
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSensitive determination of trace Cd(II) and Pb(II) in soil by an improved stripping voltammetry method using two different in situ plated bismuth-film electrodes based on a novel electrochemical measurement system†
Guo Zhao ab,
Hui Wangab and
Gang Liu*ab
ab,
Hui Wangab and
Gang Liu*ab
aKey Lab of Modern Precision Agriculture System Integration Research, Ministry of Education of China, China Agricultural University, Beijing 100083, P. R. China. E-mail: pac@cau.edu.cn; 1252624780@qq.com; Fax: +86-010-62736741; Tel: +86-010-62736741
bKey Lab of Agricultural Information Acquisition Technology, Ministry of Agricultural of China, China Agricultural University, Beijing 100083, P. R. China
First published on 30th January 2018
Abstract
In this study, a simple but effective electrochemical method was developed to minimize the interference from real soil samples and increase the sensitivity of Pb(II) and Cd(II) detection by square-wave anodic stripping voltammetry (SWASV) using a novel electrochemical measurement system, which can be used for the on-site determination of trace Cd(II) and Pb(II) in real soil samples. The method involved performing SWASV following double deposition and stripping steps at two in situ plated bismuth-film electrodes with drastically different surface properties. Pb(II) and Cd(II) were first deposited on an in situ plated bismuth-film graphite carbon paste electrode (Bi/GCPE). When the first deposition was finished, the GCPE was moved to a micro-electrolytic cell to perform the first stripping step. The following measurements were performed with the other deposition and stripping steps using a highly sensitive in situ plated bismuth-film multiwalled carbon nanotube–Nafion composite modified glassy carbon electrode (Bi/MWCNT–Nafion/GCE) as the working electrode. Pb(II), Cd(II) and Bi(III) stripped from the GCPE in the micro-electrolytic cell were partially deposited on the MWCNT–Nafion/GCE, and the stripping current signals were obtained from their oxidation during the second stripping step. Considering the small volume of the micro-electrolytic cell, the concentrations of Cd(II) and Pb(II) were drastically higher than those in the bulk solution, and therefore, the detection limits were reduced. Under the optimized conditions, the concentrations in the linear range spanned from 1.0 to 45.0 μg L−1 for both Pb(II) and Cd(II), with a detection limit of 0.03 μg L−1 for Pb(II) and 0.02 μg L−1 for Cd(II) (S/N = 3). Finally, analyses of real samples were performed to detect trace levels of Pb(II) and Cd(II) in soil with satisfactory results.
Introduction
Cadmium and lead are well-known toxic pollutants in the environment, and even low levels of these pollutants have teratogenic, nervous, reproductive and carcinogenic toxicity to humans.1–3 Cadmium and lead can enter soil from natural deposits in the earth or from agricultural and industrial practices and can subsequently accumulate in vital organs, exhibiting harmful effects to the endocrine system, brain, heart, kidneys and nervous system of species throughout the food chain.4 The contamination of soil with cadmium and lead has been reported in various regions of the world, particularly in developing countries.5,6 Therefore, the quantitative analysis of Pb(II) and Cd(II) in the soil is very important.Over the past several decades, the development of highly effective analytical methods, such as inductively coupled plasma atomic emission spectroscopy (ICP-AES),7 atomic fluorescence spectrometry,8 atomic absorption spectrometry (AAS),7 colourimetric analysis,9 inductively coupled plasma mass spectrometry (ICP-MS)10,11 and ultraviolet-visible (UV-vis) spectrometry,12,13 for the determination of toxic heavy metals (HMs) at trace levels has attracted increasing interest. Although these methods are sensitive for the determination of HMs at trace levels, long analysis times and tedious sample preparation methods are needed to meet requirements for real sample analysis. Consequently, these techniques are not suitable for on-field analysis and emergency detection.
Electrochemical methods, which have the unique advantages of ease of use, low cost, short analysis time and high sensitivity,14,15 have gained significant interest in the detection of HMs as alternatives to spectroscopic-based detection methods. Anodic stripping voltammetry (ASV)16–18 is one of the most commonly used methods to determine different types of HMs because of the unique advantages of ASV, such as low detection limit, high sensitivity and wide linear dynamic range. Square-wave ASV (SWASV) has been widely used in the analysis of HMs at trace levels with high sensitivity and stability.19–21
However, soil samples contain many complicated interferents and complex matrices, which result in enormous gaps between real conditions and lab measurements. The presence of natural organic compounds in soil samples and the interference caused by other HMs, which have negative impacts on the stripping procedure, are the main problems associated with electrochemical methods. These drawbacks increase the electrolyte background to high levels and interfere with detection of the target HMs. To the best of our knowledge, few reports22,23 of HM detection in soil samples using ASV methods have been published in recent years. Therefore, a method with a high anti-interference ability and high sensitivity would be a significant advancement to improve the measurement performance of stripping voltammetry methods for the detection of trace levels of HMs in soil. Chemically modified electrodes (CMEs) are being actively studied to minimize interference from other metal ions, which can effectively improve the detection performances of electrodes by providing effective preconcentration, a selective reaction surface, good repeatability, and low background current over a wide potential range.24–27
For many years, carbon paste electrodes (CPEs) have experienced significant popularity for use in electrochemical analysis because of their low residual currents, low cost and renewable surfaces. The presence of monolayer and/or multilayer graphene provide graphite carbon paste electrodes (GCPEs) with numerous excellent characteristics, such as remarkable accumulation ability and dramatically large specific surface area,28 which are formed with the help of a grinding process.29 Multiwalled carbon nanotubes (MWCNTs) have been widely used as modifiers for electrodes due to their unique features, such as highly efficient catalytic activity towards certain target analytes, unique electrical conductivity and large electrochemically active surface area. Modified electrodes based on bismuth films and carbon nanotubes are excellent tools for the electrochemical analysis of trace HMs.30–33 Nafion (NA) has gained significant attention as a type of electrode modifier for HM determination due to its special characteristics, such as good HM preconcentration ability and excellent mechanical stability.34 Moreover, NA has been successfully applied to modify electrodes in combination with mercury and bismuth films.35,36
In the investigations described in this paper, a novel method is developed to simultaneously minimize the interference of other HMs on the target HMs and improve the determination sensitivity for Pb(II) and Cd(II) by SWASV. The procedure was performed with an electrochemical measurement system containing an electrochemical workstation, a micro-electrolytic cell and two combined working electrodes (i.e., Bi/GCPE and Bi/MWCNT–Nafion/glassy carbon electrode (Bi/MWCNT–Nafion/GCE); Bi/GCPE = in situ plated bismuth-film GCPE and Bi/MWCNT–Nafion/GCE = in situ plated bismuth-film MWCNT–Nafion/GCE), which possessed significantly different surface area properties. Under optimized conditions, the application of ASV and the electrochemical measurement system following the simultaneous deposition and stripping using two different combined electrodes in one measurement contributed to a decrease in the detection limits of Cd(II) and Pb(II) due to concentrating effects. Moreover, the interference of other HMs on the determination of Pb(II) and Cd(II) by SWASV was effectively improved by skillfully transferring the location of the detection of HMs by SWASV from soil extracts to acetate buffer solutions, which supplied a detection environment without interference from other HMs. Finally, the analysis of Cd(II) and Pb(II) in soil samples was performed to validate the feasibility and practicality of this method.
Experimental
Reagents and chemicals
Graphite powder (size: <30 μm, spectrum pure) and paraffin oil were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). MWCNTs (length: 10–30 mm; diameter: <8 nm; purity: >95 wt%) were purchased from Shenzhen Nanotech Port Co., Ltd. (Shenzhen, China). Nafion was obtained from Aldrich (Sigma-Aldrich, USA) and then diluted to 1 wt% with pure ethanol. Stock solutions of Pb(II), Cd(II) and Bi(III) (1000 mg L−1) were obtained from the National Standard Reference Materials Center of China and then diluted as required. Acetate buffer solution (0.1 M) was used as the supporting electrolyte for the determination of Pb(II) and Cd(II). All other chemicals were used without further purification and were of analytical grade. Millipore-Q (18.2 MΩ) water was used in all experiments.Apparatus
A JEOL JSM-6701F (Japan) field-emission scanning electron microscope was used to preform scanning electron microscopy (SEM) and energy dispersive spectroscopy (EDS). Additionally, a CHI660D electrochemical workstation (Shanghai CH Instruments, China) was used to perform SWASV.Fabrication of the GCPE and MWCNT–Nafion/GCE
The GCPE was fabricated according to the following procedure. Paraffin oil and graphite powder were mixed together in a mortar. Then, the mixture was firmly packed into the cavity of a Teflon holder (10.0 mm in diameter), and a copper wire was used to establish electrical contact. The prepared electrode was polished on a piece of weighing paper. A fresh surface was obtained by smoothing the surface of the electrode onto the very smooth weighing paper.The MWCNT–Nafion/GCE composite was prepared according to the following procedure. The GCE surface was polished by 0.05 mm alumina powder and then rinsed with HNO3–H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by volume), absolute ethanol, and water. The MWCNT dispersion was prepared by adding 1 mg of MWCNTs into 4 mL of a dimethyl formamide (DMF) solution to obtain a 0.25 mg mL−1 black suspension with the help of ultrasonic agitation for 2.5 h. Then, 500 μL of 1 wt% Nafion was added to the MWCNT dispersion to form a composite solution of MWCNT–Nafion. Next, a 6 μL suspension of the MWCNT–Nafion composite was coated onto the GCE surface and solidified by irradiating for 10 min using an infrared lamp to obtain the MWCNT–Nafion/GCE.
:1 by volume), absolute ethanol, and water. The MWCNT dispersion was prepared by adding 1 mg of MWCNTs into 4 mL of a dimethyl formamide (DMF) solution to obtain a 0.25 mg mL−1 black suspension with the help of ultrasonic agitation for 2.5 h. Then, 500 μL of 1 wt% Nafion was added to the MWCNT dispersion to form a composite solution of MWCNT–Nafion. Next, a 6 μL suspension of the MWCNT–Nafion composite was coated onto the GCE surface and solidified by irradiating for 10 min using an infrared lamp to obtain the MWCNT–Nafion/GCE.
Soil sample preparation
The soil samples were collected from a farm in China. Briefly, the samples were first ground by a mortar before being sieved with a 200 μm sieve. A portion (12.5 g) of the treated soil samples was placed into an extraction cell, and 500 mL of 0.11 M acetic acid was added to obtain extracts of the soil samples. An ultrasonic processor was used to further process the soil sample solutions at room temperature for 10 min to obtain the preliminary soil extracts. Then, the obtained extracts were centrifuged using a portable centrifuge with a rotational speed of 2000 rpm for 2 min to achieve phase separation, and then 20 mL aqueous phase (i.e., the extract solution used for detection) was loaded into the 25 mL electrolyte cell for further HMs detection. The ultrasonic-assisted extraction for soil samples has been verified by Krasnodêbska-Ostrêga et al.,37 which indicate that the ultrasonic-assisted extraction is comparable to 16 h processing of BCR made by European Union Community. According to our previous research,38,39 the pH values of the soil sample extracts were adjusted to 5.0 by adding NaOH solution (0.11 M) before the HMs in real samples could be determined.Construction of the electrochemical measurement system
The electrochemical measurement system presented in this paper for the sensitive detection of trace levels of Pb(II) and Cd(II) consisted of an electrochemical workstation, a micro-electrolytic cell and two combined working electrodes (i.e., GCPE and MWCNT–Nafion/GCE), which differed significantly in their surface area properties. The first combined electrode system (three-electrode system), as shown in Fig. S1A ESI,† was composed of the GCPE, a Pt-wire counter electrode and a Ag/AgCl reference electrode, which was used for the first deposition and the stripping step, and the second combined electrode system (three-electrode system), as shown in Fig. S1B ESI,† was composed of the MWCNT–Nafion/GCE, a Pt-wire counter electrode and a Ag/AgCl reference electrode, which was used for the second deposition and stripping step. A magnetic stir bar was placed into the stirring chamber to stir the extract solution used for detection during the deposition steps. The system was computer controlled using the CHI660D controlling program.As shown in Fig. S1 ESI,† the height and diameter of the stirring chamber were 48 mm and 22 mm, respectively, which were determined by the height of the composite electrode and the overall size of the system. The body of the composite electrode was made of PTFE and had a height of 10 mm. The volume of acetate buffer solution added to the stirring chamber during the second deposition and stripping step greatly influences the detection performance of this method. On the one hand, if the volume is too large, it would reduce the concentration performance of HMs, and then reduce the detection sensitivity. On the other hand, if the volume is too small, a vortex would be formed in the stirring chamber, which would affect the detection results. In addition, the volume of acetate buffer solution added to the stirring chamber is also determined by the size of the three-electrode system. Considering the above situation, 1 mL of acetate buffer solution (0.1 M, pH 5.0) was added to the stirring chamber for the second deposition and stripping step.
Electrochemical determination procedure
The procedure for the developed method, following the double deposition and stripping steps, for the preconcentration and detection of trace levels of Cd(II) and Pb(II) using an electrochemical measurement system is shown in Fig. 1. The SWASV detection of Pb(II) and Cd(II) was performed as follows. First, 500 mL of a real soil sample extract solution (0.1 M acetate buffer solution) was added into a beaker, and Bi(III) and potassium ferrocyanide were added to achieve a solution containing 600 μg L−1 Bi(III) and 120 μg L−1 ferrocyanide ions. Then, the first combined electrode, which contained the GCPE working electrode, was placed in the beaker containing the bulk solution with 600 μg L−1 Bi(III) to perform the first deposition step. The deposition of Cd(II) and Pb(II) was performed at a potential of −1.2 V for 700 s while stirring the solution. Cd(II) and Pb(II) were in situ electrodeposited onto the bismuth-film-modified GCPE. During the deposition process, a magnetic stir bar was used to stir the solution in the beaker. After the first deposition step, the first combined electrode was quickly transferred to the micro-electrolytic cell containing 1 mL of testing buffer solution (0.1 M, pH 5.0 acetate buffer solution), and then, the first stripping step was performed under stirring.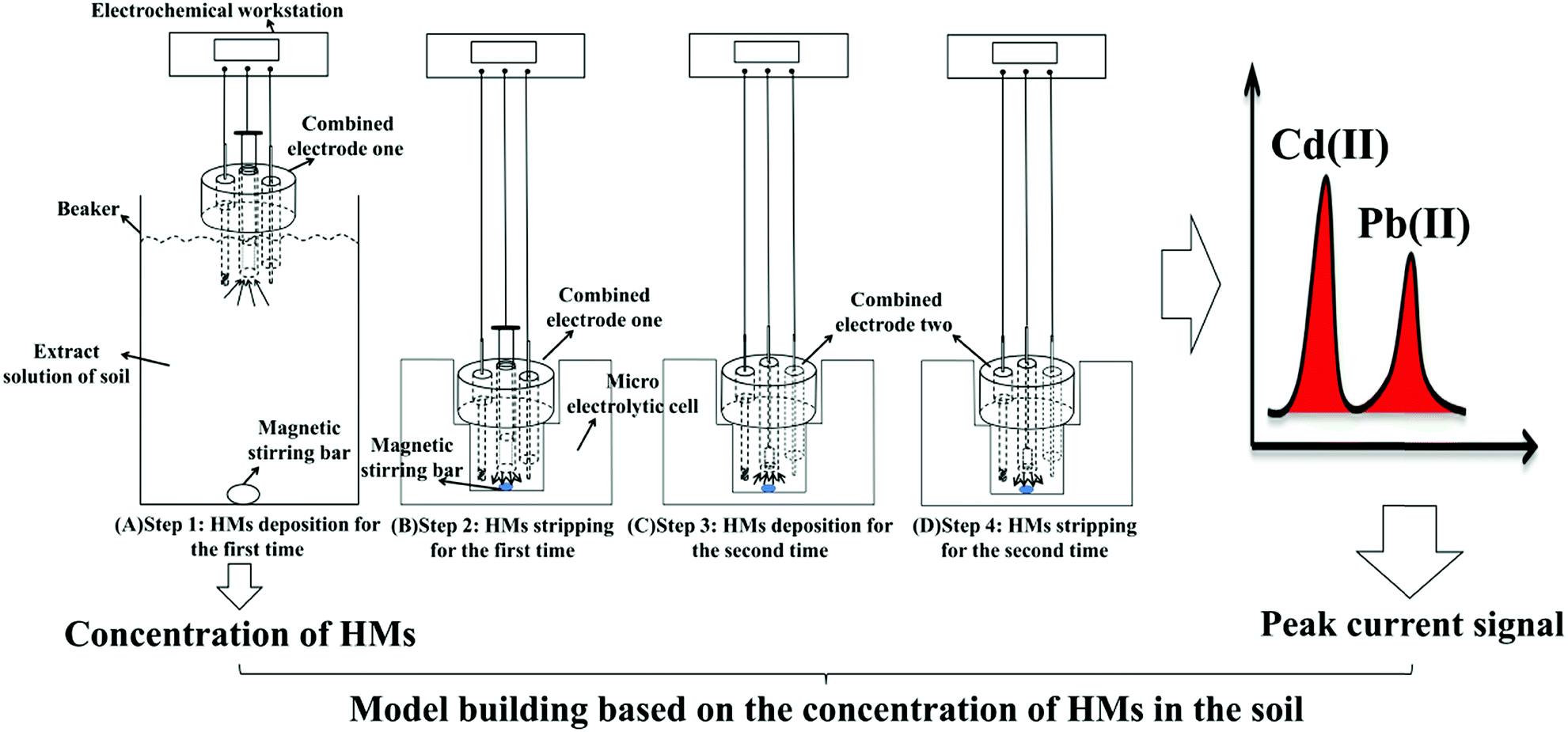 | ||
| Fig. 1 Schematic illustration of the sensitive determination of Cd(II) and Pb(II) using the double deposition and stripping steps with an electrochemical measurement system. | ||
The first combined electrode was disconnected from the electrochemical workstation, and then, the electrochemical workstation was connected to the second combined electrode. The second deposition step was performed at the second combined electrode (i.e., Bi/MWCNT–Nafion/GCE) at a potential of −1.2 V for 280 s. Then, following the second stripping step at a frequency of 25 Hz, a square-wave voltammogram was recorded as the potential was changed from −1.2 to +0.2 V without stirring. The frequency, step amplitude and pulse amplitude were 25 Hz, 5 mV and 25 mV, respectively. During the stripping step, most of heavy metals would strip from the surface of the electrode, but maybe a small amount of HMs still on the electrode surface, which would be effect the next detection. Moreover, the stripping potential (oxidation potential) of cadmium, lead and bismuth were ∼−0.8 V, ∼−0.57 V and ∼−0.15 V. In order to remove the residual HMs and Bi film from the GCE surface, a more positive potential +0.31 V using potentiostatic method was performed on the second combined electrode for 120 s to remove the residual bismuth film and metals on the surface of the MWCNT–Nafion/GCE after the second stripping step.
Results and discussion
Basic characteristics of the modified electrodes
The HM preconcentration and stripping performances of the Bi/GCPE and Bi/MWCNT–Nafion/GCE were investigated by the following procedures. First, the GCPE and MWCNT–Nafion/GCE were placed in 20 mL of acetate buffer solution (pH 5.0, 0.1 M) containing 600 μg L−1 Bi(III) and 30 μg L−1 Pb(II) and Cd(II) for 500 s at a deposition potential of −1.2 V under stirring. Then, the electrodes (i.e., GCPE and MWCNT–Nafion/GCE) were removed from the preconcentration cell. Subsequently, a bare glassy carbon electrode (GCE) was placed in the preconcentration cell and then two complete SWASV procedures were performed both with a deposition time of 140 s after the preconcentration process using the Bi/GCPE and Bi/MWCNT–Nafion/GCE, respectively. Fig. 2 shows the stripping peak signals of 30 μg L−1 Pb(II) and Cd(II) at the GCE to compare the preconcentration performance of the Bi/GCPE and Bi/MWCNT–Nafion/GCE. As shown in Fig. 2A, the stripping peak signals of Pb(II) and Cd(II) at the in situ plated bismuth-film GCE (Bi/GCE) without preconcentration are 6.342 μA and 10.27 μA, respectively. The stripping peak signals of Pb(II) and Cd(II) at the Bi/GCE after preconcentration for 500 s using the Bi/MWCNT–Nafion/GCE were 5.806 μA and 9.766 μA, respectively, as shown in Fig. 2B. In contrast, the stripping peak signals of Pb(II) and Cd(II) at the Bi/GCE after preconcentration for 500 s using Bi/GCPE were 5.367 μA and 5.682 μA (Fig. 2C), respectively, suggesting that ∼45% Cd(II) and ∼15% Pb(II) were collected on the surface of the GCPE in the preconcentration cell, potentially because of the presence of monolayer and/or multilayer graphene on the surface of the GCPE.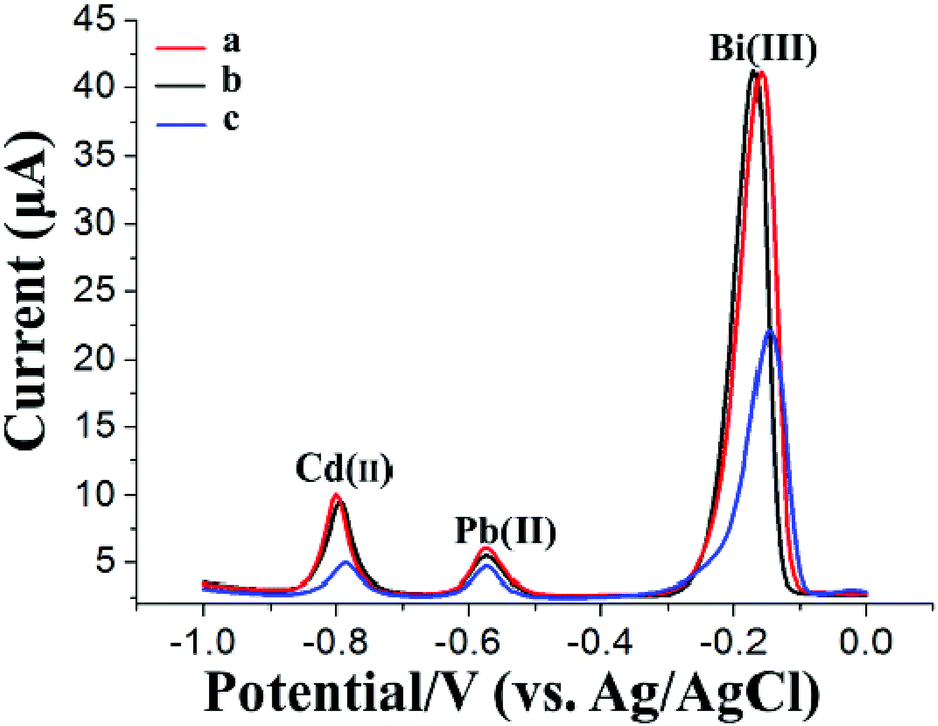 | ||
| Fig. 2 SWASV of 30 μg L−1 Cd(II) and Pb(II) in 20 mL of a 0.1 M acetate buffer solution (pH 4.5) (A) at the Bi/GCE without a preconcentration step, (B) at the Bi/GCE after a 500 s preconcentration using Bi/MWCNT–Nafion/GCE and (C) at the Bi/GCE after a 500 s preconcentration using Bi/GCPE. Preconcentration time: 500 s; preconcentration potential: −1.2 V; deposition time: 140 s; deposition potential: −1.2 V; Bi(III) concentration: 600 μg L−1. | ||
The surface area of the GCPE played a key role in the determination of HMs using the developed method, by influencing the preconcentration of the HMs, such as Pb(II) and Cd(II), on the electrode. Therefore, SEM was performed to characterize the surface morphology of the GCPE. The SEM images of the GCPE at two different magnifications are shown in Fig. 3A and B, displaying a multilayer structure of graphite flakes with clearly visible layers, which may supply more deposition sites for HMs and then improve the preconcentration efficiency of the HMs on the surface of the electrode. This observation is highly consistent with the above experimental results shown in Fig. 2. Moreover, EDS mapping acquired across representative areas of the corresponding GCPE with the deposition of Pb(II) and Cd(II) reveals that a large amount of Pb(II) and Cd(II) was electrodeposited on the electrode surface, as shown in Fig. 3C and D, likely in the form of intermetallic compounds. However, the multilayer structure of graphite flakes may reduce the conductivity of the electrode surface. In addition, because of the large surface area and adsorption property of GCPE, a large number of HMs deposited on the electrode surface may hard to strip from the surface of the electrode easily, which would reduce the rapid oxidation efficiency of HMs.
 | ||
| Fig. 3 (A and B) SEM images of the GCPE at two different magnifications. (C and D) EDS mappings of the GCPE. | ||
The stripping voltammetry behaviours of Cd(II) and Pb(II) on the bismuth-film-modified GCE were characterized by a CHI660D electrochemical workstation. The bismuth film was in situ modified19,22 with a Bi(III) concentration of 600 μg L−1. As probe metal ions, the concentrations of both Cd(II) and Pb(II) were 20 μg L−1. As shown in Fig. S2A ESI†, the stripping peak signals of Pb(II) and Cd(II) at the GCE were weak and unobvious. Comparatively, the Bi/GCE exhibited higher stripping peak signals for Cd(II) and Pb(II), suggesting that the presence of Bi could promote the reduction of Pb(II) and Cd(II) due to the unique advantages of the bismuth-film-modified electrodes, such as the ability to form alloys with the HMs. Eight repeated measurements of 20 μg L−1 Cd(II) and Pb(II) in acetate buffer solution (pH 5.0, 0.1 M) were performed to verify the reproducibility and stability of the bismuth-film-modified GCE, as shown in Fig. S2B ESI.† The results indicate that the stripping peak signals of Pb(II) and Cd(II) on the bismuth-film-modified GCE were reproducible. The relative standard deviation (RSD) of the eight repeated measurements was 2.39% and 1.68% for Pb(II) and Cd(II), respectively. Under the optimum experimental conditions, the bismuth-film-modified GCE exhibited remarkable stability and reproducibility in the stripping analysis of trace levels of Pb(II) and Cd(II), providing a reliable and stable substrate for the modification of the MWCNT–Nafion composite.
Fig. 4 shows the SWASV responses of 20 μg L−1 Pb(II) and Cd(II) at the GCE (curve a) using a general SWASV technique with single deposition and stripping steps, at the Bi/GCE using double deposition and stripping steps (curve b) (the first electrode is the Bi/GCPE, and the second electrode is the Bi/GCE) and at the Bi/MWCNT–Nafion/GCE using double deposition and stripping steps (curve c) (the first electrode is the Bi/GCPE, and the second electrode is the Bi/MWCNT–Nafion/GCE). The stripping peak signals obtained from the SWASV measurements of 20 μg L−1 Pb(II) and Cd(II) at the Bi/MWCNT–Nafion/GCE using double deposition and stripping steps were higher than those in the other two cases. These results may be because of the exceptional characteristics of the MWCNT–Nafion/GCE, such as the unique electrical conductivity, large specific surface area and the good HM preconcentration and stripping ability of the MWCNT–Nafion composite film on the surface of the electrode. The excellent preconcentration ability of the synthesized GCPE was again highlighted by these experimental results. Moreover, the reproducibility of the Bi/GCPE and Bi/MWCNT–Nafion/GCE was validated by five repeated measurements of 20 μg L−1 Cd(II) and Pb(II) with 600 μg L−1 Bi(III) in acetate buffer solution (pH 5.0, 0.1 M). The RSD of the five repeated measurements was 2.28% and 2.23% for Pb(II) and Cd(II) using the Bi/GCPE, respectively, and 2.04% and 1.95% using the Bi/MWCNT–Nafion/GCE, respectively.
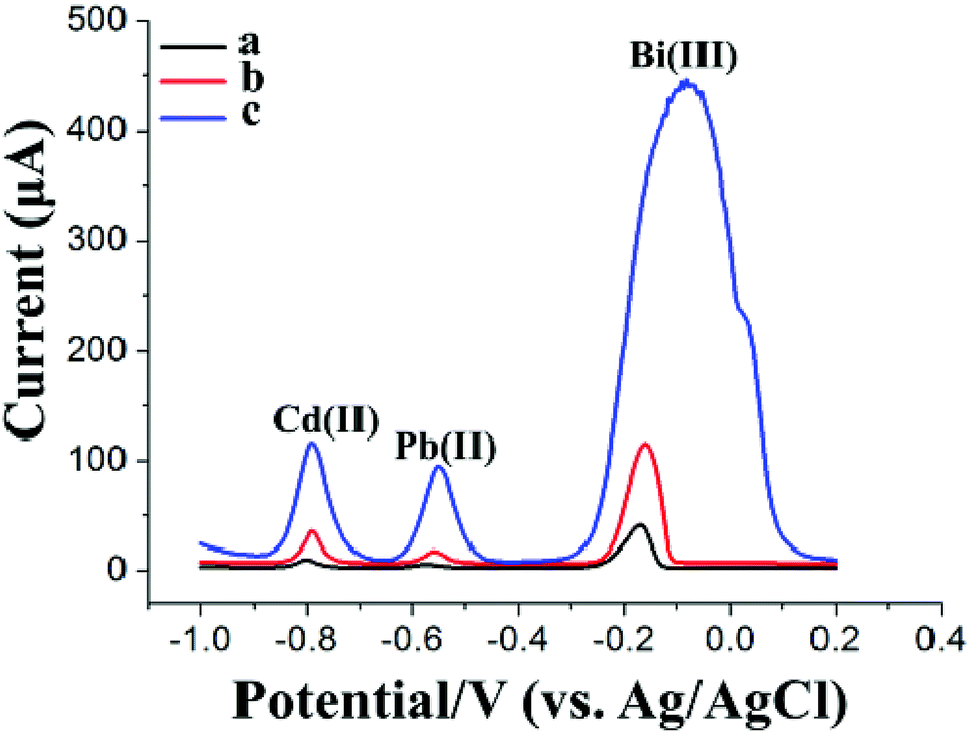 | ||
| Fig. 4 (A) SWASV of 20 μg L−1 Cd(II) and Pb(II) in 20 mL of acetate buffer solution (0.1 M, pH 4.5) at the Bi/GCE using a single deposition and stripping step (deposition time: 140 s; deposition potential: −1.2 V; Bi(III) concentration: 600 μg L−1). (B) SWASV of 20 μg L−1 Cd(II) and Pb(II) in 1 mL of acetate buffer solution (0.1 M, pH 5.0) at the Bi/GCE using double deposition and stripping steps (the first electrode is the GCPE, and the second electrode is the GCE). (C) SWASV of 20 μg L−1 Cd(II) and Pb(II) in 1 mL of acetate buffer solution (0.1 M, pH 5.0) at the Bi/MWCNT–Nafion/GCE using double deposition and stripping steps (the first electrode is the Bi/GCPE, and the second electrode is the Bi/MWCNT–Nafion/GCE). First deposition step: deposition time: 700 s; deposition potential: −1.2 V; Bi(III) concentration: 600 μg L−1. Second deposition step: deposition time: 140 s; deposition potential: −1.2 V. | ||
Effects of the deposition and stripping conditions
The double-stripping voltammetry method was designed and developed to improve the sensitivity and anti-interference ability for the detection of HMs based on SWASV with double deposition and stripping steps. The experimental conditions of the developed double-stripping voltammetry method, such as the pH, Bi(III) concentration, deposition potential and deposition time, play important roles in the simultaneous determination of Pb(II) and Cd(II). Therefore, these parameters were optimized to obtain better sensitivity.The following experiments were performed to optimize the first deposition step and stripping step by setting the parameters of second deposition step and stripping step as following: pH = 5.0; deposition time = 280 s; deposition potential = −1.2 V. The parameters of the first deposition step and the stripping step were determined by the magnitude of stripping currents on the Bi/MWCNT–Nafion/GCE. As shown in Fig. 5A, double-stripping voltammetry was performed using the supporting electrolyte with different pH values for the first deposition step and stripping step. The maximum value of the stripping peak signal on the Bi/MWCNT–Nafion/GCE occurred at a pH of 5.0, and therefore, a pH of 5.0 was chosen for the first deposition step. Fig. 5B displays the influence of the Bi(III) concentration on the stripping peak signals of Pb(II) and Cd(II) without the additional addition of Bi(III) to the micro-electrolytic cell during the second deposition and stripping steps. A Bi(III) concentration of 600 μg L−1 was shown to be the optimal value to obtain the maximal values of the stripping responses of Cd(II) and Pb(II). The effect of the deposition potential on the stripping responses of Pb(II) and Cd(II) in the first deposition was investigated in the potential range of −0.8 to −1.4 V for a deposition of 700 s, as shown in Fig. 5C. The maximal stripping peak signals for both Pb(II) and Cd(II) appeared at a deposition potential of −1.2 V. Therefore, a deposition potential of −1.2 V was adopted in the first deposition step for further measurements. In the double-stripping voltammetry method based on SWASV, the deposition time plays a key role in the improvement of the HM detection sensitivity. Thus, the optimal deposition times for the first deposition step were determined by the following experiments, as shown in Fig. 5D. The deposition time for the first deposition step was varied over the range of 100–1000 s, the peak currents of the target Pb(II) and Cd(II) at the Bi/MWCNT–Nafion/GCE increased with an increase of the deposition time for the first deposition. However, when the deposition time was longer than 700 s, the increase in the peak currents levelled off. Considering both of the efficiency and sensitivity, a deposition time of 700 s was adopted for the first deposition step. Additionally, the effect of the parameters i. e., pH, deposition time, deposition potential and Bi(III) concentration, for the second deposition step and stripping step was also investigated by setting the parameters of first deposition step and fist stripping step as following: pH = 5.0; deposition time = 700 s; deposition potential = −1.2 V and Bi(III) concentration = 600 μg L−1.
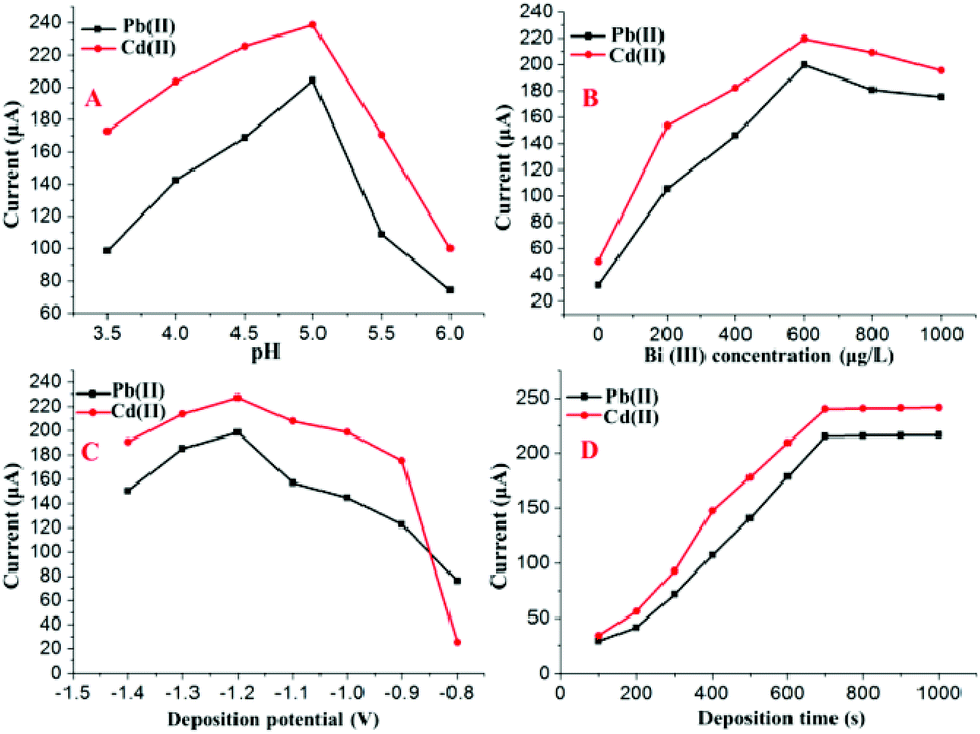 | ||
| Fig. 5 Effects of the (A) pH, (B) Bi(III) concentration, (C) deposition potential and (D) deposition time for the first deposition step on the stripping peak currents of 30 μg L−1 Cd(II) and Pb(II). | ||
According to the optimization results of the parameters for second deposition step and stripping step, a pH of 5.0, deposition time of 280 s and a deposition potential of −1.2 V were chosen for the second deposition step and stripping step. Additionally, the experimental results indicated that further addition of Bi(III) to the testing solution in the micro-electrolytic cell for the second deposition step and stripping step did not improve the peak current signals, indicating that the amount of Bi(III) stripped from the GCPE after the first stripping step was sufficient to meet requirements for detection. Therefore, the optimal Bi(III) concentration in the bulk solution was 600 μg L−1 for the first deposition step, and additional Bi(III) was not added to the testing solutions for the second deposition step.
Analytical performance of the double-stripping voltammetry method
Under the optimum conditions, the stripping voltammetry method was used for the simultaneous determination of trace levels of Cd(II) and Pb(II) following the double deposition and stripping steps. A series of stripping responses for different concentrations of Pb(II) and Cd(II) is illustrated in Fig. 6. The stripping peak signals and the concentrations displayed linear relationships for both Cd(II) and Pb(II) in the concentration range of 1.0 to 45 μg L−1, as shown in the insets of Fig. 6A and B, respectively. The calibration curves of Cd(II) and Pb(II) had the equations y = 7.06x + 8.32 (x: μg L−1, y: μA, 1.0 to 45 μg L−1) and y = 6.01x + 8.60 (x: μg L−1, y: μA, 1.0 to 45 μg L−1), respectively, and the correlation coefficients were 0.996 for Cd(II) and 0.995 for Pb(II). The standard errors in the slope and intercept of the calibration curves were 0.140 and 3.75 for Pb(II) and 0.141 and 3.75 for Cd(II). Moreover, the “Prob > F” values of the calibration curves were all less than 0.01, which indicated the high significance of the calibration curves of Cd(II) and Pb(II). To complete the methodology and extend its application to lower Cd(II) and Pb(II) concentrations in real soil samples, the voltammetric signal was also studied in the range of 0.05–1.0 μg L−1. By increasing the accumulation time for the second deposition step to 350 s, the detection limit, defined as DL = 3Sb/m,54 where Sb is the standard deviation of the blank (n = 5) and m is the slope of the calibration curve, was found to be 0.02 μg L−1 for Cd(II) and 0.03 μg L−1 for Pb(II), which are far lower than the maximum contaminant levels for total cadmium and lead in soil than those set by US EPA guidelines.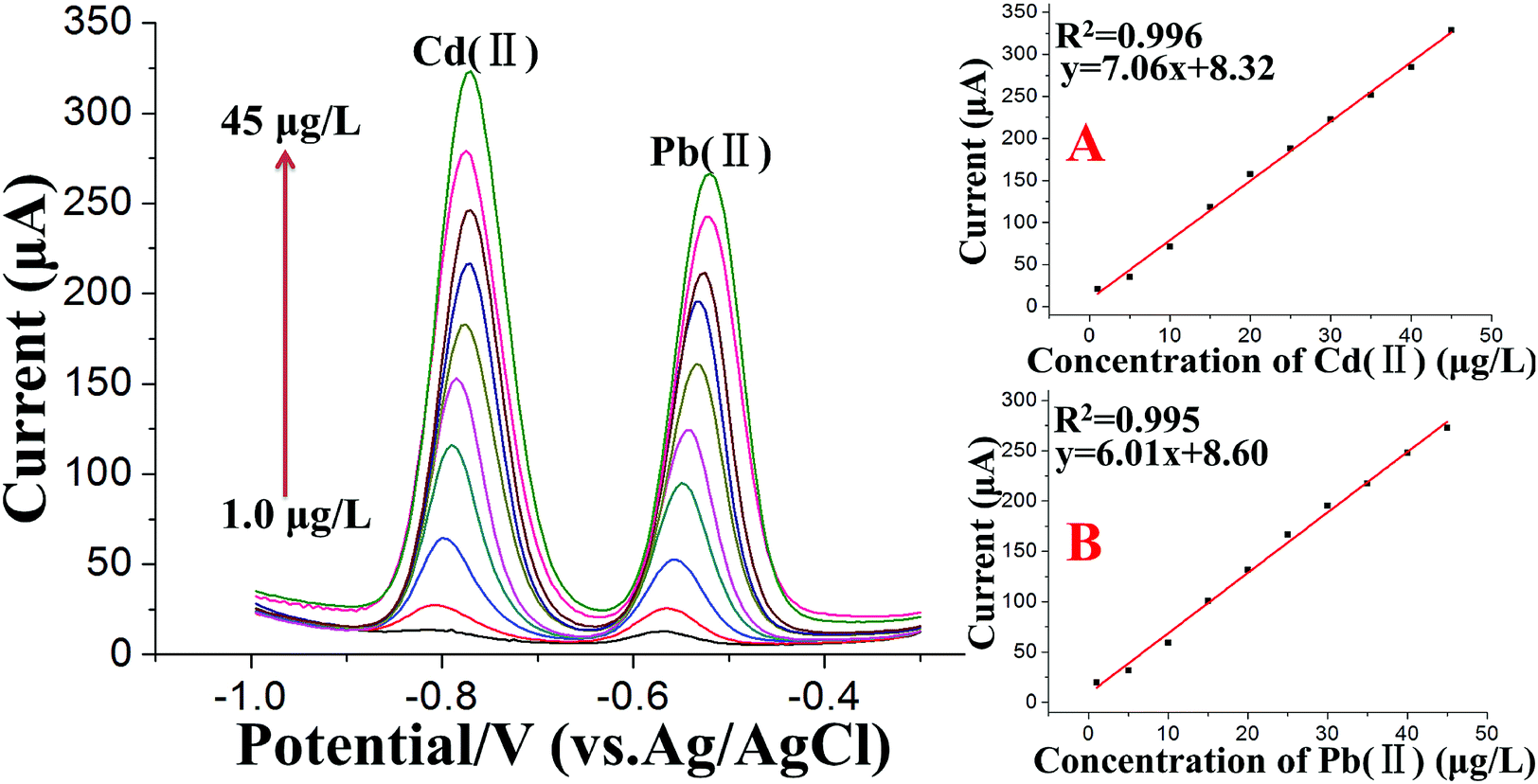 | ||
| Fig. 6 SWASV of different concentrations of Cd(II) and Pb(II) at the Bi/MWCNT–Nafion/GCE using double deposition and stripping steps. From bottom to top: 1, 5, 10, 15, 20, 25, 30, 35, 40 and 45 μg L−1. The right section of the figure shows the calibration curves for (A) Cd(II) and (B) Pb(II). First deposition step: deposition time: 700 s; deposition potential: −1.2 V; Bi(III) concentration: 600 μg L−1. Second deposition step: deposition time: 280 s; deposition potential: −1.2 V. | ||
To assess the effectiveness of the SWASV measurements following the double deposition and stripping steps, the calibration curves of Pb(II) and Cd(II) using a general SWASV technique following a single deposition and stripping step at a Bi/MWCNT–Nafion/GCE were also investigated. Under the optimized conditions, in the concentration range of 1.0 to 45 μg L−1, the calibration curve was y = 1.08x − 1.56 with a correlation coefficient of 0.991 for Cd(II) and y = 0.404x + 0.885 with a correlation coefficient of 0.998 for Pb(II). The limit of detection (S/N = 3) was 2.3 μg L−1 for Pb(II) and 1.2 μg L−1 for Cd(II).
The new method clearly shows a higher sensitivity (slope of the calibration curve) than the single-step method. The sensitivity was over 7 times higher than that obtained by the general stripping voltammetry technique, which resulted in a two orders of magnitude difference between the reported DLs. Considering the small volume of the micro-electrolytic cell, the concentrations of Cd(II) and Pb(II) were dramatically higher than those in the bulk solution, therefore reducing the detection limits.
The comparative analysis of the double-stripping voltammetry method and the general stripping voltammetry technique shows that the analytical performance of this method using double deposition and stripping steps is comparable to and even better than the general stripping voltammetry technique, which offered lower detection limits. Compared with the general stripping voltammetry technique, the developed double-stripping voltammetry method exhibited a higher sensitivity, potentially because of the preconcentration of HMs, which reduces the volume of the testing buffer solution. Moreover, the excellent electrochemical properties, such as the unique electrical conductivity, the large specific surface area and the good HM preconcentration and stripping ability, of the Bi/MWCNT–Nafion/GCE also contributed to this remarkable sensitivity.
Propagating errors will directly affect the stability and reproducibility of the developed method. The stability and reproducibility of the improved stripping voltammetry method were tested via five repeated measurements of 30 μg L−1 Pb(II) and Cd(II) in 0.1 M acetate buffer solution (pH 5.0). The stripping peak currents of Pb(II) and Cd(II) in the second stripping step after a complete testing process indirectly reflect the stability and reproducibility of the improved stripping voltammetry method. The stripping peak currents of Pb(II) and Cd(II) in the second stripping step after a complete testing process were reproducible, with RSDs of 2.24% for Cd(II) and 2.37% for Pb(II), which indicated the proposed method has a good stability and reproducibility. The proposed method was composed of two deposition and stripping steps, which a GCPE was used for the first deposition and stripping step and the MWCNT–Nafion/GCE was used for the second deposition and stripping step. The good stability and reproducibility of MWCNT–Nafion/GCE and GCPE play an important role in the stability and reproducibility of the proposed method. However, in the first deposition step, in addition to the target HMs, other HM ions in real samples may also be deposited on the surface of the electrode. The influence of various possible interferents, such as Cu(II), Na(I), As(III), Cr(II), K(I), Ca(II) and Zn(II), on the determination of Pb(II) and Cd(II) was examined under the optimized conditions. At higher concentrations of interference ions, no significant changes were observed in the stripping peak currents of either HM (changes in peak currents <10%), except in the presence of Cu(II), which negatively affected the stripping peak currents. Nevertheless, this Cu(II) interference could be reduced by the addition of 0.1 mM ferrocyanide to the sample extract solutions to form an insoluble and stable copper–ferrocyanide complex with the help of a ligand.40–43
Moreover, HMs in the double electrode layer may be lost in the transfer process of the GCPE after the first deposition step. In addition, target HMs on the electrode surface may also be oxidized during the transfer process. Therefore, after the first deposition step, the GCPE must be rapidly transferred to the micro-electrolytic cell (in no more than 2 s); however, partial oxidation is unavoidable. The results show that the stripping signals of the HMs after the second deposition step are higher than those obtained from conventional single-stripping voltammetry. This may be because the GCPE has a large specific surface area and can adsorb large amounts of HMs.28,29 Furthermore, the micro-electrolytic cell is able to concentrate the HM ions, which improved the voltammetric signal. In other words, the loss of HMs during the transfer process after the first deposition step does not affect the final detection performance. Moreover, detection of the target HMs was performed using the relationship established between the stripping voltammetry signals from the second stripping step and the HM concentrations in the samples. Interference in the transfer process does not affect the detection results. However, after the first deposition step, the GCPE transfer process is performed manually, during which the GCPE is exposed to the air. If the transfer rate is slow, the detection results will be negatively affected, which is a key issue to be addressed in our future research.
Table 1 shows a comparison of the analytical performances obtained in this work and previous works. The detection limits of Pb(II) and Cd(II) obtained by the developed method are lower than those in most previous reports. Moreover, CMEs used for HM detection were most often used in aqueous samples, while methods using CMEs for soil sample analysis are rarely reported. In addition, the strategy present in this paper consists of a deposition step on a “real” sample, followed by transfer of the electrode to a micro-electrolysis cell where the contents are stripped from the electrode, and then re-deposited on a different electrode which enhances the signal level and reduces the impact of interfering matrix effects (e.g. by using an optimized acetate buffer and optimized conditions), since it allows to preconcentrate the sample and to perform the analysis under more controlled and interference-free conditions, which effectively reduces the interference of background matrices and the pH, indicating the excellent performance of the developed method for the detection of Pb(II) and Cd(II).
| Electrode | Technique | Linear range (μg L−1) | Detection limit (μg L−1) | pH effects | Interferents | Matrix effects | Applicability to specific samples | Reference | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Cd(II) | Pb(II) | Cd(II) | Pb(II) | |||||||
| GO-MWCNT/GCE | DPASV | 0.5–30 | 0.5–30 | 0.1 | 0.2 | Presence | Zn(II) and Cu(II) | Presence | Effluent | 44 |
| AG-NA/GCE | DPASV | 5–100 | 5–100 | 0.07 | 0.05 | Presence | No description | Presence | Tap water | 45 |
| Bi-D24C8/Nafion/SPCE | SWASV | 0.5–60 | 0.5–60 | 0.27 | 0.11 | Presence | Cu(II) | Presence | Rice | 46 |
| GR/PSS/Bi/SPE | DPASV | 0.5–120 | 0.5–120 | 0.04 | 0.09 | Presence | Zn(II), Sn(II), Ni(II) and Cu(II) | Presence | Water | 47 |
| ERGO–TH–MES/GCE | SWASV | 1–40 | 1–40 | 0.1 | 0.05 | Presence | No description | Presence | Water | 48 |
| BiOCl/MWCNT/GCE | SWASV | 5–50 | 5–50 | 1.2 | 0.57 | Presence | Cu(II) | Presence | Pore water | 49 |
| Bi-GCPE | SWASV | 0.1–50 | 0.1–50 | 0.07 | 0.04 | Presence | No description | Presence | Tap water | 50 |
| Bi-xerogel/Nafion/GCE | SWASV | 0.56–11.24 | 1.04–20.72 | 0.37 | 1.3 | Presence | Cu(II) | Presence | Tap, table and lake water | 51 |
| Nafion-G/GCE | DPASV | 1.5–30 | 0.5–50 | 0.02 | 0.02 | Presence | Triton X-100 | Presence | Water | 52 |
| MWCNT/poly(PCV)/GCE | DPASV | 1–300 | 1–200 | 0.2 | 0.4 | Presence | Fe(III), Ag(I), Ni(II) and Cu(II) | Presence | Water | 53 |
| Bi/GCPE and Bi/MWCNT-Nafion/GCE | SWASV | 1–45 | 1–45 | 0.02 | 0.03 | Not obvious | Cu(II) | Not obvious | Soil | This work |
Application to real sample analysis
To evaluate the feasibility and practicality of the developed stripping voltammetry method, the double-stripping voltammetry method was used to determine the Pb(II) and Cd(II) concentrations in real soil samples. The soil samples were prepared according to section 2.3. An electrodeposition method was used to remove Cd(II) and Pb(II) naturally present in the real soil sample extracts until the stripping current signals of the HM residuals were very weak and undetectable with both AAS and the developed stripping voltammetry method. Additionally, the soil samples were treated with 0.1 mM ferrocyanide ions before determination to reduce the interference of Cu(II) on the stripping responses of Cd(II) and Pb(II).40–43Under the optimized conditions, in the concentration range of 1.0 to 45 μg L−1, the peak current signals of Cd(II) and Pb(II) increased linearly with increasing concentration, and the calibration curves were y = 5.57x + 9.59 with a correlation coefficient of 0.997 for Cd(II) and y = 4.54x + 5.33 with a correlation coefficient of 0.997 for Pb(II). These results showed that the performance of the developed method also depended on the supporting electrolyte due to complicated influences from the dissolved constituents in the soil, as the real samples showed lower sensitivity and selectivity than the laboratory samples. Specifically, the calibration curves obtained from the pure acetate buffer solution were not suitable for evaluation of the real samples. Thus, these calibration curves were selected for the analyses of trace Pb(II) and Cd(II) in real samples soil. Four soil samples obtained from a farm in China were used for real sample analysis by the developed double-stripping voltammetry method. Additionally, the detection results were compared with those obtained by AAS, as summarized in Table 2. The experimental results showed no significant difference between the two sets of data, indicating the good reliability of the developed method. Recovery tests were performed to verify the feasibility of the developed method. The average recovery was 98.68% for Pb(II) and 102.42% for Cd(II), respectively. The recovery tests further demonstrate that the developed method can determine trace levels of Pb(II) and Cd(II) in real samples.
| Sample no. | Added (μg L−1) | Found by SWASVa (μg L−1) | Found by AASa (μg L−1) | Recovery (%) | |||
|---|---|---|---|---|---|---|---|
| Cd(II) | Pb(II) | Cd(II) | Pb(II) | Cd(II) | Pb(II) | ||
| a Mean value ± standard deviation. | |||||||
| 1 | — | 3.56 ± 0.18 | 5.42 ± 0.25 | 3.68 ± 0.15 | 5.49 ± 0.18 | — | — |
| 2.00 | 5.74 ± 0.22 | 7.38 ± 0.16 | 109.00 | 98.00 | |||
| 4.00 | 7.62 ± 0.10 | 9.40 ± 0.15 | 101.50 | 99.50 | |||
| 2 | — | 4.32 ± 0.12 | 4.78 ± 0.21 | 4.75 ± 0.20 | 4.96 ± 0.12 | — | — |
| 3.00 | 7.48 ± 0.23 | 7.65 ± 0.16 | 105.33 | 95.67 | |||
| 6.00 | 10.53 ± 0.18 | 10.64 ± 0.24 | 103.50 | 97.67 | |||
| 3 | — | 5.74 ± 0.16 | 4.93 ± 0.12 | 5.93 ± 0.16 | 5.12 ± 0.14 | — | — |
| 4.00 | 9.85 ± 0.23 | 8.86 ± 0.26 | 102.75 | 98.25 | |||
| 8.00 | 13.60 ± 0.14 | 13.04 ± 0.25 | 98.25 | 101.38 | |||
| 4 | — | 3.60 ± 0.21 | 4.15 ± 0.13 | 3.84 ± 0.22 | 4.26 ± 0.18 | — | — |
| 5.00 | 8.49 ± 0.22 | 9.06 ± 0.20 | 97.80 | 98.20 | |||
| 10.00 | 13.72 ± 0.18 | 14.23 ± 0.25 | 101.20 | 100.80 | |||
Conclusions
In the present work, an improved stripping voltammetry method using double deposition and stripping steps was developed for the sensitive determination of trace levels of Cd(II) and Pb(II). In addition, an electrochemical measurement system was introduced to perform the double deposition and stripping steps for the detection of Pb(II) and Cd(II). Due to the preconcentration effect by transferring HMs got from a 500 mL sample solution into a micro-electrolytic cell, the concentrations of Cd(II) and Pb(II) were drastically higher than those in the bulk solution, and therefore, the detection limits were reduced. Moreover, SWASV analysis of HMs was carried out in the ideal acetate buffer solution instead of real sample exacts, which would effectively reduce the interference caused by complicated interferents and complex matrices in the real samples. However, the operation of double stripping voltammetric method is more complicated and also needs a longer analysis time than traditional stripping voltammetric method to ensure its detection precision, which would be improved in our future research. The feasibility of the developed method was further confirmed by the analysis of Pb(II) and Cd(II) in real soil samples. The SWASV current peaks exhibited good linearities from 1.0 μg L−1 to 45 μg L−1 for Pb(II) and Cd(II) in the soil samples. The concentrations of Cd(II) and Pb(II) in the soil samples were calculated using the proposed method, and the results were compared with AAS results to validate the reliability of the developed method, which has great promise for application in the fields of food safety and environmental monitoring.Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
This work was supported by the General Program of the National Natural Science Foundation of China (No. 31671578) and the National High Technology Research and Development Program of China (No. 2013AA102302).Notes and references
- H. Bagheri, A. Afkhami, M. Saber-Tehrani and H. Khoshsafar, Preparation and characterization of magnetic nanocomposite of Schiff base/silica/magnetite as a preconcentration phase for the trace determination of heavy metal ions in water, food and biological samples using atomic absorption spectrometry, Talanta, 2012, 97, 87–95, DOI:10.1016/j.talanta.2012.03.066.
- G. Lee, H. M. Lee, Y. R. Uhm, M. K. Lee and C. K. Rhee, Square-wave voltammetric determination of thallium using surface modified thick-film graphite electrode with Bi nanopowder, Electrochem. Commun., 2008, 10, 1920–1923, DOI:10.1016/j.elecom.2008.10.015.
- H. Bagheri, A. Afkhami, A. Shirzadmehr and H. Khoshsafar, A new nano-composite modified carbon paste electrode as a high performance potentiometric sensor for nanomolar Tl(I) determination, J. Mol. Liq., 2014, 197, 52–57, DOI:10.1016/j.molliq.2014.04.023.
- Z. Gu, M. Zhao, Y. Sheng, L. A. Bentolila and Y. Tang, Detection of mercury ion by infrared fluorescent protein and its hydrogel-based paper assay, Anal. Chem., 2011, 83, 2324–2329, DOI:10.1021/ac103236g.
- A. A. Meharg, Venomous Earth: How Arsenic Caused the World's Worst Mass Poisoning, 2005 Search PubMed.
- World Health Organization, Exposure to Arsenic: A Major Public Health Concern, WHO Public Health and Environment, Geneva, 2010 Search PubMed.
- M. D. Ioannidou, G. A. Zachariadis, A. N. Anthemidis and J. A. Stratis, Direct determination of toxic trace metals in honey and sugars using inductively coupled plasma atomic emission spectrometry, Talanta, 2005, 65, 92–97, DOI:10.1016/j.talanta.2004.05.018.
- K. Leopold, L. Harwardt, M. Schuster and G. Schlemmer, A new fully automated on-line digestion system for ultra trace analysis of mercury in natural waters by means of FI-CV-AFS, Talanta, 2008, 76, 382–388, DOI:10.1016/j.talanta.2008.03.010.
- A. Abbaspour, M. A. Mehrgardi, A. Noori, M. A. Kamyabi, A. Khalafi-Nezhad and M. N. Soltani Rad, Speciation of iron(II), iron(III) and full-range pH monitoring using paptode: a simple colorimetric method as an appropriate alternative for optodes, Sens. Actuators, B, 2006, 113, 857–865, DOI:10.1016/j.snb.2005.03.119.
- R. O. Kadara and I. E. Tothill, Stripping chronopotentiometric measurements of lead(II) and cadmium(II) in soils extracts and wastewaters using a bismuth film screen-printed electrode assembly, Anal. Bioanal. Chem., 2004, 378, 770–775, DOI:10.1007/s00216-003-2351-x.
- D. Manivannan and V. M. Biju, Determination of toxic heavy metals in sea water by FAAS after preconcentration with a novel chelating resin, Water Sci. Technol., 2011, 64, 803–808, DOI:10.2166/wst.2011.501.
- J. P. Cornard, A. Caudron and J. C. Merlin, UV-visible and synchronous fluorescence spectroscopic investigations of the complexation of Al(III) with caffeic acid, in aqueous low acidic medium, Polyhedron, 2006, 25, 2215–2222, DOI:10.1016/j.poly.2006.01.013.
- A. Torreggiani, M. Tamba, A. Trinchero and S. Bonora, Copper(II)–Quercetin complexes in aqueous solutions: spectroscopic and kinetic properties, J. Mol. Struct., 2005, 744–747, 759–766, DOI:10.1016/j.molstruc.2004.11.081.
- Kh. Z. Brainina, N. A. Malakhova and N. Y. Stojko, Stripping voltammetry in environmental and food analysis, Fresenius. J. Anal. Chem., 2000, 368, 307–325, DOI:10.1007/s002160000525.
- H. Bagheri, A. Afkhami, H. Khoshsafar, M. Rezaei and A. Shirzadmehr, Simultaneous electrochemical determination of heavy metals using a triphenylphosphine/MWCNTs composite carbon ionic liquid electrode, Sens. Actuators, B, 2013, 186, 451–460, DOI:10.1016/j.snb.2013.06.051.
- J. Wang, Stripping Analysis: Principles, Instrumentation, and Applications, VCH Publishers, Weinheim, Germany, 1985 Search PubMed.
- G. Zhao, H. Wang, G. Liu and Z. Wang, Box–Behnken response surface design for the optimization of electrochemical detection of cadmium by Square Wave Anodic Stripping Voltammetry on bismuth film/glassy carbon electrode, Sens. Actuators, B, 2016, 235, 67–73, DOI:10.1016/j.snb.2016.05.051.
- V. Guzsvány, H. Nakajima and N. Soh, et al. Anodic Stripping Voltammetry Combined with Sequential Injection Analysis for Measurements of Trace Metal Ions with Bismuth and Antimony Film Electrodes under Comparable Conditions, Electroanalysis, 2011, 23, 1593–1601 CrossRef.
- G. Zhao, H. Wang and G. Liu, Electrochemical determination of trace cadmium in soil by a bismuth film/graphene–beta–cyclodextrin–nafion composite modified electrode, Int. J. Electrochem. Sci., 2016, 11, 1840–1851 CAS.
- G. Zhao, H. Wang, G. Liu and Z. Wang, Optimization of stripping voltammetric sensor by a back propagation artificial neural network for the accurate determination of Pb(II) in the presence of Cd(II), Sensors, 2016, 16, 1540, DOI:10.3390/s16091540.
- A. Wong, C. A. Razzino, T. A. Silva and O. Fatibello-Filho, Square-wave voltammetric determination of clindamycin using a glassy carbon electrode modified with graphene oxide and gold nanoparticles within a crosslinked chitosan film, Sens. Actuators, B, 2016, 231, 183–193, DOI:10.1016/j.snb.2016.03.014.
- Z. Bi, C. S. Chapman, P. Salaün and C. M. G. van den Berg, Determination of lead and cadmium in sea- and freshwater by anodic stripping voltammetry with a vibrating bismuth electrode, Electroanalysis, 2010, 22, 2897–2907, DOI:10.1002/elan.201000429.
- Z. Bi, P. Salaün and C. M. G. van den Berg, Study of bare and mercury-coated vibrated carbon, gold and silver microwire electrodes for the determination of lead and cadmium in seawater by anodic stripping voltammetry, Electroanalysis, 2013, 25, 357–366, DOI:10.1002/elan.201200446.
- L. Xiao, H. Xu, S. Zhou, T. Song, H. Wang, S. Li, W. Gan and Q. Yuan, Simultaneous detection of Cd(II) and Pb(II) by differential pulse anodic stripping voltammetry at a nitrogen-doped microporous carbon/nafion/bismuth-film electrode, Electrochim. Acta, 2014, 143, 143–151, DOI:10.1016/j.electacta.2014.08.021.
- L. Chen, Z. H. Su, X. H. He, Y. Liu, C. Qin, Y. P. Zhou, Z. Li, L. H. Wang, Q. J. Xie and S. Z. Yao, Square wave anodic stripping voltammetric determination of Cd and Pb ions at a Bi/nafion/thiolated polyaniline/glassy carbon electrode, Electrochem. Commun., 2012, 15, 34–37, DOI:10.1016/j.elecom.2011.11.021.
- J. Wang, J. M. Lu, Ü. A. Kirgöz, S. B. Hocevar and B. Ogorevc, Insights into the anodic stripping voltammetric behavior of bismuth film electrodes, Anal. Chim. Acta, 2001, 434, 29–34, DOI:10.1016/S0003-2670(01)00818-2.
- C. W. Foster, A. P. de Souza, J. P. Metters, M. Bertotti and C. E. Banks, Metallic modified (bismuth, antimony, tin and combinations thereof) film carbon electrodes, Analyst, 2015, 140, 7598–7612, 10.1039/c5an01692d.
- X. Zhang, M. Li, Y. Cui, J. Zhu, J. Zhao, B. Chen and K. Qu, Investigation on high performance of graphite-ionic liquid paste electrode: characterization and new hypothesis, Int. J. Electrochem. Sci., 2013, 8, 4839–4849 CAS.
- G. Zhao, H. Wang and G. Liu, et al., Simultaneous and sensitive detection of Cd (II) and Pb (II) using a novel bismuth film/ordered mesoporous carbon-molecular wire modified graphite carbon paste electrode, Electroanalysis, 2017, 29, 497–505, DOI:10.1002/elan.201600430.
- G. H. Hwang, W. K. Han, J. S. Park and S. G. Kang, Determination of trace metals by anodic stripping voltammetry using a bismuth-modified carbon nanotube electrode, Talanta, 2008, 76, 301–308, DOI:10.1016/j.talanta.2008.02.039.
- N. Wang and X. Dong, Stripping voltammetric determination of Pb(II) and Cd(II) based on the multiwalled carbon nanotubes-nafion-bismuth modified glassy carbon electrodes, Anal. Lett., 2008, 41, 1267–1278, DOI:10.1080/00032710802052817.
- G. Liu, Y. Lin, Y. Tu and Z. Ren, Ultrasensitive voltammetric detection of trace heavy metal ions using carbon nanotube nanoelectrode array, Analyst, 2005, 130, 1098–1101, 10.1039/b419447k.
- K. Wu, S. Hu, J. Fei and W. Bai, Mercury-free simultaneous determination of cadmium and lead at a glassy carbon electrode modified with multi-wall carbon nanotubes, Anal. Chim. Acta, 2003, 489, 215–221, DOI:10.1016/S0003-2670(03)00718-9.
- S. Legeai and O. Vittori, A Cu/nafion/Bi electrode for on-site monitoring of trace heavy metals in natural waters using anodic stripping voltammetry: an alternative to mercury-based electrodes, Anal. Chim. Acta, 2006, 560, 184–190, DOI:10.1016/j.aca.2005.12.010.
- J. Murimboh, M. T. Lam, N. M. Hassan and C. L. Chakrabarti, A study of nafion-coated and uncoated thin mercury film-rotating disk electrodes for cadmium and lead speciation in model solutions of fulvic acid, Anal. Chim. Acta, 2000, 423, 115–126, DOI:10.1016/S0003-2670(00)01075-8.
- H. Xu, L. Zeng, D. Huang, Y. Xian and L. Jin, A nafion-coated bismuth film electrode for the determination of heavy metals in vegetable using differential pulse anodic stripping voltammetry: an alternative to mercury-based electrodes, Food Chem., 2008, 109, 834–839, DOI:10.1016/j.foodchem.2007.12.065.
- B. Krasnodêbska-Ostrêga and J. Kowalska, Ultrasound-assisted acetic acid extraction of metals from soils, Chem. Anal., 2003, 48, 967–974 Search PubMed.
- R. O. Kadara and I. E. Tothill, Development of disposable bulk-modified screen-printed electrode based on bismuth oxide for stripping chronopotentiometric analysis of lead (II) and cadmium (II) in soil and water samples, Anal. Chim. Acta, 2008, 623, 76–81, DOI:10.1016/j.aca.2008.06.010.
- J. Cooper, J. A. Bolbot, S. Saini and S. J. Setford, Electrochemical method for the rapid on site screening of cadmium and lead in soil and water samples, Water Air Soil. Pollut, 2007, 179, 183–195, DOI:10.1007/s11270-006-9223-x.
- K. Crowley and J. Cassidy, Trace analysis of lead at a nafion-modified electrode using square-wave anodic stripping voltammetry, Electroanalysis, 2002, 14, 1077–1082, DOI:10.1002/1521-4109(200208)14.
- C. Kokkinos, A. Economou, I. Raptis and C. E. Efstathiou, Lithographically fabricated disposable bismuth-film electrodes for the trace determination of Pb(II) and Cd(II) by anodic stripping voltammetry, Electrochim. Acta, 2008, 53, 5294–5299, DOI:10.1016/j.electacta.2008.02.079.
- B. L. Li, Z. L. Wu, C. H. Xiong, H. Q. Luo and N. B. Li, Anodic stripping voltammetric measurement of trace cadmium at tin-coated carbon paste electrode, Talanta, 2012, 88, 707–710, DOI:10.1016/j.talanta.2011.11.070.
- N. Lezi, A. Economou, P. A. Dimovasilis, P. N. Trikalitis and M. I. Prodromidis, Disposable screen-printed sensors modified with bismuth precursor compounds for the rapid voltammetric screening of trace Pb(II) and Cd(II), Anal. Chim. Acta, 2012, 728, 1–8, DOI:10.1016/j.aca.2012.03.036.
- H. Huang, T. Chen, X. Liu and H. Ma, Ultrasensitive and simultaneous detection of heavy metal ions based on three-dimensional graphene–carbon nanotubes hybrid electrode materials, Anal. Chim. Acta, 2014, 852, 45–54, DOI:10.1016/j.aca.2014.09.010.
- S. Lee, S. Bong, J. Ha, M. Kwak, S. Park and Y. Piao, Electrochemical deposition of bismuth on activated graphene-nafion composite for anodic stripping voltammetric determination of trace heavy metals, Sens. Actuators, B, 2015, 215, 62–69, DOI:10.1016/j.snb.2015.03.032.
- K. Keawkim, S. Chuanuwatanakul, O. Chailapakul and S. Motomizu, Determination of lead and cadmium in rice samples by sequential injection/anodic stripping voltammetry using a bismuth film/crown ether/nafion modified screen-printed carbon electrode, Food Control, 2013, 31, 14–21, DOI:10.1016/j.foodcont.2012.09.025.
- C. Huangfu, L. Fu, Y. Li, X. Li, H. Du and J. Ye, Sensitive stripping determination of cadmium(II) and lead(II) on disposable graphene modified screen-printed electrode, Electroanalysis, 2013, 25, 2238–2243, DOI:10.1002/elan.201300239.
- Z. Li, L. Chen, F. He, L. Bu, X. Qin, Q. Xie, S. Yao, X. Tu, X. Luo and S. Luo, Square wave anodic stripping voltammetric determination of Cd2+ and Pb2+ at bismuth-film electrode modified with electroreduced graphene oxide-supported thiolated thionine, Talanta, 2014, 122, 285–292, DOI:10.1016/j.talanta.2014.01.062.
- S. Cerovac, V. Guzsvány, Z. Kónya, A. M. Ashrafi, I. Švancara, S. Rončević, Á. Kukovecz, B. Dalmacija and K. Vytřas, Trace level voltammetric determination of lead and cadmium in sediment pore water by a bismuth-oxychloride particle-multiwalled carbon nanotube composite modified glassy carbon electrode, Talanta, 2015, 134, 640–649, DOI:10.1016/j.talanta.2014.12.002.
- W. Wonsawat, S. Chuanuwatanakul, W. Dungchai, E. Punrat, S. Motomizu and O. Chailapakul, Graphene-carbon paste electrode for cadmium and lead ion monitoring in a flow-based system, Talanta, 2012, 100, 282–289, DOI:10.1016/j.talanta.2012.07.045.
- P. A. Dimovasilis and M. I. Prodromidis, Bismuth-dispersed xerogel-based composite films for trace Pb(II) and Cd(II) voltammetric determination, Anal. Chim. Acta, 2013, 769, 49–55, DOI:10.1016/j.aca.2013.01.040.
- J. Li, S. Guo, Y. Zhai and E. Wang, High-sensitivity determination of lead and cadmium based on the nafion-graphene composite film, Anal. Chim. Acta, 2009, 649, 196–201, DOI:10.1016/j.aca.2009.07.030.
- M. A. Chamjangali, H. Kouhestani, F. Masdarolomoor and H. Daneshinejad, A voltammetric sensor based on the glassy carbon electrode modified with multi-walled carbon nanotube/poly(pyrocatechol violet)/bismuth film for determination of cadmium and lead as environmental pollutants, Sens. Actuators, B, 2015, 216, 384–393, DOI:10.1016/j.snb.2015.04.058.
- H. El-Mai, E. Espada-Bellido and M. Stitou, et al., Determination of ultra-trace amounts of silver in water by differential pulse anodic stripping voltammetry using a new modified carbon paste electrode, Talanta, 2016, 151, 14–22 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra12767g |
| This journal is © The Royal Society of Chemistry 2018 |