
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced catalytic performance of reduced graphene oxide–TiO2 hybrids for efficient water treatment using microwave irradiation
Aashu Anshuman ,
Sina Saremi-Yarahmadi and
Bala Vaidhyanathan*
,
Sina Saremi-Yarahmadi and
Bala Vaidhyanathan*
Department of Materials, Loughborough University, Loughborough, LE11 3TU, UK. E-mail: b.vaidhyanathan@lboro.ac.uk; Tel: +44(0) 1509 223152
First published on 16th February 2018
Abstract
Towards achieving efficient waste water treatment, the degradation of a common water pollutant, Orange G azo dye, was studied using a new hybrid catalyst and microwave irradiation. The fabrication of a hybrid catalyst based on reduced graphene oxide–titania (rGO–TiO2), was first achieved in a single mode microwave cavity by reducing the precursor consisting of graphene oxide (GO) and titania. Catalytic performance was then assessed in both microwave assisted and conventional heat treatment conditions. The hybrid catalyst showed significant improvement under microwave irradiation, with more than 88% dye degradation after 20 minutes of treatment at 120 °C. The microwave effect was found to be more dominant in the early stages of the catalysis – the hybrid catalyst decomposed ∼65% of the dye in just 5 minutes of microwave treatment compared to only 18% degradation obtained during conventional heating. The improved performance with microwaves is mainly attributed to the formation of the hot spots at the surface of the hybrid catalyst which ultimately results in higher degradation rates. The morphological and catalytic properties of the hybrid catalyst are investigated using High Resolution Transmission Electron Microscopy (HRTEM) and UV-Vis Spectroscopy, respectively. Successful reduction of GO to rGO was confirmed using Raman spectroscopy and X-ray diffraction. The outstanding performance of microwave irradiated hybrids offers a viable low energy, low carbon footprint process with a new catalyst for wastewater treatment and for highly polluted wastewater conditions where photocatalysis is deemed not feasible.
1. Introduction
Dyes are a major source of industrial water pollution. Azo dyes in particular constitute more than half of all the dyes and colorants used on the industrial scale,1 the majority of which will end up in wastewater entering aquatic ecosystems and are resistant to microbial degradation.2 As a result of rapid industrialisation, the problem of dealing with large volumes of wastewater contaminated by dyes has received worldwide attention.3,4 A variety of methods have been used to remove organic pollutants from wastewater including membrane separation processes and adsorption which function on the basis of transferring the contaminated phase from water to another phase.5,6 However, these methods do not satisfactorily answer the challenge as secondary pollution in the new phase still persists. A more versatile approach is through advanced oxidation processes (AOPs) for wastewater treatment which have garnered significant interest due to several advantages over conventional methods.3,7–10 AOPs have demonstrated faster treatment rates and the ability to treat a wide variety of contaminants including industrial dyes. Recently, microwave assisted AOPs have shown promise in further improving treatment speed as well as allowing for the treatment of non-transparent wastewater where photocatalytic AOP processes lose their effectiveness.2,11,12 The composite catalysts used for these processes usually consist of activated carbon (AC) and an oxide semiconductor such as TiO2 and ZnO where the organic pollutants are adsorbed on the nanocomposite. The thermal degradation mechanism proposed, especially for the carbon-based materials, is the local heating of a region close to the surface of the catalyst under microwave heating, rather than the dielectric heating of the entire solution. This local heating upon microwave (MW) irradiation leads to the accumulation of heat at the surface of the catalyst, i.e. formation of “hot spots” on the surface, hence enabling microwave assisted rate enhancements.2,13–15Reduced graphene oxide rGO–TiO2 composites have shown improvement over TiO2 in the photocatalytic decomposition of dyes and water contaminants.16–19 Enhanced generation of hydroxyl radicals in solution has also been reported when microwave radiation was used with TiO2 which can improve the removal of organic contaminants in wastewater.1 Here, we report the synthesis of hybrid rGO–TiO2 catalyst and demonstrate for the first time significant improvements in the degradation of commonly used industrial azo dye, Orange G (OG), under microwave irradiation using this hybrid system and, in particular, on shorter time scales and low temperatures.
2. Experimental
2.1. Microwave heating system
Microwave heating was carried out using a single mode microwave cavity (Monowave 300, Anton Paar) using closed glass vessels of 10 ml and 30 ml capacity. The temperature was controlled using a dual monitoring system of an infra-red sensor as well as an in situ ruby thermometer. Microwave power (0 to 300 W) was manipulated automatically so that the temperature was maintained at the required values. The pressure inside the vessel was monitored through an in-built pressure sensor.2.2. Preparation of rGO–TiO2 catalyst
rGO was prepared using a modified Hummer's method.20 In a typical process, 1 g of graphite was dissolved in 25 ml of H2SO4. The mixture was stirred for 1 h and then 0.5 g NaNO3 was added to the mixture. After 6 hours, 3 g KMnO4 was added to the mixture and the mixture was stirred for a further 24 hours. The reaction was terminated by adding 140 ml of deionised water to the mixture followed by 10 ml of H2O2. The mixture was then washed and filtered 3 times to obtain GO powders. Microwave-assisted reduction of GO was carried out using the procedure adopted from Zhao et al.21 In a typical preparation, TiO2 (Aeroxide® P25, Sigma Aldrich) was added to the as-prepared GO to make a 1 wt% suspension. Ethanol was then added to the suspension at a ratio of 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :65 vol% (ethanol:rGO–TiO2 aqueous suspension). Microwave reduction was carried out in a closed glass vessel of 30 ml capacity in several batches; for every batch of 5 ml of suspension, the mixture was thermally reduced in the microwave cavity at 180 °C for 1 hour to obtain rGO–TiO2.
:65 vol% (ethanol:rGO–TiO2 aqueous suspension). Microwave reduction was carried out in a closed glass vessel of 30 ml capacity in several batches; for every batch of 5 ml of suspension, the mixture was thermally reduced in the microwave cavity at 180 °C for 1 hour to obtain rGO–TiO2.
2.3. Characterisation of hybrid catalysts
The morphologies of the TiO2 powder, GO and rGO were studied using transmission electron microscopy. The surface of the rGO–TiO2 catalyst was further observed using high resolution transmission electron microscopy (HRTEM). Raman spectroscopy was performed using a HORIBA Jobin Yvon LabRAM HR (with 532 nm laser). The surface area and pore size distribution of powders were measured by using an automated Brunauer–Emmett–Teller (BET) nitrogen gas adsorption analyser (Tristar™ 3000, Micromeritics Instrument Corporation, Norcross, USA). X-ray diffraction (XRD) results were obtained using a Bruker D2 Phaser X-ray diffractometer (Bruker AXS, Karlsruhe, Germany, 2000), using Ni filtered Cu Kα radiation (λ = 1.542 Å) run at 30 kV and 10 mA. Patterns were recorded from 5–80° 2θ, using a step size of 0.02° and an equivalent time per step of 2 seconds.2.4. Orange G (OG) dye degradation
The dye degradation was carried out using a 10 μM solution of OG dye in water and fixed amount of the catalyst. The samples were then placed in a 10 ml closed glass tube and degradation was performed by applying microwave and conventional heating, respectively. The experiments were carried out at 4 different temperatures, 30, 60, 90, and 120 °C and 3 different times, 5, 10, and 20 minutes, for both types of heating methods. In microwave experiments, the temperature was measured in situ using a ruby thermometer. The concentration of the OG dye in the solutions before and after degradation was measured using UV spectrophotometry (Lambda 35, Perkin Elmer, UV/Vis Spectrophotometer) from the optical absorption at 478 nm using the Lambert–Beer's law. In addition, microwave control experiments were conducted at 120 °C with the addition of H2O2 alone, to assess the intrinsic effect of radical auto-generation by decomposition and its impact on the oxidation of the dye.3. Results and discussions
3.1. rGO–TiO2 hybrid morphology and structure
The morphology of the nanoparticles and composites was studied at every step of the process using TEM and HRTEM. Fig. 1(a) shows the morphology of the TiO2 nanoparticles with an average size of 20 nm suggesting a degree of agglomeration. Fig. 1(b) depicts the GO layers formed whilst the formation of rGO–TiO2 hybrids was confirmed by HR-TEM as shown in Fig. 1(c). The surface area and porosity of the materials used were measured at 49 m2 g−1 with a pore size of 15 nm for TiO2, and surface area of 44 m2 g−1 and a pore size of 7 nm for GO, respectively, which is in agreement with previously reported literature.22,23 | ||
| Fig. 1 (a) TEM image of the TiO2 nanoparticles, (b) TEM image of the GO layer, (c) HR-TEM image of the TiO2 in contact with rGO. | ||
In order to establish the formation of rGO, the reduction of GO using the microwave irradiation was confirmed by Raman spectroscopy and XRD (Fig. 2 and 3). Raman spectroscopy has been used extensively to study carbon-based materials24,25 and in particular to investigate the ratio of sp2/sp3 bonding in graphene based materials.25–29 Traditionally, to identify defective graphene oxide, either GO or rGO, the Raman peaks of ∼1350 cm−1, D peak, and ∼1580 cm−1, G peak, were studied. The D peak is due to the disorder resulting from defects created in the pristine graphene structure which is sp2 hybridised. The G peak is a first-order Raman peak which is characteristic of pristine graphite. It has been proposed that oxidation of graphite to GO would result in the shift of G peaks to higher wavenumbers as compared to that of the graphite, from ∼1589 to 1610 cm−1, whilst reduction of GO to rGO restored the G peak to its original position of ∼1580 cm−1 corresponding to the recovery of hexagonal network of carbon atoms with defects.27,30 Furthermore, the ratio of the intensity of D and G peaks (ID/IG) in the Raman spectra of carbon based materials has been used as an indication of the degree of the reduction of defective graphene where an increase in this ratio was attributed to the increased defect concentration present in rGO relative to that of in the GO.30,31 However, this approach is not reliable and contradicting ratios could be observed depending on the reduction processes, crystallinity, laser wavelength, etc.32–34 Recently, King et al.35 introduced a new Raman tool to distinguish between GO and rGO mainly due to the unreliability of ID/IG in defective graphene. Using the same approach by considering the Gapp peak resulting from the superposition of G peak and D′ peak as shown in Fig. 2, we confirm the extensive reduction of GO due to significant degree of G peak intensity contribution in the formation of the Gapp peak.
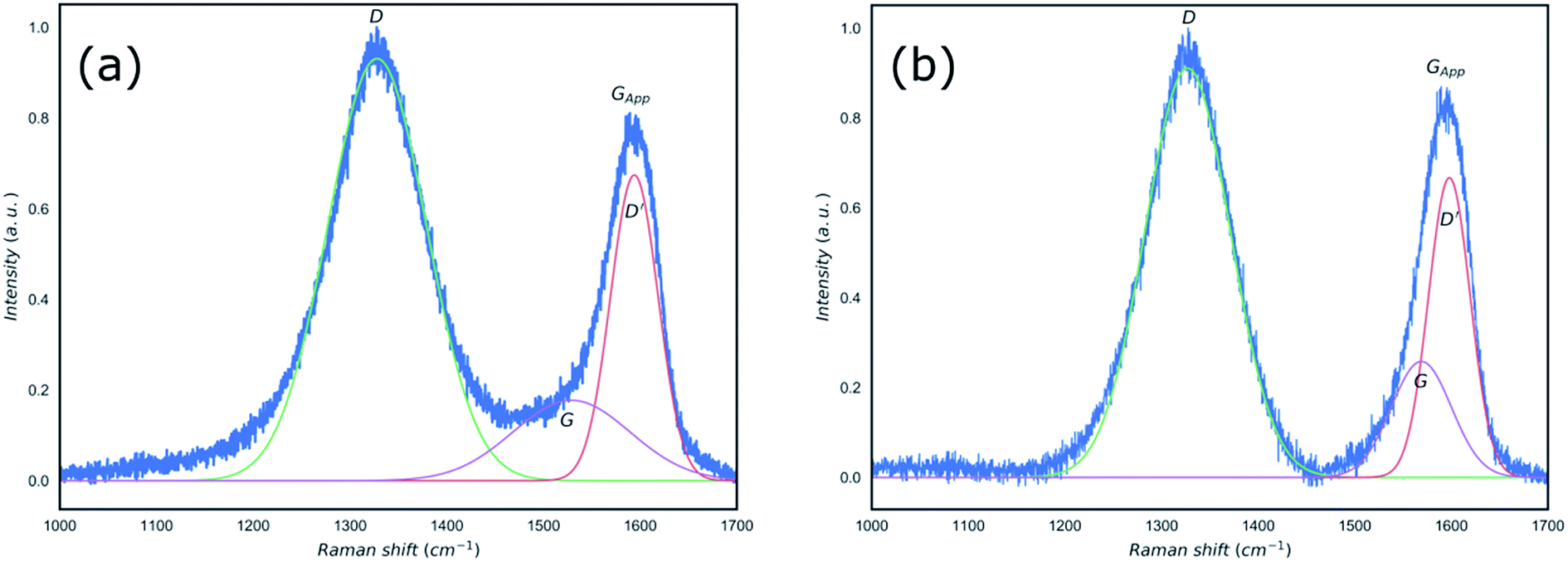 | ||
| Fig. 2 Raman spectra of the GO (a) and microwave reduced rGO (b) with peak fits applied and position of D, D′, G and Gapp clearly identified. | ||
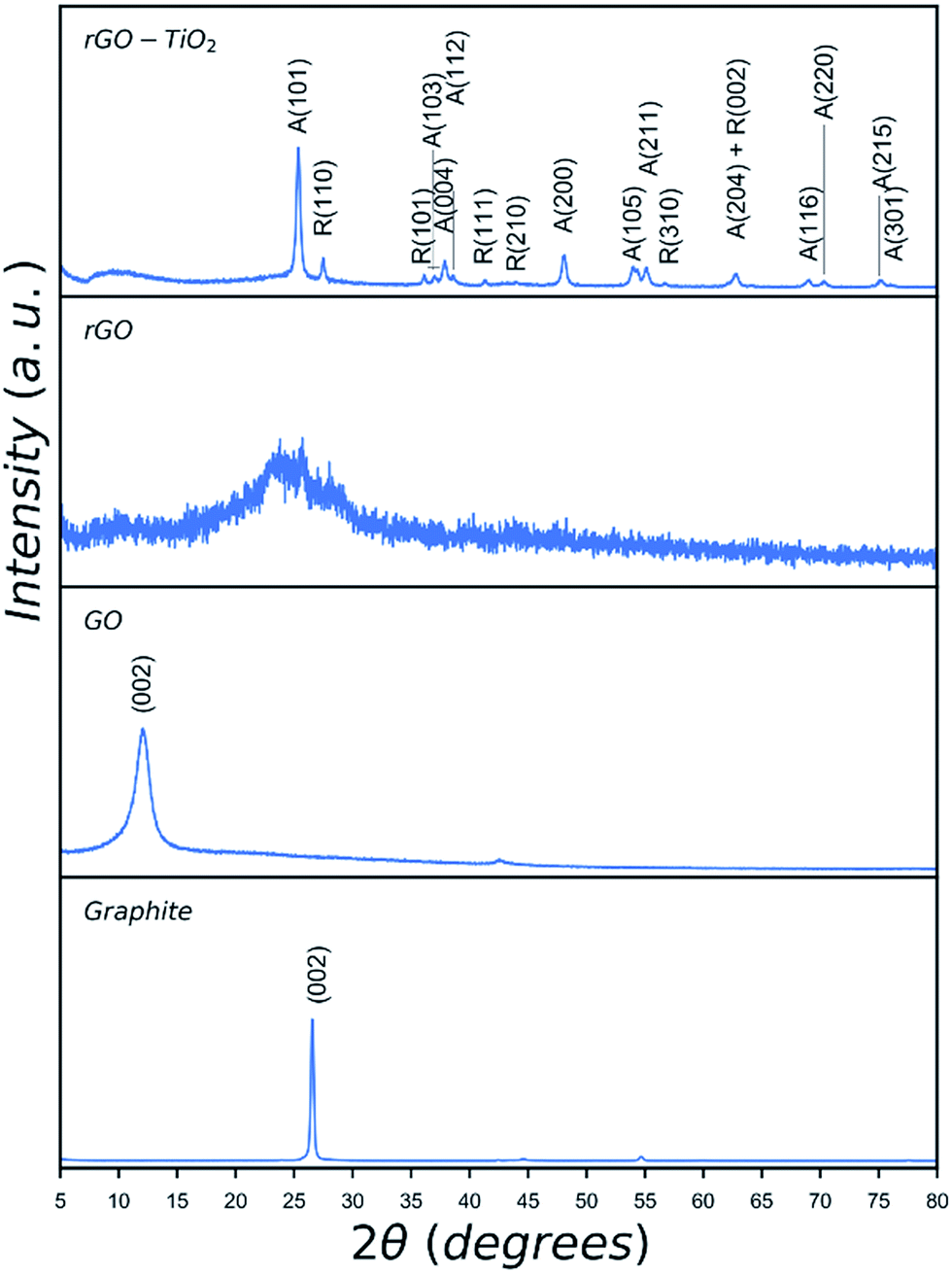 | ||
| Fig. 3 XRD patterns of graphite, GO, rGO and rGO–TiO2 hybrid catalyst. Peaks of anatase are denoted as A and those of rutile are denoted as R. | ||
The measured and calculated values of specific spectral peaks before and after microwave radiation are listed in Table 1. The proposed method by King et al.35 takes into account the value of the “D′ − Gapp” to define the boundary between GO and rGO. Values smaller than zero are representative of GO and “D′ − Gapp < 25” is used as an indicator of rGO formation. Based on the results shown in Table 1 and Fig. 2, whilst the position of the D peak did not change as a result of the microwave reduction, both D′ and G peaks shifted from 1594 and 1528 cm−1 to 1598 and 1569 cm−1, respectively. The microwave reduction of GO is confirmed by comparing D′ − Gapp obtained which showed the value of −2 for the GO and 4 for the rGO, in agreement with the method proposed by King et al.35
| Sample | G | D′ | Gapp | D′ − Gapp |
|---|---|---|---|---|
| GO | 1528 | 1594 | 1596 | −2 |
| rGO | 1569 | 1598 | 1594 | 4 |
XRD patterns of graphite, GO, rGO, and rGO–TiO2 hybrid are given in Fig. 3. The as-received graphite shows a characteristic peak at 2θ = 26.7°. Oxidation and exfoliation of graphite and formation of GO is confirmed by shifting of this characteristic peak to lower 2θ values due to introduction of oxygen functionalities in GO. Depending on the degree of oxidation, oxygen content and exfoliation of graphite precursor the shift could be as large as approximately 16°.36 The observed peak for GO at 12.2° suggests an oxygen level of 40–50 wt% which shows some remnant graphitic structure in the synthesised GO.36 In the case of rGO, the sharp peak at 12.2° has completely disappeared and a broad peak appears indicating the reduction of oxygen rich functionalities and formation of rGO.37 The peaks observed in the pattern of rGO–TiO2 hybrid catalyst result from the presence of titania as indexed, which agrees well with reported values for nanocrystalline P25 TiO2.38
3.2. Dye degradation studies
To investigate the efficiency of the catalyst systems under microwave heating, comparable experiments were conducted using microwave as well as conventional heating. This was done to establish if there were any significant non-thermal improvements in the degradation rates when microwave was applied. Fig. 4(a) compares the decomposition of the dye after different durations for different catalyst systems and heating methods at a constant temperature of 120 °C. The UV-Vis spectra of the OG dye solution after 20 minutes of microwave irradiation for different catalyst systems and the control experiment (H2O2 alone) have been compared in Fig. 4(b). While H2O2 alone is responsible for some decomposition of the dye (∼4%), the performance is inferior to both TiO2 (59%) and rGO–TiO2 (88%) hybrid catalyst systems. Similar trends of degradation were observed at other temperatures tested. As shown in Fig. 4(a), microwave process exhibited enhanced decomposition of the OG dye compared to the conventional heating process irrespective of the catalyst system.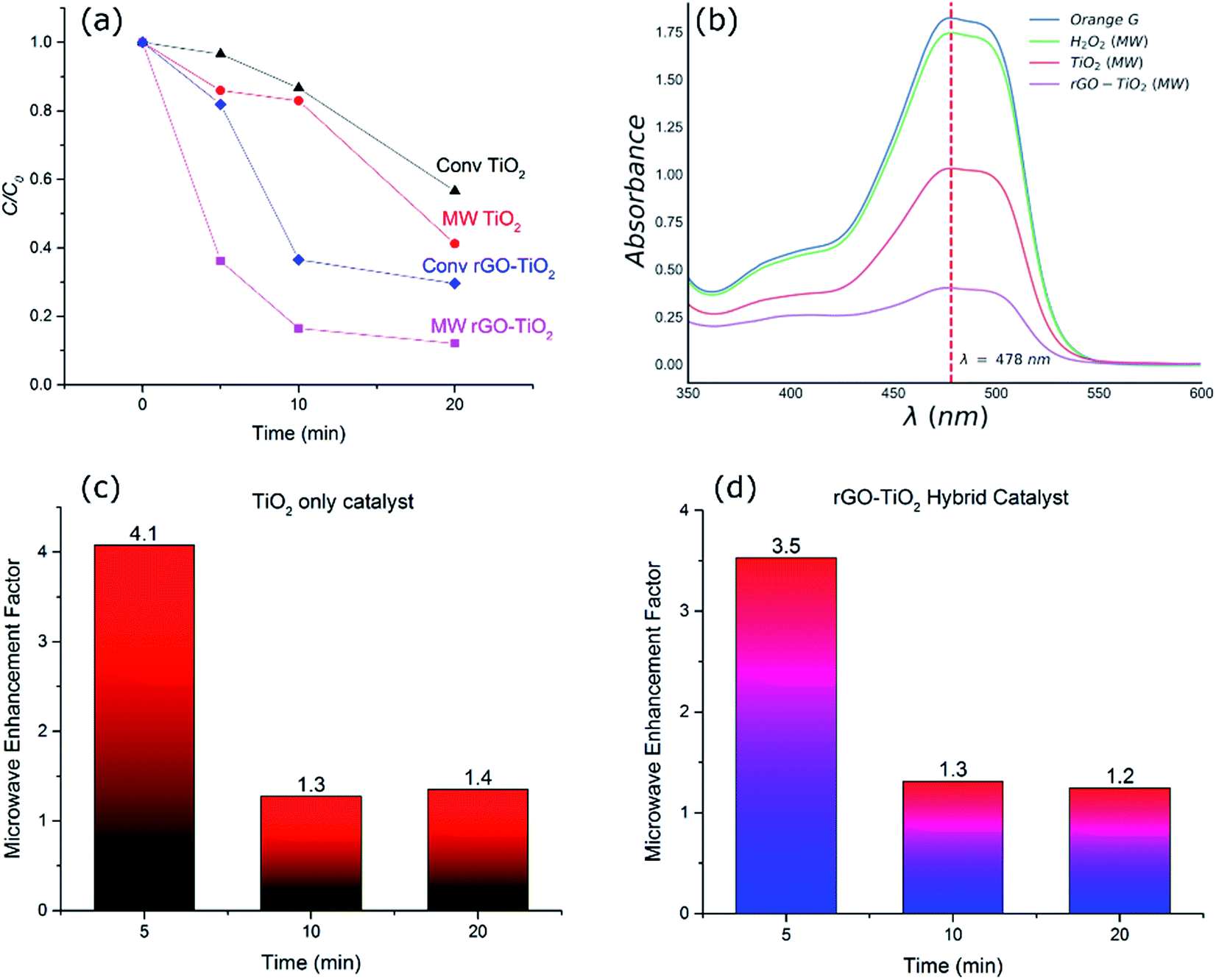 | ||
| Fig. 4 (a) Dye degradation graph of different TiO2 and rGO–TiO2 nanocomposite catalysts under conventional and microwave heating. (b) UV-Vis spectra of OG dye solution as prepared, after 20 minutes of microwave irradiation with catalysts and with only H2O2. (c) Microwave enhancement factor in the case of TiO2 catalyst at an isothermal condition of 120 °C at different times. (d) Microwave enhancement factor in the case of hybrid catalyst at an isothermal condition of 120 °C at different times. | ||
The microwave enhancement factor of the process is depicted in Fig. 4(c and d) where the catalytic performance enhancement is given by the following relation:
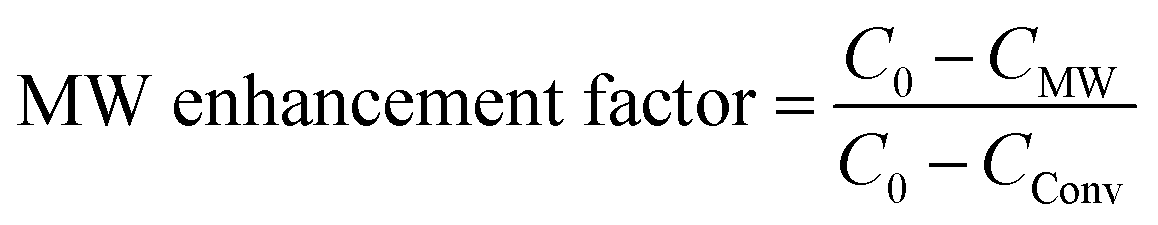
There are a series of contributing factors which result in the improvement of the hybrid catalysts and could explain the performance with and without microwave irradiation. In the case of TiO2 nanoparticle catalyst system, it has been shown that microwave irradiation alone could facilitate the generation of electron/hole pairs in the titania semiconductor and help produce hydroxyl radicals and enhance reaction kinetics.1 Moreover, it has also been shown that microwave irradiation could enhance the generation of hydroxyl radicals from H2O2.39,40 This is in addition to the role of oxidative H2O2 which is present in all systems and at the same level in the experiments carried out in this research. We observed similar improvements in the performance of titania catalyst upon irradiation of microwave and enhanced degradation of the dye. The fact that this improvement was consistent over a range of time scales under isothermal conditions (Fig. 4) suggests the genuine effect of microwaves in improving the performance of catalysts. The increased catalytic decomposition of the dye under microwave irradiation in the TiO2 system is attributed to the enhanced generation of hydroxyl radicals from H2O2 as well as improved electron/hole pair generation in TiO2 as a result of microwave irradiation, leading to faster kinetics and higher degree of decomposition overall.
To elucidate the effect of thermal degradation in the performance of the catalysts, a comparison of the decomposition of the dye using titania and the hybrid rGO–TiO2 catalyst was investigated at a range of different temperatures. The addition of the rGO and formation of the hybrid catalyst drastically changes the catalytic performance (Fig. 5). At higher temperatures, hydrogen peroxide irreversibly decomposes to produce water and oxygen. This contributes to the slowing down of the reaction rates with time along with the consumption of the hydroxyl radicals directly involved in the dye degradation process. Microwaves have been observed to enhance the formation of hydroxyl radicals in the reaction solution to an extent which cannot be fully explained by the increase in temperature alone.41 The precise nature of the phenomenon is not completely understood, but non-thermal interactions between the catalyst and microwaves have been suggested as being responsible for the improvement in radical generation.41
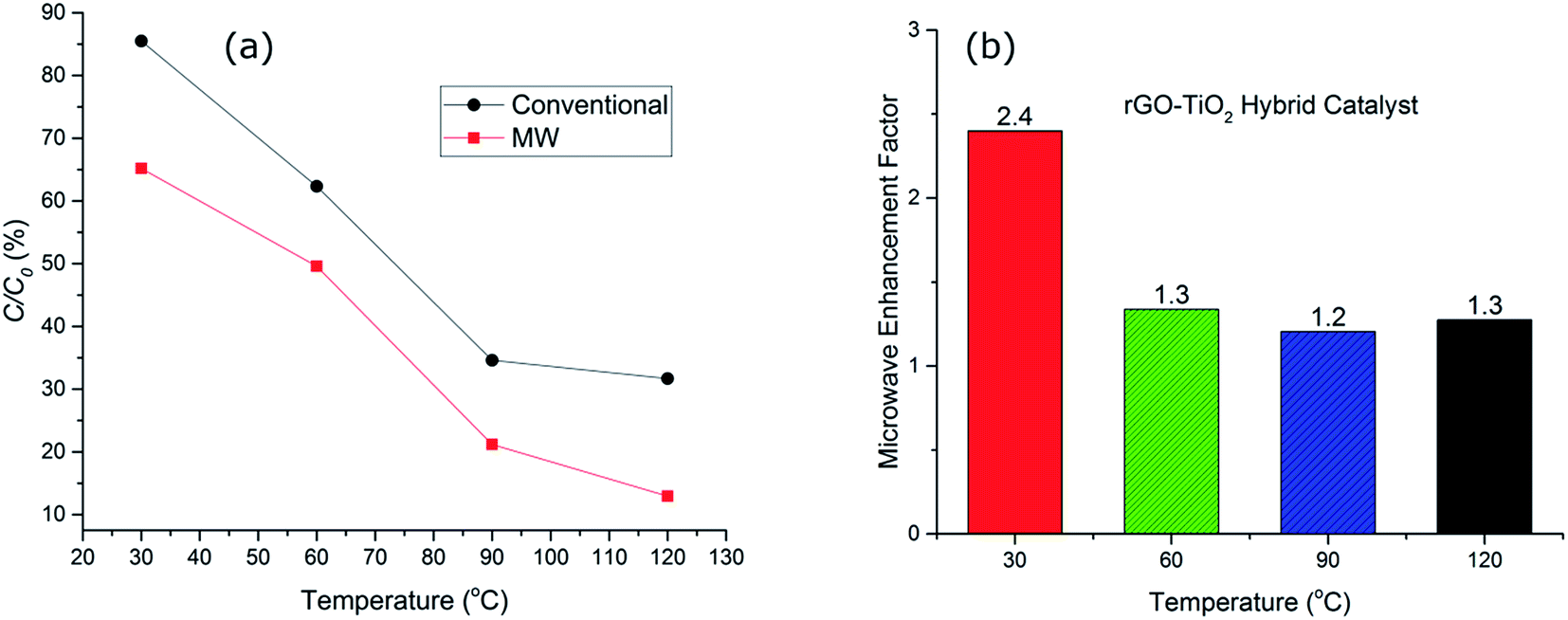 | ||
| Fig. 5 (a) Dye degradation graph of rGO–TiO2 hybrid catalyst under conventional and microwave heating at a fixed time of 20 minutes across a range of temperatures. (b) Microwave enhancement factor for the hybrid catalyst at different temperatures and 20 minutes of treatment time. | ||
Fig. 5(b), compares the MW enhancement factor for the hybrid catalyst at the range of temperature from 30–120 °C. This clearly signifies the fact that microwave contribution is more significant at lower temperature regimes where the catalyst system is normally expected to be inefficient. The enhancement factor was ∼2.4 at 30 °C whilst it was reduced to half, ∼1.2–1.3, for higher temperatures. This implies that the hot spot mechanism of dye degradation is more prevalent at lower temperature regions while at higher temperatures other decomposition mechanisms such as peroxide oxidation and thermal excitation of electron/hole pairs start to compete with hot spot formation and the impact of microwaves is reduced. Nevertheless, microwave irradiation at 120 °C for 20 minutes results in only ∼12% non-degraded dye whilst conventional heating showed >30% of non-degraded dye. This confirms the effectiveness of microwave radiation in enhancing the performance of the hybrid catalysts. The microwave enhancement capability of rGO–TiO2 hybrid catalyst for dye degradation has not been reported before, however similar improvements have been reported for photocatalytic oxidation processes with the addition of GO and/or rGO.42,43 For example, Jiang et al.42 reported 22% OG dye degradation efficiency for a UV irradiated heat treated GO–TiO2 hybrid catalyst after 12 minutes of irradiation at room temperature. We have demonstrated that microwave irradiated hybrid catalyst used in this study resulted in 35% OG dye degradation at 30 °C after 20 minutes at significantly low microwave powers (less than 10 W). Further detailed discussion of microwave AOPs is provided in a recent review by Karayannis et al.44
In addition to the enhanced performance of TiO2 itself, rGO provides larger number of active sites for the adsorption of the dye molecules closer to the titania catalyst whilst reducing the extent of aggregation of titania nanoparticles. It has been demonstrated that rGO performs as an excellent microwave absorber45 hence leading to the formation of the hot spots at the surface of the hybrid catalyst. The formation of such local hot spots results in excitation of more electron/hole pairs at the surface of the hybrid catalysts which consequently would react with O2 and H2O to form ˙OH and superoxide radical anion (O2˙−) and ultimately the degradation rates are enhanced. Similar mechanisms have also been observed for TiO2/activated carbon catalysts in microwave degradation of methyl orange dye.1 This is followed by significant improvements in the conductivity and enhanced electron scavenging performance of the rGO4,46 inhibiting recombination of the charge carriers formed at the hybrid catalysts. The availability of the larger number of the charge carriers would also result in improved decomposition of the dye molecules. More work is required to optimise the loading of the hybrid catalyst as well as utilising more efficient oxidative agents such as peroxymonosulfate (PMS) to replace the H2O2 and obtain even more efficient decomposition pathways.
4. Conclusions
rGO–TiO2 hybrid catalyst was prepared using a microwave assisted process and the catalyst demonstrated significant improvements in the azo dye degradation efficiency when irradiated by microwaves across a range of temperatures from 30 to 120 °C. The combination of hybrid catalyst and microwave irradiation resulted in more than 88% dye degradation within 20 minutes of treatment at high temperatures. The improved performance of the hybrid catalyst system is attributed to (i) excellent microwave absorption of rGO leading to the formation of the hot spots at the surface of the hybrid catalyst (ii) larger number of active sites available for the adsorption of the dye molecules, and (iii) consequent formation of ˙OH and superoxide radical anion (O2˙−) which ultimately result in higher degradation rates. The outstanding wastewater treatment performance of microwave irradiated hybrid catalyst offers a viable low energy, low carbon footprint alternative to conventional thermally activated catalytic processes of waste treatment.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors would like to thank Loughborough Materials Characterisation Centre (LMCC). Engineering and Physical Sciences Research Council (EPSRC) and Innovate UK are acknowledged for research funding and AA's studentship.References
- Z. Zhang, Y. Xu, X. Ma, F. Li, D. Liu, Z. Chen, F. Zhang and D. D. Dionysiou, J. Hazard. Mater., 2012, 209–210, 271–277 CrossRef CAS PubMed.
- Z. Zhang, D. Xu, M. Shen, D. Wu, Z. Chen, X. Ji, F. Li and Y. Xu, Water Sci. Technol., 2011, 63, 424–431 CrossRef CAS PubMed.
- S. Rajendran, M. M. Khan, F. Gracia, J. Qin, V. K. Gupta and S. Arumainathan, Sci. Rep., 2016, 6, 1–11 CrossRef PubMed.
- R. Giovannetti, E. Rommozzi, M. Zannotti and C. A. D'Amato, Catalysts, 2017, 7, 305 CrossRef.
- F. Harrelkas, A. Azizi, A. Yaacoubi, A. Benhammou and M. N. Pons, Desalination, 2009, 235, 330–339 CrossRef CAS.
- Y. Ying, W. Ying, Q. Li, D. Meng, G. Ren, R. Yan and X. Peng, Appl. Mater. Today, 2017, 7, 144–158 CrossRef.
- N. Tripathy, R. Ahmad, J. E. Song, H. A. Ko, Y.-B. Hahn and G. Khang, Mater. Lett., 2014, 136, 171–174 CrossRef CAS.
- S. Ameen, M. Shaheer Akhtar, H.-K. Seo and H.-S. Shin, Mater. Lett., 2013, 113, 20–24 CrossRef CAS.
- J. Han, H. Li, X. Xu, L. Yuan, N. Wang and H. Yu, Mater. Lett., 2016, 166, 71–74 CrossRef CAS.
- K. Pingmuang, A. Nattestad, W. Kangwansupamonkon, G. G. Wallace, S. Phanichphant and J. Chen, Appl. Mater. Today, 2015, 1, 67–73 CrossRef.
- U. Riaz, S. M. Ashraf and M. Farooq, Colloid Polym. Sci., 2015, 293, 1035–1042 CAS.
- U. Riaz and S. M. Ashraf, RSC Adv., 2014, 4, 47153–47162 RSC.
- F. Marken, U. K. Sur, B. A. Coles and R. G. Compton, Electrochim. Acta, 2006, 51, 2195–2203 CrossRef CAS.
- P. K. Chen, M. R. Rosana, G. B. Dudley and A. E. Stiegman, J. Org. Chem., 2014, 79, 7425–7436 CrossRef CAS PubMed.
- N. Wang and P. Wang, Chem. Eng. J., 2016, 283, 193–214 CrossRef CAS.
- K. Zhang, K. C. Kemp and V. Chandra, Mater. Lett., 2012, 81, 127–130 CrossRef CAS.
- P. Benjwal, M. Kumar, P. Chamoli and K. K. Kar, RSC Adv., 2015, 5, 73249 RSC.
- D. Wang, X. Li, J. Chen and X. Tao, Chem. Eng. J., 2012, 198–199, 547–554 CrossRef CAS.
- J. Zhang, Z. Xiong and X. S. Zhao, J. Mater. Chem., 2011, 21, 3634 RSC.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- C. Zhao, W.-J. Wang, D. Sun, X. Wang, J.-R. Zhang and J.-J. Zhu, Chem.–Eur. J., 2014, 20, 7091–7097 CrossRef CAS PubMed.
- T. Szabó, E. Tombácz, E. Illés and I. Dékány, Carbon, 2006, 44, 537–545 CrossRef.
- K. Suttiponparnit, J. Jiang, M. Sahu, S. Suvachittanont, T. Charinpanitkul and P. Biswas, Nanoscale Res. Lett., 2010, 6, 27 Search PubMed.
- F. Tuinstra and J. L. Koenig, J. Chem. Phys., 1970, 53, 1126–1130 CrossRef CAS.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095 CrossRef CAS.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS PubMed.
- G. K. Ramesha and S. Sampath, J. Phys. Chem. Lett., 2009, 113, 7985 CrossRef CAS.
- S. Sahoo, G. Khurana, S. K. Barik, S. Dussan, D. Barrionuevo and R. S. Katiyar, J. Phys. Chem. C, 2013, 117, 5485–5491 CAS.
- A. Kaniyoor and S. Ramaprabhu, Appl. Phys. Lett., 2012, 2, 032183 Search PubMed.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon N. Y., 2007, 45, 1558–1565 CrossRef CAS.
- N. M. Latiff, C. C. Mayorga-Martinez, L. Wang, Z. Sofer, A. C. Fisher and M. Pumera, Appl. Mater. Today, 2017, 9, 204–211 CrossRef.
- D. Voiry, J. Yang, J. Kupferberg, R. Fullon, C. Lee, H. Y. Jeong, H. S. Shin and M. Chhowalla, Science, 2016, 353, 1413–1416 CrossRef CAS PubMed.
- D. Loez-Díaz, M. Loez Holgado, J. García-Fierro and M. Mercedes Velaquez, J. Phys. Chem. C, 2017, 121, 20489–20497 Search PubMed.
- A. Wróblewska, A. Dużyńska, J. Judek, L. Stobiński, K. Żerańska, A. P. Gertych and M. Zdrojek, J. Phys.: Condens. Matter, 2017, 29, 475201 CrossRef PubMed.
- A. A. K. King, B. R. Davies, N. Noorbehesht, P. Newman, T. L. Church, A. T. Harris, J. M. Razal and A. I. Minett, Sci. Rep., 2016, 6, 19491 CrossRef CAS PubMed.
- N. Morimoto, T. Kubo and Y. Nishina, Sci. Rep., 2016, 6, 4–11 CrossRef PubMed.
- J. Xu, S. Gai, F. He, N. Niu, P. Gao, Y. Chen and P. Yang, Dalton Trans., 2014, 43, 11667 RSC.
- M. N. Khan and J. Bashir, J. Mod. Phys., 2011, 2, 962–965 CrossRef CAS.
- Y. Ju, S. Yang, Y. Ding, C. Sun, C. Gu, Z. He, C. Qin, H. He and B. Xu, J. Hazard. Mater., 2009, 171, 123–132 CrossRef CAS PubMed.
- M. Ravera, A. Buico, F. Gosetti, C. Cassino, D. Musso and D. Osella, Chemosphere, 2009, 74, 1309–1314 CrossRef CAS PubMed.
- S. Horikoshi, H. Hidaka and N. Serpone, Chem. Phys. Lett., 2003, 376, 475–480 CrossRef CAS.
- G. Jiang, Z. Lin, C. Chen, L. Zhu, Q. Chang, N. Wang, W. Wei and H. Tang, Carbon N. Y., 2011, 49, 2693–2701 CrossRef CAS.
- H. Sun, S. Liu, S. Liu and S. Wang, Appl. Catal., B, 2014, 146, 162–168 CrossRef CAS.
- V. Karayannis, K. Moustakas, A. Vatalis, P. Sapalidis and A. Domopoulou, Desalin. Water Treat., 2017, 91, 138–145 CrossRef.
- A. A. Amer, S. M. Reda, M. A. Mousa and M. M. Mohamed, RSC Adv., 2017, 7, 826 RSC.
- R. K. Upadhyay, N. Soin and S. S. Roy, RSC Adv., 2014, 4, 3823–3851 RSC.
| This journal is © The Royal Society of Chemistry 2018 |