
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An unexpected re-arrangement of the antibiotic carbapenem core to new 1,4-diazepin-5-one scaffolds†
Byron K. Petersa,
Rufaro Razuwikaa,
Marivel Samipillaia,
Per I. Arvidsson ab,
Hendrik G. Krugera,
Thavendran Govender*a and
Tricia Naicker*a
ab,
Hendrik G. Krugera,
Thavendran Govender*a and
Tricia Naicker*a
aCatalysis and Peptide Research Unit, University of KwaZulu Natal, Durban, South Africa 4001. E-mail: govenderthav@ukzn.ac.za; naickert1@ukzn.ac.za
bScience for Life Laboratory, Drug Discovery & Development Platform, Division of Translational Medicine and Chemical Biology, Department of Medical Biochemistry and Biophysics, Karolinska Institutet, Stockholm, Sweden
First published on 19th December 2017
Abstract
Herein we report a peculiar organocatalyzed domino-reaction on the carbapenem core structure. As previously reported, 30 mol% of proline yields diazabicyclo[4.2.1]nonane analogues, while we currently report the formation of novel 1,4-diazepin-5-ones from the same starting material in the presence of 100 mol% proline. The inherent chirality of the starting material led to the stereochemical preference of the products with excellent diastereoselectivity (>99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The diazepinone products were confirmed using X-ray diffraction and 2D-NMR structure elucidation. A plausible mechanism of the re-arrangement, involving an unactivated retro-Dieckmann condensation, is also presented.
:1). The diazepinone products were confirmed using X-ray diffraction and 2D-NMR structure elucidation. A plausible mechanism of the re-arrangement, involving an unactivated retro-Dieckmann condensation, is also presented.
Diazepinone analogues have become essential scaffolds due to their pharmacological properties and peptidomimetic functions.1–3 Preparation of these derivatives are currently gaining momentum in modern day organic synthesis with focus on the discovery and optimization of bioactive compounds.4–6 There has been an increase in fragment-based drug discovery and lead optimization which focuses on the fragments' structural shape or complexity.7,8 Fragments of a drug containing a saturated core, with sp3 carbon atoms, are commonly considered to have a 3D character (protruding diffusely in all directions), as opposed to planar aromatic rings (flat and protruding mainly in two planes). Chan and co-worker's review article exemplifies that utilizing more 3D character in drug design would increase the diversity of fragment screening libraries, improve ADMET properties, and provide better starting points for lead generation as well as possibly address ‘difficult’ targets.9 Therefore, moving towards sp3-rather than sp2-character drug scaffolds is now a major focus in the organic/medicinal chemistry community.10,11 Diazepinone containing molecules, aside from being rigid and readily tuneable, possess a high degree of 3D structure. Therefore, the development of catalytic methods for their preparation will be beneficial for expanding the scope of fused heterocycles.
Our group has recently reported several organocatalyzed transformations on the bicyclic carbapenem core as a route to unexplored β-lactam derivatives.12–15 The carbapenem-based drugs have a wide range of activities and also the greatest potencies against Gram positive and negative bacteria.16–20 The high reactivity of β-lactams makes them an important class of compounds in organic synthesis as chiral building blocks for molecules of potential pharmaceutical importance.21,22 Our findings on the Mannich reaction on the carbapenem core has led to some interesting and unexpected products (Scheme 1, A and B).13 Previously, we reported the 2- and 3-component Mannich reactions respectively; the diazabicyclo[4.2.1]nonane formed from reaction B was explained by an intramolecular nucleophilic cyclization re-arrangement due to its close proximity to the lactam ring.13 Herein we report an entirely different re-arrangement induced by increasing the amount of proline used, wherein a new class of fused diazepinones (C) are formed from 1.
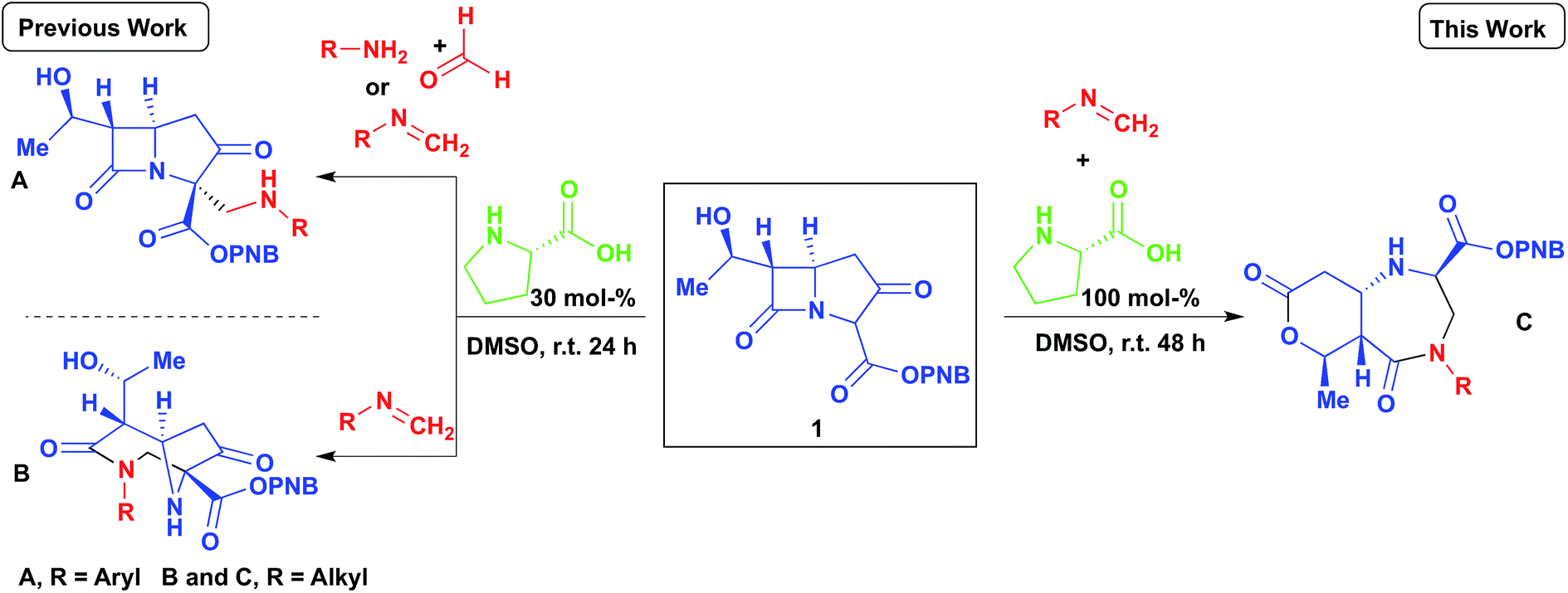 | ||
| Scheme 1 The organocatalyzed transformations of p-nitrobenzyl-6-(1-hydroxyethyl)-1-azabicyclo(3.2.0)heptane-3,7-dione-2-carboxylate (carbapenem), by 3-component Mannich reaction (reaction A) and by 2-component Mannich reactions to bicyclic diazanonane (reaction B) and to 1,4-diazepin-5-one (reaction C) respectively. | ||
Results and discussion
As an extension of our previous work,13 the PNB protected carbapenem was smoothly converted into diazabicyclo[4.2.1]nonane derivatives (via the enamine intermediate), bearing a variety of groups (Scheme 2). Phenyl, methyl, and COOMe were all tolerated and the products furnished in good yields. Moreover, crude imines generated from the reaction of paraformaldehyde and the corresponding amine in the presence of anhydrous MgSO4 could be used without any negative impact on the yield of 2. Diastereomeric ratios were >99:1 from the crude 1H NMR spectrum. This was attributed to the inherent chirality on the carbapenem scaffold.14
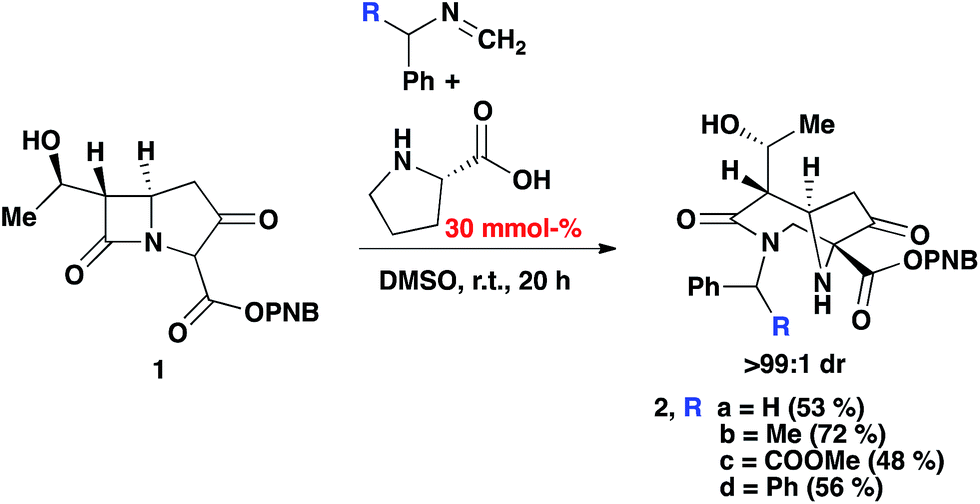 | ||
| Scheme 2 2-Component Mannich reaction of an imine and carbapenem, with 30% mmol (S)-proline in DMSO. | ||
The reaction was subjected to an increased amount of proline while maintaining the same concentration of carbapenem. Unexpectedly, at 100 mol% proline it was observed that after 48–72 hours, an entirely new product was formed (Scheme 3). The differences in the transformation of the carbapenem-based products were detected in the 1H NMR, LC-MS retention time and by analysis of a single crystal with X-ray diffraction (vide infra). These analyses revealed that a new heterocyclic type structure had formed (3). Expanding the scope of the reaction further produced diazepinones for both R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Me and COOMe (3b and 3c). Even with a bulky 1-naphthyl group the product was formed smoothly in 72 hours in good yield (3d). Addition of 200 mol% proline or even a larger excess brought about the same result. Reducing the amount of proline to 30% generated a product mixture after 48 hours, with characteristic signals for both 2 and 3 being detected at a ratio of 61:38 in the crude 1H NMR spectrum. Upon isolation of product 2b followed by subjection to the aforementioned reaction conditions with an excess of proline after 24 hours resulted in the formation of product 3b, confirming that 2b is a necessary intermediate to 3b. A linear type relationship was observed for the formation of 3b over a 12–48 hour period with increasing amounts of proline. For time studies using fixed amounts of proline, a steady state was observed after 12 hours, indicating that the formation of product 3b is strongly dependent on the amount of proline that is present.
Me and COOMe (3b and 3c). Even with a bulky 1-naphthyl group the product was formed smoothly in 72 hours in good yield (3d). Addition of 200 mol% proline or even a larger excess brought about the same result. Reducing the amount of proline to 30% generated a product mixture after 48 hours, with characteristic signals for both 2 and 3 being detected at a ratio of 61:38 in the crude 1H NMR spectrum. Upon isolation of product 2b followed by subjection to the aforementioned reaction conditions with an excess of proline after 24 hours resulted in the formation of product 3b, confirming that 2b is a necessary intermediate to 3b. A linear type relationship was observed for the formation of 3b over a 12–48 hour period with increasing amounts of proline. For time studies using fixed amounts of proline, a steady state was observed after 12 hours, indicating that the formation of product 3b is strongly dependent on the amount of proline that is present.
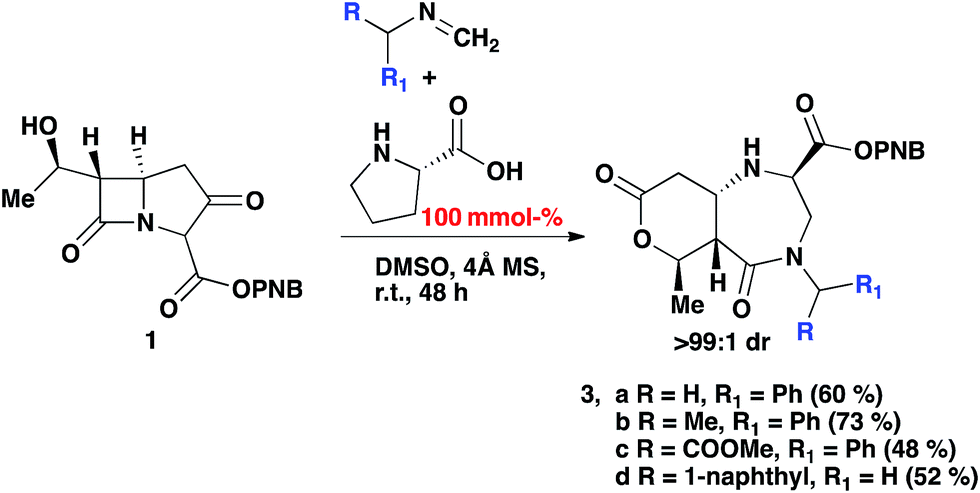 | ||
| Scheme 3 2-Component Mannich reaction of an imine and carbapenem, with 100% mmol (S)-proline and 4 Å MS in DMSO. | ||
Diazepinone heterocycles are frequently encountered in natural products and pharmaceutically active molecules.1,23–25 Studies have also shown that their biological activities can be heightened when fused with aromatic rings. Therefore, these products boast high value to the pharmaceutical industry as novel core structures for drug candidates and as synthetic intermediates. The stereocenters of the rearranged products were assigned to be (R)C2, (S)C7, (R)C8 and (S)C14 based on the X-ray structure of 3b (Fig. 1).26 The absolute configuration of the other products were assigned by the analogy. The six membered tetrahydro-2H-pyran-2-one ring of undec-6,10-dione appeared in a twisted boat conformation while the seven membered diazepane ring is in a chair conformation.
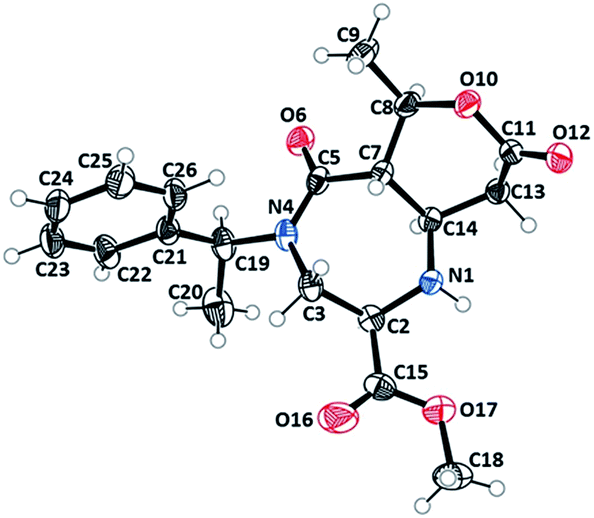 | ||
| Fig. 1 X-ray crystal structure (ORTEP) of 3b analogue.26 | ||
In Scheme 4 we propose a mechanism to rationalize the origins of this useful re-arrangement. First 2 is formed as suggested previously (i, 2)13 via an intermediate (ib, 6), then an excess of proline participates in the formation of an iminium intermediate (ii, 4). This highly electrophilic species can then undergo an intermolecular nucleophilic attack from the alcohol (iii, 5). Hydration to regenerate the proline followed by a retro-Dieckmann generates the diazepinone scaffold (iv, 3).
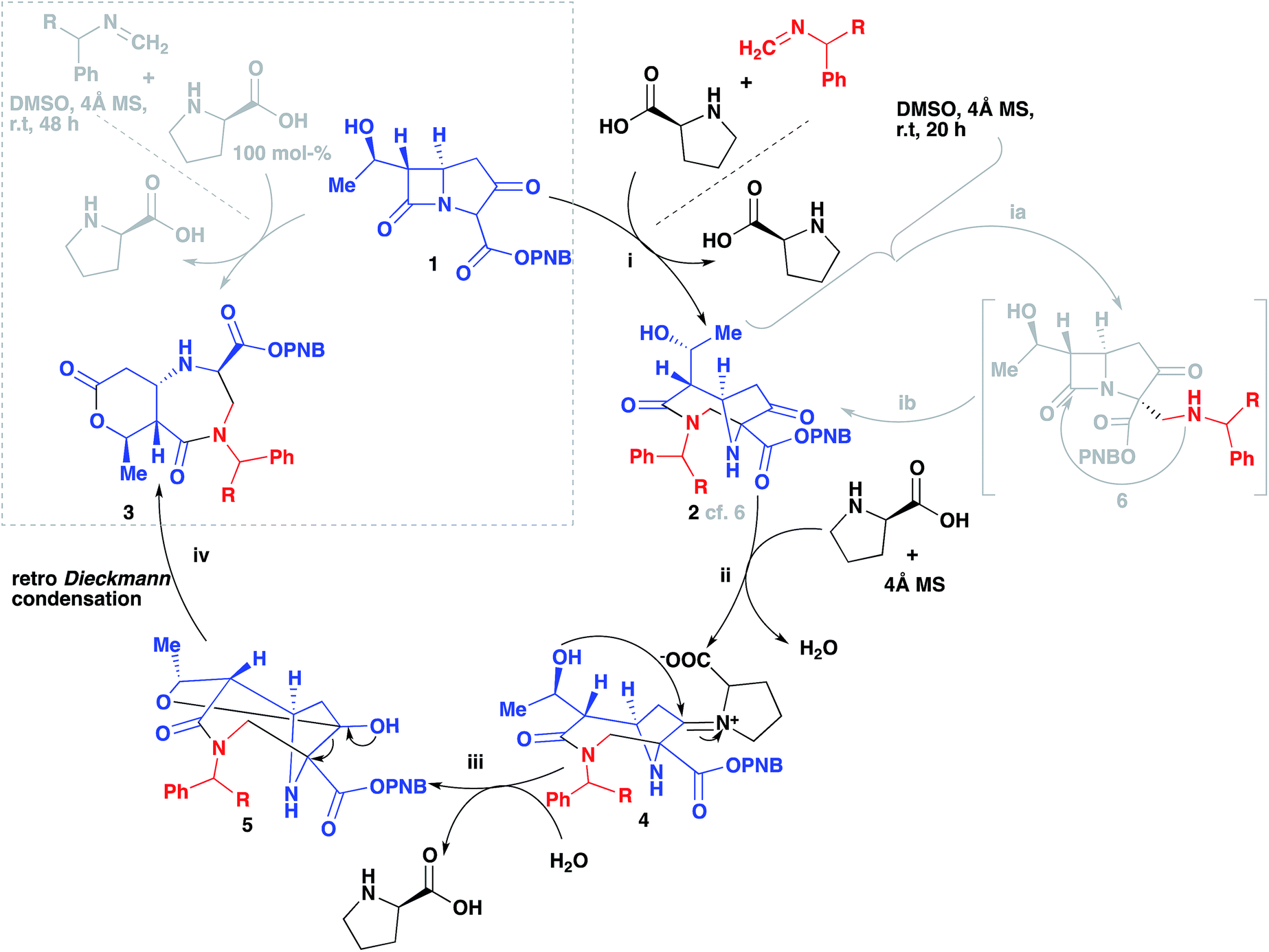 | ||
| Scheme 4 The proposed re-arrangement mechanism of 3a–d in the presence of excess (S)-proline. | ||
Conclusions
This methodology demonstrated that an organocatalyzed Mannich reaction could alter the reactivity of the carbapenem core into a series of novel diazepinone compounds. High diastereoselectivities were observed in all of the reaction products (>99:1). The structure of the fused diazepinone scaffold was also supported by a single X-ray crystal structure as well as full 2D NMR elucidation (see ESI†). In both cases, multiple bond formation takes place in one-pot, an otherwise rare and challenging synthetic operation. To our knowledge this is the first time that such a re-arrangement has been observed by any β-lactam analogue. The new highly functionalised scaffolds can be further transformed to allow for the preparation of unexplored fused diazepinone libraries containing 7 or 8-membered heterocyclic rings that could be used to address the ever-increasing intricacy of biological targets/systems from various diseases.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Research Foundation (South Africa), the University of KwaZulu Natal and Aspen Pharmacare for financial support.Notes and references
- A. A. Dörr and W. D. Lubell, Org. Lett., 2015, 17, 3592–3595 CrossRef PubMed.
- C. G. Joseph, K. R. Wilson, M. S. Wood, N. B. Sorenson, D. V. Phan, Z. Xiang, R. M. Witek and C. Haskell-Luevano, J. Med. Chem., 2008, 51, 1423–1431 CrossRef CAS PubMed.
- K. Upadhyay, A. Manvar, K. Rawal, S. Joshi, J. Trivedi, R. Chaniyara and A. Shah, Chem. Biol. Drug Des., 2012, 80, 1003–1008 CAS.
- A. A. Fesenko, M. S. Grigoriev and A. D. Shutalev, J. Org. Chem., 2017, 82, 8085–8110 CrossRef CAS PubMed.
- M. Viviano, C. Milite, D. Rescigno, S. Castellano and G. Sbardella, RSC Adv., 2015, 5, 1268–1273 RSC.
- R. Costil, Q. Lefebvre and J. Clayden, Angew. Chem., Int. Ed., 2017, 56, 14602–14606 CrossRef CAS PubMed.
- C. W. Murray, M. L. Verdonk and D. C. Rees, Trends Pharmacol. Sci., 2012, 33, 224–232 CrossRef CAS PubMed.
- A. W. Hung, A. Ramek, Y. Wang, T. Kaya, J. A. Wilson, P. A. Clemons and D. W. Young, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6799–6804 CrossRef CAS PubMed.
- M. Aldeghi, S. Malhotra, D. L. Selwood and A. W. E. Chan, Chem. Biol. Drug Des., 2014, 83, 450–461 CAS.
- O. E. Jerry and Z. Weifan, Curr. Top. Med. Chem., 2010, 10, 669–679 CrossRef.
- F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed.
- Z. E. D. Cele, P. I. Arvidsson, H. G. Kruger, T. Govender and T. Naicker, Eur. J. Org. Chem., 2015, 2015, 638–646 CrossRef CAS.
- Z. E. D. Cele, S. A. Pawar, T. Naicker, G. E. M. Maguire, P. I. Arvidsson, H. G. Kruger and T. Govender, Eur. J. Org. Chem., 2014, 2014, 2253–2260 CrossRef CAS.
- S. A. Pawar, S. Alapour, S. Khanyase, Z. E. D. Cele, S. Chitti, H. G. Kruger, T. Govender and P. I. Arvidsson, Org. Biomol. Chem., 2013, 11, 8294–8297 CAS.
- S. Khanyase, T. Naicker, G. E. M. Maguire, H. G. Kruger, P. I. Arvidsson and T. Govender, Tetrahedron: Asymmetry, 2014, 25, 969–973 CrossRef CAS.
- A. Batirel, I. Balkan, O. Karabay, C. Agalar, S. Akalin, O. Alici, E. Alp, F. Altay, N. Altin and F. Arslan, Eur. J. Clin. Microbiol. Infect. Dis., 2014, 33, 1311–1322 CrossRef CAS PubMed.
- J. S. Bradley, J. Garau, H. Lode, K. V. I. Rolston, S. E. Wilson and J. P. Quinn, Int. J. Antimicrob. Agents, 1999, 11, 93–100 CrossRef CAS PubMed.
- N. Heinrich, R. Dawson, J. du Bois, K. Narunsky, G. Horwith, A. J. Phipps, C. A. Nacy, R. E. Aarnoutse, M. J. Boeree, S. H. Gillespie, A. Venter, S. Henne, A. Rachow, P. P. J. Phillips, M. Hoelscher and A. H. Diacon, J. Antimicrob. Chemother., 2015, 70(5), 1558–1566 CrossRef CAS PubMed.
- A. C. Rodloff, E. J. C. Goldstein and A. Torres, J. Antimicrob. Chemother., 2006, 58, 916–929 CrossRef CAS PubMed.
- P. M. Shah and R. D. Isaacs, J. Antimicrob. Chemother., 2003, 52, 538–542 CrossRef CAS PubMed.
- C. T. Brain, A. Chen, A. Nelson, N. Tanikkul and E. J. Thomas, Tetrahedron Lett., 2001, 42, 1247–1250 CrossRef CAS.
- A. Chen, A. Nelson, N. Tanikkul and E. J. Thomas, Tetrahedron Lett., 2001, 42, 1251–1254 CrossRef CAS.
- G. I. Yranzo and E. L. Moyano, in Comprehensive Heterocyclic Chemistry III, ed. C. A. Ramsden, E. F. V. Scriven and R. J. K. Taylor, Elsevier, Oxford, 2008, pp. 399–431, DOI:10.1016/B978-008044992-0.01214-1.
- D. Y. Wang, M. I. Abboud, M. S. Markoulides, J. Brem and C. J. Schofield, Future Med. Chem., 2016, 8, 1063–1084 CrossRef CAS PubMed.
- O. Monasson, M. Ginisty, J. Mravljak, G. Bertho, C. Gravier-Pelletier and Y. Le Merrer, Tetrahedron: Asymmetry, 2009, 20, 2320–2330 CrossRef CAS.
- See ESI† for details. CCDC 1455806 contains the supplementary crystallographic data for this paper.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1455806. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra12688c |
| This journal is © The Royal Society of Chemistry 2018 |