
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electronic and steric effects of substituents in 1,3-diphenylprop-2-yn-1-one during its reaction with Ru3(CO)12†
Lei Xu ,
Shasha Li,
Liping Jiang,
Guofang Zhang*,
Weiqiang Zhang and
Ziwei Gao
,
Shasha Li,
Liping Jiang,
Guofang Zhang*,
Weiqiang Zhang and
Ziwei Gao
Key Laboratory of Applied Surface and Colloid Chemistry, MOE/School of Chemistry and Chemical Engineering, Shaanxi Normal University, Xi'an 710062, China. E-mail: gfzhang@snnu.edu.cn
First published on 24th January 2018
Abstract
Thermal reaction of Ru3(CO)12 with alkynyl ketones PhC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CC(O)R (R
CC(O)R (R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Ph (1); 2-Cl-Ph (2); 4-NO2-Ph (3); 2-NH2-Ph (4); and 2-CH3COO-Ph (5)) proceeds in toluene with the formation of Ru3(CO)9(μ-CO)(η4-triruthenium) derivatives (1a–5a), Ru(CO)3(η4-ruthenole) derivatives (1b–4b, 1c–5c and 4d) and cyclotrimerization products (1e–2e and 1f–3f). Compounds 1a–5a were isolated from the reaction of Ru3(CO)12 with one equivalent of 1–5, respectively. Ruthenoles 1b–3b and 1c–3c were collected by adding 1–3 to the corresponding 1a–3a in a molar ratio of 1
Ph (1); 2-Cl-Ph (2); 4-NO2-Ph (3); 2-NH2-Ph (4); and 2-CH3COO-Ph (5)) proceeds in toluene with the formation of Ru3(CO)9(μ-CO)(η4-triruthenium) derivatives (1a–5a), Ru(CO)3(η4-ruthenole) derivatives (1b–4b, 1c–5c and 4d) and cyclotrimerization products (1e–2e and 1f–3f). Compounds 1a–5a were isolated from the reaction of Ru3(CO)12 with one equivalent of 1–5, respectively. Ruthenoles 1b–3b and 1c–3c were collected by adding 1–3 to the corresponding 1a–3a in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Cyclotrimerization products 1e–2e and 1f–3f were obtained when 1–3 were added to their corresponding 1b–2b and 1c–3c, respectively. 4b, 4c and 4d were afforded during the reaction of 4 with 4a, but only 5c was collected during the reaction of 5 with 5a. All compounds were characterized by NMR, FT-IR, and MS-ESI and most of them were structurally confirmed by single crystal X-ray diffraction. The results suggest that electronic and steric effects of the substituents in the phenyl ring of 1,3-diphenylprop-2-yn-1-one play important roles in regulating the reaction pathways. An electron-withdrawing group is beneficial to the formation of b, c and further formation of e and f; an electron-donating group favors the production of ruthenoles b, c and d, but disfavors the formation of e and f; a substituent with large steric-hindrance prefers only the formation of c.
:1. Cyclotrimerization products 1e–2e and 1f–3f were obtained when 1–3 were added to their corresponding 1b–2b and 1c–3c, respectively. 4b, 4c and 4d were afforded during the reaction of 4 with 4a, but only 5c was collected during the reaction of 5 with 5a. All compounds were characterized by NMR, FT-IR, and MS-ESI and most of them were structurally confirmed by single crystal X-ray diffraction. The results suggest that electronic and steric effects of the substituents in the phenyl ring of 1,3-diphenylprop-2-yn-1-one play important roles in regulating the reaction pathways. An electron-withdrawing group is beneficial to the formation of b, c and further formation of e and f; an electron-donating group favors the production of ruthenoles b, c and d, but disfavors the formation of e and f; a substituent with large steric-hindrance prefers only the formation of c.
Introduction
Ru3(CO)12 has attracted the great interest of researchers in inorganic, organic and catalytic chemistry due to its abundant reactivity and unique catalytic activity.1 It has been highly effective in the activation and conversion of chemical bonds in the construction of diverse C–X (X = C, N, O, Si, etc.) bonds,2 and widely used in reactions such as hydroamination,3 hydroesterification,4 silylation,5 cycloaddition,6 transfer hydrogenation,7 carbonylation8 and hydrohydroxyalkylation.9 In order to understand the mechanisms of Ru3(CO)12 activated C–C and C–H bonds, reactions of Ru3(CO)12 with NHCs, arenes, alkenes and alkynes were extensively investigated.10 Some carbonyl ruthenium compounds formed via Ru3(CO)12 and unsaturated hydrocarbons have been used in catalytic systems.11 In these reactions, coordination atoms play a very important role in the activation of neighboring chemical bonds and construction of compound skeletons,12 such as the interaction between oxygen atoms and ruthenium atoms in the activation of open-cage fullerenes with ruthenium carbonyl clusters.13 These conclusions provide more possibilities for Ru3(CO)12 to become a potential catalyst for the conversion of alkyne compounds.The reactions of Ru3(CO)12 with alkynes have been studied for many years. A variety of coordination modes were reported.14 There are usually relevant to many catalytic processes involving polynuclear species and unsaturated organic molecules.15 In 2005, compound (μ-H)2Ru3(CO)9{μ3-η2[H2CC(H)CCC(O)OCH3]} was isolated by the activation of methanol and CO in the reaction of 1,4-dichloro-but-2-yne (DCB) and Ru3(CO)12.16 Then, two types of complexes Ru3(CO)9(μ-CO){μ3-η2-(‖)-HCCR} and Ru3(CO)9(μ-CO){μ3-η2-(⊥)-HCCR}(RC6H4-4-CH3, C6H3-2,5-(CH3)2, etc.) were collected by M. Hernandez-Sandoval in the reaction of activated cluster [Ru3(CO)10(NCMe3)2] with terminal alkynes.17 Recently, P. Mathur demonstrated that one CC bond of FcC2C2Ph (Fc = ferrocenyl) was activated by Ru3(CO)12, and a series of novel mixed Ru/Pt clusters were isolated.18 In general, the reaction of Ru3(CO)12 with functionalized acetylenes can afford a variety of unexpected products,19 that means alkyne ligands always experienced sophisticated transformations, these properties limited our understanding on activation of CC bonds.
Alkynyl ketones are important structural units with unsaturated functional groups. They are often employed as key templates in modern chemical synthesis.20 The CC bonds of ynones are good π-acceptors,21 whilst the adjacent carbonyl groups can coordinate as 2e σ donors.22 For understanding the reactive activity of 1,3-ynones with Ru3(CO)12, thermolytic reaction of Ru3(CO)12 with 1-(4-methoxyphenyl)-3-phenylprop-2-yn-1-one have been investigated systematically in our group, and a series of Ru2–Ru4 framework clusters including a triruthenium and a tetraruthenium intermediates and three ruthenoles were isolated.23 After a detailed analysis of the results, we proposed that the electron-donating methoxy group in the phenyl ring of the 1-(4-methoxyphenyl)-2-propyn-1-one may play an important role in the transformation reaction. As a continuing work in the chemistry of Ru3(CO)12 with alkynyl ketones, we investigated the electronic and steric effects of the substituents in the phenyl ring of 1,3-diphenylprop-2-yn-1-one on the structures of the intermediates and final products during its thermolytic reactions with Ru3(CO)12.
In this paper, we examined in detail the reaction process of Ru3(CO)12 with 1,3-diphenylprop-2-yn-1-one derivatives (1–5) containing in its phenyl ring, for example, no substituent (1), electron-withdrawing groups (2–3), an electron-donating group (4) and a sterically hindered group (5). The coordination and coupling of the alkynyl ketone molecules formed a series of intermediate ruthenium clusters and cyclotrimerization products. Through a detailed examination of the reaction processes, we revealed that substituents in the phenyl rings of the acetylenic ketones are vital for the reaction process and that the formation of products has been affected comprehensively by the steric hindrance and/or electronic properties of the substituents.
Results and discussion
Syntheses and characterization
Reactions of Ru3(CO)12 and the 1,3-ynones were performed in toluene at 90 °C under nitrogen atmosphere and monitored consistently by TLC technique. It was found that color of each reaction solution gradually changed from orange to red-brown and the non-reacted Ru3(CO)12 precipitated after cooling. All isolated compounds were completely characterized by FT-IR, NMR, ESI mass spectrometry and most of them were additionally characterized by single crystal X-ray diffraction. The reaction process is illustrated in Scheme 1 according to the experimental results and TLC technique as well as the structural characterization analyses.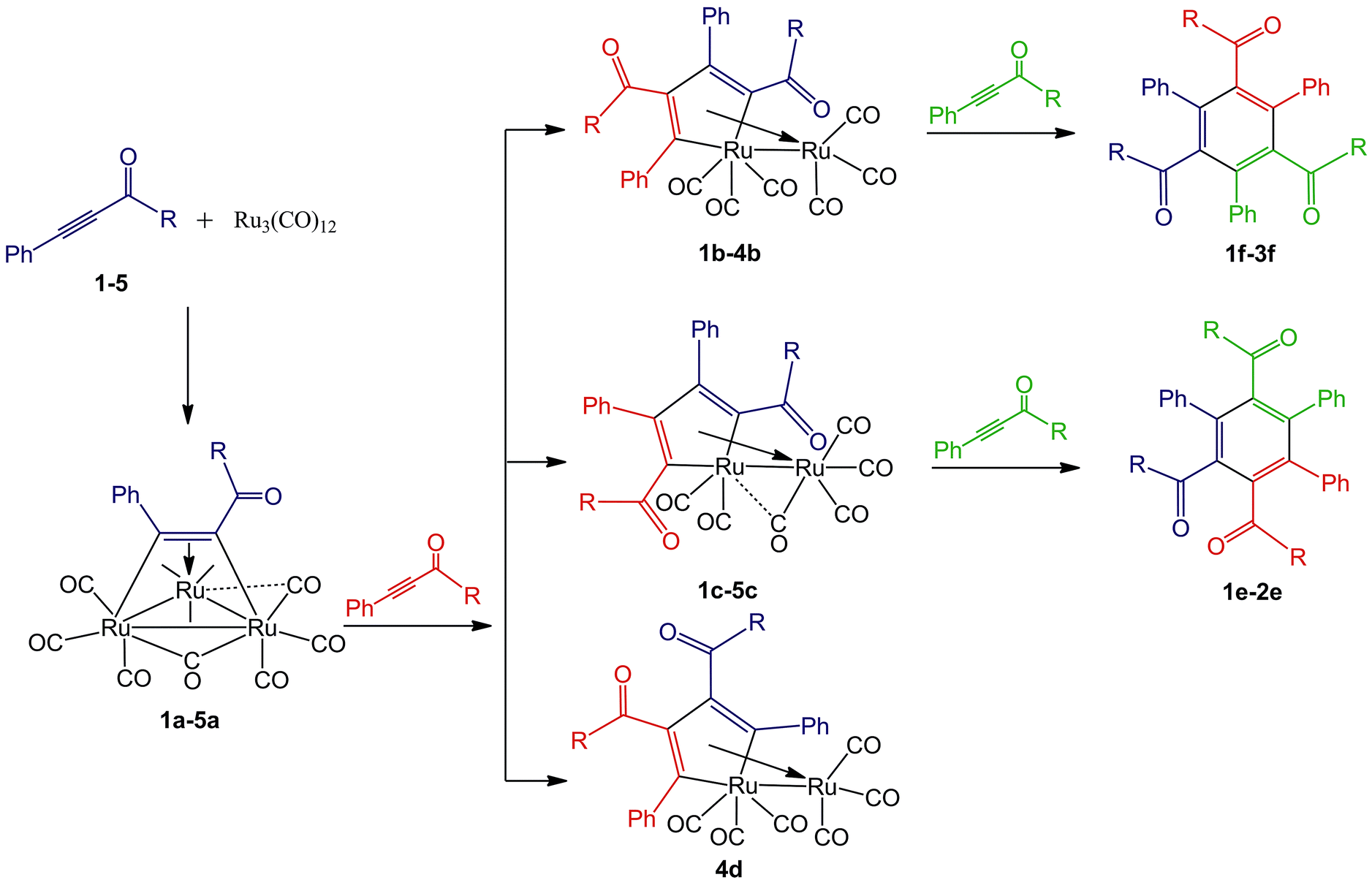 | ||
| Scheme 1 The reaction process of Ru3(CO)12 with 1,3-ynones (1–5). RPh (1); 2-Cl-Ph (2); 4-NO2-Ph (3); 2-NH2-Ph (4); 2-CH3COO-Ph (5). | ||
Synthesis of Ru3(CO)9(μ-CO)(η4-triruthenium) derivatives 1a–5a
When a toluene solution of an 1,3-ynone (1–5) (0.03 mmol) and Ru3(CO)12 (0.01 mmol) was stirred at 90 °C for 30 min under nitrogen atmosphere, Ru3(CO)9(μ-CO)(η4-triruthenium) derivatives (1a–5a) were formed due to binding of the CC bonds of 1–5 and the Ru metal skeletons. Each of the new clusters has a similar structural framework according to the characterization results and the crystal structure of 5a is shown in Fig. 1 was taken as an example. The IR spectrum of 5a was similar to those observed in the compounds M3(CO)9(μ-CO)L (M = Ru, Os).23,24 The absorption bands peaked in the range of 2017–2104 cm−1 and at 1878 cm−1 were assigned to the terminal and bridging CO groups, respectively. The dramatic red shift of the stretch vibration of the CC bond in 5a from 2191 cm−1 to ca. 1446 cm−1 reveals further that the coordination interaction between the carbon atoms of the CC bond and the Ru central metals is very strong. The chemical shifts of the CC bond moved downwards to 150.8 and 148.2 ppm, confirming again the strong interaction between the CC bond and the Ru atoms. The chemical shifts of the carbonyl carbon atoms directly with Ru atoms cannot be distinguished, indicating that in solution there are fluxional for the terminal and bridging CO on the 13C{1H} NMR time scale.
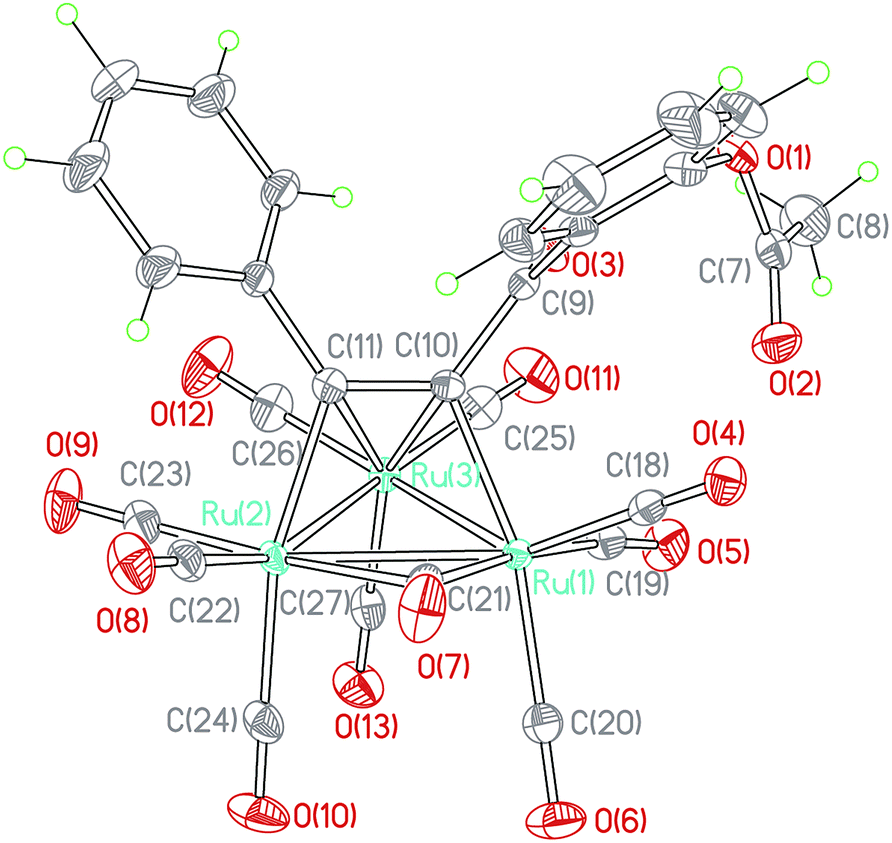 | ||
| Fig. 1 ORTEP view of cluster 5a showing 50% ellipsoids. Selected bond lengths (Å) and bond angles (°): Ru1–Ru2 = 2.845(2); Ru2–Ru3 = 2.712(2); Ru1–Ru3 = 2.749(2); Ru1–C19 = 1.939(3); Ru1–C21 = 2.029(2); Ru1–C10 = 2.126(2); Ru2–C11 = 2.091(2); Ru2–C21 = 2.337(2); Ru3–C11 = 2.317(2); Ru3–C10 = 2.211(2); Ru3–C19 = 2.873(2); C10–C11 = 1.396(3); C9–C10 = 1.498(3); C9–O3 = 1.219(2); C19–O5 = 1.337(3); C21–O7 = 1.149(3); Ru1–C19–Ru3 = 66.4(1); Ru1–C21–Ru2 = 81.0(1). | ||
The compound 5a consists of a triangular arrangement of the ruthenium atoms, in which the Ru–Ru bond distances are in the range of 2.712(2)–2.845(2) Å. The coordination of the alkynyl ketone (5) with Ru3 is in a η2 mode, but with Ru1 and Ru2 is in a η1 mode. The C10–C11 bond length is 1.396(3) Å, located between the typical C–C single bond and double bond, indicative again of the strong coordination strength of the Ru–C bonds.25 The distances of Ru1–C19 and Ru3–C19 are 1.939(3) and 2.873(2) Å, respectively. The angle of Ru1–C19–O5 is 169.2(2)°. In addition, there is a carbonyl exists between Ru1 and Ru3 atoms.
Synthesis of Ru(CO)3(η4-ruthenole) derivatives 1b–4b, 1c–5c and 4d
When the reaction time of the above reaction was prolonged from 0.5 h to 1 h, Ru(CO)3(η4-ruthenole) derivatives b (1b–4b), c (1c–5c) and d (4d) were separated by flash chromatography. The structural characterizations showed that structures of the new ruthenoles are of diverse coupling combinations of the CC units in the 1,3-ynones, such as modes b (head to tail coupling), c (head to head coupling) and d (tail to tail coupling).23 Each ruthenole contains a metallacyclopentadienyl framework, which is alike in structure to the carbonyl complexes [M(CO)(η4-metallole)] (M = Fe, Ru, Os) formed by combination of alkynes with group 8 metals.23,26 The molecular structure of 1b, as an example, was depicted in Fig. 2. The structure of 1b showed an eclipsed conformation of the carbonyls on the two ruthenium centers, with the dihedral angle of R1–Ru2–C35plane and R1–Ru2–C32plane being 2.1(1)°. The special arrangement geometrically prevents an apical carbonyl to be close enough to the ring metal Ru1 for a CO bridging conformation. Hence, all carbonyls are terminal with almost parallel Ru–C–O angle.
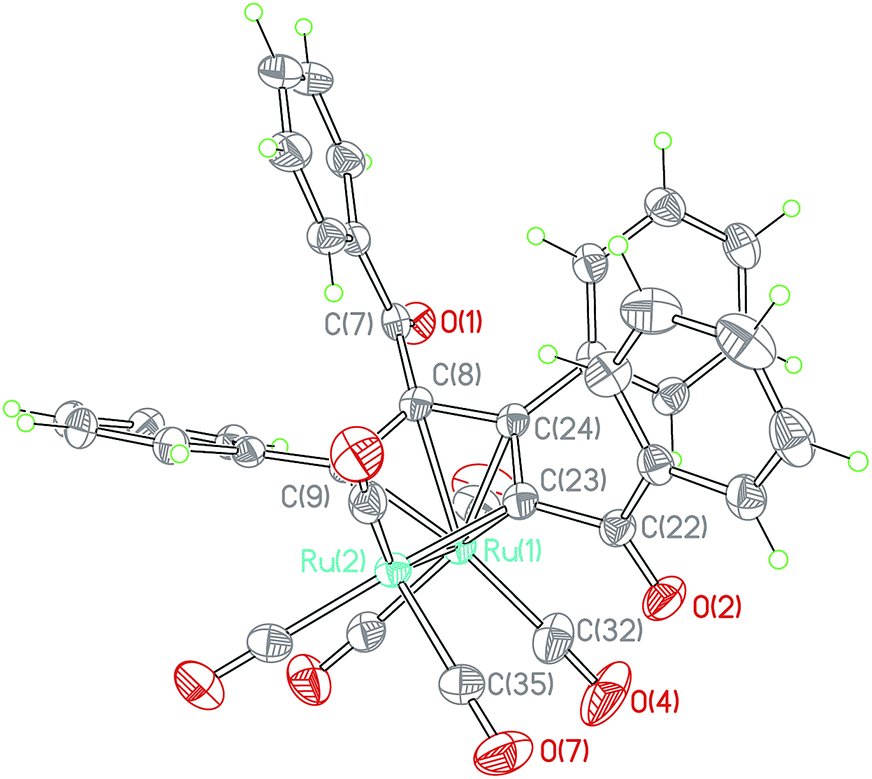 | ||
| Fig. 2 ORTEP view of cluster 1b showing 50% ellipsoids. Selected bond lengths (Å) and bond angles (°): Ru1–Ru2 = 2.734(2), Ru1–C8 = 2.254(2), Ru1–C9 = 2.316(2), Ru1–C24 = 2.243(2), Ru1–C23 = 2.252(2), Ru2–C9 = 2.098(2), Ru2–C23 = 2.068(2), C7–C8 = 1.515(3), C7–O1 = 1.214(2), C8–C9 = 1.411(3), C8–C24 = 1.458(3), C23–C24 = 1.420(3), C22–C23 = 1.491(3), C22–O2 = 1.217(2), C9–Ru2–C23 = 77.6(1), Ru1–C32–O4 = 177.0(2), Ru2–C35–O7 = 179.6(2). | ||
Compared the structures of b and c, each of the crystal structures of ruthenoles c (taking 2c·1/2C6H14 shown in Fig. 3 as an example) appeared a routine staggered conformation of the carbonyls. The dihedral angles between Ru1–Ru2–C35plane and Ru1–Ru2–C31plane, Ru1–Ru2–C32plane are 58.4(2)° and 41.0(2)°, respectively. The distances of Ru1–C35 is 2.742(5) Å. The staggered orientation in 2c·1/2C6H14 allows the C35 close to the Ru1 metal for a weak interaction, which formed a semi-bridging carbonyl across the Ru–Ru bond. The IR absorption at 1944 cm−1 also confirms its presence.
 | ||
| Fig. 3 ORTEP view of cluster 2c·1/2C6H14 showing 50% ellipsoids (solvent molecules have been omitted for clarity). Selected bond lengths (Å) and bond angles (°): Ru1–Ru2 = 2.712(1), Ru1–C8 = 2.097(4), Ru1–C23 = 2.096(5), Ru1–C35 = 2.742(5), Ru2–C8 = 2.223(4), Ru2–C9 = 2.300(5), Ru2–C23 = 2.232(5), Ru2–C24 = 2.310(5), Ru2–C35 = 1.908(5), C7–C8 = 1.496(7), C7–O1 = 1.213(7), C8–C9 = 1.436(6), C9–C24 = 1.435(7), C23–C24 = 1.440(5), C22–C23 = 1.495(7), C22–O2 = 1.215(5), C8–Ru1–C23 = 53.2(1), Ru2–C35–O7 = 169.7(4), Ru2–C35–Ru1 = 68.7(1). | ||
The crystal structure of 4d·3/4CH2Cl2 (Fig. 4) also showed an eclipsed conformation of carbonyls on the two ruthenium centers, the dihedral angle of R1–Ru2–C35plane and R1–Ru2–C33plane is 1.0(1)° between the equatorial Ru2–CO and Ru1–CO bonds, indicating that the Ru1, Ru2, C33 and C35 atoms are almost coplanar. All carbonyls are terminal with almost linear Ru–C–O angle, similar to those in compounds 1b–4b.
 | ||
| Fig. 4 ORTEP view of cluster 4d·3/4CH2Cl2 showing 50% ellipsoids (solvent molecules have been omitted for clarity). Selected bond lengths (Å) and bond angles (°): Ru1–Ru2 = 2.704(4), Ru1–C9 = 2.085(3), Ru1–C24 = 2.083(3), Ru2–C9 = 2.319(4), Ru2–C8 = 2.224(3), Ru2–C23 = 2.234(2), Ru2–C24 = 2.339(3), C7–C8 = 1.518(5), C7–O1 = 1.227(5), C8–C9 = 1.417(5), C8–C23 = 1.460(5), C23–C24 = 1.412(5), C22–C23 = 1.523(5), C22–O2 = 1.233(5), C9–Ru1–C24 = 79.2(1), Ru1–C33–O5 = 176.9(3), Ru2–C35–O7 = 174.8(4). | ||
Synthesis of the cyclotrimerization products e (1e–2e) and f (1f–3f)
When Ru3(CO)12 (0.1 mmol) reacted with 3 equivalent of an 1,3-ynone (1–3) (0.3 mmol) in toluene (90 °C) for 2 h under nitrogen atmosphere, two types of cyclotrimerization products e (1e–2e) and f (1f–3f) were formed. The structures of 1e and 3f·CH2Cl2 are illustrated in Fig. 5, taking as examples. The reaction of Ru3(CO)12 with 4 or 5 afforded, however, no cyclotrimerization products in the same reaction conditions and even extended the reaction time. M. Kawatsura also isolated the alkyne cyclotrimerization products using Ru3(CO)12 as catalyst.11c | ||
| Fig. 5 ORTEP view of cluster 1e (left) and 3f·CH2Cl2 (right) showing 50% ellipsoids (solvent molecules have been omitted for clarity). Selected bond lengths (Å) and bond angles (°): 1e (the left): C1–C2 = 1.403(3), C2–C3 = 1.405(2), C3–C4 = 1.400(2), C4–C5 = 1.409(3), C5–C6 = 1.403(3), C1–C6 = 1.398(2), C1–C7 = 1.519(3), C3–C20 = 1.516(3), C6–C39 = 1.513(3), C7–O1 = 1.220(2), C20–O2 = 1.222(3), C39–O3 = 1.222(3). 3f·CH2Cl2 (the right): C1–C2 = 1.399(5), C2–C3 = 1.399(6), C3–C4 = 1.397(4), C4–C5 = 1.398(5), C5–C6 = 1.402(6), C1–C6 = 1.399(4), C1–C7 = 1.507(6), C3–C20 = 1.512(6), C5–C33 = 1.505(4), C7–O1 = 1.217(5), C20–O4 = 1.208(5), C33–O = 1.215(4). | ||
Step-wise conversion from ynones to final products
To clarify the formation process of the products at each step, the reaction of Ru3(CO)12 with ynones 1–5 were studied in detail and monitored by TLC technique. It was observed that the triruthenium clusters 1a–5a are unstable, decomposing slowly in toluene. The mixing of 1a–5a with Ru3(CO)12 in toluene did not give any new ruthenium clusters. By adding the corresponding 1,3-ynones 1–3 in the clusters 1a–3a in toluene at 90 °C, ruthenoles b (1b–3b) and c (1c–3c) were detected. If 4 was added in 4a in toluene, three ruthenoles 4b, 4c and 4d were found. However, the addition of 5 in 5a in toluene only 5c was monitored. Compared with our previous work,23 no corresponding tetraruthenium clusters were detected in the reaction, however. It is possible that the corresponding tetraruthenium clusters is extremely unstable in the reaction system and cannot be found. The formation of the diverse ruthenoles through reaction of the triruthenium clusters a with the corresponding 1,3-ynones justify that the triruthenium cluster a is the key intermediates in the generation of ruthenoles.Then reactions of the ruthenoles with the corresponding ynones were studied to explore how the cyclotrimerization products e and f were formed. It was found that e (1e–2e) was detected when ruthenoles 1c and 2c were mixed with 1 and 2, respectively, in toluene at 90 °C. The cyclotrimerization products 1f–3f were found in the reaction of ruthenoles 1b–3b with the corresponding 1–3, respectively. Therefore, we believed that ruthenoles are intermediates during the formation of the cyclotrimerization products.
The electronic effect of substituents on the acetylene cyclotrimerization has been reported by T. Takahashi.27 He found that acetylenes with electron-withdrawing groups (CN, CONMe2 or COPh) could be used for cycloaddition reaction, and unactivated alkynes such as 4-octyne did not give the desired benzene derivatives even at higher temperature.27 According to the explanation of T. Takahashi, 1,3-diphenylprop-2-yn-1-one is an electron-withdrawing group activated alkyne, so it is feasible that the reaction of 1 with the corresponding ruthenole can afford the cyclotrimerization products 1e and 1f. Therefore, it is easier that 2e, 2f and 3f can be produced in the similar reactions. Compound 4 is a derivative of 1 with a strong electron-donating group in the phenyl ring, which weakens the reactivity of 4 in the cyclotrimerization reaction. However, an electron-donating group in the phenyl ring of 1 favors the formation of ruthenole d, which can explain the formation of 4d and a similar compound we reported previously.23 An ortho-acetyl group in the phenyl ring of 5 has certain electron-withdrawing ability, but its larger steric hindrance results in the forming of 5c as the unique ruthenole in the reaction of 5a with 5, and this strong steric effect further prevents the formation of cyclotrimerization products.
Experimental
Materials and equipment
All reactions and manipulations were performed using standard Schlenk line techniques under dry nitrogen. Ru3(CO)12 and the 1,3-ynones were synthesized according to the literature procedure.28 Solvents were purified, dried and distilled under nitrogen atmosphere prior to use. FT-IR spectra were recorded on a Bruker Tensor 27 Fourier-transform spectrometer. 1H and 13C{1H} NMR spectra were performed on a Bruker Avance 400 MHz spectrometer unless indicated. ESI was recorded on a Thermo DecaMax (LC-MS) mass spectrometer with an ion-trap mass detector. While high-resolution mass spectra were recorded in ESI mode on a Waters UPLC-Q-TOF mass spectrometer. The structural measurements of single crystals were carried out with a Bruker SMART APEX-II CCD detector.Synthesis
1,3-diphenylprop-2-yn-1-one (1), 1-(2-chloro-phenyl)-3-phenylprop-2-yn-1-one (2), 1-(4-nitro-phenyl)-3-phenylprop-2-yn-1-one (3), 1-(2-amino-phenyl)-3-phenylprop-2-yn-1-one (4) and 2-(3-phenylpropioloyl)phenylacetate (5) were used to react with Ru3(CO)12. Since the reaction processes are similar, the reaction procedure of 1 with Ru3(CO)12 was taken as an example. Both 1 (0.1856 g, 0.9 mmol) and Ru3(CO)12 (0.1918 g, 0.3 mmol) were added in 15 mL toluene and heated at 90 °C for 0.5 h, during which the color of the reaction solution gradually changed from orange to red-brown. The solvent was removed and the residue was chromatographed (the chromatographic column is of 305 mm length and 32 mm internal diameter) on silica gel with dichloromethane and petroleum ether, five main products were eluted in the sequence of 1a, 1b, 1c, 1e and 1f, with the eluent dichloromethane/petroleum ether being (v/v) 1:5, 3:5, 1:1, 5:3 and 3:1, respectively. And then the products were recrystallized by dichloromethane and hexane.
C), 132.8, 132.3, 129.9, 128.6, 128.3, 128.2, 127.7 (C6H5). MS (m/z, ESI−) 790.737 (M−). Anal. calcd for C25H10O11Ru3: 790.738.C), 135.5, 134.4, 134.1, 133.9, 133.6, 133.3, 132.8, 132.6, 130.2, 129.8, 129.7, 129.6, 129.1, 128.6, 128.4, 128.0, 127.9, 127.7, 127.6, 123.5 (C6H5). MS (m/z, ESI−) 783.926 (M−). Anal. calcd for C36H20O8Ru2: 783.925.C), 135.2, 134.2, 132.7, 131.5, 130.9, 130.0, 129.6, 129.4, 129.1, 128.8, 128.7, 128.3, 128.1, 127.8, 123.9, 121.3 (C6H5). MS (m/z, ESI−) 780.923 (M−). Anal. calcd for C36H20O8Ru2: 780.926.![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) ), 133.0, 132.8, 132.7, 131.0, 130.0, 129.6, 129.5, 129.4, 128.0, 127.8, 127.7, 127.6, 127.3, 127.1, 126.9 (C6H5). MS (m/z, ESI−) 618.219 (M−). Anal. calcd for C45H30O3: 618.219.), 132.9, 130.7, 129.3, 128.0, 127.7, 127.4 (C6H5). MS (m/z, ESI−) 618.219 (M−). Anal. calcd for C45H30O3: 618.219.C), 135.1, 133.0, 132.0, 131.1, 131.0, 128.1 (C6H4), 127.9, 127.86, 126.4 (C6H5). MS (m/z, ESI−) 824.628 (M−). Anal. calcd for C25H9ClO11Ru3: 824.699.C), 140.9, 135.0, 134.8, 134.6, 133.4, 133.2, 133.1, 133.0, 132.8, 132.2, 132.2, 131.4 (C6H4), 130.8, 130.6, 128.2, 128.1, 127.7, 127.5, 126.8, 125.7, 124.1, 123.9 (C6H4). MS (m/z, ESI−) 851.862 (M−). Anal. calcd for C36H18Cl2O8Ru2: 851.847.C), 135.5, 134.0, 133.4, 132.2, 131.6, 131.5 (C6H4), 130.2, 128.1, 127.7, 125.8 (C6H5). MS (m/z, ESI−) 851.845 (M−). Anal. calcd for C36H18Cl2O8Ru2: 851.847.), 133.3, 133.1, 133.0, 132.8, 132.4, 132.3, 132.2, 131.6, 130.8, 130.4 (C6H4), 130.1, 129.5, 127.9, 127.7, 127.6, 127.4, 127.1, 127.0, 126.3, 126.0 (C6H5). MS (m/z, ESI−) 722.099 (M−). Anal. calcd for C45H27Cl3O3: 722.100.), 136.3, 133.0, 132.6, 132.0, 130.7 (C6H4), 129.9, 127.8, 127.7, 126.9, 126.3 (C6H5). MS (m/z, ESI−) 722.100 (M−). Anal. calcd for C45H27Cl3O3: 722.100.C), 147.3, 136.6, 130.0, 129.5, 127.8 (C6H4), 127.4, 126.8, 126.5, 122.7, 122.1 (C6H5), 122.0 (C6H4). MS (m/z, ESI−) 835.724 (M−). Anal. calcd for C25H9NO13Ru3: 835.723.C), 145.3, 139.4, 139.0, 133.5, 130.4, 130.2, 129.7, 129.6, 129.1, 128.7 (C6H4), 128.5, 128.1, 123.9, 123.5 (C6H5), 123.4 (C6H4). MS (m/z, ESI−) 872.910 (M−). Anal. calcd for C36H18N2O12Ru2: 872.896.C), 149.9, 146.9, 143.1, 139.7, 134.3, 133.5, 131.3, 131.2, 130.7, 130.3 (C6H4), 129.4, 129.1, 128.9, 128.7, 128.6, 128.2, 123.9 (C6H5), 123.5, 123.4, 121.3 (C6H4). MS (m/z, ESI−) 872.893 (M−). Anal. calcd for C36H18N2O12Ru2: 872.896.), 134.9, 130.9, 130.0, 129.9 (C6H4), 128.7, 128.1, 127.9, 123.4, 123.2, 123.1 (C6H5). MS (m/z, ESI−) 753.175 (M−). Anal. calcd for C45H27N3O9: 753.175.C), 132.5, 128.1 (C6H4), 128.0, 127.7 (C6H5), 113.9 (C6H4). MS (m/z, ESI−) 805.673 (M−). Anal. calcd for C25H11NO11Ru3: 805.749.C), 134.8, 134.4, 134.3, 133.9, 133.8, 130.1, 128.7, 128.1 (C6H4), 127.8, 127.4, 127.3, 123.2, 117.4, 117.0, 115.9 (C6H5), 114.8 (C6H4). MS (m/z, ESI−) 812.948 (M−). Anal. calcd for C36H22N2O8Ru2: 812.948.C), 134.8, 128.5, 125.1, 120.7 (C6H4), 118.5, 115.9, 112.0 (C6H5), 111.5 (C6H4). MS (m/z, ESI−) 812.948 (M−). Anal. calcd for C36H22N2O8Ru2: 812.948.C), 135.3, 134.6, 134.5, 134.3, 133.2, 131.4, 131.0, 130.1 (C6H4), 129.0, 128.9, 128.3, 127.6, 127.3, 123.2, 117.3, 116.3 (C6H5), 115.9, 115.3 (C6H4). MS (m/z, ESI−) 815.947 (M−). Anal. calcd for C36H22N2O8Ru2: 815.947.C), 133.5, 131.7, 128.3, 127.9 (C6H4), 127.7, 125.9, 125.6 (C6H5), 124.4 (C6H4), 21.0 (CH3). MS (m/z, ESI−) 846.728 (M−). Anal. calcd for C27H12O13Ru3: 846.742.C), 134.1, 133.9, 133.3, 133.1, 132.7, 132.5, 132.2, 131.9 (C6H4), 131.4, 130.0, 128.6, 128.4, 128.2, 128.1, 127.8, 127.6, 127.4, 125.7, 125.0, 124.9 (C6H5), 124.5, 123.9 (C6H4), 20.9 (CH3). MS (m/z, ESI−) 899.931 (M−). Anal. calcd for C40H24O12Ru2: 899.935.
), 133.0, 132.8, 132.7, 131.0, 130.0, 129.6, 129.5, 129.4, 128.0, 127.8, 127.7, 127.6, 127.3, 127.1, 126.9 (C6H5). MS (m/z, ESI−) 618.219 (M−). Anal. calcd for C45H30O3: 618.219.), 132.9, 130.7, 129.3, 128.0, 127.7, 127.4 (C6H5). MS (m/z, ESI−) 618.219 (M−). Anal. calcd for C45H30O3: 618.219.C), 135.1, 133.0, 132.0, 131.1, 131.0, 128.1 (C6H4), 127.9, 127.86, 126.4 (C6H5). MS (m/z, ESI−) 824.628 (M−). Anal. calcd for C25H9ClO11Ru3: 824.699.C), 140.9, 135.0, 134.8, 134.6, 133.4, 133.2, 133.1, 133.0, 132.8, 132.2, 132.2, 131.4 (C6H4), 130.8, 130.6, 128.2, 128.1, 127.7, 127.5, 126.8, 125.7, 124.1, 123.9 (C6H4). MS (m/z, ESI−) 851.862 (M−). Anal. calcd for C36H18Cl2O8Ru2: 851.847.C), 135.5, 134.0, 133.4, 132.2, 131.6, 131.5 (C6H4), 130.2, 128.1, 127.7, 125.8 (C6H5). MS (m/z, ESI−) 851.845 (M−). Anal. calcd for C36H18Cl2O8Ru2: 851.847.), 133.3, 133.1, 133.0, 132.8, 132.4, 132.3, 132.2, 131.6, 130.8, 130.4 (C6H4), 130.1, 129.5, 127.9, 127.7, 127.6, 127.4, 127.1, 127.0, 126.3, 126.0 (C6H5). MS (m/z, ESI−) 722.099 (M−). Anal. calcd for C45H27Cl3O3: 722.100.), 136.3, 133.0, 132.6, 132.0, 130.7 (C6H4), 129.9, 127.8, 127.7, 126.9, 126.3 (C6H5). MS (m/z, ESI−) 722.100 (M−). Anal. calcd for C45H27Cl3O3: 722.100.C), 147.3, 136.6, 130.0, 129.5, 127.8 (C6H4), 127.4, 126.8, 126.5, 122.7, 122.1 (C6H5), 122.0 (C6H4). MS (m/z, ESI−) 835.724 (M−). Anal. calcd for C25H9NO13Ru3: 835.723.C), 145.3, 139.4, 139.0, 133.5, 130.4, 130.2, 129.7, 129.6, 129.1, 128.7 (C6H4), 128.5, 128.1, 123.9, 123.5 (C6H5), 123.4 (C6H4). MS (m/z, ESI−) 872.910 (M−). Anal. calcd for C36H18N2O12Ru2: 872.896.C), 149.9, 146.9, 143.1, 139.7, 134.3, 133.5, 131.3, 131.2, 130.7, 130.3 (C6H4), 129.4, 129.1, 128.9, 128.7, 128.6, 128.2, 123.9 (C6H5), 123.5, 123.4, 121.3 (C6H4). MS (m/z, ESI−) 872.893 (M−). Anal. calcd for C36H18N2O12Ru2: 872.896.), 134.9, 130.9, 130.0, 129.9 (C6H4), 128.7, 128.1, 127.9, 123.4, 123.2, 123.1 (C6H5). MS (m/z, ESI−) 753.175 (M−). Anal. calcd for C45H27N3O9: 753.175.C), 132.5, 128.1 (C6H4), 128.0, 127.7 (C6H5), 113.9 (C6H4). MS (m/z, ESI−) 805.673 (M−). Anal. calcd for C25H11NO11Ru3: 805.749.C), 134.8, 134.4, 134.3, 133.9, 133.8, 130.1, 128.7, 128.1 (C6H4), 127.8, 127.4, 127.3, 123.2, 117.4, 117.0, 115.9 (C6H5), 114.8 (C6H4). MS (m/z, ESI−) 812.948 (M−). Anal. calcd for C36H22N2O8Ru2: 812.948.C), 134.8, 128.5, 125.1, 120.7 (C6H4), 118.5, 115.9, 112.0 (C6H5), 111.5 (C6H4). MS (m/z, ESI−) 812.948 (M−). Anal. calcd for C36H22N2O8Ru2: 812.948.C), 135.3, 134.6, 134.5, 134.3, 133.2, 131.4, 131.0, 130.1 (C6H4), 129.0, 128.9, 128.3, 127.6, 127.3, 123.2, 117.3, 116.3 (C6H5), 115.9, 115.3 (C6H4). MS (m/z, ESI−) 815.947 (M−). Anal. calcd for C36H22N2O8Ru2: 815.947.C), 133.5, 131.7, 128.3, 127.9 (C6H4), 127.7, 125.9, 125.6 (C6H5), 124.4 (C6H4), 21.0 (CH3). MS (m/z, ESI−) 846.728 (M−). Anal. calcd for C27H12O13Ru3: 846.742.C), 134.1, 133.9, 133.3, 133.1, 132.7, 132.5, 132.2, 131.9 (C6H4), 131.4, 130.0, 128.6, 128.4, 128.2, 128.1, 127.8, 127.6, 127.4, 125.7, 125.0, 124.9 (C6H5), 124.5, 123.9 (C6H4), 20.9 (CH3). MS (m/z, ESI−) 899.931 (M−). Anal. calcd for C40H24O12Ru2: 899.935.Crystallography
X-ray structural measurements were carried out with a Bruker SMART APEX-II CCD detector using graphite monochromated MoKα radiation (λ = 0.71073). The data were collected by the ω–2θ scan mode, and absorption correction was applied by using Multi-Scan. The structure was solved by direct methods (SHELXS-97) and refined by full-matrix least squares against F2 using SHELXL-97 software.29 Non-hydrogen atoms were refined with anisotropic thermal parameters. All hydrogen atoms were geometrically fixed and refined using a riding model.The single crystals of compounds 1a, 1b, 1e, 1f, 2a, 2c, 3b, 3f, 4b, 4d, 5a and 5c suitable for single crystal X-ray diffraction were successfully grown up from slow evaporation of their dichloromethane/hexane solutions at 4 °C. Relevant crystallographic data were given in Table S1 in the ESI.† Crystallographic data for the structural analysis have been deposited with the Cambridge Crystallographic Data Centre: 1553521 (1a), 1553523 (1b), 1553526 (1e), 1553527 (1f), 1553511 (2a), 1553512 (2c), 1545197 (3b), 1545199 (3f), 1553513 (4b), 1553516 (4d), 1553504 (5a) and 1553509 (5c).†
Conclusions
We isolated a series of new ruthenium clusters and cyclotrimerization products by investigating reactions of five 1,3-diphenylprop-2-yn-1-one derivatives with Ru3(CO)12. The activation of the 1,3-ynones and the transformation of the cluster skeletons demonstrated that the triruthenium clusters and ruthenoles are important intermediates during the formation of the final products. More importantly, the substituents in the phenyl ring of 1,3-diphenylprop-2-yn-1-one exerted great effects on the reaction pathways during the transformation reactions. We revealed that the product distribution is dependent strongly on both the electronic and steric effects of the substituents on the acetylenic ketones. This discovery allows us to control the direction of the reaction by adjusting the substituents in the phenyl ring of 1,3-diphenylprop-2-yn-1-one. Moreover, further reactions between ruthenoles and the corresponding alkyne ketones proceed if a group can activate the carbon–carbon triple bond of an alkyne ketone, it brings us a new idea about applications of ruthenoles in the field of organic synthesis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (21401124, 21171112).Notes and references
- (a) T. C. Johnson, W. G. Totty and M. Wills, Org. Lett., 2012, 14, 5230 CrossRef CAS PubMed; (b) A. Lennartson, A. Lundin, K. Boerjesson, V. Gray and K. Moth-Poulsen, Dalton Trans., 2016, 45, 8740 RSC; (c) M. Chen, M. Zhang, B. Xiong, Z. Tan, W. Lv and H. Jiang, Org. Lett., 2014, 16, 6028 CrossRef CAS PubMed; (d) D. C. Schmitt, J. Lee, A. R. Dechert-Schmitt, E. Yamaguchi and M. J. Krische, Chem. Commun., 2013, 49, 6096 RSC; (e) L. Wang and L. Ackermann, Chem. Commun., 2014, 50, 1083 RSC; (f) F. Xie, M. Zhang, H. Jiang, M. Chen, W. Lv, A. Zheng and X. Jian, Green Chem., 2015, 17, 279 RSC; (g) J. A. Cabeza, I. del Rio, E. Perez-Carreno and V. Pruneda, Organometallics, 2011, 30, 1148 CrossRef CAS.
- (a) J. Liu, C. Kubis, R. Franke, R. Jackstell and M. Beller, ACS Catal., 2016, 6, 907 CrossRef CAS; (b) Z. Fan, J. Ni and A. Zhang, J. Am. Chem. Soc., 2016, 138, 8470 CrossRef CAS PubMed; (c) Y. Yuki, K. Takahashi, Y. Tanaka and K. Nozaki, J. Am. Chem. Soc., 2013, 135, 17393 CrossRef CAS PubMed; (d) N. Armanino, M. Lafrance and E. M. Carreira, Org. Lett., 2014, 16, 572 CrossRef CAS PubMed; (e) S. T. Tan, J. W. Kee and W. Y. Fan, Organometallics, 2011, 30, 4008 CrossRef CAS; (f) X. Guo and C. J. Li, Org. Lett., 2011, 13, 4977 CrossRef CAS PubMed.
- L. Wu, I. Fleischer, R. Jackstell and M. Beller, J. Am. Chem. Soc., 2013, 135, 3989 CrossRef CAS PubMed.
- (a) I. Profir, M. Beller and I. Fleischer, Org. Biomol. Chem., 2014, 12, 6972 RSC; (b) W. J. Park, C. H. Lee, D. S. Kim and C. H. Jun, Chem. Commun., 2015, 51, 14667 RSC; (c) D. S. Kim, W. J. Park, C. H. Lee and C. H. Jun, J. Org. Chem., 2014, 79, 12191 CrossRef CAS PubMed; (d) M. Nishiumi, H. Miura, K. Wada, S. Hosokawa and M. Inoue, Adv. Synth. Catal., 2010, 352, 3045 CrossRef CAS.
- W. Li, X. Huang and J. You, Org. Lett., 2016, 18, 666 CrossRef CAS PubMed.
- (a) A. Saxena, F. Perez and M. J. Krische, Angew. Chem., Int. Ed., 2016, 55, 1493 CrossRef CAS PubMed; (b) B. Groell, M. Schnuerch and M. D. Mihovilovic, J. Org. Chem., 2012, 77, 4432 CrossRef CAS PubMed; (c) A. Saxena, F. Perez and M. J. Krische, J. Am. Chem. Soc., 2015, 137, 5883 CrossRef CAS PubMed; (d) M. Kawatsura, M. Yamamoto, J. Namioka, K. Kajita, T. Hirakawa and T. Itoh, Org. Lett., 2011, 13, 1001 CrossRef CAS PubMed.
- (a) B. Y. Park, T. Luong, H. Sato and M. J. Krische, J. Am. Chem. Soc., 2015, 137, 7652 CrossRef CAS PubMed; (b) T. C. Johnson, W. J. Totty and M. Wills, Org. Lett., 2012, 14, 5230 CrossRef CAS PubMed.
- (a) K. Takahashi, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2012, 134, 18746 CrossRef CAS PubMed; (b) E. L. McInturff, J. Mowat, A. R. Waldeck and M. J. Krische, J. Am. Chem. Soc., 2013, 135, 17230 CrossRef CAS PubMed; (c) B. Li, Y. Park and S. Chang, J. Am. Chem. Soc., 2014, 136, 1125 CrossRef CAS PubMed.
- (a) B. Y. Park, T. P. Montgomery, H. J. Garza and M. J. Krische, J. Am. Chem. Soc., 2013, 135, 16320 CrossRef CAS PubMed; (b) S. E. Denmark and Z. D. Matesich, J. Org. Chem., 2014, 79, 5970 CrossRef CAS PubMed.
- (a) D. Chen, C. Zhang, S. Xu, H. Song and B. Wang, Organometallics, 2011, 30, 676 CrossRef CAS; (b) C. Zhang, B. Li, H. Song, S. Xu and B. Wang, Organometallics, 2011, 30, 3029 CrossRef CAS; (c) W. K. Tsui, L. H. Chung, W. H. Tsang, C. F. Yeung, C. H. Chiu, H. S. Lo and C. Y. Wong, Organometallics, 2015, 34, 1005 CrossRef CAS; (d) B. F. G. Johnson, J. M. Matters, P. E. Gaede, S. L. Ingham, N. Choi, M. Mcpartlin and M. A. Pearsall, Dalton Trans., 1997, 18, 3251 RSC; (e) M. I. Bruce, M. L. Cole, R. S. C. Fung, C. M. Forsyth, P. C. Junk and K. Konstas, Dalton Trans., 2008, 31, 4118 RSC.
- (a) H. Sato, M. Bender, W. J. Chen and M. J. Krische, J. Am. Chem. Soc., 2016, 138, 16244 CrossRef CAS PubMed; (b) J. P. Hopewell, J. E. D. Martins, T. C. Johnson, J. Godfrey and M. Wills, Org. Biomol. Chem., 2012, 10, 134 RSC; (c) M. Kawatsura, M. Yamamoto, J. Namioka, K. Kajita, T. Hirakawa and T. Itoh, Org. Lett., 2011, 13, 1001 CrossRef CAS PubMed.
- S. V. Osintseva, F. M. Dolgushin, N. A. Shtel'tser, P. V. Petrovskii, A. S. Peregudov, A. Z. Kreindlin and M. Y. Antipin, Organometallics, 2010, 29, 1012 CrossRef CAS.
- C. S. Chen, Y. F. Lin and W. Y. Yeh, Chem.–Eur. J., 2014, 20, 936 CrossRef CAS PubMed.
- (a) M. Krempe, R. Lippert, F. Hampel, I. Ivanovic-Burmazovic, N. Jux and R. R. Tykwinski, Angew. Chem., Int. Ed., 2016, 55, 14802 CrossRef CAS PubMed; (b) C. Cesari, L. Sambri, S. Zacchini, V. Zanotti and R. Mazzoni, Organometallics, 2014, 33, 2814 CrossRef CAS; (c) H. Masai, J. Terao, S. Seki, S. Nakashima, M. Kiguchi, K. Okoshi, T. Fujihara and Y. Tsuji, J. Am. Chem. Soc., 2014, 136, 1742 CrossRef CAS PubMed; (d) O. F. Koentjoro, P. J. Low, R. Rousseau, C. Nervi, D. S. Yufit, J. A. K. Howard and K. A. Udachin, Organometallics, 2005, 24, 1284 CrossRef CAS.
- P. J. Low, J. Cluster Sci., 2008, 19, 5 CrossRef CAS.
- G. Gervasio, D. Marabello, E. Sappa and A. Secco, J. Organomet. Chem., 2005, 690, 1594 CrossRef CAS.
- M. Hernandez-Sandoval, G. Sanchez-Cabrera, M. J. Rosales-Hoz, M. A. Leyva, V. Salazar, J. G. Alvarado-Rodriguez and F. J. Zuno-Cruz, Polyhedron, 2013, 52, 170 CrossRef CAS.
- P. Mathur, D. K. Rai, R. K. Joshi, B. Jha and S. M. Mobin, Organometallics, 2014, 33, 3857 CrossRef CAS.
- (a) S. V. Osintseva, F. M. Dolgushin, N. A. Shtel'tser, P. V. Petrovskii, A. Z. Kreindlin, L. V. Rybin and M. Y. Antipin, Organometallics, 2005, 24, 2279 CrossRef CAS; (b) W. K. Tsui, L. H. Chung, W. H. Tsang, C. F. Yeung, C. H. Chiu, H. S. Lo and C. Y. Wong, Organometallics, 2015, 34, 1005 CrossRef CAS.
- (a) B. H. Xu, G. Kehr, R. Froehlich, B. Wibbeling, B. Schirmer, S. Grimme and G. Erker, Angew. Chem., Int. Ed., 2011, 50, 7183 CrossRef CAS PubMed; (b) W. P. Unsworth, J. D. Cuthbertson and R. J. K. Taylor, Org. Lett., 2013, 15, 3306 CrossRef CAS PubMed; (c) J. Shen, G. Cheng and X. Cui, Chem. Commun., 2013, 49, 10641 RSC; (d) J. D. Kirkham, S. J. Edeson, S. Stokes and J. P. A. Harrity, Org. Lett., 2012, 14, 5354 CrossRef CAS PubMed; (e) M. Yoshida, K. Saito, Y. Fujino and T. Doi, Chem. Commun., 2012, 48, 11796 RSC; (f) N. Arai, H. Satoh, N. Utsumi, K. Murata, K. Tsutsumi and T. Ohkuma, Org. Lett., 2013, 15, 3030 CrossRef CAS PubMed.
- G. G. Melikyan, R. Davis, B. Anker, D. Meron and K. Duncan, Organometallics, 2016, 35, 4060 CrossRef CAS.
- (a) S. V. Osintseva, F. M. Dolgushin, N. A. Shtel'tser, P. V. Petrovskii, A. S. Peregudov, A. Z. Kreindlin and M. Y. Antipin, Organometallics, 2010, 29, 1012 CrossRef CAS; (b) C. S. Chen, Y. F. Lin and W. Y. Yeh, Chem.–Eur. J., 2014, 20, 936 CrossRef CAS PubMed.
- J. Yang, W. Zhang, G. Zhang and Z. Gao, J. Organomet. Chem., 2015, 799, 166 CrossRef.
- D. Delgado, E. Hernandez, A. Martin and M. Menacho, Organometallics, 2006, 25, 2960 CrossRef.
- X. M. Chen and J. W. Cai, in Single-Crystal Structural Analysis-Principles and Practices, Science China Press, Beijing, China, 2nd edn, 2004, p. 114 Search PubMed.
- (a) A. J. Arce, P. Arrojo, A. J. Deeming and Y. De Sanctis, Dalton Trans., 1992, 15, 2423 RSC; (b) A. J. Arce, R. Machado, C. Rivas, Y. De Sanctis and A. J. Deeming, J. Organomet. Chem., 1991, 419, 63 CrossRef CAS.
- T. Takahashi, Z. Xi, A. Yamazaki, Y. Liu, K. Nakajima and M. Kotora, J. Am. Chem. Soc., 1998, 120, 1672 CrossRef CAS.
- B. Yu, H. Sun, Z. Xie, G. Zhang, L. Xu, W. Zhang and Z. Gao, Org. Lett., 2015, 17, 3298 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystal structures of 1a, 1f, 2a, 3b, 4b, 5c, 2f, 3c and 1c, CIF; checkcif, 1H NMR spectra, 13C{1H} NMR spectra, FT-IR spectra and other electronic format. CCDC 1553521, 1553523, 1553526, 1553527, 1553511, 1553512, 1545197, 1545199, 1553513, 1553516, 1553504 and 1553509. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra13626a |
| This journal is © The Royal Society of Chemistry 2018 |