
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAg3PO4 electrocatalyst for oxygen reduction reaction: enhancement from positive charge†
Yong Qin‡
a,
Fan Li‡a,
Peng Tu‡b,
Yanling Maa,
Wenlong Chena,
Fenglei Shia,
Qian Xianga,
Hao Shana,
Lifu Zhanga,
Peng Tao a,
Chengyi Songa,
Wen Shanga,
Tao Denga,
Hong Zhu*abc and
Jianbo Wu*ac
a,
Chengyi Songa,
Wen Shanga,
Tao Denga,
Hong Zhu*abc and
Jianbo Wu*ac
aState Key Laboratory of Metal Matrix Composites, School of Materials Science and Engineering, Shanghai Jiao Tong University, 800 Dongchuan Road, Shanghai, 200240, People's Republic of China. E-mail: jianbowu@sjtu.edu.cn; hong.zhu@sjtu.edu.cn
bUniversity of Michigan – Shanghai Jiao Tong University Joint Institute, Shanghai Jiao Tong University, 800 Dongchuan Road, Shanghai, 200240, P. R. China
cMaterials Genome Initiative Center, Shanghai Jiao Tong University, 800 Dongchuan Road, Shanghai, 200240, P. R. China
First published on 31st January 2018
Abstract
We have demonstrated Ag3PO4 as an active non-Pt electrocatalyst with enhanced activity compared with Ag for oxygen reduction reaction (ORR). Density functional theory reveals that better ORR performance of Ag atoms on Ag3PO4 surface than that on pure silver surface originates from more appropriate oxygen adsorption on positively charged Ag atoms. Further study of the surface geometry of Ag3PO4 including tetrahedron, rhombic dodecahedron and cube indicates that the highest density of Ag and appropriate oxygen adsorption on {110} surface of rhombic dodecahedral Ag3PO4 lead to the highest ORR activity, which is about 12 times that of Pt catalysts from a commercial perspective. It may be applicable for developing low-cost and highly active non-Pt catalytic materials from a broader range of material systems.
Introduction
Oxygen reduction reaction (ORR) is one of the crucial limiting factors in commercialization of proton-exchange membrane fuel cells due to its sluggish kinetics.1–5 Platinum group metal has been demonstrated to be the most practical ORR catalyst because of its appropriate adsorption of oxygen, which facilitates the adsorption of oxygen on Pt surface for reduction and the desorption of the intermediate OH for turnover.6 In the past few years, great efforts have been made to design active ORR catalysts by shape control, composition engineering and modulation of element distribution.7–16 However, Pt-based electrocatalysts are usually of high cost and from limited reserves, which bring limitations for the commercialization of these catalysts, especially for mass production of vehicles.17 Thus, a large number of substitutes for Pt have been considered including metals (Pd, Fe, Co, Mn, etc.),18,19 metal oxides,20–22 carbon materials,23 Fe–N–C,24 and perovskites.25,26Non-platinum monometallic catalysts such as Pd,27,28 Au,29–31 and Ag32 are capable of catalyzing the ORR. However, most of these cannot resist the oxidative etching in the working condition of the ORR, including the electrolyte and high potential.33,34 For example, Ag is the most abundant precious metal, which is 38 times that of Pt in terms of global reserves,35 illustrating that Ag is much easier to obtain and use for mass production. However, the standard redox potential of Ag is 0.7993 V (ref. 36), which results in the oxidation and degradation of Ag-based catalysts at high potential under working conditions.
In this paper, we have demonstrated that the oxidized Ag+ ion in the lattice of Ag3PO4 can still electro-catalytically reduce oxygen, which makes Ag3PO4 nanoparticles a non-Pt ORR catalyst, and also avoid the aforementioned oxidation limitation. Moreover, Ag+ ions of Ag3PO4 are even more active than Ag atoms. Density functional theory calculation reveals that the positively charged Ag in the lattice of Ag3PO4 leads to a more appropriate oxygen adsorption, and therefore to enhanced ORR activity compared to pure Ag. Further study of the surface geometry of Ag3PO4 by altering Ag3PO4 nanoparticles from tetrahedron to rhombic dodecahedron and cube shows that the highest density of Ag and appropriate oxygen adsorption on {110} surface of rhombic dodecahedral Ag3PO4 lead to the improved ORR activity, which is about 12 times that of Pt catalysts from a commercial perspective.
Experimental section
Materials
Silver acetate (CH3COOAg, 99.5%), sodium phosphate dibasic (Na2HPO4, 99.99%), potassium hydroxide (KOH, 99.99%) and short multi-walled carbon nanotubes (CNT) were purchased from Aladdin. Silver nitrate (AgNO3, 99.8%) and ammonia solution (NH3·H2O) were purchased from Shanghai Ling Feng Chemical Reagent Co. Ltd. Phosphoric acid (H3PO4, 85.0%) was purchased from Sinopharm Chemical Reagent Co. Ltd. Ethanol (CH3CH2OH, 99.7%) was purchased from Changshou City Yangyuan Chemical Co. Ltd. Carbon (C, Vulcan®XC-72R) purchased from Cabot was used as support material. Oxygen (O2, 99.999%) and argon (Ar, 99.999%) were purchased from Shanghai Weichuang Standard Gas Analytical Technology Co. Ltd.Synthesis of Ag3PO4 particles
Ag3PO4 particles were synthesized by a simple method.37 For Ag3PO4 tetrahedron, 12 mmol AgNO3 was dissolved in 80 ml ethanol with strong magnetic stirring. Meanwhile, 20 ml H3PO4 was blended with 80 ml ethanol under rapid magnetic stirring. After AgNO3 was completely dissolved in ethanol solution and these two solutions were transparent, the AgNO3-ethanol mixed solution was added to the H3PO4-ethanol solution drop by drop. Sediment appeared quickly and then disappeared under strong stirring. AgNO3-ethanol mixture was added dropwise continuously until H3PO4-ethanol mixed solution became cloudy; at this moment, the whole H3PO4-ethanol and AgNO3-ethanol suspension were added back to the AgNO3-ethanol solution. There appeared a yellow precipitate immediately. After stirring for 1 hour, the mixed solution turned a bright green colour, indicating the nucleation and growth of Ag3PO4 with specific shapes. Finally, the product was washed with deionized water and ethanol three times each.37For Ag3PO4 rhombic dodecahedron, 0.2 g CH3COOAg was dissolved in 100 ml deionized water. And then an as-prepared Na2HPO4 solution (0.15 M) was added to the above solution dropwise. A golden yellow precipitate was formed. After 1 hour of stirring, the obtained products were washed with deionized water and ethanol three times to remove CH3COO−.
With regard to the Ag3PO4 cube, 0.2 g AgNO3 was dissolved in 100 ml deionized water. Ammonia aqueous solution (0.1 M) was added drop by drop to the AgNO3-water solution. When the above solution became transparent, an as-prepared Na2HPO4 solution (0.15 M) was added to the mixed solution. After stirring for 1 hour, the samples were washed with deionized water and ethanol to remove impurities.38
Finally, these three kinds of obtained samples were dried in the atmosphere, and it was ensured that all moisture was removed from the catalyst.
Material characterization
Scanning electron microscopy (SEM) micrographs were obtained using an LV UHR FE-SEM. The SEM was operated at an accelerating voltage of 5 kV. Transmission electron microscopy (TEM) images were obtained using a JEOL JEM-2100F microscope with an accelerating voltage 200 kV. X-ray diffraction (XRD) patterns were obtained using a Shimadzu LabX XRD-6100 X-ray diffractometer with Cu source. The XRD patterns were recorded from 10° to 90° with a scanning rate of 0.167° s−1.Preparation of catalyst
These three kinds of Ag3PO4 with different morphologies can be used as catalysts for the ORR. In order to improve the ORR performance, Ag3PO4 was mixed with carbon. Vulcan XC-72 carbon and CNT were chosen as the carbon support, becoming Ag3PO4/C and Ag3PO4/CNT catalysts. The carbon and CNT were each dissolved in methanol. After sonicating for 1 hour, Ag3PO4 with different facets were added. And the mass ratio of Ag3PO4 particles to carbon was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4. The mixture was stirred for at least 24 hours. Afterwards, the solid products were obtained by centrifugation. Subsequently, the samples were dried by freeze-drying for ORR tests.
:4. The mixture was stirred for at least 24 hours. Afterwards, the solid products were obtained by centrifugation. Subsequently, the samples were dried by freeze-drying for ORR tests.
Electrochemical measurements
A three-electrode cell was used to test the electrochemical performance. A glassy-carbon rotating disk electrode (RDE), whose area was 0.196 cm2, was used as the working electrode. The counter electrode was a 5 cm platinum wire. And a reversible hydrogen electrode (RHE) was used as the reference, which was put in a separate compartment connected with the main cell through a salt bridge. The electrolyte for Ag3PO4-related ORR tests was 0.1 M KOH. In order to prepare a catalyst for the working electrode, 5 mg of Ag3PO4, Ag3PO4/C and Ag3PO4/CNT were dissolved in 5 ml solution, which was composed of 4 ml deionized water, 1 ml isopropanol and 25 μl Nafion (5%). The mixed solution was sonicated for 15 min, forming a uniform ink. So as to ensure the mass of Ag3PO4 used for the catalyst was constant, we selected 10 μl solutions in pure Ag3PO4 ink. Meanwhile, it is necessary to select 50 μl solutions in Ag3PO4/C and Ag3PO4/CNT ink. This ink was added to the RDE and dried under a warm flow of air. The Pt/C and Ag were designed as 40.82 g cm−2 and 40.82 g cm−2 on the RDE by following a similar procedure. The electrochemical active surface area was obtained from cyclic voltammetry curves, which were measured at room temperature in argon-saturated 0.1 M KOH solution. The potential scan rate was 50 mV s−1. And then the ORR properties were tested in 0.1 M KOH solution that was pumped with oxygen for 30 min in advance. The scan rate for ORR measurements was 10 mV s−1.Results and discussion
Fig. 1a shows a typical SEM micrograph of the as-synthesized Ag3PO4 tetrahedrons. It can be seen that single crystalline Ag3PO4 tetrahedrons with smooth surfaces have been fabricated in large quantities. The average edge length of the Ag3PO4 tetrahedrons is 1140 nm (Fig. S2a†). The as-prepared Ag3PO4 tetrahedron was enclosed completely by four well-defined {111} planes (Fig. 1a and S1a†). Fig. 1b shows a representative SEM image of the Ag3PO4 rhombic dodecahedrons, and the average edge length of these is 478.2 nm with regular rhombic dodecahedral morphology. Furthermore, the SEM and TEM micrographs (Fig. S1b†) indicate that the Ag3PO4 rhombic dodecahedron was enclosed completely by twelve {110} facets. Interestingly, when using [Ag(NH3)2]+ complex replacing silver acetate in this synthetic system, we can obtain uniform Ag3PO4 cubes (Fig. 1c). In addition, the enlarged SEM and TEM (Fig. S1c†) micrographs indicate that the as-prepared Ag3PO4 cube was enclosed by six square {100} planes, whose average edge length is 676.7 nm. Moreover, the XRD patterns (Fig. 1d) clearly show that all of the diffraction peaks of the tetrahedral, rhombic dodecahedral and cubic particles could be indexed to that of body-centered cubic crystalline Ag3PO4 (JCPDS no. 06-0505). And there are no other impurity peaks or trace peaks associated with the precursors found in the patterns of as-synthesized samples.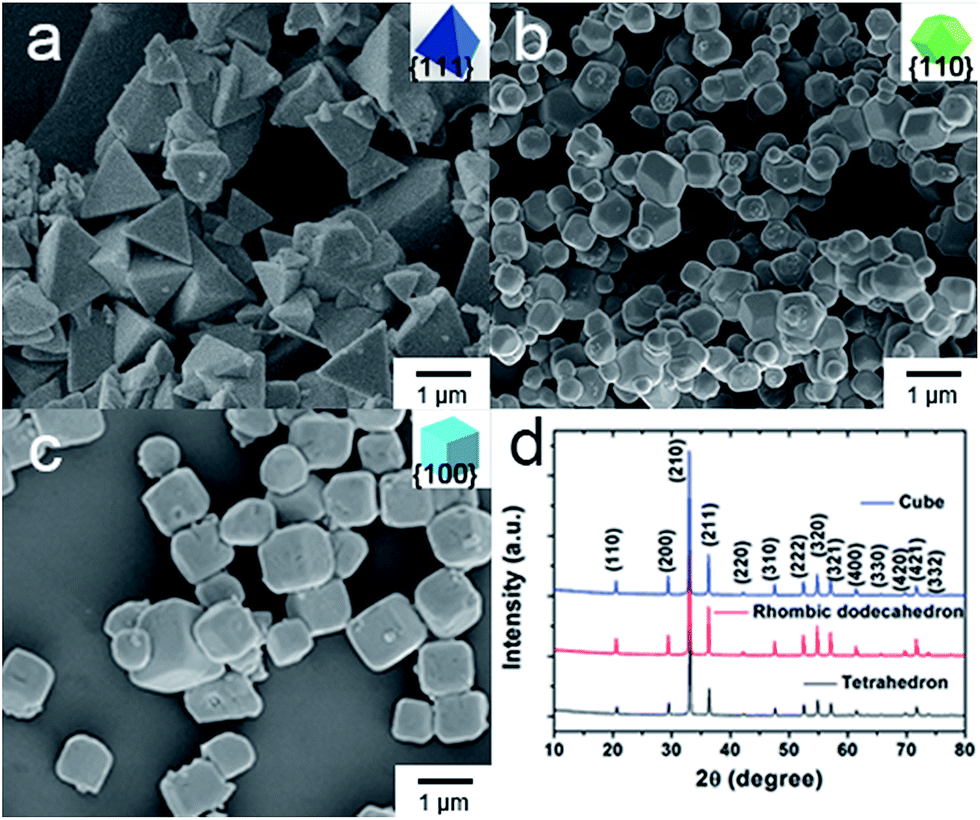 | ||
| Fig. 1 SEM micrographs of Ag3PO4 crystals: (a) tetrahedron, (b) rhombic dodecahedron, (c) cube. (d) XRD patterns of faceted Ag3PO4 crystals. | ||
The catalytic performance of the three Ag3PO4/C catalysts towards the ORR was investigated using the three-electrode method (Fig. 2a–c). The polarization curves of the tetrahedron and rhombic dodecahedron have positive shifts of 5 mV and 30 mV compared with that of the cube. The corresponding ORR mass activity (im) of tetrahedral, rhombic dodecahedral and cubic Ag3PO4 is 4.65 mA mgAg−1, 5.57 mA mgAg−1 and 3.49 mA mgAg−1, respectively. The mass activities are obtained from the following equation:
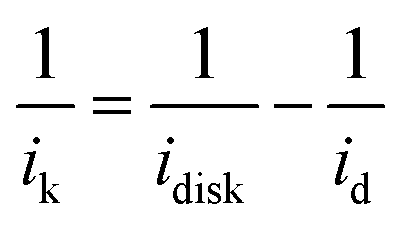 | (1) |
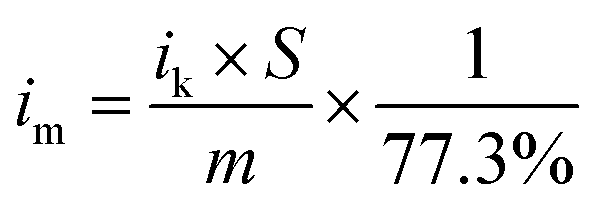 | (2) |
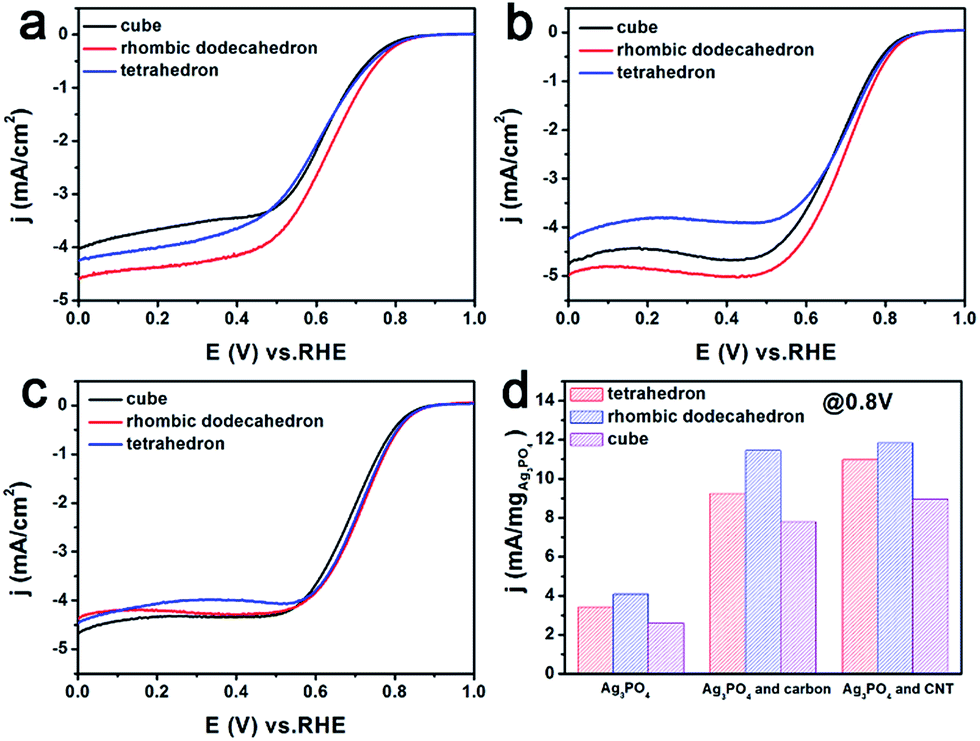 | ||
| Fig. 2 ORR polarization curves of tetrahedral, rhombic dodecahedral and cubic Ag3PO4 catalysts: (a) pure Ag3PO4, (b) Ag3PO4 and carbon, (c) Ag3PO4 and CNT. The mass of Ag3PO4 is 10 μg. (d) Comparison of the mass current density based on 0.8 V (vs. RHE), ORR properties, of cubic, rhombic dodecahedral and tetrahedral Ag3PO4. | ||
When Ag3PO4 is mixed with carbon, the corresponding ORR performances of all Ag3PO4 particles show great improvement, which is 193% for the tetrahedron, 201% for the rhombic dodecahedron, and 216% for the cube. The ORR performance is further boosted when Ag3PO4 is combined with CNT, for which the improvement is 256% for the tetrahedron, 220% for the rhombic dodecahedron, and 271% for the cube.
From Fig. S3,† we can find that the Ag3PO4 crystals were completely dispersed in carbon and CNT. Carbon material such as Vulcan XC-72 and CNT is not only a silver phosphate crystal support, but also plays a role of collaborative catalyst.39 The incorporation of carbon can increase the electrochemical accessible surface areas and electron conductivity.40 And CNT was discovered as a promising support material, due to its specific surface area, electrical conductivity and electrochemical stability.41–43 So the dramatic improvements of catalytic activity are because Ag3PO4 with CNT has better electrical conductivity and greater specific surface area than Ag3PO4 and Ag3PO4 with carbon.
Indeed, the rhombic dodecahedral Ag3PO4/CNT exhibits a mass activity of 13.76 mA mgAg3PO4−1 (0.0178 mA μgAg−1), which is about 12% of that of a commercial Pt/C catalyst (0.1495 mA μgPt−1; Fig. S10†). This result of Pt/C is similar to reported data.44 In fact, considering that the price of Ag is only 1/100 of that of Pt,45,46 the rhombic dodecahedral Ag3PO4/CNT catalyst is about 12 times as active as Pt catalysts from a commercial perspective. Moreover, the global reserve of Pt metal is about 38 times less than that of Ag, illustrating that Ag is much easier to acquire and use for mass production.
The long-term stability of the catalysts was evaluated through accelerated durability tests (Fig. S7 and S8†). The mass activity of rhombic dodecahedral Ag3PO4/CNT shows 12.5% loss after 5000 cycles, while the reported loss for Pt/C catalyst was 31.3% after 5000 cycles.47 Therefore, the rhombic dodecahedral Ag3PO4/CNT catalyst shows better durability than commercial Pt/C.
The ORR performance of the Ag3PO4/C catalysts was further examined as a function of the rotation speed of RDE from 400 to 2500 rpm (Fig. 3a–c). Fig. 3d shows the Koutecky–Levich plots as a function of potential for Ag3PO4/C catalysts bounded with each of {111}, {110} and {100}, which shows different slopes, indicating the different reaction kinetics occurring on the three faceted Ag3PO4/C catalysts. The number of transferred electrons during the ORR has also been investigated via changing the rotation speed of the RDE ranging from 400 to 2500 rpm (Fig. 3a–c).
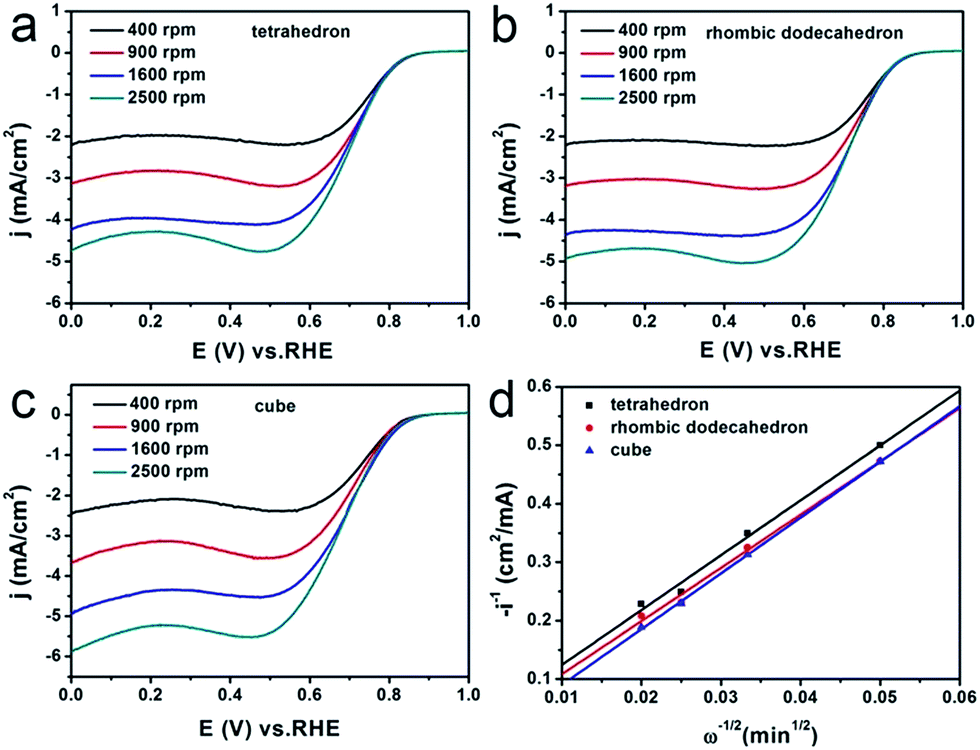 | ||
| Fig. 3 RDE voltammograms for the ORR of (a) tetrahedral, (b) rhombic dodecahedral and (c) cubic Ag3PO4/C electrocatalysts at various rotation rates. (d) Koutecky–Levich plots of the rotating disk current at 0.3 V (vs. RHE). The tests were conducted in O2-saturated 0.1 M KOH solution and the scan rate was kept at 10 mV s−1. | ||
Then, we calculated the slopes of the three Koutecky–Levich plots from the following equation:
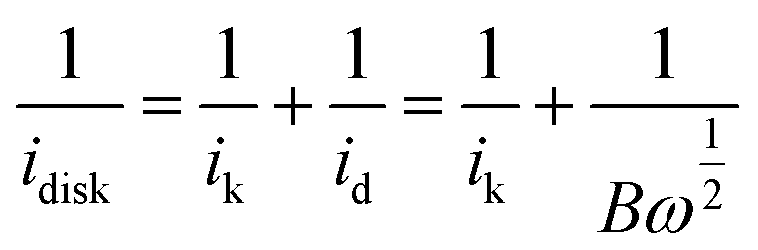 | (3) |
The relationship between the Levich constant B and the number of transferred electrons n during ORR could be obtained as follows:
| B = 0.2nFD02/3C0ν−1/6 = 3.5 × 10−5n | (4) |
Therefore, from the slopes of the three curves shown in Fig. 3d, we can obtain the value of n, which is 3.1, 3.2, and 3.0, respectively, for the tetrahedral, the rhombic dodecahedral, and the cubic Ag3PO4/C catalysts. This indicates that the reduction from O2 to OH− on these Ag3PO4/C catalysts can be regarded as mixed pathways of four-electron and two-electron processes. But the {110} of Ag3PO4 shows a higher portion of the four-electron pathway than the other two facets.
Fig. 4a shows the crystal structure of Ag3PO4 which possesses a body-centered cubic structure. Fig. 4b–d shows the surface atomic configuration of the {111}, {110}, and {100} close-packed planes of Ag3PO4 crystals. Obviously, all the facets including {111}, {110} and {100} have different terminations composed of Ag atoms, O atoms or P atoms. However, the outermost Ag atomic ratio, that is the Ag atomic ratio involved in the reaction, and its atomic activity (Table S2†) are different on the three surfaces (Table S2,† see the calculation details in ESI†).
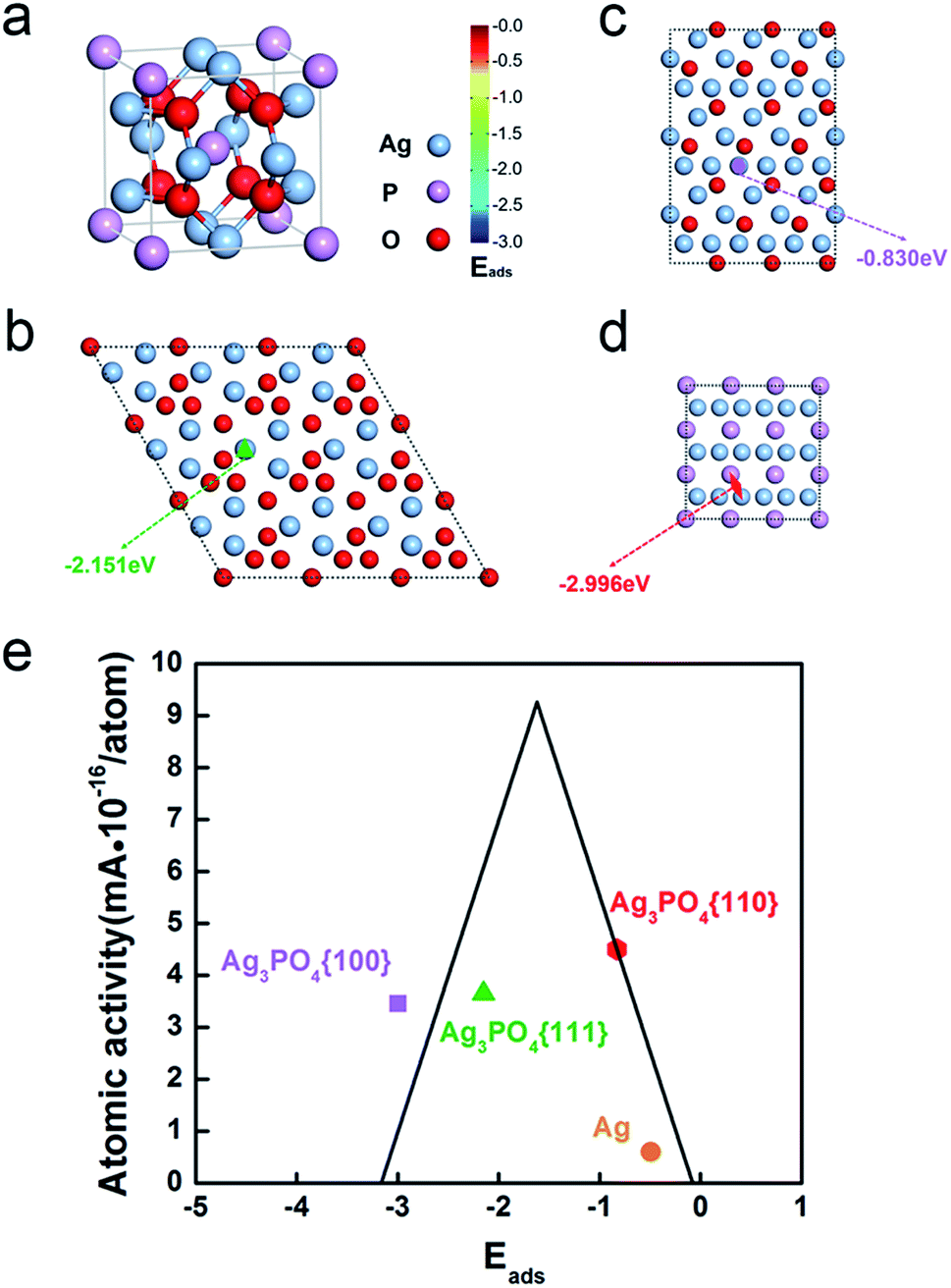 | ||
| Fig. 4 (a) The crystal structure of Ag3PO4. Illustration that the favored adsorption site on (b) the {111} planes of Ag3PO4 crystals is Ag-top site with Eads = −2.151 eV, (c) the {110} planes of Ag3PO4 crystals is Ag-top site with Eads = −0.83 eV, and (d) the {100} planes of Ag3PO4 crystals is Ag–P bridge with Eads = −2.996 eV. (e) Volcano plot of pure Ag and different surfaces of Ag3PO4. Eads is the adsorption energy of oxygen. | ||
We calculated the adsorption of oxygen at various sites on these three surfaces (Table S4†). The most stable oxygen adsorption energies, Eads, on different faceted Ag3PO4 are shown in Fig. 4, namely, −2.151 eV, −0.830 eV and −2.996 eV for {111}, {110} and {100} facets, respectively (Table S4†). The rhombic dodecahedral Ag3PO4 with {110} planes shows the best ORR performance of the three different morphologies of Ag3PO4 because of the appropriate adsorption energy of oxygen based on the Sabatier principle (the volcano plot in Fig. 4e).48
Moreover, the Ag atoms in the lattice of Ag3PO4 also show a better atomic ORR performance than Ag atoms in pure silver. If only counting the surface Ag atoms participating in the ORR, Ag of Ag3PO4 (4.50425 × 10−16 mA per atomAg for Ag3PO4{110}) is 7.5 times better than pure Ag (5.99961 × 10−17 mA per atomAg) (Table S3†). Based on the calculation, the Ag atoms on Ag3PO4 have a stronger binding with oxygen than pure Ag (−0.49 eV in Fig. 4e and Table S5†) due to the positive charge on Ag of Ag3PO4, which leads to a downshift of density of states of Ag, and hence the 7.5 times enhancement of ORR activity.48
Conclusions
In summary, we have demonstrated Ag3PO4 nanoparticles as an active non-Pt ORR catalyst. The Ag+ ions in the lattice of Ag3PO4 show very good ORR performance compared with the Ag atoms of pure Ag due to more appropriate oxygen adsorption on positively charged Ag atoms. Among the three geometric structures of Ag3PO4, namely tetrahedron, rhombic dodecahedron, and cube, the highest density of Ag atoms and appropriate oxygen adsorption on {110} surface of rhombic dodecahedral Ag3PO4 lead to the highest ORR activity, which is about 12 times that of Pt catalysts from a commercial perspective. This fundamental discovery may be applicable for developing low-cost and highly active non-Pt catalytic materials. This demonstration provides an alternative strategy to develop low-cost and highly active non-Pt catalytic materials from a broader range of material candidates.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the thousand talents program for distinguished young scholars from Chinese government and the National Science Foundation of China (91333115 and 51420105009), and startup fund (J. B. W.) and the Zhi-Yuan Endowed fund (T. D.) from Shanghai Jiao Tong University. H. Z. is grateful for the financial support from the Shanghai Sailing Program (16YF1406000) and the computing resources from Shanghai Jiao Tong University Supercomputer Center. The authors thank the Instrumental Analysis Center of Shanghai Jiao Tong University for access to SEM and XRD. The authors also thank the State Key Laboratory of Metal Matrix Composites for access to TEM.Notes and references
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS PubMed.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56, 9–35 CrossRef CAS.
- F. T. Wagner, B. Lakshmanan and M. F. Mathias, J. Phys. Chem. Lett., 2010, 1, 2204–2219 CrossRef CAS.
- M. H. Shao, Q. W. Chang, J. P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594–3657 CrossRef CAS PubMed.
- A. A. Gewirth and M. S. Thorum, Inorg. Chem., 2010, 49, 3557–3566 CrossRef CAS PubMed.
- J. B. Wu and H. Yang, Acc. Chem. Res., 2013, 46, 1848–1857 CrossRef CAS PubMed.
- L. Zhang, L. T. Roling, X. Wang, M. Vara, M. F. Chi, J. Y. Liu, S. I. Choi, J. Park, J. A. Herron, Z. X. Xie, M. Mavrikakis and Y. N. Xia, Science, 2015, 349, 412–416 CrossRef CAS PubMed.
- L. Z. Bu, N. Zhang, S. J. Guo, X. Zhang, J. Li, J. L. Yao, T. Wu, G. Lu, J. Y. Ma, D. Su and X. Q. Huang, Science, 2016, 354, 1410–1414 CrossRef CAS PubMed.
- T. Bian, H. Zhang, Y. Y. Jiang, C. H. Jin, J. B. Wu, H. Yang and D. R. Yang, Nano Lett., 2015, 15, 7808–7815 CrossRef CAS PubMed.
- C. Chen, Y. J. Kang, Z. Y. Huo, Z. W. Zhu, W. Y. Huang, H. L. L. Xin, J. D. Snyder, D. G. Li, J. A. Herron, M. Mavrikakis, M. F. Chi, K. L. More, Y. D. Li, N. M. Markovic, G. A. Somorjai, P. D. Yang and V. R. Stamenkovic, Science, 2014, 343, 1339–1343 CrossRef CAS PubMed.
- D. L. Wang, H. L. L. Xin, R. Hovden, H. S. Wang, Y. C. Yu, D. A. Muller, F. J. DiSalvo and H. D. Abruna, Nat. Mater., 2013, 12, 81–87 CrossRef CAS PubMed.
- J. B. Wu, J. L. Zhang, Z. M. Peng, S. C. Yang, F. T. Wagner and H. Yang, J. Am. Chem. Soc., 2010, 132, 4984–4985 CrossRef CAS PubMed.
- J. B. Wu, A. Gross and H. Yang, Nano Lett., 2011, 11, 798–802 CrossRef CAS PubMed.
- J. B. Wu, L. Qi, H. J. You, A. Gross, J. Li and H. Yang, J. Am. Chem. Soc., 2012, 134, 11880–11883 CrossRef CAS PubMed.
- Y. C. Yan, H. Shan, G. Li, F. Xiao, Y. Y. Jiang, Y. Y. Yan, C. H. Jin, H. Zhang, J. B. Wu and D. R. Yang, Nano Lett., 2016, 16, 7999–8004 CrossRef CAS PubMed.
- Y. L. Xiong, H. Shan, Z. N. Zhou, Y. C. Yan, W. L. Chen, Y. X. Yang, Y. F. Liu, H. Tian, J. B. Wu, H. Zhang and D. R. Yang, Small, 2017, 13, 1603423 CrossRef PubMed.
- Y. N. Xia, Y. J. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS PubMed.
- M. Lefevre, E. Proietti, F. Jaouen and J. P. Dodelet, Science, 2009, 324, 71–74 CrossRef CAS PubMed.
- E. Antolini, Energy Environ. Sci., 2009, 2, 915–931 CAS.
- J. B. Xu, P. Gao and T. S. Zhao, Energy Environ. Sci., 2012, 5, 5333–5339 CAS.
- H. T. Chung, J. H. Won and P. Zelenay, Nat. Commun., 2013, 4, 1922 CrossRef PubMed.
- Y. Y. Liang, Y. G. Li, H. L. Wang, J. G. Zhou, J. Wang, T. Regier and H. J. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS PubMed.
- C. G. Hu and L. M. Dai, Adv. Mater., 2017, 29, 1604942 CrossRef PubMed.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443–447 CrossRef CAS PubMed.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. Shao-Horn, Nat. Chem., 2011, 3, 546–550 CrossRef CAS PubMed.
- R. Bashyam and P. Zelenay, Nature, 2006, 443, 63–66 CrossRef CAS PubMed.
- L. Xiao, L. Zhuang, Y. Liu, J. T. Lu and H. D. Abruna, J. Am. Chem. Soc., 2009, 131, 602–608 CrossRef CAS PubMed.
- C. L. Lee and H. P. Chiou, Appl. Catal., B, 2012, 117, 204–211 CrossRef.
- C. Jeyabharathi, S. S. Kumar, G. V. M. Kiruthika and K. L. N. Phani, Angew. Chem., Int. Ed., 2010, 49, 2925–2928 CrossRef CAS PubMed.
- W. Chen and S. W. Chen, Angew. Chem., Int. Ed., 2009, 48, 4386–4389 CrossRef CAS PubMed.
- S. S. Kim, Y. R. Kim, T. D. Chung and B. H. Sohn, Adv. Funct. Mater., 2014, 24, 2764–2771 CrossRef CAS.
- B. B. Blizanac, P. N. Ross and N. M. Markovic, J. Phys. Chem. B, 2006, 110, 4735–4741 CrossRef CAS PubMed.
- J. B. Wu, W. P. Gao, H. Yang and J. M. Zuo, ACS Nano, 2017, 11, 1696–1703 CrossRef CAS PubMed.
- Y. Y. Jiang, G. M. Zhu, F. Lin, H. Zhang, C. H. Jin, J. Yuan, D. R. Yang and Z. Zhang, Nano Lett., 2014, 14, 3761–3765 CrossRef CAS PubMed.
- Wikipedia, Precious metal, https://en.wikipedia.org/wiki/, accessed 6, 1980.
- W. M. Haynes, CRC handbook of chemistry and physics, CRC Press, Boca Raton, FL, USA, 95th edn, 2014 Search PubMed.
- D. J. Martin, N. Umezawa, X. W. Chen, J. H. Ye and J. W. Tang, Energy Environ. Sci., 2013, 6, 3380–3386 CAS.
- Y. Bi, S. Ouyang, N. Umezawa, J. Cao and J. Ye, J. Am. Chem. Soc., 2011, 133, 6490–6492 CrossRef CAS PubMed.
- W. L. Xu, X. C. Zhou, C. P. Liu, W. Xing and T. H. Lu, Electrochem. Commun., 2007, 9, 1002–1006 CrossRef CAS.
- G. Wu, L. Li, J. H. Li and B. Q. Xu, Carbon, 2005, 43, 2579–2587 CrossRef CAS.
- G. Che, B. B. Lakshmi, C. R. M. And and E. R. Fisher, Langmuir, 1999, 15, 750–758 CrossRef CAS.
- T. Matsumoto, T. Komatsu, K. Arai, T. Yamazaki, M. Kijima, H. Shimizu, Y. Takasawa and J. Nakamura, Chem. Commun., 2004, 840–841, 10.1039/b400607k.
- W. Li, C. Liang, W. Zhou, J. Qiu, H. Li, G. Sun and Q. Xin, Carbon, 2004, 42, 436–439 CrossRef CAS.
- J. B. Wu and H. Yang, Nano Res., 2011, 4, 72–82 CrossRef CAS.
- A. E. S. Sleightholme, J. R. Varcoe and A. R. Kucernak, Electrochem. Commun., 2008, 10, 151–155 CrossRef CAS.
- M. J. Simpson, A. Svendsen and P. L. Chan, Phys. Rev. C: Nucl. Phys., 2007, 30, 463–478 Search PubMed.
- J. Zhu, C. He, Y. Li, S. Kang and P. K. Shen, J. Mater. Chem. A, 2013, 1, 14700–14705 CAS.
- J. K. Norskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra12643c |
| ‡ These authors contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |