
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFullerene derivatives act as inhibitors of leukocyte common antigen based on molecular dynamics simulations†
Yi Yu,
Huiyong Sun,
Tingjun Hou,
Suidong Wang* and
Youyong Li *
*
Institute of Functional Nano & Soft Materials (FUNSOM), Soochow University, Suzhou 215123, China. E-mail: yyli@suda.edu.cn; wangsd@suda.edu.cn; Tel: +86-512-65882037
First published on 16th April 2018
Abstract
Fullerene-based molecules are being studied as potential inhibitors of protein tyrosine phosphatases due to their unique properties and low toxicity. However, the underlying molecular mechanism remains elusive. In this study, molecular dynamics (MD) simulations in conjunction with molecular docking calculations were utilized to investigate the binding effects of C60, C60(NH2)30, and C60(OH)30 on the enzymatic activity of CD45 (a receptor-like protein tyrosine phosphatase). Our results show that all the investigated molecules can be docked into the region between D1 and D2 domains of CD45, and stabilize the protein structure. The average number of residues that directly interact with the C60(NH2)30 is two more than that of C60(OH)30, F819 and F820 (located in the loop connects α3 and β12), resulting in different effects of C60(NH2)30 and C60(OH)30 on protein activity. Detailed MD simulation analyses show that transformation of the interaction network caused by C60(NH2)30 is completely different from that of the control simulation due to the misfolding of α3. Furthermore, the movement of D1 active pocket and KNRY motif are most severely impaired by docking with C60(NH2)30. Our simulation results illustrate that fullerene derivatives modified with amino groups exhibit conspicuous tumor inhibition to protein tyrosine phosphatases, and can act as effective inhibitors. Our results give insight into the inhibitory effects of fullerene-based molecules on protein tyrosine phosphatases and providing a theoretical basis for the design of effective inhibitors.
Introduction
The fast developing field of nanotechnology has led to applications in different Frontier technologies, such as medicine, catalysts, chemicals and coatings. However, the effects of nanomaterials on the structure and activity of proteins remain mostly unclear.1,2 In order to facilitate their biological applications, such as gene delivery,3 cellular imaging,4 tumor therapy,5 and biological experimental technology,6 it is essential to understand the interactions between nanomaterials and proteins.Carbon-based nanomaterials, one of the most prevalent types of nanomaterials, such as fullerene, graphene and carbon nanotubes, have attracted considerable attention because of their excellent chemical and physical properties.7–10 Fullerene, with the 7.1 Å wide cage, contains 20 hexagons and 12 pentagons, and all the double bonds within its geodesic structure are conjugated.11 The presence of both five- and six-membered rings might introduce more specific reactive sites for covalent derivatization,12,13 and an anchor point for non-covalent interactions, respectively. Several investigators have developed fullerene-based inhibitors for the therapy of HIV-1P for AIDS.14–16 Additionally, fullerene-based molecules have also been found to bind and inhibit the activity of glutathione-s-transferase (GST),17,18 where subsequent docking simulations revealed that C60 bound at the interface of GST dimer. Blocking this binding site can suppress the function of GST, thus lead to cytotoxicity to cells. Park et al. have used MD simulations to show that fullerenes with an average diameter of 0.7–0.9 nm could bind onto the entrance of a potassium channel, leading to the blocking of K+ ions entering into the protein channel.19 Further theoretical analyses (based on molecular docking and MD simulations) suggest that the specific binding of fullerenes mainly depend on the structure of different K+ channels,20 where fullerenes trigger rearrangement of proteins and inhibit the enzyme activity by allosteric effect. Recently, NMR chemical shift perturbation analysis identified the binding position of C60 in solution, which is localized at the catalytic site of the protein and with high specificity.21
In the aspect of physical properties, the pristine fullerene is highly hydrophobic and can exhibit strong hydrophobic interactions to hydrophobic binding pocket of various protein targets, which may make C60 act as effective inhibitor for these proteins. Surface modification of fullerene increases their poor solubility and low biocompatibility, leading to new functional properties for a large variety of applications ranging from imaging,22 drug delivery23 and bio-sensing,24 cancer and gene therapy,25,26 and tissue engineering,27 etc. More studies have been reported that many fullerene-based molecules with different surfaces and architectures could be used as actives or (potential actives) with low toxicity to interfere or alter the native functions of proteins,28,29 where interactions between fullerene-based actives and proteins have been identified both experimentally30–33 and computationally.34–36
Comprehensive analyses of interactions between a protein and fullerene-based nanomaterial molecules by simple models can help understanding its potential toxicity, a topic still under debate.37,38 MD simulations can provide accurate information with respect to interactions between fullerene-based molecules to proteins, and can explain the mechanism of effects of surface chemistry on inhibition efficiency as well.
Recently, some investigators have shown that fullerene-based molecules exhibit inhibitory effects on protein tyrosine phosphatases.39 Leukocyte common antigen cd (CD45) is a receptor-like protein tyrosine phosphatase (RPTP) and has been found in all nucleated hematopoietic cells.40,41 T and B cells express large amounts of CD45 on their surfaces, indicating that CD45 plays an essential role in antigen-induced activation of T and B cells.41 CD45 controls immune response by dephosphorylating a number of signal molecules.40,42 All CD45 isoforms share a same cytoplasmic region (707 amino acids), which includes a membrane proximal PTP D1 domain and a membrane distal PTP D2 domain. Experimental data that corroborate this model have been reported.43,44 There are extensively complementary surfaces and interactions between D1 and D2 domains, and thus maintain relative orientation and stability of CD45. The energy required to destroy the interactions between the two monomers is ∼16 kcal mol−1. Because of these multiple factors, the occurrence of slightly structural changes of D2 can affect the conformational stability of D1 through inter-molecular and intra-molecular interactions. In other words, the D1 and D2 domains establish extensive association, so that one requires another to maintain the reasonable structure of CD45.
Due to the encouraging effects of fullerene-based molecules to CD45, here, we studied the structural dynamics of CD45 complexed with C60, C60(OH)30, and C60(NH2)30 by performing molecular docking and MD simulations. The effects of fullerene-based molecules on the enzymatic activity of CD45 were also investigated. Our simulation results show that all the three fullerene-based compounds (C60, C60(OH)30, and C60(NH2)30) can be docked into the region between D1 and D2 domains of CD45, leading to the deformation of the originally existed stable interactions between D1 and D2 domains. D1 and D2 domains establish compact connection to the extent, where even subtle changes will destroy the maintenance of their proper structures. Thus, the active sites of D1 and D2 domains can be deformed by binding with fullerene-based molecules, in which deformation caused by binding with C60(NH2)30 to CD45 is the most serious among the three fullerene-based molecules. To our knowledge, this is the first report to investigate how fullerene-based molecules modified with different functional groups interfere or alter the functions of proteins.
Materials and methods
Systems preparation
The native structure of protein tyrosine phosphatase (PDB: 1YGR45) was obtained from Protein Data Bank (http://www.pdb.org/pdb). The structure of protein was prepared by Discover Studio version 2.5,46 which includes residues repair and geometry optimization. We also used Discover Studio 2.5 to predict protein ionization. The calculated pKa of all histidines are shown in Table S1.† Since the pKa of each histidine is lower than the isoelectric point (7.59), all histidines were treated with unprotonated model. N- and C-terminals were methylamidated and acetylated, respectively.In the current work, we simulated the structures of CD45 docked with pristine fullerene and two derivatives modified with hydroxyl and amino groups, respectively. The pristine fullerene was built by Material Studio 5.5.47 Chemical groups of –OH and –NH2 were evenly attached to carbon atoms of the pristine fullerene, where the ratio between hydroxyl groups (or amino groups) and carbon atoms was set to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, namely, C60(OH)30 and C60(NH2)30.
:2, namely, C60(OH)30 and C60(NH2)30.
Then geometry optimizations were performed for the three fullerene-based materials by the Forcite module47 with Dreiding force field48 and Gasteiger charges in Material Studio 5.5 (the maximum iterations were set to 50000 and the convergence was set to 5.0 × 10−6). The ultra fine quality was used for the energy calculation. The final structures were used as the initial structures for the following docking simulation.
Molecular docking
Recently, numerous docking protocols have been used for generating initial coordinates for complexes containing proteins and nanomaterials. Here, PatchDock49 was used for docking of the three fullerene-based molecules (C60, C60(OH)30 and C60(NH2)30) to CD45, which works by maximizing the complementarity of the surface shapes of the two molecules, while minimizing the number of steric clashes. To accurately characterize the binding modes of the three fullerene-based molecules to CD45, FireDock50 was further used to re-score the three fullerene-containing protein complexes (CD45 + C60, CD45 + C60(OH)30, CD45 + C60(NH2)30). For each complex, two thousand docking runs were performed and ranked by the docking score. The top 50 docking poses of C60, C60(OH)30, C60(NH2)30 between CD45 are shown in Fig. S3.† Binding mode with the highest docking score of the three complexes was saved as the initial structures for the following MD simulations.Molecular dynamics (MD) simulation
Particle Mesh Ewald (PME)52 method was applied to calculate electrostatics interactions, with a cutoff of 12 Å for both long-range electrostatic and van der Waal (vdW) interactions. Hydrogen atoms were maintained by SHAKE algorithm, and a time step of 1 fs was applied for all the MD runs. Langevin dynamics53 and Nosé-Hoover Langevin piston54 were adopted to control the system temperature (at 310 K) and pressure (at 1 bar), respectively.
The MD simulations were carried out with NAMD software package,55 where the proteins (CD45) were simulated with CHARMM27 force field56 and the fullerene-based molecules were performed with Dreiding force field. Visualization and data collection were done by VMD.57
000 steps of energy minimization were performed. Then, a three steps protocol was used for the equilibration of the systems: (1) 500 ps-equilibration in NPT ensemble in the absence of positional restraints of water molecules at 310 K; (2) a further 500 ps-equilibration by only restricting the heavy atoms of C60/C60(NH2)30/C60(OH)30; (3) a 500 ps-equilibration without any positional constraints. Finally, 100 ns unrestrained full atomistic MD simulations were carried out for the four equilibrated systems (CD45, CD45 + C60, CD45 + C60(NH2)30, and CD45 + C60(OH)30).Results and discussion
To investigate the binding effects of fullerene-based molecules to tyrosine phosphatase CD45, molecular docking and 100 ns unrestrained MD simulations were performed for the four systems: apo-state CD45, CD45 + C60, CD45 + C60(NH2)30, and CD45 + C60(OH)30.The rationality of C60(NH2)30 and C60(OH)30
According to experimental data, the highest water solubility and biocompatibility are obtained when the number of hydroxyl groups is 20–30.58,59 The reason we choose the ratio (1:2) is the following: (i) the results obtained in many laboratories show that polyhydroxylated fullerenes, C60(OH)n (n = 20–30) water soluble and exert mainly antioxidant activity in biological systems.60,61 In addition, the fluorescence detection of water soluble fullerene derivatives under different pH values shows that when pH = 6.5–7.5, it is a neutral state.2 Thus, the ionization of C60(OH)30 and C60(NH2)30 do not occur at the pH of our work (∼7.4).
We carried out a 20 ns MD simulation of C60(NH2)30/C60(OH)30 in a water box, and the energy was shown in the Fig. S1.† It indicates that the model is stable. We have drawn a grid of interactions between C60(NH2)30/C60(OH)30 and protein. It can be seen from the Fig. S2† that only three amino groups (two hydroxyl groups) are not involved in the interaction with proteins, which suggests that reducing the concentration of functional groups can weaken the interaction and make it ineffective. Increased concentration, fullerene derivatives are unstable at pH = 7.4.2 In addition, combined with the data in the Table S2 and Fig. S5,† the interaction energy between the –NH2 and CD45 significantly higher than that of –OH. Thus, if a part of the –NH2 is replaced by –OH, the coexistence of the two functional groups can also weaken the interaction energy with the protein, thus reducing its inhibitory effect.
Binding modes of fullerene-based molecules to CD45
We used PatchDock49 to generate the initial coordinates of the complexes of CD45 and fullerene-based molecules. The top 50 docking poses of CD45 + C60, CD45 + C60(NH2)30, and CD45 + C60(OH)30 are shown in Fig. S3.† All the fullerene-based molecules (C60, C60(NH2)30 and C60(OH)30) can be docked into the region between the membrane proximal PTP D1 domain and the membrane distal PTP D2 domain (herein after referred as D1 and D2 domain, respectively) of CD45. Interestingly, for these three docked complexes, almost ninety percent of the binding poses are concentrated in the same region, which also includes that with highest score. Thus, we identified the binding pose with the highest docking score as the binding mode of the CD45-fullerenes complexes. As a result, the top 1 docking pose was saved as the initial structure for the 100 ns MD simulations for each complex.The equilibrium of the investigated systems can be described by root mean square deviations (RMSD), calculated as following,
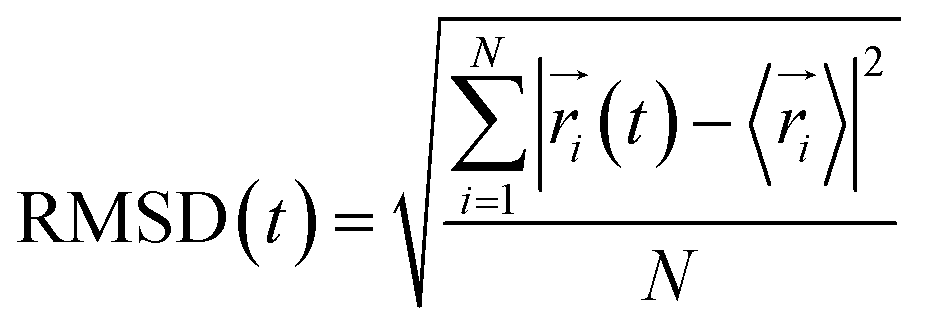 | (1) |
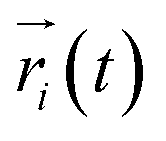 represents the position of atom i at time t,
represents the position of atom i at time t, 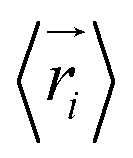 represents the average position of atom i at all simulation times.
represents the average position of atom i at all simulation times.
Fig. 1 shows the time evolutions of the root mean square deviations (RMSDs) of C60-, C60(NH2)30-, C60(OH)30-bound CD45 during 100 ns MD simulations. The RMSD values of C60, C60(NH2)30, and C60(OH)30 are 0.15 Å, 0.91 Å and 0.75 Å, respectively, indicating that the selected docking pose of C60 in CD45 is the most stable one. Although the fluctuation of C60(NH2)30 is the largest among the three molecules, it does not affect the stability of the docking structure with the average RMSD value lower than 1 Å. These results indicate that the three bound-state complexes are stable during 100 ns MD simulations. Furthermore, the convergences of the three bound-state and the control (apo-state CD45) simulations are verified by the RMSD changes of Cα atoms of CD45. As shown in Fig. 2, the average RMSD values of Cα atoms of CD45 docked with C60, C60(NH2)30, and C60(OH)30 are 2.25 Å, 2.65 Å, 2.48 Å, respectively. In addition, these three bound-state complexes are equilibrated within ∼20 ns. However, the control simulation is equilibrated after ∼60 ns with the value of RMSD stabilized at around 3.25 Å. The lower RMSDs and shorter equilibrating simulation time of the bound-state complexes (compared with the control group) mean that the docking of fullerene-based molecules makes CD45 more stable.
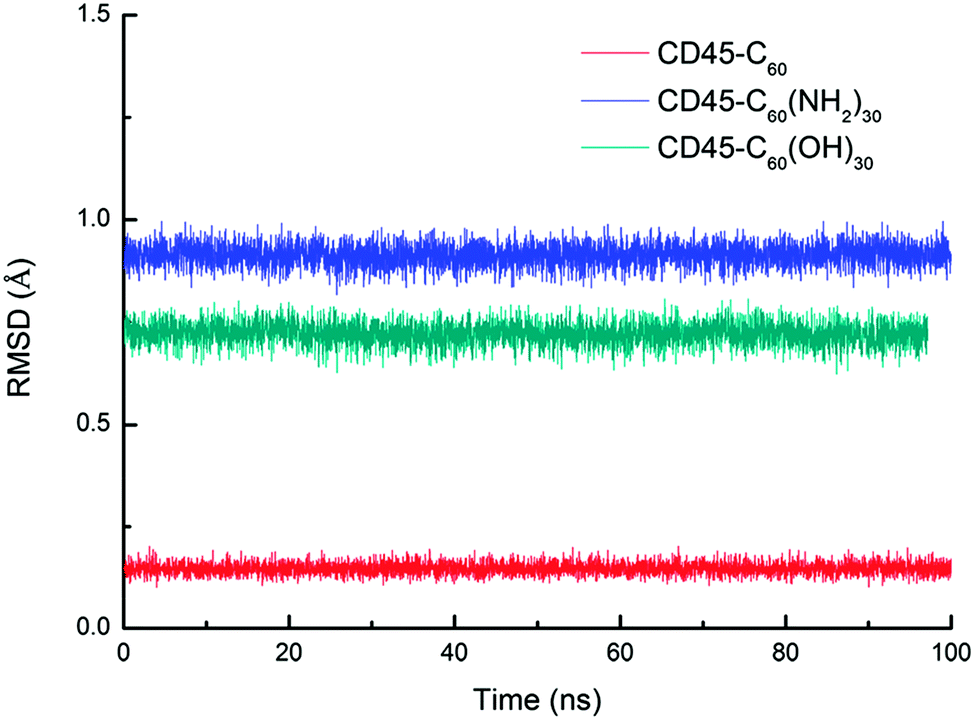 | ||
| Fig. 1 Time evolutions of RMSDs of C60 (red), C60(NH2)30 (blue), C60(OH)30 (green) in bound-state complexes. The low RMSDs (<1 Å) indicate that the three docked complexes are stable during 100 ns MD simulations. | ||
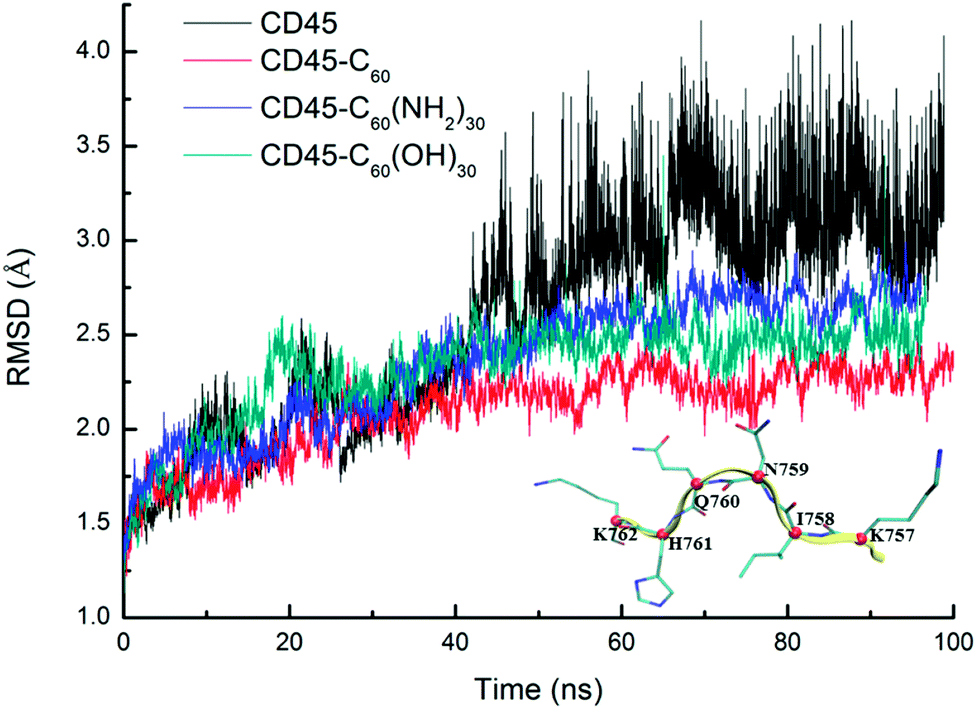 | ||
| Fig. 2 Time evolutions of RMSDs of Cα atoms of CD45 docked with C60 (red), C60(NH2)30 (blue), and C60(OH)30 (green) during 100 ns MD simulations. To give a comparison, a control group of apo-state CD45 is also shown in black line. We randomly selected a segment of CD45 (K757–K762, β9) to indicate the location of Cα atoms of every residues, which is colored by red ball. | ||
The final structures obtained from 100 ns MD simulations are shown in Fig. 3. Owing to the different surface properties of fullerene-based molecules, the compositions of their binding pockets are different in a large extent. We consider that the residues within 6 Å of fullerene-based molecules can directly interact with them. The average number of residues that interact directly with C60 is ∼20, with hydrophobic amino acids predominantly, such as P765, L805, Y1001, V1175 and A1178, etc. Amphipathic and hydrophilic residues, such as K762, K771, R812, E894, E1167, E1168 and so on, play main roles in the formation of hydrophilic interactions with C60(NH2)30 and C60(OH)30. Nevertheless, the average number of residues that interact with C60(NH2)30 is ∼22, slightly more than that of C60(OH)30 (∼20). Table S4† list the probability of these residues interacts with these two fullerene derivatives during the last 80 ns simulations. From this table, the compositions of the binding pockets of C60(NH2)30 and C60(OH)30 are almost identical except for F819 and F820, which are highlighted in Fig. 3b and c. C60(NH2)30 can interact strongly with phenylalanine because nitrogen atoms are more electrophilic than oxygen atoms. It is noteworthy that, although F816 and F819/F820 are the same types of amino acids, there is a probability of 100% for both C60(NH2)30 and C60(OH)30. The main reason is the difference in the initial position of F816 and F819/F820. As shown in Fig. S4,† F819/F820 is outwardly offset in crystal structure without docking any molecule. When docked with C60(NH2)30 and C60(OH)30, F819/F820 formed stronger interaction with the former, resulting in its proximity to C60(NH2)30 as a part of its binding pocket. However, in the crystal structure, F816 is located near the center of the binding pocket. The initial distances between F816 and C60(NH2)30/C60(OH)30 are about 5 Å, resulting in a strong hydrophobic interaction, so it is difficult to escape during the simulation process. F819 and F820 are located in the loop (from residue N814 to I824) between α3 helix and β12 strand, which connects the active pocket (from residue V826 to G836) of CD45 D1 domain. The flexibility of this loop is severely restricted by the strong interactions formed by F819/F820 and C60(NH2)30, which further affects the movement of the active pocket (detailed analysis will be discussed in the section “Conformational changes of the active site”).
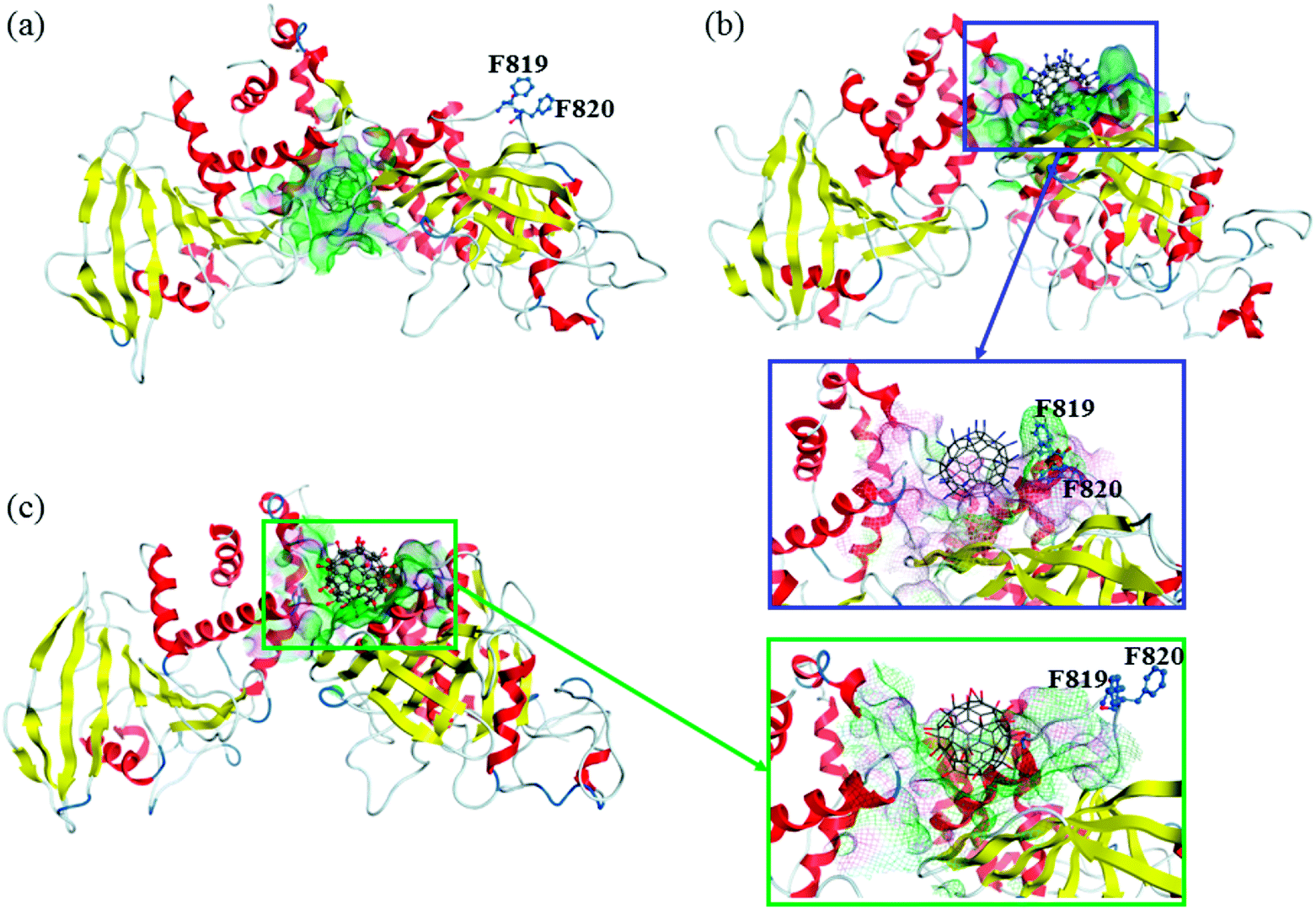 | ||
| Fig. 3 The resulted bound structures of (a) CD45 + C60, (b) CD45 + C60(NH2)30, and (c) CD45 + C60(OH)30. Water molecules are excluded for clarity. The surfaces of their binding pockets are colored by H-bonding regions (purple), hydrophobic regions (green), and polar regions (blue). Residues F819 and F820 are highlighted. | ||
In addition, the interaction probability of C764 and S817 that directly interact with C60(NH2)30 and C60(OH)30 are also different. For C764, as a sulfur-containing amino acid, the sulfhydryl groups on it are more likely to interact with amino groups. For S817, as shown in Fig. S4,† due to the intense interaction between F819/F820 and C60(NH2)30, this loop undergoes a conformational change that results in S817 being far away from C60(NH2)30, reducing the probability of its interaction with C60(NH2)30. The differences between the binding models of C60(NH2)30 and C60(OH)30 cause their different effects on CD45.
Interactions between fullerene-based molecules and CD45
For detailed analyses of the interactions between fullerene-based molecules and CD45, we firstly counted the probability of the number of H-bonds formed between C60, C60(NH2)30, C60(OH)30 and CD45. Here, the distance between a hydrogen donor and an acceptor no larger than 3.5 Å and the supplementary angle formed by the donor, hydrogen, and acceptor no larger than 35° are used as the criterion for identifying a H-bond. As shown in Fig. 4, due to the highly hydrophobic property of C60, the probability of forming H-bonds (including forming 1, 2 and 3 H-bonds) is only 12% during the 100 ns MD simulation. Nevertheless, the probability of forming H-bonds between C60(NH2)30, C60(OH)30 and CD45 are 86% and 97%, respectively. Whereas, the probability to form three or more H-bonds between C60(NH2)30 and CD45 is only 35%, which is significantly lower than that between C60(OH)30 and CD45 (65%), moreover, the average numbers of H-bonds formed between C60(NH2)30, C60(OH)30 and CD45 are 2 and 3, respectively. In summary, the ability to form H-bonds between C60(NH2)30 and CD45 is slightly weaker than that of C60(OH)30.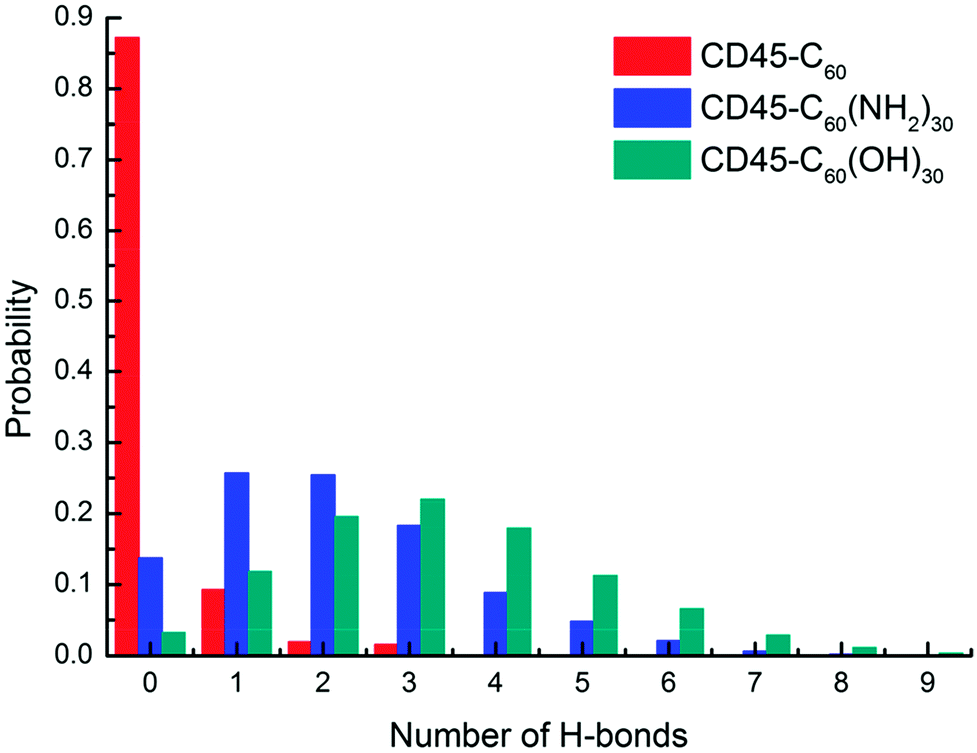 | ||
| Fig. 4 Probability of the number of H-bonds formed between C60 (red), C60(NH2)30 (blue), C60(OH)30 (green) and CD45 during 100 ns MD simulations. | ||
Furthermore, we also calculated the interaction energies between C60, C60(NH2)30, C60(OH)30 and CD45. These results are listed in Table 1. Owing to the pure hydrophobicity of C60, the electrostatic energy between it and CD45 is 0 kcal mol−1, and the vdW energy is −71.95 ± 3.42 kcal mol−1, which is the strongest among the three systems. We break down the vdW energy into every residue, the result is shown in Fig. S5.† Although the average number of residues that interact with C60 is two less than with C60(NH2)30, the vdW energy between each residue and C60 is higher than that of C60(NH2)30. So the total vdW energy of C60 and CD45 is more stronger. For the other two fullerene-based molecules, the electrostatic energy between CD45 and C60(NH2)30 is −12.08 ± 0.23 kcal mol−1, slightly less than that of C60(OH)30 complexed CD45 (−15.46 ± 0.71 kcal mol−1), which is consistent with the hydrogen bonding results obtained earlier. However, the vdW energies between CD45 and C60(NH2)30, and C60(OH)30 are −51.38 ± 3.42 and −42.54 ± 2.70 kcal mol−1, respectively, probably due to the relatively greater average number of residues that interact directly with C60(NH2)30. Totally, the interaction energy between CD45 and C60 is the strongest among the three systems, while weaker interactions is shown of system of C60(NH2)30-CD45.
| Fullerene-based molecules | vdW | Elec. | Total |
|---|---|---|---|
| C60 | −71.95 ± 3.42 | 0 | −71.95 ± 3.42 |
| C60(NH2)30 | −51.38 ± 3.42 | −12.08 ± 0.23 | −63.46 ± 3.14 |
| C60(OH)30 | −42.54 ± 2.70 | −15.46 ± 0.71 | −58.00 ± 1.93 |
Deformation of the conformations between D1 and D2
As reported in previous work,40 the cytoplasmic region of CD45 contains two homologous protein tyrosine phosphatase domains, the membrane proximal PTP D1 domain and the membrane distal PTP D2 domain. There are extensively complementary surfaces between D1 and D2 domains, including the residues of the α3 and α6 helices of D1 and the α4 and α5 helices of D2. D1 and D2 domains pack tightly against each other by an extensive network of interactions, consisting of H-bonds, van der Waals interactions, and salt bridges. Critical residues such as D766, Y767, E801, R812 (in D1 domain) and E894, K1003, E1167 (in D2 domain) play important roles in maintaining a stable inter-domain orientation between D1 and D2 domains. Among these residues, R812 and E1167 are highly conserved in most RPTP. Schematic of the complementary surfaces between D1 and D2 domains and these critical residues are shown in Fig. 5.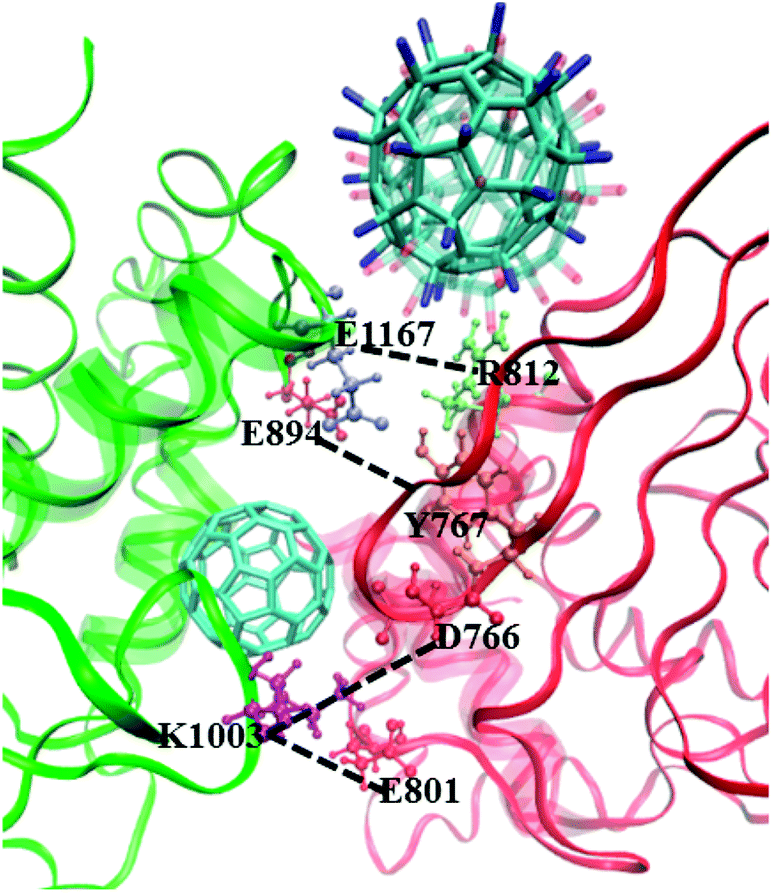 | ||
| Fig. 5 Complementary surfaces between D1 (red) and D2 (green) segments of CD45. The α3 and α6 helices of D1 and the α4 and α5 helices of D2 are highlighted by transparent NewCartoon representation. The critical residues involved in intra-molecular interactions between D1 and D2 are represented by Licorice and CPK models. C60, C60(NH2)30, and C60(OH)30 complexed CD45 are shown with Licorice representation. | ||
Our docking results show that both fullerene-based molecules can be docked into the region between D1 and D2 domains. Therefore, binding of fullerene-based molecules to CD45 can seriously impair the network of the original interactions. We elaborated the interactions by analyzing distances between critical residues in D1 and D2 domains and the results are shown in Fig. 6. Generally speaking, two H-bond formable residues can form strong H-bond or salt bridge interactions when their distance is less than ∼3.5 Å. The strength of the interaction decreases with the increase of distance, and completely disappears when the distance reaches up to ∼6 Å. Fig. 6a shows the time evolutions of the distance between R812 in D1 and E1167 in D2. We can see that the distance of the control simulation (black line) keep stable around 3.5 Å, then it is destroyed and cannot be recovered until the ending of simulation. The distance of the system of CD45 + C60 is averaged at ∼6 Å after ∼30 ns simulation, which is similar to that of the control simulation, indicating that the interaction formed between R812 and E1167 is not significantly affected by the binding of C60. However, the distances keep stable at around 3 Å throughout the simulations for both the systems of CD45 + C60(NH2)30 (blue lines) and CD45 + C60(OH)30 (green line). These results mean that R812 and E1167 can form strong and stable interactions by binding with C60(NH2)30 and C60(OH)30 in CD45. The distance between Y767 in D1 and E894 in D2 is shown in Fig. 6b. For the control simulation, there are strong variations of the distance in the early stage of the simulation, whereas keep stable at around 6 Å after ∼30 ns MD simulation, indicating that weak interaction exists between the two residues. Significantly, both of these distances are reduced to 4 Å and maintained extreme stable by binding with C60 C60(NH2)30 and C60(OH)30 in CD45, indicating that these two residues can form strong interactions mediated by the fullerene-based molecules.
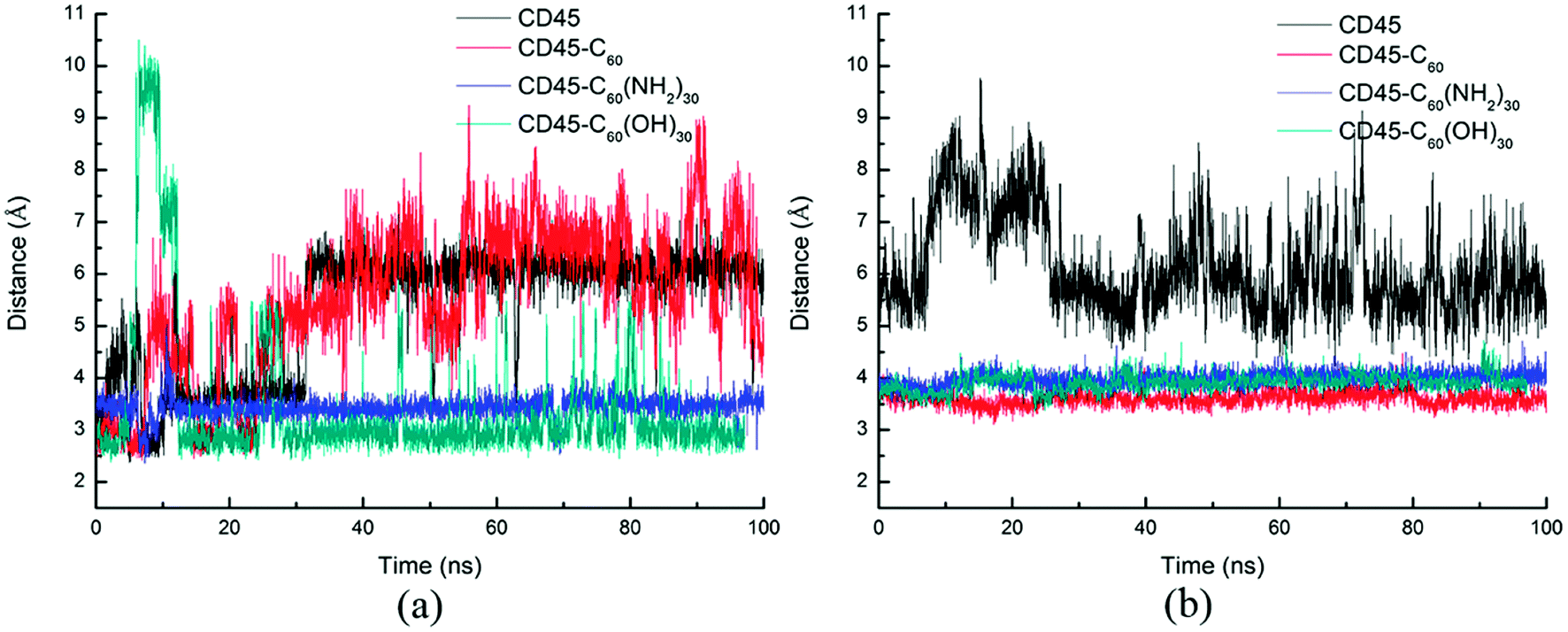 | ||
| Fig. 6 Time evolutions of the distances between critical residues (a) R812 in D1 and E1167 in D2, (b) Y767 in D1 and E894 in D2 during 100 ns MD simulations, where the interactions can be considered disappear when the distance is up to 6 Å. | ||
Fig. 7 shows the distances between D766 and E1003, and E801 and E1003, respectively. Similar tendency can be observed from Fig. 7a and b. For control simulation (black line), the distances between D766 and E1003 (in Fig. 7a), and E801 and E1003 (in Fig. 7b) keep stable at 3 Å and 3.5 Å, respectively. For the bound-state complexes of CD45 + C60 and CD45 + C60(OH)30, the average distances only increase by ∼0.1 Å and ∼0.5 Å, respectively. These results mean that the interactions are not significantly affected and remain stable throughout the simulations. Nevertheless, when CD45 binding with C60(NH2)30, these two distances become very unstable and fluctuate between 3 Å and 7 Å, implying that the interactions between D766 and E1003, and E801 and E1003, are significantly impaired by the binding of C60(NH2)30 in CD45 (blue lines in Fig. 7a and b). For a profound comprehension of the mechanisms of fullerene-based molecules to the intermolecular interactions between D1 and D2 domains, a schematic diagram is shown in Fig. 8. Compared with the control simulation (Fig. 8a), the transformation of the interaction networks of CD45 + C60 (Fig. 8b) and CD45 + C60(OH)30 (Fig. 8d) are not significant. Nevertheless, the binding of C60(NH2)30 in CD45 (Fig. 8c) completely changes the original interaction mode between D1 and D2 domains. The main reason is that F819 and F820 form strong interactions with C60(NH2)30, resulting in R812 in helix α3 to approach D2 domain and the misfolding of α3. Meanwhile, the loop region, which contains from R763 to Y767, is more flexible and detrimental to interact with K1003 in D2 domain (detailed analysis will be discussed in the section “Conformational changes of the active site”).
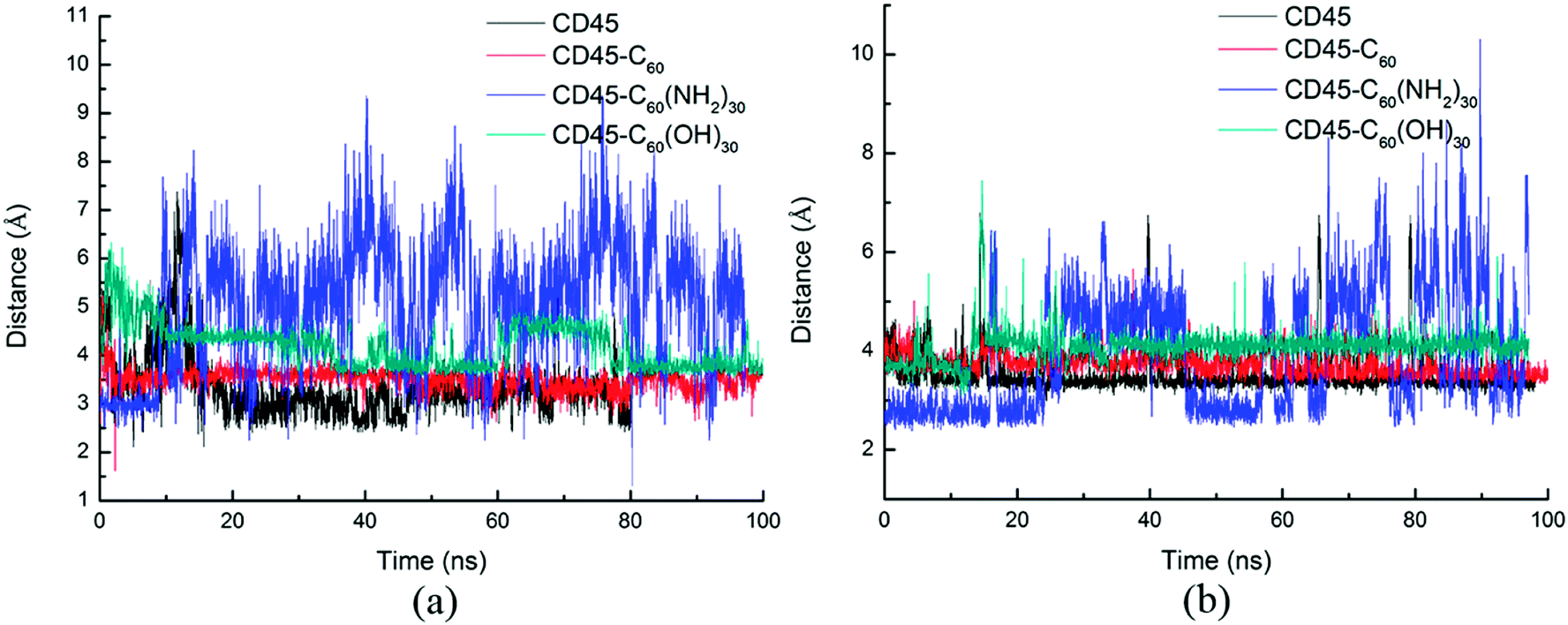 | ||
| Fig. 7 Time evolutions of the distances between critical residues (a) D766 in D1 and E1003 in D2, (b) E801 in D1 and E1003 in D2 during 100 ns MD simulations. | ||
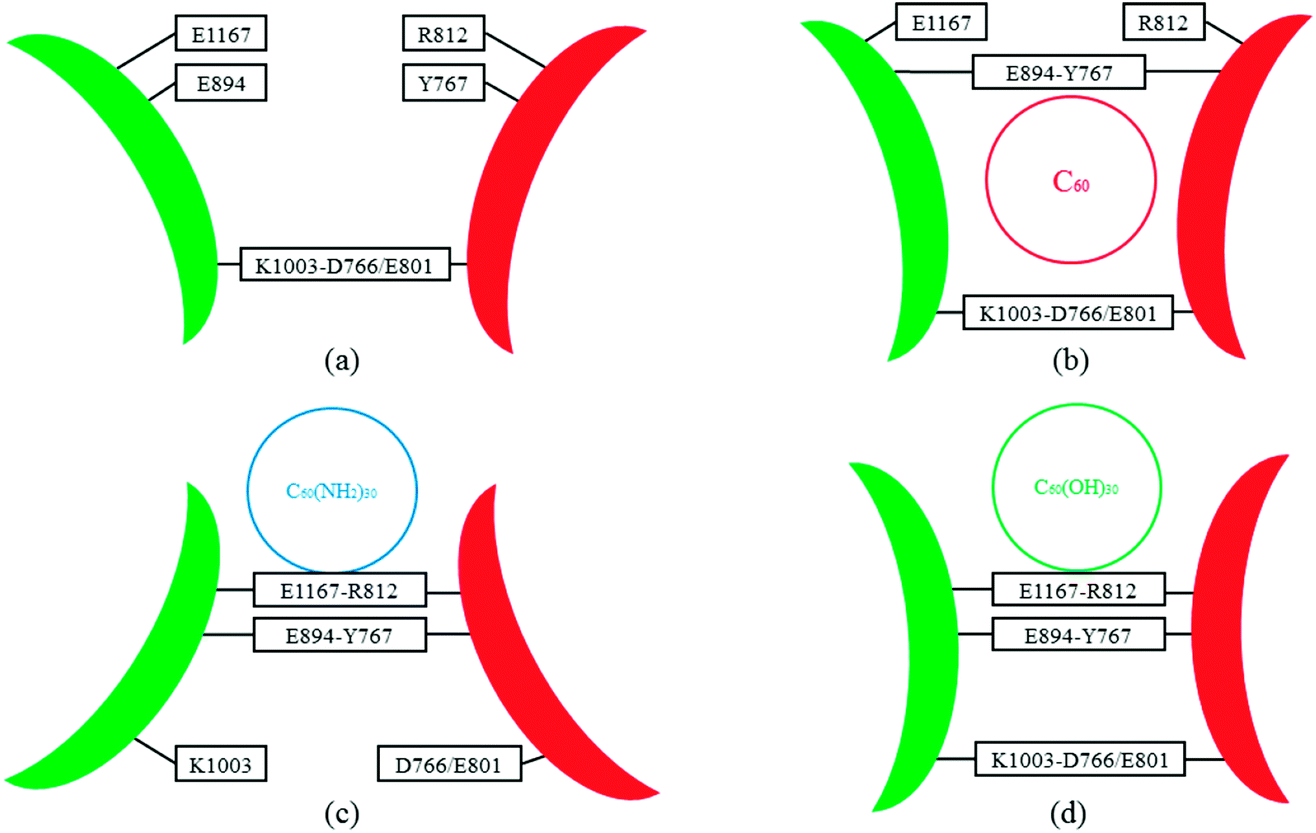 | ||
| Fig. 8 Schematic representation of intermolecular interaction networks of (a) CD45, (b) CD45 + C60, (c) CD45 + C60(NH2)30, and (d) CD45 + C60(OH)30. The D1 and D2 domains are represented in red and green arcs, respectively. The binding sites of C60, C60(NH2)30, and C60(OH)30 are represented by circles. The critical residues in D1 and D2 domains are highlighted in the square frames. They form interactions when they are in the same square frame. On the contrary, there is no interaction when the two residues are not in the same square frame. | ||
Conformational changes of D1 and D2 domains
D1 and D2 domains establish extensive association to the extent that one requires another for maintenance of its proper structure. Thus the deformation of the intermolecular interactions between D1 and D2 domains will cause structural changes of D1 and D2 domains.Fig. 9 shows the time evolutions of RMSDs of the Cα atoms of D1 (Fig. 9a) and D2 (Fig. 9b) domains. For each of the four systems, the fluctuation of RMSD values of D1 domain are coincide with those of the whole CD45 (Fig. 3), indicating that conformational changes of CD45 mainly occur in D1 domain. Significantly, the RMSD values of D2 domain are much lower and more stable, either for the control simulation or the other three bound-state complexes. These results indicate that the binding of fullerene-based molecules to CD45 have more notable influences on D1 domain than D2 domain. Our conclusions also partly confirm that the D2 domain is not biologically active, consistent with the results in the literature.42
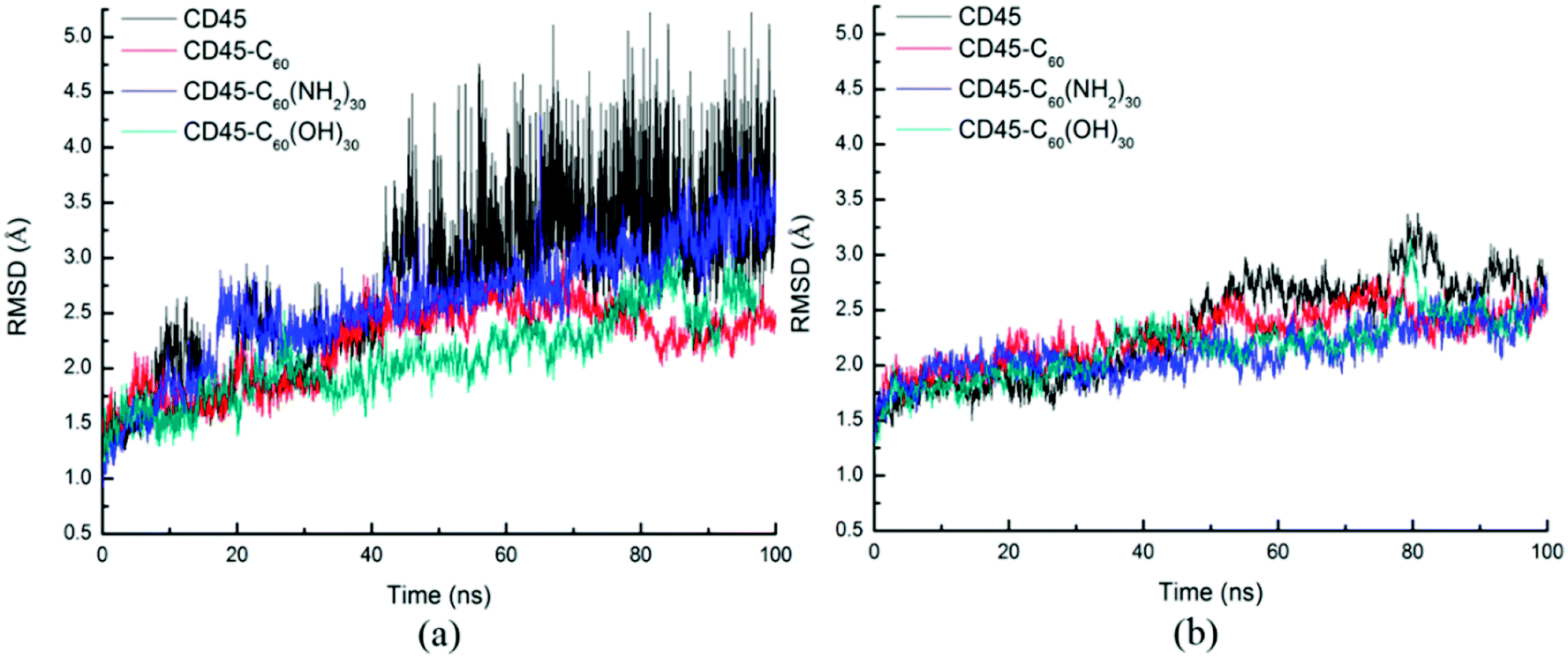 | ||
| Fig. 9 Time evolutions of RMSDs of the Cα atoms of (a) D1 and (b) D2 domains complexed with C60 (red), C60(NH2)30 (blue), and C60(OH)30 (green) during 100 ns MD simulations. The control simulation without any fullerene-based molecules is colored in black lines. | ||
Fig. 10 shows the root mean square fluctuation (RMSF) of every residue of CD45, CD45 + C60, CD45 + C60(NH2)30, and CD45 + C60(OH)30. There are several unusual active fragments in nature structure of CD45 (black line in this figure), such as residues E624, Y658, D796, S829, D1043, D1088 or E1103, indicating by the peak values in Fig. 10. These amino acids can flexibly regulate their position and orientation to accommodate substrate due to their high flexibility. However, the freedom of motion of these residues is severely restricted with the binding of fullerene-based molecules, so that they are not flexible enough to bind substrate to achieve the catalytic function of CD45. As reported in the literature,42 the active pocket of CD45 D1 domain is composed by the residues of the PTP signature motif (from V826 to G836), and surrounded by three additional segments: the KNRY motif (from K655 to Y658), the WPD motif (from W794 to D766), and the Q-rich motif (from Q872 to Q876). Importantly, as shown in the inset in Fig. 10, the RMSF values of active pocket (mainly the residues from H827 to V832) are significantly reduced after binding with fullerene-based molecules, especially for C60(NH2)30. The RMSF values of the loop region (N814–I824) are the lowest of the all investigated system, especially for F819 and F820 (RMSF are 0.928 and 0.838, respectively). The results indicate that the flexibility of the loop containing F819 and F820 is significantly reduced due to the formation of the intense interactions with C60(NH2)30, thereby inhibiting the movement of β12, that is the active pocket. Moreover, the high RMSF values of E624, Y658/D660 (in KNRF motif), D796/H797 (in WPD motif) are also significantly reduced, accompanied by the binding of fullerene-based molecules.
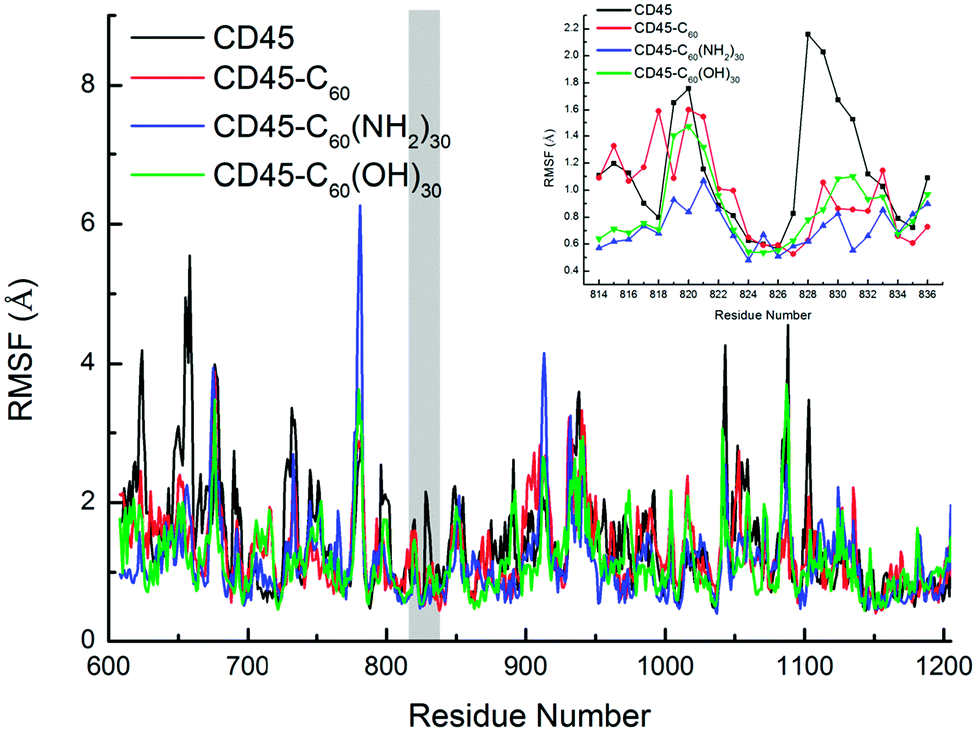 | ||
| Fig. 10 Comparison between the RMSF plots of Cα atoms for CD45 (black), CD45 + C60 (red), CD45 + C60(NH2)30 (blue), and CD45 + C60(OH)30 (green) during the MD simulations. The residues of the loop region (N814–I824) and active pocket (V826–G836) are highlighted by gray area, and the corresponding RMSF values are clearly indicated by an inset. | ||
Unexpectedly, the binding of C60(NH2)30 greatly increases the RMSF values of the loop region (residues E779 to T782) between β10 and β11. Combining the data in Table S4,† the probability of the residues which located in β10 and β11 (C764, Y767, I769, K771, T787 and I789) interact with C60(NH2)30 are greater than those interact with C60(OH)30. Furthermore, according to the data in Fig. S5b,† when C60(NH2)30 is docked, the total vdW energy of the aforementioned residues (13.08 kcal mol−1) is almost three times than that of C60(OH)30 (3.419 kcal mol−1). As these residues need to maintain strong interaction with C60(NH2)30, loop region needs to be more flexible to adjust the conformation of protein. We speculated that this is an indirect reason for the increase in the RMSF value of the loop.
We compared the crystal structure of CD45 bound with substrate (T-cell receptor: CD3 zeta ITAM-1 peptide) to our simulation structures, and the results are shown in Fig. 11. The aromatic ring of Y658 in the KNRY motif stabilizes the substrate by forming π–π interaction. However, it flips to perpendicular to the substrate by the binding of either for C60, C60(NH2)30 or C60(OH)30, which is extremely detrimental to the formation of π–π interaction with substrate. As shown in Fig. 11c, D660 moved outward ∼4.8 Å with the binding of C60(NH2)30, while it did not move along with the combination of C60, C60(OH)30. Furthermore, this positioning of these two oppositely charged residues, D660 and H797 (in WPD motif) determine the backbone configuration of the substrate. In the crystal structure, the distance between D660 and H797 is ∼8 Å, which increases to 18.1 Å after the binding of C60(NH2)30, whereas this distance increases only to 10.8 Å and 11.3 Å by the binding of C60 and C60(OH)30. Importantly, compared with the KNRY motif in the crystal structure, it moves outward by ∼6 Å in the structure bound with C60(NH2)30.
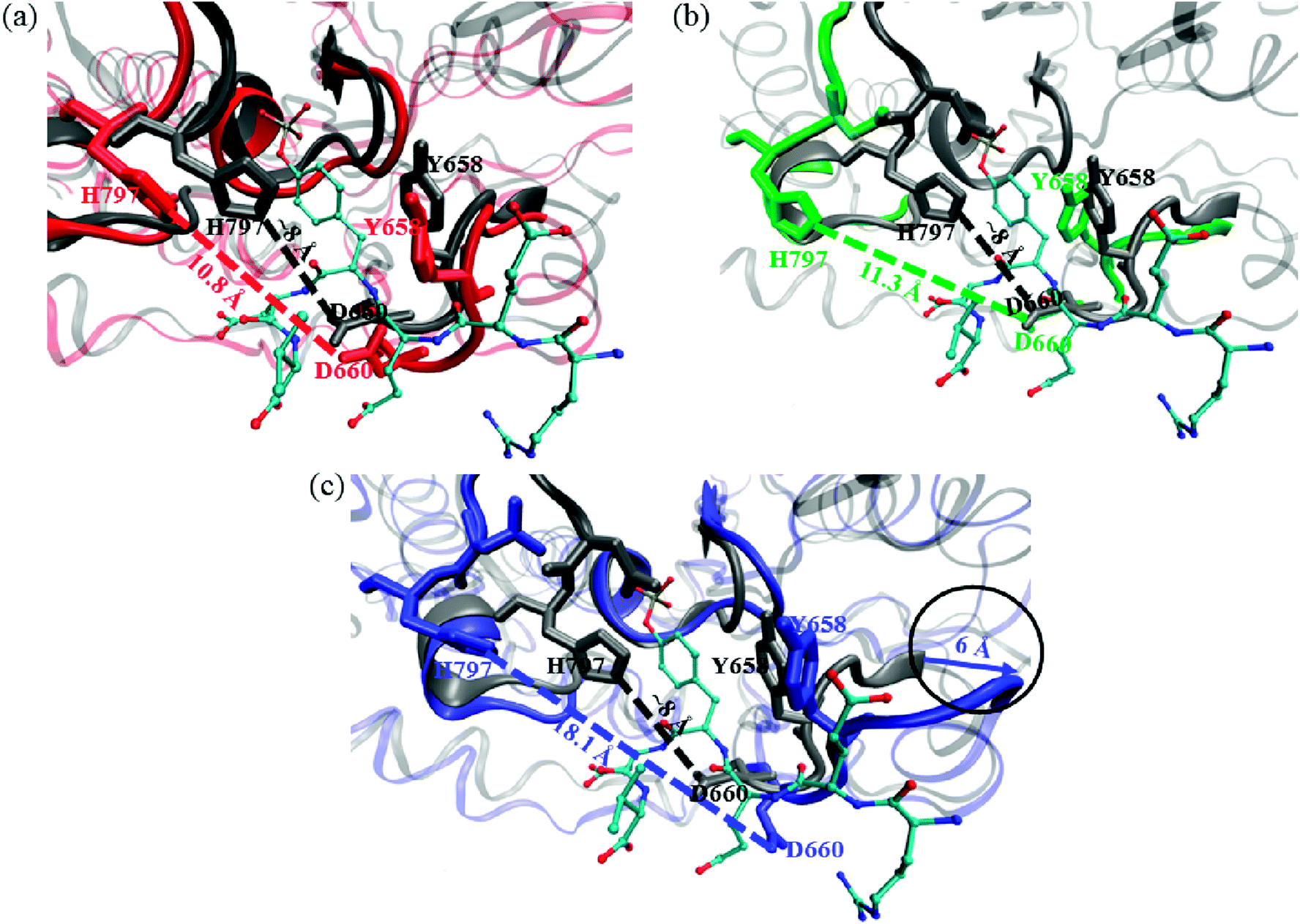 | ||
| Fig. 11 Comparison of the active pocket of CD45 D1 domain in crystal structure (gray) with the structure bound with (a) C60 (red), (b) C60(NH2)30 (blue), and (c) C60(OH)30 (green). The critical resides, Y658, D660 and H797 are also represented in relative colors. The crystal structure of substrate is represented by CPK. The circular frame in (c) indicates that the KNRY motif moves outward by ∼6 Å in the structure bound with C60(NH2)30. | ||
In summary, the binding of fullerene-based molecules affects the movement of the KNRY, and the active pocket, in which the effect of C60(NH2)30 on both is most severe.
Conclusions
Fullerene is being studied for their potential applications to be effective inhibitors for protein tyrosine phosphatases, where appropriate surface modification will enhance its inhibitory effect. However, very little is known about how fullerene-based molecules modified with different functional groups affect the activity of protein.In this study, we utilized molecular docking and atomistic MD simulations to investigate the binding modes of CD45-fullerenes complexes. Docking calculations of fullerene-based molecules (C60, C60(NH2)30, and C60(OH)30) to CD45 revealed that all the three fullerene-based molecules can be docked into the region between D1 and D2 domains. Due to the different surface properties, C60 was docked into the region mainly composed of hydrophobic and aromatic residues, while the binding positions of C60(NH2)30 and C60(OH)30 are very similar, except that the average number of residues directly interact with the former is two more than that of the latter. These two amino acids, F819 and F820, lead to different effects of C60(NH2)30 and C60(OH)30 on protein activity. Detailed MD simulation analyses of the interactions formed between D1 and D2 domains show that the behavior of C60(NH2)30 is completely different from that of the control simulation due to the misfolding of α3. Furthermore, the movement of active pocket and KNRY motif of D1 domains are most severely impaired by docking with C60(NH2)30. Succinctly, fullerene derivatives modified with amino group exhibit conspicuous tumor inhibition to CD45.
Our study and understanding of the fullerenes–protein interactions can be exploited in the view of design biologically-active hybrid materials for the inhibition of protein tyrosine phosphatases.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Basic Research Program of China (2017YFB0701600 and 2017YFA0204800), the National Natural Science Foundation of China (Grants 51761145013, 21673149), and a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD). This is also a project supported by the Fund for Innovative Research Teams of Jiangsu Higher Education Institutions, Jiangsu Key Laboratory for Carbon Based Functional Materials and Devices, Collaborative Innovation Center of Suzhou Nano Science and Technology.References
- J. Klein, Probing the Interactions of Proteins and Nanoparticles, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2029–2030 CrossRef CAS PubMed.
- X. Sun, Z. Feng, L. Zhang, T. Hou and Y. Li, The Selective Interaction between Silica Nanoparticles and Enzymes from Molecular Dynamics Simulations, PLoS One, 2014, 9, e107696 Search PubMed.
- N. L. Rosi, D. A. Giljohann, C. S. Thaxton, A. K. Lytton-Jean, M. S. Han and C. A. Mirkin, Oligonucleotide-Modified Gold Nanoparticles for Intracellular Gene Regulation, Science, 2006, 312, 1027–1030 CrossRef CAS PubMed.
- X. Michalet, F. Pinaud, L. Bentolila, J. Tsay, S. Doose, J. Li, G. Sundaresan, A. Wu, S. Gambhir and S. Weiss, Quantum Dots for Live Cells, in Vivo Imaging, and Diagnostics, Science, 2005, 307, 538–544 CrossRef CAS PubMed.
- X. Wang, L. Yang, Z. G. Chen and D. M. Shin, Application of Nanotechnology in Cancer Therapy and Imaging, Ca-Cancer J. Clin., 2008, 58, 97–110 CrossRef PubMed.
- H. Li, J. Huang, J. Lv, H. An, X. Zhang, Z. Zhang, C. Fan and J. Hu, Nanoparticle PCR: Nanogold-Assisted PCR with Enhanced Specificity, Angew. Chem., Int. Ed., 2005, 44, 5100–5103 CrossRef CAS PubMed.
- A. K. Geim, Graphene: Status and Prospects, Science, 2009, 324, 1530–1534 CrossRef CAS PubMed.
- V. C. Sanchez, A. Jachak, R. H. Hurt and A. B. Kane, Biological Interactions of Graphene-Family Nanomaterials–an Interdisciplinary Review, Chem. Res. Toxicol., 2012, 25, 15 CrossRef CAS PubMed.
- X. Sun, Z. Feng, T. Hou and Y. Li, Mechanism of Graphene Oxide as An Enzyme Inhibitor from Molecular Dynamics Simulations, ACS Appl. Mater. Interfaces, 2014, 6, 7153–7163 CAS.
- Y. Yu, H. Sun, K. Gilmore, T. Hou, S. Wang and Y. Li, Aggregated Single-Walled Carbon Nanotubes Absorb and Deform Dopamine-Related Proteins Based on Molecular Dynamics Simulations, ACS Appl. Mater. Interfaces, 2017, 9, 32452–32462 CAS.
- W. Krätschmer, L. D. Lamb, K. Fostiropoulos and D. R. Huffman, Solid C60: A New Form of Carbon, Nature, 1990, 347, 27 CrossRef.
- M. Prato, Q. C. Li, F. Wudl and V. Lucchini, Addition of Azides to Fullerene C60: Synthesis of Azafulleroids, J. Am. Chem. Soc., 1993, 115, 1148–1150 CrossRef CAS.
- H. M. Lee, M. M. Olmstead, E. Iezzi, J. C. Duchamp, H. C. Dorn and A. L. Balch, Crystallographic Characterization and Structural Analysis of the First Organic Functionalization Product of the Endohedral Fullerene Sc3N@ C80, J. Am. Chem. Soc., 2002, 124, 3494–3495 CrossRef CAS PubMed.
- S. Durdagi, T. Mavromoustakos, N. Chronakis and M. G. Papadopoulos, Computational Design of Novel Fullerene Analogues as Potential HIV-1 PR Inhibitors: Analysis of the Binding Interactions between Fullerene Inhibitors and HIV-1 PR Residues using 3D QSAR, Molecular Docking and Molecular Dynamics Simulations, Bioorg. Med. Chem., 2008, 16, 9957–9974 CrossRef CAS PubMed.
- S. Friedman, D. Decamp, R. Sijbesma, G. Srdanov, F. Wudl and G. Kenyon, Bucky-Ball Based Inhibitors of the HIV Protease-2nd-Generation Design and Model Studies, FASEB J., 1993, 1184 Search PubMed.
- A. A. Toropov, A. P. Toropova, E. Benfenati, D. Leszczynska and J. Leszczynski, SMILES-Based Optimal Descriptors: QSAR Analysis of Fullerene-Based HIV-1 PR Inhibitors by Means of Balance of Correlations, J. Comput. Chem., 2010, 31, 381–392 CAS.
- N. Miyata, Y. Yamakoshi, H. Inoue, M. Kojima, K. Takahashi, N. Iwata, Abstracts of Papers of the American Chemical Society, in Inhibition of glutathione S-transferase by [60] fullerene, Amer. Chemical Soc., 1155 16th St, NW, Washington, DC 20036 USA, 1998, p. U882 Search PubMed.
- N. Iwata, T. Mukai, Y. N. Yamakoshi, S. Haraa, T. Yanase, M. Shoji, T. Endo and N. Miyata, Effects of C60, A Fullerene, on the Activities of Glutathione S-Transferase and Glutathione-Related Enzymes in Rodent and Human Livers, Fullerene Sci. Technol., 1998, 6, 213–226 CrossRef CAS.
- K. H. Park, M. Chhowalla, Z. Iqbal and F. Sesti, Single-Walled Carbon Nanotubes are a New Class of Ion Channel Blockers, J. Biol. Chem., 2003, 278, 50212–50216 CrossRef CAS PubMed.
- S. Kraszewski, M. Tarek, W. Treptow and C. Ramseyer, Affinity of C60 Neat Fullerenes with Membrane Proteins: a Computational Study on Potassium Channels, ACS Nano, 2010, 4, 4158–4164 CrossRef CAS PubMed.
- M. Calvaresi, F. Arnesano, S. Bonacchi, A. Bottoni, V. Calo, S. Conte, G. Falini, S. Fermani, M. Losacco and M. Montalti, C60@ Lysozyme: Direct Observation by Nuclear Magnetic Resonance of a 1:1 Fullerene Protein Adduct, ACS Nano, 2014, 8, 1871–1877 CrossRef CAS PubMed.
- H. Hong, T. Gao and W. Cai, Molecular Imaging with Single-Walled Carbon Nanotubes, Nano Today, 2009, 4, 252–261 CrossRef CAS PubMed.
- C. Ménard-Moyon, E. Venturelli, C. Fabbro, C. Samorì, T. Da Ros, K. Kostarelos, M. Prato and A. Bianco, The Alluring Potential of Functionalized Carbon Nanotubes in Drug Discovery, Expert Opin. Drug Discovery, 2010, 5, 691–707 CrossRef PubMed.
- M. B. Lerner, J. D'Souza, T. Pazina, J. Dailey, B. R. Goldsmith, M. K. Robinson and A. C. Johnson, Hybrids of a Genetically Engineered Antibody and a Carbon Nanotube Transistor for Detection of Prostate Cancer Biomarkers, ACS Nano, 2012, 6, 5143–5149 CrossRef CAS PubMed.
- C. Samorì, H. Ali-Boucetta, R. Sainz, C. Guo, F. M. Toma, C. Fabbro, T. Da Ros, M. Prato, K. Kostarelos and A. Bianco, Enhanced Anticancer Activity of Multi-Walled Carbon Nanotube–Methotrexate Conjugates using Cleavable Linkers, Chem. Commun., 2010, 46, 1494–1496 RSC.
- W. Wu, S. Wieckowski, G. Pastorin, M. Benincasa, C. Klumpp, J. P. Briand, R. Gennaro, M. Prato and A. Bianco, Targeted Delivery of Amphotericin B to Cells by using Functionalized Carbon Nanotubes, Angew. Chem., Int. Ed., 2005, 44, 6358–6362 CrossRef CAS PubMed.
- S. L. Edwards, J. A. Werkmeister and J. A. Ramshaw, Carbon Nanotubes in Scaffolds for Tissue Engineering, Expert Rev. Med. Devices, 2009, 6, 499–505 CrossRef CAS PubMed.
- J. Kolosnjaj, H. Szwarc, F. Moussa, Toxicity Studies of Fullerenes and Derivatives, Bio-Applications of Nanoparticles, Springer, 2007, pp. 168–180 Search PubMed.
- K. Aschberger, H. J. Johnston, V. Stone, R. J. Aitken, C. L. Tran, S. M. Hankin, S. A. Peters and F. M. Christensen, Review of Fullerene Toxicity and Exposure–Appraisal of a Human Health Risk Assessment, Based on Open Literature, Regul. Toxicol. Pharmacol., 2010, 58, 455–473 CrossRef CAS PubMed.
- Y. Pan, L. Wang, S.-g. Kang, Y. Lu, Z. Yang, T. Huynh, C. Chen, R. Zhou, M. Guo and Y. Zhao, Gd–Metallofullerenol Nanomaterial Suppresses Pancreatic Cancer Metastasis by Inhibiting the Interaction of Histone Deacetylase 1 and Metastasis-Associated Protein 1, ACS Nano, 2015, 9, 6826–6836 CrossRef CAS PubMed.
- Y. Miao, J. Xu, Y. Shen, L. Chen, Y. Bian, Y. Hu, W. Zhou, F. Zheng, N. Man and Y. Shen, Nanoparticle as Signaling Protein mMmic: Robust Structural and Functional Modulation of CaMKII upon Specific Binding to Fullerene C60 Nanocrystals, ACS Nano, 2014, 8, 6131–6144 CrossRef CAS PubMed.
- T. A. Ratnikova, P. Nedumpully Govindan, E. Salonen and P. C. Ke, In Vitro Polymerization of Microtubules with a Fullerene Derivative, ACS Nano, 2011, 5, 6306–6314 CrossRef CAS PubMed.
- S. Zanzoni, A. Ceccon, M. Assfalg, R. K. Singh, D. Fushman and M. D'Onofrio, Polyhydroxylated [60] Fullerene Binds Specifically to Functional Recognition Sites on a Monomeric and a Dimeric Ubiquitin, Nanoscale, 2015, 7, 7197–7205 RSC.
- M. Calvaresi and F. Zerbetto, Baiting Proteins with C60, ACS Nano, 2010, 4, 2283–2299 CrossRef CAS PubMed.
- M. Calvaresi, S. Furini, C. Domene, A. Bottoni and F. Zerbetto, Blocking the Passage: C60 Geometrically Clogs K+ Channels, ACS Nano, 2015, 9, 4827–4834 CrossRef CAS PubMed.
- S. Radic, P. Nedumpully-Govindan, R. Chen, E. Salonen, J. M. Brown, P. C. Ke and F. Ding, Effect of Fullerenol Surface Chemistry on Nanoparticle Binding-Induced Protein Misfolding, Nanoscale, 2014, 6, 8340–8349 RSC.
- A. Nel, T. Xia, L. Mädler and N. Li, Toxic Potential of Materials at the Nanolevel, Science, 2006, 311, 622–627 CrossRef CAS PubMed.
- A. D. Maynard, R. J. Aitken, T. Butz, V. Colvin, K. Donaldson, G. Oberdörster, M. A. Philbert, J. Ryan, A. Seaton and V. Stone, Safe Handling of Nanotechnology, Nature, 2006, 444, 267–269 CrossRef CAS PubMed.
- O. L. Kobzar, V. V. Trush, V. Y. Tanchuk, A. V. Zhilenkov, P. A. Troshin and A. I. Vovk, Fullerene Derivatives as a New Class of Inhibitors of Protein Tyrosine Phosphatases, Bioorg. Med. Chem. Lett., 2014, 24, 3175–3179 CrossRef CAS PubMed.
- M. L. Hermiston, Z. Xu and A. Weiss, CD45: a Critical Regulator of Signaling Thresholds in Immune Cells, Annu. Rev. Immunol., 2003, 21, 107–137 CrossRef CAS PubMed.
- J. T. Pingel and M. L. Thomas, Evidence that the Leukocyte-Common Antigen is Required for Antigen-Induced T Lymphocyte Proliferation, Cell, 1989, 58, 1055–1065 CrossRef CAS PubMed.
- T. Mustelin, K. M. Coggeshall and A. Altman, Rapid Activation of the T-cell Tyrosine Protein Kinase pp56lck by the CD45 Phosphotyrosine Phosphatase, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 6302–6306 CrossRef CAS.
- P. Johnson, H. Ostergaard, C. Wasden and I. S. Trowbridge, Mutational Analysis of CD45. A Leukocyte-Specific Protein Tyrosine Phosphatase, J. Biol. Chem., 1992, 267, 8035–8041 CAS.
- M. Streuli, N. X. Krueger, T. Thai, M. Tang and H. Saito, Distinct Functional Roles of the Two Intracellular Phosphatase Like Domains of the Receptor-Linked Protein Tyrosine Phosphatases LCA and LAR, EMBO J., 1990, 9, 2399 CAS.
- H.-J. Nam, F. Poy, H. Saito and C. A. Frederick, Structural Basis for the Function and Regulation of the Receptor Protein Tyrosine Phosphatase CD45, J. Exp. Med., 2005, 201, 441–452 CrossRef CAS PubMed.
- P. Abreu, W. Adam, T. Adye, E. Agasi, I. Ajinenko, R. Aleksan, G. Alekseev, R. Alemany, P. Allport and S. Almehed, Performance of the DELPHI Detector, Nucl. Instrum. Methods Phys. Res., Sect. A, 1996, 378, 57–100 CrossRef CAS.
- I. Accelrys, Materials Studio, Accelrys Software Inc, 2010 Search PubMed.
- M. D. Gourlay, J. Kendrick and F. J. Leusen, Rationalization of Racemate Resolution: Predicting Spontaneous Resolution Through Crystal Structure Prediction, Cryst. Growth Des., 2007, 7, 56–63 CAS.
- C. Zhang, G. Vasmatzis, J. L. Cornette and C. DeLisi, Determination of Atomic Desolvation Energies from the Structures of Crystallized Proteins, J. Mol. Biol., 1997, 267, 707–726 CrossRef CAS PubMed.
- N. Andrusier, R. Nussinov and H. J. Wolfson, FireDock: Fast Interaction Refinement in Molecular Docking, Proteins: Struct., Funct., Bioinf., 2007, 69, 139–159 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, Comparison of Simple Potential Functions for Simulating Liquid Water, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, Particle Mesh Ewald: An N·log(N) Method for Ewald Sums in Large Systems, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- G. S. Grest and K. Kremer, Molecular Dynamics Simulation for Polymers in the Presence of a Heat Bath, Phys. Rev., 1986, 33, 3628 CAS.
- G. J. Martyna, D. J. Tobias and M. L. Klein, Constant Pressure Molecular Dynamics Algorithms, J. Chem. Phys., 1994, 101, 4177–4189 CrossRef CAS.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kale and K. Schulten, Scalable Molecular Dynamics with NAMD, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS PubMed.
- A. D. MacKerell Jr, D. Bashford, M. Bellott, R. L. Dunbrack Jr, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo and S. Ha, All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins, J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef PubMed.
- W. Humphrey, A. Dalke and K. Schulten, VMD: Visual Molecular Dynamics, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- Y. C. Long, F. J. Lu and J. T. Lin, Free radical scavenging activity of water-soluble fullerenol, J. Chem. Soc., Chem. Commun., 1995, 12, 1283–1284 Search PubMed.
- R. Singh and T. Goswami, Synthesis and evaluation of thermal, photophysical and magnetic properties of novel starlike fullerene–organosilane macromolecules, J. Organomet. Chem., 2008, 693, 2021–2032 CrossRef CAS.
- X. Cai, J. Hao, X. Zhang, B. Yu, J. Ren, C. Luo, Q. Li, Q. Huang, X. Shi and W. Li, The polyhydroxylated fullerene derivative C60(OH)24 protects mice from ionizing-radiation-induced immune and mitochondrial dysfunction, Toxicol. Appl. Pharmacol., 2010, 243, 27–34 CrossRef CAS PubMed.
- A. Krokosz, J. Grebowski, A. Rodacka, B. Pasternak and M. Puchala, The effect of fullerenol C60(OH)∼30 on the alcohol dehydrogenase activity irradiated with X-rays, Radiat. Phys. Chem., 2014, 97, 102–106 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1: the energy of C60(NH2)30/C60(OH)30 in a water box during 20 ns simulation, Fig. S2: the grid of interactions (gray region) between C60(NH2)30, C60(OH)30 and CD45, Fig. S3 shows docking poses of CD45 + C60, CD45 + C60(NH2)30, and CD45 + C60(OH)30, Fig. S4 shows comparison of the segment (C764 to D766, P800 to I824) of CD45 + C60, CD45 + C60(NH2)30, and CD45 + C60(OH)30, Fig. S5 shows the vdW energy between every residue of binding pocket and C60 C60(NH2)30, C60(OH)30, Table S1 shows the calculated pKa values of all histidines, Table S2 shows the probility of residues that directly interact with C60(NH2)30 and C60(OH)30, Table S3 shows the full name and the abbreviation of amino acids. See DOI: 10.1039/c7ra13543b |
| This journal is © The Royal Society of Chemistry 2018 |