
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect regioselective C–H borylation of [5]helicene†
R. P. Kaiser a,
J. Ulča,
I. Císařováb and
D. Nečas*a
a,
J. Ulča,
I. Císařováb and
D. Nečas*a
aDepartment of Organic Chemistry, Faculty of Science, Charles University, Albertov 6, 12843 Praha 2, Czech Republic. E-mail: david.necas@natur.cuni.cz
bDepartment of Inorganic Chemistry, Faculty of Science, Charles University, Albertov 6, 12843, Praha 2, Czech Republic
First published on 2nd January 2018
Abstract
Ir-catalyzed borylation of [5]helicene was studied for the first time. The obtained results indicate that borylation proceeded preferentially at the 2- and 3-positions. By using an appropriate catalytic system, 3-borylated [5]helicene can be formed as the major product in a high yield and regioselectivity (up to 89%, 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of isomers). The monoborylated [5]helicenes were further utilized in a Suzuki–Miyaura cross-coupling reaction to produce 2- or 3-arylated helicenes in very good isolated yields (80–94%).
:1 ratio of isomers). The monoborylated [5]helicenes were further utilized in a Suzuki–Miyaura cross-coupling reaction to produce 2- or 3-arylated helicenes in very good isolated yields (80–94%).
Helicenes represent a unique class of polycyclic aromatic hydrocarbons where the benzene rings are all ortho-fused, fully conjugated, and with a non-planar topology. They have attracted increasing attention owing to their unrivalled structural features1–3 and many potential applications in chiral materials,4–8 self-assembly,9–11 asymmetric synthesis,12–14 and optoelectronic materials.15–18 Of importance, unique properties of helicenes can be addressed by introducing substituents at the periphery of the helical core. From a synthetic point of view, such modifications are not trivial, therefore the selectively substituted helicenes are usually made from pre-functionalized substrates.1,2 These methods are usually not general enough to produce a large library of congeners. This can be explained by the lack of compatibility of some functional groups or their deactivation effects on the key reaction for producing helicenes. Although a post-functionalization of helicenes appears attractive and would greatly accelerate the development of new functional molecules, it has been underdeveloped in the history of helicene chemistry,19–21 and restricts, to some extent, practical applications of helicenes.
Based on our previous results on borylation of [4]helicene21 we envisioned that [5]helicene (Fig. 1) could be selectively functionalized by an iridium catalyzed C–H bond activation/borylation process. Since regioselectivity of direct borylation of aromatic compounds under the standard conditions ([Ir(OMe)(cod)]2/dtbpy) is sterically driven, it is generally accepted that C–H bonds ortho to the substituent and ortho to ring junction (peri-position) do not usually react.22,23 Hence, [5]helicene should be borylated at sterically more accessible positions 2 and 3. Out of these two, position 2 is more sterically hindered (overlapping by the other end of the molecule), therefore the appropriate choice of catalytic system and the third dimension (helicity) of the molecule should contrive an additional level of regioselectivity to distinguish between these two positions.
 | ||
| Fig. 1 Molecular structure of [5]helicene. | ||
According to our previous study of [4]helicene21 and the preceding studies of iridium-catalyzed borylation of arenes22–27 and fused polyarenes,28–40 we subjected [5]helicene 1 to standard borylation conditions. Thus, equimolar quantities of 1 and B2pin2 (pin = pinacolato), a catalytic amount of [Ir(OMe)(cod)]2 (5 mol%) and 4,4′-di-tert-butyl-2,2′-bipyridine (dtbpy) (10 mol%) were allowed to react in cyclohexane. A reaction carried out at 23 °C for 16 h afforded only the starting material and traces of monoborylated products (according to EI/MS analysis). An increase of the reaction temperature to 50 °C and subsequently to 80 °C resulted in better conversion of 1 and a mixture of two monoborylated compounds in slightly better yield (∼10%) was obtained. These results clearly showed that C–H activation/borylation of [5]helicene is possible but requires harsher reaction conditions than sterically distinct [4]helicene. In view of the aforementioned, borylation of [5]helicene at 100 °C for 24 h was attempted (Scheme 1). After removal of the volatiles and the subsequent column chromatography of the residue on silica gel (hexane/DCM from 100:0 to 0:100) three colorless fractions were obtained: unreacted 1 (79%), a mixture of 2- and 3-borylated [5]helicenes (16%) and a small fraction containing a trace amount of bisborylated [5]helicene (∼1%). The subsequent separation of the second fraction by non-aqueous reverse phase chromatography (NARP) afforded two regioisomers: 2-borylated [5]helicene 2a (4%) and 3-borylated [5]helicene 2b (12%). The structure of the major product 2b was unequivocally confirmed by a single-crystal X-ray diffraction analysis (Fig. 2). The third fraction contained only one compound, structure of which was later on assigned to symmetrical 3,12-bisborylated [5]helicene 2c. The formation of unsymmetrical 2,12-bisborylated and symmetrical 3,13-bisborylated [5]helicene was not observed under these conditions.
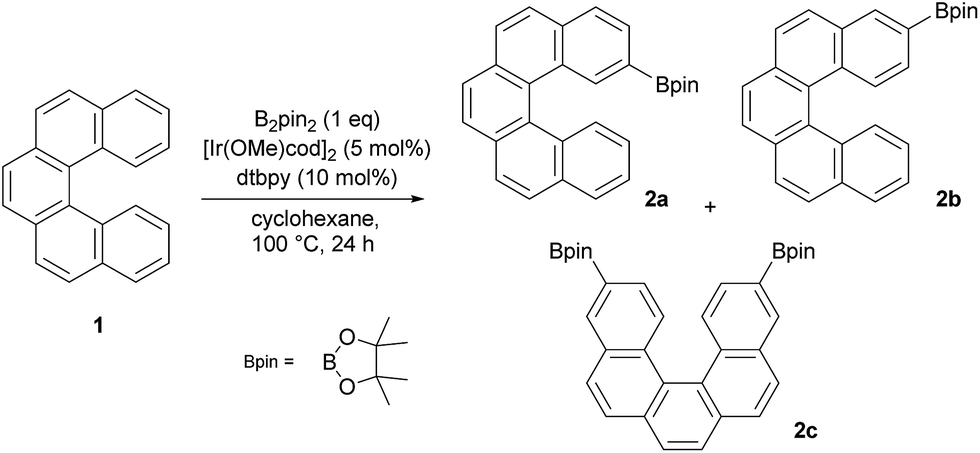 | ||
| Scheme 1 Ir-catalyzed C–H borylation of [5]helicene. | ||
 | ||
| Fig. 2 ORTEP drawing of 2b with 30% thermal ellipsoids. | ||
These results prompted us to find conditions that will increase the yields of borylated products and enable us to control regioselectivity of borylation as well (Table 1). First, 2/1 ratio of 1 to B2Pin2 was used to suppress the formation of bisborylated product 2c. Second, the reaction temperature was increased to 120 °C. Under these conditions 1:3 mixture of 2a and 2b was obtained in 26% isolated yield (based on 1 equiv. of 1) after 24 hours (entry 1). Reactions performed in 1,4-dioxane, dibutyl ether, ethyl acetate, 2-methyl tetrahydrofurane or mesitylene gave products in low yields, if any (entries 2–6). The use of microwave conditions41,42 resulted in low conversion and lower selectivity, affording a complex mixture of products in which the presence of tris-borylated [5]helicenes was observed by EI/MS (entry 7). Change of the ligand to a more rigid and electron-rich 3,4,7,8-tetramethyl-1,10-phenanthroline (tmphen), which can enhance the activity of the resulting catalyst and often overperforms dtbpy,43–46 gave 2a and 2b in a higher yield of 56% with a considerably increased regioselectivity of 1:5 (entry 8). To fully employ the shape of the molecule and improve the regioselectivity of the borylation, we also screened sterically demanding ligands L successfully used for para-borylation of benzene derivatives47,48 or regioselective borylation of [4]helicene.21 These bis(phosphine) type ligands in combination with [Ir(cod)OH]2 afforded the products 2a and 2b with ratios in the range of 1:4.7–6. DM-MeO-BIPHEP L1, reported as the best ligand for para-borylation, gave rise to 1:5 mixture of 2a and 2b in a low combined yield of 14% (entry 9). DM-Segphos L2 (entry 10) or DM-Garphos L3 (entry 11), ligands of choice for borylation of [4]helicene, provided 2a:2b in combined yields of 21% and 18% and regioselectivity of 1:4.7 and 1:6, respectively (see the ESI† for the complete list of conditions tested).
|
||||
|---|---|---|---|---|
| Entry | Liganda | Solvent | Yield 2a + 2bb (%) | Ratio of 2a:2bc |
| a dtbpy: 4,4′-di-tert-butyl-2,2′-dipyridyl combined with [Ir(OMe)cod]2; tmphen: 3,4,7,8-tetramethyl-1,10-phenanthroline combined with [Ir(OMe)cod]2; L1, L2 and L3 combined with [Ir(OH)cod]2.b Isolated combined yield of 2a + 2b based on 1 equiv. of 1.c Ratio determined by 1H NMR.d Microwave reactor was used.e Complex mixture – not determined. | ||||
| 1 | dtbpy | Cyclohexane | 26 | 1:3 |
| 2 | dtbpy | 1,4-Dioxane | 0 | — |
| 3 | dtbpy | Bu2O | 20 | 1:3.6 |
| 4 | dtbpy | EtOAc | 19 | 1:3.2 |
| 5 | dtbpy | 2-Me-THF | 0 | — |
| 6 | dtbpy | Mesitylene | 9 | 1:3.1 |
| 7d | dtbpy | MTBE | n.d.e | — |
| 8 | tmphen | Cyclohexane | 56 | 1:5 |
| 9 | L1 | Cyclohexane | 14 | 1:5 |
| 10 | L2 | Cyclohexane | 21 | 1:4.7 |
| 11 | L3 | Cyclohexane | 18 | 1:6 |

These unsatisfactory results turned our attention back to rigid phenanthrene type ligands. We prepared several iridium complexes49 and carried out the reactions in cyclohexane at 120 °C (Table 2). In general, these pre-prepared bench stable complexes afforded highest yields of 2a and 2b and also highest regioselectivity. The use of Ir[(dtbpy)(cod)Cl] C1 afforded 2a and 2b in 60% yield and 1:5.4 ratio (entry 1). Complexes based on neocuproin C2, 4,7-dimethoxy-1,10-phenanthroline C3, bathophenanthroline C4 and 3,8-bis[3,5-bis(trifluoro-methyl)phenyl]-1,10-phenanthroline C5 gave 2a and 2b in a moderate yields (∼50%) and lower regioselectivity in the range of 1:2.3–4.3 (entries 2–5). A complex with a simple 1,10-phenanthroline C6 gave better regioselectivity of 1:6 in a moderate yield 52% (entry 6). The best result, in terms of the yield and selectivity (89%, 1:8), was obtained with the Ir-tmphen complex C7 (entry 7). The use of other solvents did not result in any improvement and the use of a [Ir(cod)Cl]2/tmphen mixture resulted in a lower yield (70%) and a drop in selectivity to 1:6.4 (see the ESI† for the complete list of conditions tested).

Our effort to also produce bis-borylated [5]helicenes in high yield was not successful. Although reaction carried out under standard conditions only produced selectively the symmetric 3,12-bisborylated [5]helicene 2c, the yields were negligible even when a big excess of B2pin2 was used together with a longer reaction time and a higher temperature. The use of more potent catalyst Ir[(tmphen)(cod)Cl] with an excess of B2pin2 resulted in loss of selectivity and produced an inseparable complex reaction mixture where the presence of mono-, bis-, tris- and tetra-borylated [5]helicenes was observed by EI/MS.
To demonstrate the synthetic applicability of 2a and 2b, Suzuki–Miyaura cross-coupling reactions of both isomers with selected aryl iodides were carried out (Scheme 2). Both monoborylated [5]helicenes showed a good reactivity and the respective arylated products 3a–3b and 4a–4c were obtained in good isolated yields (86–94%).
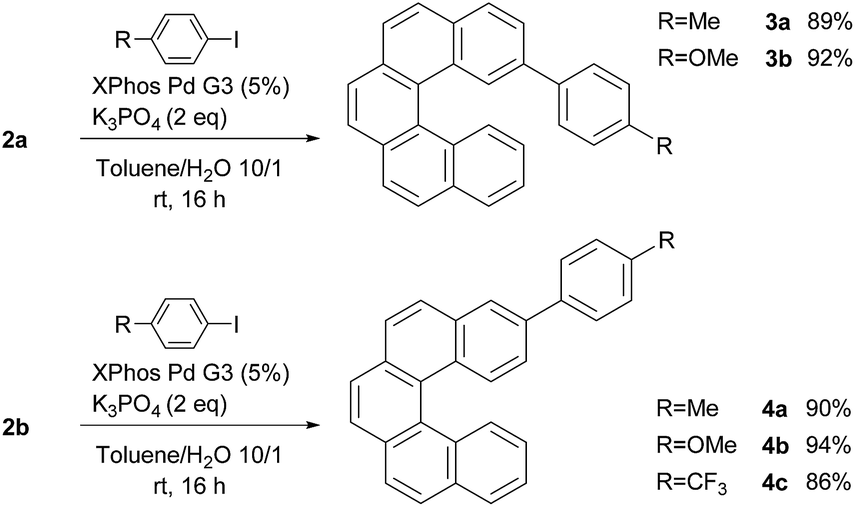 | ||
| Scheme 2 Pd-catalyzed coupling reactions of 2a and 2b. | ||
Conclusions
In summary, we have shown that the Ir-tmphen complex C7 can be successfully used for selective monoborylation of [5]helicene to 3-Bpin-[5]helicene 2b. The reaction proceeded with a high regioselectivity (2a:2b = 1:8) and a high yield (89%). In addition, both formed borylated [5]helicenes were stable and were successfully used in Suzuki–Miyaura cross-coupling reactions to furnish the corresponding 2-aryl and 3-aryl[5]helicenes. Application of this chemistry and extension for higher helicenes are underway in our laboratory.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors are grateful for financial support from the Czech Science Foundation (reg. No. 14-16391P).Notes and references
- M. Gingras, Chem. Soc. Rev., 2013, 42, 968–1006 RSC.
- M. Gingras, G. Félix and R. Peresutti, Chem. Soc. Rev., 2013, 42, 1007–1050 RSC.
- M. Gingras, Chem. Soc. Rev., 2013, 42, 1051–1095 RSC.
- M. Ferreira, G. Naulet, H. Gallardo, P. Dechambenoit, H. Bock and F. Durola, Angew. Chem., Int. Ed., 2017, 56, 3379–3382 CrossRef CAS PubMed.
- Z. Y. Wang, E. K. Todd, X. S. Meng and J. P. Gao, J. Am. Chem. Soc., 2005, 127, 11552–11553 CrossRef CAS PubMed.
- J. N. Moorthy, S. Mandal, A. Mukhopadhyay and S. Samanta, J. Am. Chem. Soc., 2013, 135, 6872–6884 CrossRef CAS PubMed.
- D. Schweinfurth, M. Zalibera, M. Kathan, C. Shen, M. Mazzolini, N. Trapp, J. Crassous, G. Gescheidt and F. Diederich, J. Am. Chem. Soc., 2014, 136, 13045–13052 CrossRef CAS PubMed.
- L. Pospíšil, L. Bednárová, P. Štěpánek, P. Slavíček, J. Vávra, M. Hromadová, H. Dlouhá, J. Tarábek and F. Teplý, J. Am. Chem. Soc., 2014, 136, 10826–10829 CrossRef PubMed.
- C. Nuckolls, T. J. Katz and L. Castellanos, J. Am. Chem. Soc., 1996, 118, 3767–3768 CrossRef CAS.
- W. Ichinose, J. Ito and M. Yamaguchi, Angew. Chem., Int. Ed., 2013, 52, 5290–5294 CrossRef CAS PubMed.
- X.-Y. Wang, T. Dienel, M. Di Giovannantonio, G. B. Barin, N. Kharche, O. Deniz, J. I. Urgel, R. Widmer, S. Stolz, L. H. De Lima, M. Muntwiler, M. Tommasini, V. Meunier, P. Ruffieux, X. Feng, R. Fasel, K. Müllen and A. Narita, J. Am. Chem. Soc., 2017, 139, 4671–4674 CrossRef CAS PubMed.
- M. T. Reetz, E. W. Beuttenmüller and R. Goddard, Tetrahedron Lett., 1997, 38, 3211–3214 CrossRef CAS.
- N. Takenaka, J. Chen, B. Captain, R. S. Sarangthem and A. Chandrakumar, J. Am. Chem. Soc., 2010, 132, 4536–4537 CrossRef CAS PubMed.
- K. Yavari, P. Aillard, Y. Zhang, F. Nuter, P. Retailleau, A. Voituriez and A. Marinetti, Angew. Chem., Int. Ed., 2014, 53, 861–865 CrossRef CAS PubMed.
- S. Sahasithiwat, T. Sooksimuang, L. Kangkaew and W. Panchan, Dyes Pigm., 2017, 136, 754–760 CrossRef CAS.
- J. R. Brandt, X. Wang, Y. Yang, A. J. Campbell and M. J. Fuchter, J. Am. Chem. Soc., 2016, 138, 9743–9746 CrossRef CAS PubMed.
- Y. Yang, R. C. da Costa, D.-M. Smilgies, A. J. Campbell and M. J. Fuchter, Adv. Mater., 2013, 25, 2624–2628 CrossRef CAS PubMed.
- Y. Yang, R. C. da Costa, M. J. Fuchter and A. J. Campbell, Nat. Photonics, 2013, 7, 634–638 CrossRef CAS.
- P. M. op den Brouw and W. H. Laarhoven, Recl. Trav. Chim. Pays-Bas, 1978, 97, 265–268 CrossRef CAS.
- J. W. Diesveld, J. H. Borkent and W. H. Laarhoven, Recl. Trav. Chim. Pays-Bas, 1980, 99, 391–394 CrossRef CAS.
- D. Nečas, R. P. Kaiser and J. Ulč, Eur. J. Org. Chem., 2016, 5647–5652 CrossRef.
- T. Ishiyama, J. Takagi, K. Ishida, N. Miyaura, N. R. Anastasi and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 390–391 CrossRef CAS PubMed.
- J. F. Hartwig, Chem. Soc. Rev., 2011, 40, 1992–2002 RSC.
- T. Ishiyama, Y. Nobuta, J. F. Hartwig and N. Miyaura, Chem. Commun., 2003, 2924–2925 RSC.
- J.-Y. Cho, M. K. Tse, D. Holmes, R. E. Maleczka and M. R. Smith, Science, 2002, 295, 305–308 CrossRef CAS PubMed.
- I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS PubMed.
- T. Ishiyama and N. Miyaura, Pure Appl. Chem., 2006, 78, 1369–1375 CrossRef CAS.
- G. Zhang, F. Rominger and M. Mastalerz, Chem.–Eur. J., 2016, 22, 3084–3093 CrossRef CAS PubMed.
- D. N. Coventry, A. S. Batsanov, A. E. Goeta, J. A. K. Howard, T. B. Marder and R. N. Perutz, Chem. Commun., 2005, 2172–2174 RSC.
- R. Ozawa, K. Yoza and K. Kobayashi, Chem. Lett., 2011, 40, 941–943 CrossRef CAS.
- T. Kimoto, K. Tanaka, Y. Sakai, A. Ohno, K. Yoza and K. Kobayashi, Org. Lett., 2009, 11, 3658–3661 CrossRef CAS PubMed.
- M. N. Eliseeva and L. T. Scott, J. Am. Chem. Soc., 2012, 134, 15169–15172 CrossRef CAS PubMed.
- S. D. Ros, A. Linden, K. K. Baldridge and J. S. Siegel, Org. Chem. Front., 2015, 2, 626–633 RSC.
- A. G. Crawford, Z. Liu, I. A. I. Mkhalid, M.-H. Thibault, N. Schwarz, G. Alcaraz, A. Steffen, J. C. Collings, A. S. Batsanov, J. A. K. Howard and T. B. Marder, Chem.–Eur. J., 2012, 18, 5022–5035 CrossRef CAS PubMed.
- Z. Liu, Y. Wang, Y. Chen, J. Liu, Q. Fang, C. Kleeberg and T. B. Marder, J. Org. Chem., 2012, 77, 7124–7128 CrossRef CAS PubMed.
- S. Hitosugi, Y. Nakamura, T. Matsuno, W. Nakanishi and H. Isobe, Tetrahedron Lett., 2012, 53, 1180–1182 CrossRef CAS.
- L. Ji, K. Fucke, S. K. Bose and T. B. Marder, J. Org. Chem., 2015, 80, 661–665 CrossRef CAS PubMed.
- H. Shinokubo, Proc. Jpn. Acad., Ser. B, 2014, 90, 1–11 CrossRef CAS PubMed.
- Y. Koyama, S. Hiroto and H. Shinokubo, Angew. Chem., Int. Ed., 2013, 52, 5740–5743 CrossRef CAS PubMed.
- Y. Takaki, K. Yoza and K. Kobayashi, Chem. Lett., 2017, 46, 655–658 CrossRef CAS.
- P. Harrisson, J. Morris, T. B. Marder and P. G. Steel, Org. Lett., 2009, 11, 3586–3589 CrossRef CAS PubMed.
- L. Zeqing, L. Zhibin, W. Yuqiang and Y. Pei, Res. Chem. Intermed., 2012, 39, 1917–1926 CrossRef.
- S. M. Preshlock, B. Ghaffari, P. E. Maligres, S. W. Krska, R. E. Maleczka and M. R. Smith, J. Am. Chem. Soc., 2013, 135, 7572–7582 CrossRef CAS PubMed.
- C. W. Liskey and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 12422–12425 CrossRef CAS PubMed.
- T. Ohmura, T. Torigoe and M. Suginome, J. Am. Chem. Soc., 2012, 134, 17416–17419 CrossRef CAS PubMed.
- G. Zhang, F. Rominger, U. Zschieschang, H. Klauk and M. Mastalerz, Chem.–Eur. J., 2016, 22, 14840–14845 CrossRef CAS PubMed.
- Y. Saito, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2015, 137, 5193–5198 CrossRef CAS PubMed.
- B. E. Haines, Y. Saito, Y. Segawa, K. Itami and D. G. Musaev, ACS Catal., 2016, 7536–7546 CrossRef CAS.
- C. C. C. J. Seechurn, V. Sivakumar, D. Satoskar and T. J. Colacot, Organometallics, 2014, 33, 3514–3522 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, compounds characterization data, screened ligands and conditions, 1H and 13C NMR spectra of obtained compounds. CCDC 1587494 (2b). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra13021j |
| This journal is © The Royal Society of Chemistry 2018 |