part of Fr. C was subjected to silica gel CC eluted with a gradient of CHCl3–MeOH–H2O (90:10:1 to 30:70:5, v/v) to afford Fr. Ca–i. Fr. Ca was chromatographed on an ODS column with a gradient of acetone–H2O (20:80 to 100:0, v/v) as the mobile phase to afford four sub-fractions (Fr. Ca1–4). Fr. Ca1 was further purified using a Waters Xbridge OBD Prep C-18 column, eluted with CH3CN–H2O (47:53, v/v) to yield compounds 3 (12 mg) and 4 (17 mg). Compound 1 (25 mg) was obtained from Fr. Ca3 using a Waters Xbridge OBD Prep C-18 column, eluted with CH3CN–H2O (50:50, v/v).
:100, 30:70, 70:30, 95:5, v/v) to afford four sub-fractions (Fr. A–D). After concentration under vacuum, Fr. B and Fr. C were suspended in H2O and extracted successively with ethyl acetate (10 L × 3) and n-butyl alcohol (10 L × 3). The n-butyl alcohol extract of Fr. B was subjected to silica gel CC eluted with a gradient of CHCl3–MeOH–H2O (90:10:1 to 10:90:5, v/v) to afford five sub-fractions (Fr. Ba–e). Fr. Ba was further purified using a Waters Xbridge OBD Prep C-18 column, eluted with CH3CN–H2O (38:62, v/v) to yield compounds 2 (23 mg), 5 (9 mg), 6 (11 mg) and 7 (35 mg). The n-butyl alcohol part of Fr. C was subjected to silica gel CC eluted with a gradient of CHCl3–MeOH–H2O (90:10:1 to 30:70:5, v/v) to afford Fr. Ca–i. Fr. Ca was chromatographed on an ODS column with a gradient of acetone–H2O (20:80 to 100:0, v/v) as the mobile phase to afford four sub-fractions (Fr. Ca1–4). Fr. Ca1 was further purified using a Waters Xbridge OBD Prep C-18 column, eluted with CH3CN–H2O (47:53, v/v) to yield compounds 3 (12 mg) and 4 (17 mg). Compound 1 (25 mg) was obtained from Fr. Ca3 using a Waters Xbridge OBD Prep C-18 column, eluted with CH3CN–H2O (50:50, v/v).
Characterization of new compounds
Acid hydrolysis
Compounds 1–6 (2 mg each) were individually refluxed with 2 mL of 2 M HCl (HCl–H2O–dioxane, 1:2:3) at 95 °C for 4 h in a water bath and then dioxane was removed by evaporation. Each solution was then diluted with H2O (2 mL) and further extracted with EtOAc (2 mL), with the aqueous layer repeatedly evaporated until a neutral residue was obtained (10–15 times). The residue was then analyzed by TLC over silica gel together with authentic sugar samples using EtOAc–pyridine-absolute EtOH–H2O (8:1:1:2) and aniline–diphenylamine–phosphoric acid (5:5:1) as developing solution and detection solution, respectively. The remaining residue in pyridine (300 μL) was mixed with L-cysteine methyl ester hydrochloride (4 mg) and then heated at 60 °C in an oil bath for 1.5 h. Then 300 μL of hexamethyldisilazane–trimethylchlorosilane (2:1) was added and the mixture was kept at 60 °C for a further 30 min. After centrifugation, the supernatant was analyzed by GC under the following conditions: capillary column, HP-5 (0.32 mm × 30 m × 0.5 μm); flame ionization detection; detector temperature, 280 °C; injection temperature, 250 °C; initial temperature, 150 °C and an initial time of 5 min, 1.1 °C min−1 to 260 °C and then held for 10 min; carrier, N2; split ratio, 1/25. The monosaccharides of compound 1 were identified as D-glucose, L-rhamnose and D-apiofuranose by comparing the retention times (TR) of monosaccharide derivatives with the derivatives prepared in the same procedure from standard sugars. Similarly, D-glucose, D-xylose, L-rhamnose and L-arabinose were identified from compounds 2, 3 and 4, D-glucose, D-xylose and L-rhamnose were identified from compounds 5 and 6.
Cytotoxicity assay
The in vitro cytotoxic activities of the isolates against MDA-MB-435, HepG2 and A549 cell lines were measured by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay as described in the literature32 and 5-fluorouracil was used as positive control. All cells lines were purchased from the Cell Bank of the Shanghai Institute of Chinese Academy of Sciences and incubated at 37 °C in a humidified atmosphere of 95% air and 5% CO2, with high glucose DMEM medium (Gibco, Grand Island, NY, USA) containing 10% fetal bovine serum (Life Technologies Corporation, Carlsbad, CA, USA) used as culture medium. The cells in logarithmic phase were seeded in 96-well plates for 12 h, then the medium was replaced by fresh medium containing test compounds at various concentrations and treated for an additional 48 h. At the end of the treatment period, the medium was replaced by 100 μL of fresh medium containing 0.5 mg mL−1 MTT and the cells were cultured for a further 3 h. The medium solution was then removed and 150 μL of DMSO was added to dissolve the MTT reduction product (formazan crystals). The optical density was measured by detection of the absorbance at 570 nm and reference wavelength of 650 nm on a microplate reader.Conclusions
Steroidal constituents exhibit various biological activities due to diversity of their chemical structures. The clear antitumorigenic properties of steroidal constituents have attracted the attention of pharmaceutical researchers for the development of anticancer drugs. In this work, six new steroidal saponins (1–6) and one known steroidal saponin (7) were obtained and characterized from the root of O. japonicas. Cytotoxicity data of these compounds against MDA-MB-435, HepG2 and A549 cancer cell lines in vitro indicated that compound 1 exhibited significant cytotoxicity with IC50 values ranging from 1.69 to 4.39 μM, compounds 5 and 7 showed moderate cytotoxicity with IC50 values ranging from 9.13 to 29.12 μM. These compounds may have a possibility for use in anticancer drug development.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the National Natural Science Foundation of China (No. 81473317 and 81673555), the Major National Science and Technology Project of China for Significant New Drugs Development (No. 2012ZX09102201-015), Qing Lan Project, the Priority Academic Program Development of Jiangsu Higher Education Institutions, the 2011' Program for Excellent Scientific and Technological Innovation Team of Jiangsu Higher Education, and the Major Project Program of State Key Laboratory of Natural Medicines, China Pharmaceutical University (No. SKLNMZZ201203).Notes and references
- Editorial Committee of Flora of China, Flora of China, Science Press, Beijing, 2014 Search PubMed.
- National Pharmacopoeia Committee, Pharmacopoeia of Peoples Republic of China 2015, Part 1, China Medical Science Press, Beijing, 2015 Search PubMed.
- X. Lu, W. Tong, S. Wang, J. Li, J. Zheng and X. Fan, J. Pharm. Biomed. Anal., 2016, 134, 60–70 CrossRef PubMed.
- Y. Wang, Y. Zhu, K. Ruan, H. Wei and Y. Feng, Carbohydr. Polym., 2014, 114, 183–189 CrossRef CAS PubMed.
- M. Zhao, W. F. Xu, H. Y. Shen, P. Q. Shen, J. Zhang and D. D. Wang, J. Pharm. Biomed. Anal., 2017, 138, 134 CrossRef CAS PubMed.
- Y. L. Zhang, M. Z. Xi, Y. B. Choi and B. H. Lee, J. Med. Food, 2017, 20, 637–645 CrossRef CAS PubMed.
- J. Chen, J. Yuan, L. Zhou, M. Zhu, Z. Shi, J. Song, Q. Xu, G. Yin, Y. Lv, Y. Luo, X. Jia and L. Feng, Biomed. Pharmacother., 2017, 87, 118–126 CrossRef CAS PubMed.
- A. Tada, R. Kasai, T. Saitoh and J. Shoji, Chem. Pharm. Bull., 1980, 28, 2039–2044 CrossRef CAS.
- A. Tada, R. Kasai, T. Saitoh and J. Shoji, Chem. Pharm. Bull., 1980, 28, 1477–1484 CrossRef CAS.
- J. M. Chang, C. C. Shen, Y. L. Huang, M. Y. Chien, J. C. Ou, B. J. Shieh and C. C. Chen, J. Nat. Prod., 2002, 65, 1731–1733 CrossRef CAS.
- N. Li, J. Y. Zhang, K. W. Zeng, L. Zhang, Y. Y. Che and P. F. Tu, Fitoterapia, 2012, 83, 1042–1045 CrossRef CAS PubMed.
- C. L. Duan, Z. Y. Kang, C. R. Lin, Y. Jiang, J. X. Liu and P. F. Tu, J. Asian Nat. Prod. Res., 2009, 11, 876–879 CrossRef PubMed.
- C. Zhou, L. Zou, J. Mo, X. Wang, B. Yang and Q. He, Helv. Chim. Acta, 2013, 96, 1397–1405 CrossRef CAS.
- Z. Iqbal, S. Isojima, A. Noda and Y. Fujii, J. Weed Sci. Technol., 2003, 47, 160–161 CrossRef.
- C. Jiang, Z. H. Liu, L. Li, B. B. Lin, F. Yang and M. J. Qin, J. Asian Nat. Prod. Res., 2012, 14, 491–495 CrossRef CAS PubMed.
- C. L. Duan, Y. Jiang, Y. Jiang, C. R. Lin, J. X. Liu and P. F. Tu, J. Chin. Pharm. Sci., 2009, 18, 236–239 CAS.
- M. Tomoda and S. Kato, Jpn. J. Pharmacogn., 1966, 20, 6–9 Search PubMed.
- M. Tomoda and S. Kato, Chem. Pharm. Bull., 1968, 16, 113–116 CrossRef CAS.
- N. Li, L. Zhang, K. W. Zeng, Y. Zhou, J. Y. Zhang, Y. Y. Che and P. F. Tu, Steroids, 2013, 78, 1–7 CrossRef CAS PubMed.
- C. L. Duan, Y. J. Li, P. Li, Y. Jiang, J. X. Liu and P. F. Tu, Helv. Chim. Acta, 2010, 93, 227–232 CrossRef CAS.
- J. Qi, Z. F. Hu, Y. F. Zhou, Y. J. Hu and B. Y. Yu, Chem. Pharm. Bull., 2015, 63, 187–194 CrossRef CAS PubMed.
- Z. H. Cheng, T. Wu and B. Y. Yu, J. Asian Nat. Prod. Res., 2006, 8, 555–559 CrossRef CAS PubMed.
- Y. F. Zhou, J. Qi, D. N. Zhu and B. Y. Yu, Chin. Chem. Lett., 2008, 19, 1086–1088 CrossRef CAS.
- M. Kuroda, Y. Mimaki, K. Ori, H. Sakagami and Y. Sashida, J. Nat. Prod., 2004, 67, 1690–1696 CrossRef CAS PubMed.
- L. Tang, Z. Wang, H. Wu, A. Yokosuka and Y. Mimaki, Phytochemistry, 2014, 107, 102–110 CrossRef CAS PubMed.
- J. Z. Wang, L. M. Ye and X. B. Chen, Chin. Chem. Lett., 2008, 19, 82–84 CrossRef CAS.
- M. A. Fernandez-Herrera, M. G. Hernandez-Linares, G. Guerrero-Luna, S. Meza-Reyes, S. Montiel-Smith and J. Sandoval-Ramirez, Lett. Org. Chem., 2011, 8, 341–346 CrossRef CAS.
- K. W. Wang, H. Zhang, L. Q. Shen and W. Wang, Carbohydr. Res., 2011, 346, 253–258 CrossRef CAS PubMed.
- J. J. Chen, Z. L. Zhu and S. D. Luo, Acta Bot. Yunnanica, 2000, 22, 97–102 CAS.
- X. Zhou, X. He, G. Wang, H. Gao, G. Zhou, W. Ye and X. Yao, J. Nat. Prod., 2006, 69, 1158–1163 CrossRef CAS PubMed.
- M. Sugiyama, K. Nakano, T. Tomimatsu and T. Nohara, Chem. Pharm. Bull., 2008, 32, 1365–1372 CrossRef.
- Y. Wu, X. M. Wang, S. X. Bi, W. Zhang, R. M. Li, R. J. Wang, B. Y. Yu and J. Qi, RSC Adv., 2017, 7, 13696–13706 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra12363a |
| ‡ Co-first author. |
| This journal is © The Royal Society of Chemistry 2018 |