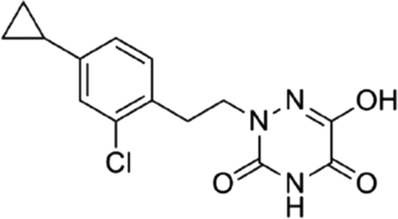
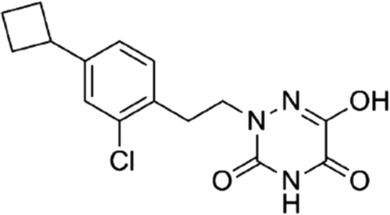
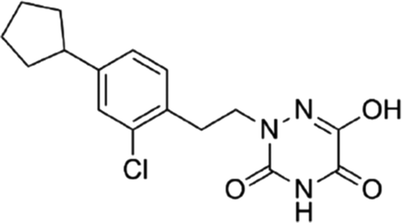
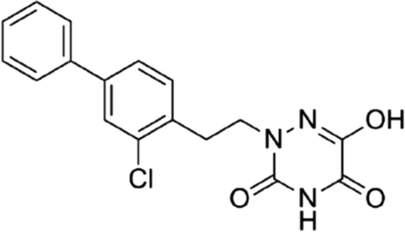
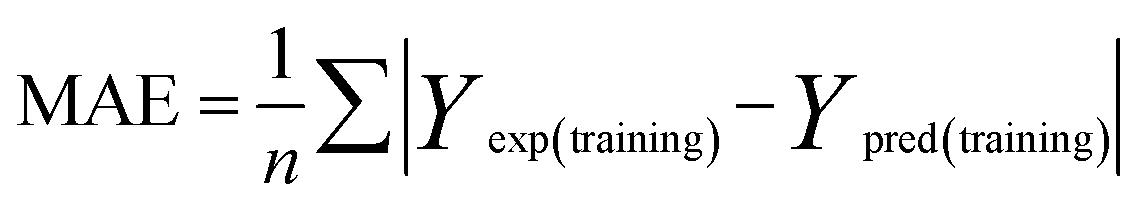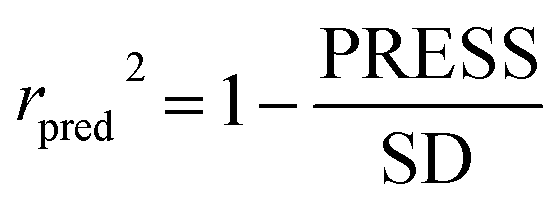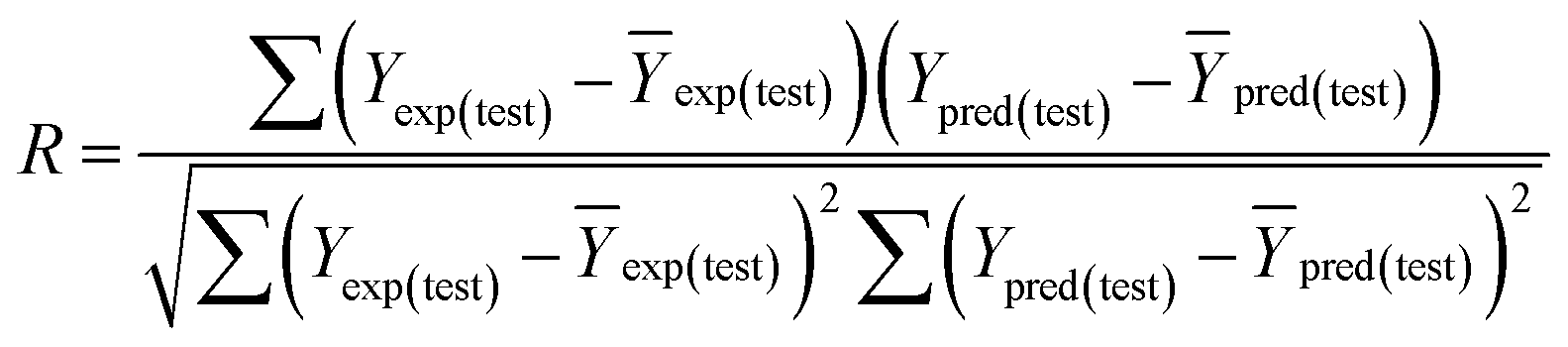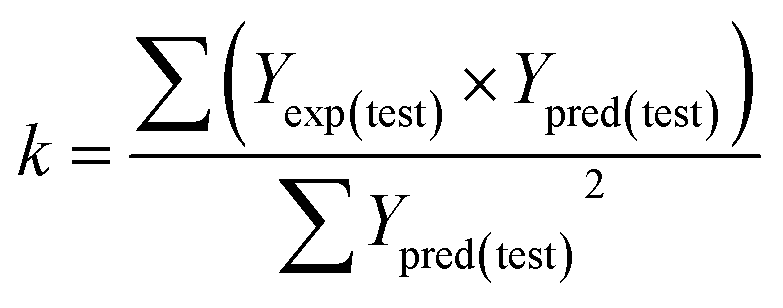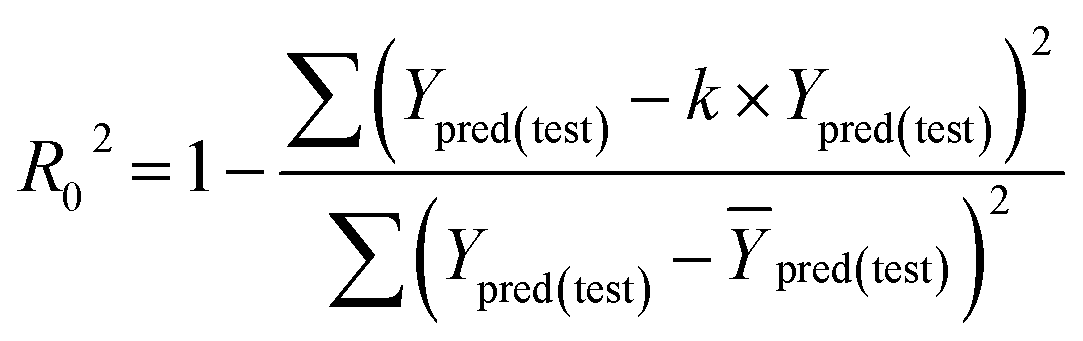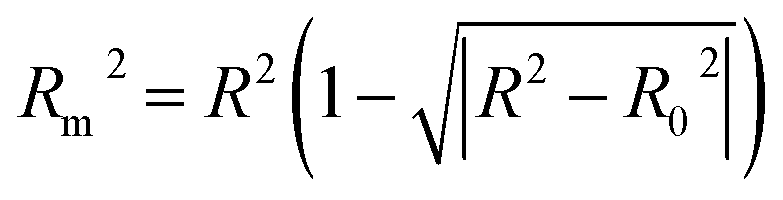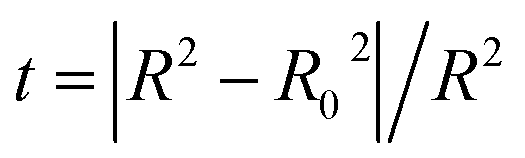DOI:
10.1039/C8RA00094H
(Paper)
RSC Adv., 2018,
8, 14311-14327
Molecular modeling studies of 1,2,4-triazine derivatives as novel h-DAAO inhibitors by 3D-QSAR, docking and dynamics simulations†
Received
4th January 2018
, Accepted 30th March 2018
First published on 17th April 2018
Abstract
Human D-amino acid oxidase (h-DAAO) can effectively act on D-serine, which has been actively explored as a novel therapeutic target for treating schizophrenia. In this study, 37 h-DAAO inhibitors based on a 6-hydroxy-1,2,4-triazine-3,5(2H,4H)-dione scaffold were obtained to construct the optimal comparative molecular field analysis (CoMFA, q2 = 0.613, r2 = 0.966) and comparative molecular similarity index analysis (CoMSIA, q2 = 0.669, r2 = 0.985) models. The results indicate that the models have good predictability and strong stability. Furthermore, contour maps of the three-dimensional quantitative structure–activity relationship (3D-QSAR) revealed the relationships between the structural features and inhibitory activity. A total of nine new h-DAAO inhibitors were designed, which exhibited good predicted pIC50 values. Through molecular docking and molecular dynamics simulation, four essential residues (i.e., Gly313, Arg283, Tyr224 and Tyr228) were considered to interact with the inhibitor. The hydrogen bonds produced by the triazine structure with protein and the hydrophobic interactions with the residues (i.e., Leu51, His217, Gln53 and Leu215) play an important role in the stability of the inhibitor at the binding site of the protein. Additionally, the compounds D1, D3 and D8, with higher predicted activities, were selected by ADME and bioavailability prediction. The present study could offer a reliable theoretical basis for future structural optimisation, design and synthesis of effective antipsychotics.
Introduction
Schizophrenia can be literally interpreted as a ‘shattered mind’. It is among the top 10 causes of long-term disability that affects approximately 24 million people worldwide.1 Symptoms of schizophrenia mainly include hallucinations, delusions, and disordered thoughts and emotions. Schizophrenia also poses the risk of suicide or even murder. Additionally, young people are prone to suffering from schizophrenia, and it causes an enormous burden to society.2 In recent years, developing countries have increasingly reported that the deaths of schizophrenics, including undergraduates, postgraduates, PhD students, white-collar workers, laid-off workers, etc., result from education and employment pressures. Furthermore, most patients with schizophrenia are not appropriately treated. The current methods for treating schizophrenia include electroconvulsive therapy and positive psychotherapy,3,4 but drug treatment remains the most effective and widely accepted.5 Therefore, there is an urgent need for effective and safe antipsychotic drugs.
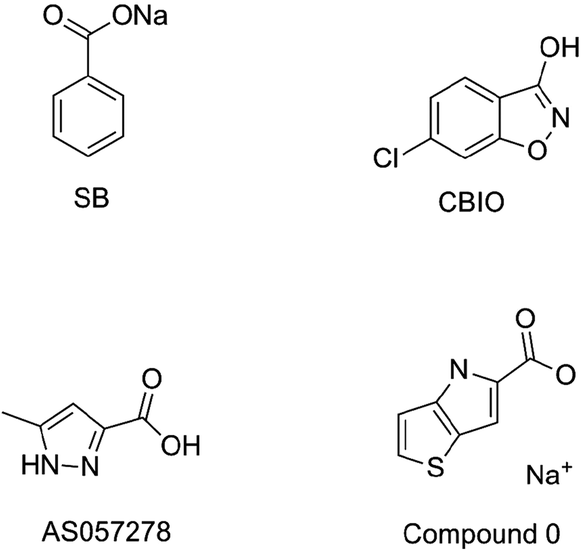
Over the past few decades, all typical and second-generation antipsychotics that act on the D2 dopamine receptor (e.g., perphenazine,6 clozapine,7 olanzapine,8 risperidone,9 ziprasidone10 and aripiprazole11) have caused considerable metabolic and negative side effects.12 In addition, only a small number of patients have completely responded to these currently available antipsychotics, which cannot satisfy the requirements of patients with different etiologies. The situations necessitate new antipsychotics that will facilitate the antipsychotic target from the D2 dopamine receptor to the N-methyl-D-aspartate (NMDA) receptor. D-Serine, which is a full agonist at the glycine modulatory site of the NMDA receptor, has recently been actively explored as a therapeutic target for the treatment of schizophrenia.13 Human D-amino acid oxidase (h-DAAO) is a peroxidase that catalyzes the oxidation of D-serine to H2O2 and the corresponding α-keto acids.14 There is compelling evidence that the inhibition of h-DAAO could provide dual beneficial effects of D-serine therapy, namely, the weakening of D-serine induced nephrotoxicity and the enhancement of D-serine bioavailability. Thus far, researchers have developed several representative h-DAAO inhibitors, including sodium benzoate (SB),15 5-chloro-benzo[d]isoxazol-3-ol (CBIO),16,17 5-methylpyrazole-3-carboxylic acid (AS057278)18 and 4H-thieno [3,2-b]pyrrole-5-carboxylic acid (compound 0)19 (Fig. 1); however, some demerits still exist. For example, SB exerts relatively low antipsychotic efficiency and requires high doses. In addition, although CBIO and AS057278 exhibit acceptable inhibitory potency against h-DAAO, their high acidity and low hydrophobicity hamper cell permeability. To overcome these drawbacks, researchers are continuously performing further studies on h-DAAO inhibitors, and many h-DAAO inhibitors with high activities have been reported in recent years.20–25 Hin et al.25 reported that some potent h-DAAO inhibitors (IC50 in the nanomolar range) based on the 1,2,4-triazine scaffold appear to be metabolically resistant to O-glucuronidation, which is contrary to other structurally-related h-DAAO inhibitors. Compared to other h-DAAO inhibitors, these potent h-DAAO inhibitors not only exhibit high efficiency but also have improved metabolic stability.
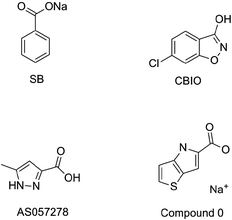 |
| | Fig. 1 Representative h-DAAO inhibitors. | |
Computer-aided drug design (CADD) has greatly improved the success rate of drug design and provides new ideas for overcoming persistent ailments. It has been developed and widely used for anticancer drugs, anti-HCV drugs, anti-inflammatory drugs and anti-AIDS drugs,26–29 but few studies on antipsychotic drugs have been conducted. Moreover, there has never been a study on h-DAAO inhibitors with a simultaneous combination of modeling, prediction, design, molecular docking and dynamics simulation. In the present work, 37 reported triazine compounds were used to obtain the optimal three-dimensional quantitative structure–activity relationship (3D-QSAR) model, and the contour maps of the 3D-QSAR revealed the relationships between structural features and inhibitory activity; a ‘crab’ conformation was initially proposed. The binding mode between the inhibitor and receptor protein was explored by molecular docking and molecular dynamics simulation. Finally, the newly designed compounds with higher predicted activities were selected by ADME and bioavailability prediction. This work was aimed at studying the effects of the substituents (type and position) of the benzene ring and the 1,2,4-triazine structure on the potency of h-DAAO inhibitors, which can be useful for further discovery, design and synthesis of new and effective antipsychotics. Moreover, the interactions between h-DAAO and their inhibitors can be calculated, which can greatly shorten the development cycle. This study therefore provides a vital reference and guide for the emergence and development of novel, broad-spectrum and highly active h-DAAO inhibitors in the future.
Materials and methods
Computational approach
All calculations in this study were conducted using the SYBYL-X 2.0 software package (Tripos Inc., St. Louis, USA) in the Windows 7 OS. Three-dimensional structures of 1,2,4-triazine derivatives as h-DAAO inhibitors were built within the 2D sketch module and the initial conformations of molecules for 3D-QSAR were decided according to the conformation of the ligand extracted from the protein (PDB code: 3W4K). Energy optimization of all compounds was performed using Gasteiger–Hückel30 charges under the impact of Tripos force field31 parameters. During the optimization process, the Powell gradient algorithm with a maximum of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 iterations was adopted, and the energy convergence criterion was limited to 0.005 kcal (mol−1 Å−1). Both comparative molecular field analysis (CoMFA)32 and the comparative molecular similarity index analysis (CoMSIA)33 models were then developed by the QSAR module in SYBYL-X 2.0. In this calculation, all other parameters were set by default (except where noted).
000 iterations was adopted, and the energy convergence criterion was limited to 0.005 kcal (mol−1 Å−1). Both comparative molecular field analysis (CoMFA)32 and the comparative molecular similarity index analysis (CoMSIA)33 models were then developed by the QSAR module in SYBYL-X 2.0. In this calculation, all other parameters were set by default (except where noted).
Dataset
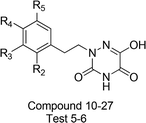
The total set of h-DAAO inhibitors used for the molecular modeling study was reported by Hin et al.25 The structures and bioactivities of 1,2,4-triazine derivatives are described in Table 1 and the IC50 values were converted into pIC50 values (pIC50 = −logIC50), then they were used as the dependent variables. The range of pIC50 values was from 4 to 7.398, indicating that a broad date set with uniform density was used for the 3D-QSAR study. All the experimental data were divided into a training set of 31 compounds for model generation and a test set of the rest for model external validation without constraints. The accuracy of the selection criteria for the training and test set compounds was checked in the form of Uni-Column statistics,34 as shown in Table 1S (see ESI data†). The maximum and minimum pIC50 values in the training and test sets meet the following requirements: (i) max (test) ≤ max (training) and (ii) min (test) ≥ min (training). The data set was output on the basis of substituent diversity and a well-proportioned distribution of biological data was taken (Fig. 1S†).
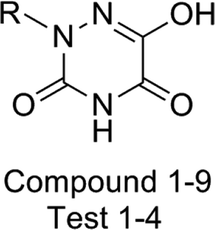
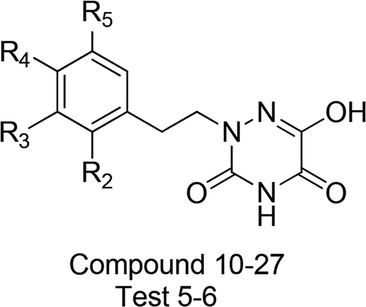
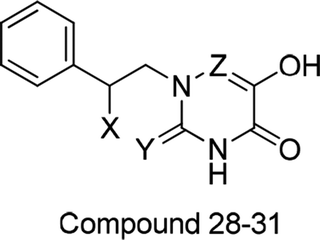
Table 1 Molecular structures of all compounds involved in the study, their actual and predicted bioactivity for CoMFA and CoMSIA
|
| No. |
Substituents |
Actual pIC50 |
Predicted CoMFA |
Residual |
Predicted CoMSIA |
Residual |
| R2 |
R3 |
R4 |
| 10 |
H |
H |
H |
7.155 |
7.144 |
−0.011 |
6.962 |
−0.193 |
| 11 |
Cl |
H |
H |
7.000 |
6.997 |
−0.003 |
7.180 |
0.180 |
| 12 |
H |
Cl |
H |
7.222 |
7.062 |
−0.159 |
7.137 |
−0.085 |
| 13 |
H |
H |
Cl |
7.398 |
7.118 |
−0.280 |
7.228 |
−0.170 |
| 14 |
F |
H |
H |
7.097 |
7.137 |
0.040 |
7.081 |
−0.015 |
| 15 |
H |
CH3 |
H |
7.155 |
6.955 |
−0.200 |
7.021 |
−0.134 |
| 16 |
H |
H |
CH3 |
7.046 |
7.092 |
0.046 |
7.094 |
0.049 |
| 17 |
H |
CF3 |
H |
7.000 |
6.883 |
−0.117 |
7.173 |
0.173 |
| 18 |
H |
H |
CF3 |
7.097 |
7.088 |
−0.008 |
7.247 |
0.150 |
| 19 |
OCH3 |
H |
H |
6.569 |
6.741 |
0.172 |
6.664 |
0.095 |
| 20 |
H |
OCH3 |
H |
6.921 |
6.865 |
−0.056 |
6.909 |
−0.012 |
| 21 |
H |
H |
OCH3 |
6.921 |
7.025 |
0.104 |
6.952 |
0.031 |
| 22 |
OH |
H |
H |
7.046 |
7.027 |
−0.018 |
7.042 |
−0.004 |
| 23 |
H |
OH |
H |
6.796 |
6.796 |
0.000 |
6.804 |
0.008 |
| 24 |
H |
H |
OH |
6.959 |
6.918 |
−0.041 |
6.927 |
−0.032 |
| 25 |
H |
OPh |
H |
6.620 |
6.751 |
0.131 |
6.684 |
0.064 |
| 26 |
H |
H |
OPh |
7.097 |
7.087 |
−0.010 |
7.068 |
−0.029 |
| 27 |
H |
H |
Ph |
7.301 |
7.307 |
0.006 |
7.315 |
0.014 |
| Test 5 |
H |
F |
H |
7.222 |
7.148 |
−0.074 |
7.003 |
−0.219 |
| Test 6 |
H |
H |
F |
7.301 |
7.151 |
−0.150 |
7.052 |
−0.249 |
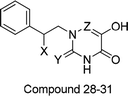
|
| No. |
X |
Y |
Z |
Actual pIC50 |
Predicted CoMFA |
Residual |
Predicted CoMSIA |
Residual |
| 28 |
CH3 |
O |
N |
5.481 |
5.481 |
0.000 |
5.483 |
0.002 |
| 29 |
OH |
O |
N |
6.328 |
6.383 |
0.055 |
6.324 |
−0.003 |
| 30 |
H |
S |
N |
7.301 |
7.302 |
0.001 |
7.239 |
−0.062 |
| 31 |
H |
O |
CH |
7.097 |
7.043 |
−0.054 |
7.146 |
0.049 |
Alignment and generation of the 3D-QSAR models
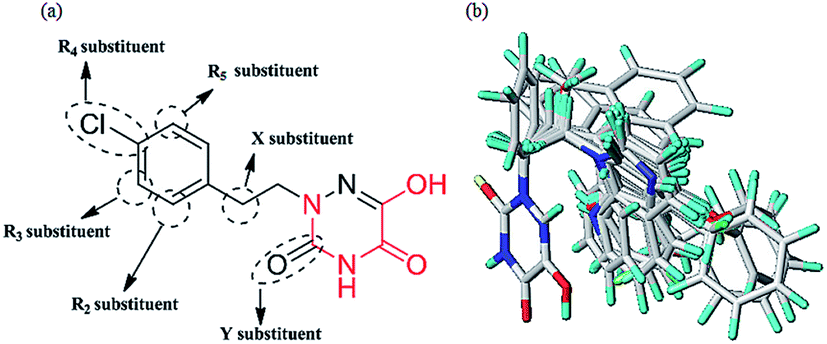
Molecular alignment is regarded as the most important factor for 3D-QSAR analysis. In this study, the ligand-based alignment rule was adopted. The highest biological activity of compound 13 as the template molecule was employed and all the remaining compounds had to be well aligned with it for further analysis. Fig. 2a shows the structure of compound 13 and the red atoms represent the common structure. Fig. 2b illustrates the alignment results based on the common substructure of compound 13.
 |
| | Fig. 2 (a) The common substructure (red) used in database alignment. (b) The alignment result based on the common substructure of compound 13. Molecules are colored in white for common C, blue for N, red for O, yellow for S, cyan for H, green for F and Cl. | |
For CoMFA analysis, steric and electrostatic fields were considered and computed on a spaced grid using a hybridized sp3 carbon atom probe. In the case of CoMSIA analysis, five similarity index descriptors consisting of steric (S), electrostatic (E), hydrophobic (H), H-bond donor (D), and H-bond acceptor (A) fields, were obtained in the same manner as described for CoMFA. Whether the five descriptors in CoMSIA are completely independent has been discussed in some papers.35,36 Different combinations of these five fields were used to build the optimal CoMSIA model in this paper, and the cross-validated correlation coefficient (q2) values of all combinations are shown in Fig. 2S.† The probable models with higher q2 values (q2 > 0.6) were selected (in green and red), including SE, SD, SEH, SED, SEA, SDA, SEHD, SEHA, SEDA and SEHDA (0.666, 0.663, 0.671, 0.668, 0.685, 0.667, 0.659, 0.646, 0.700 and 0.669, respectively). By comparison, the different combinations all have the steric field (S), illustrating that this is necessary for consideration in CoMSIA analysis. According to the entire statistical parameter results of the ten combinations in Table 2, the five fields should all be considered for CoMSIA analysis. Finally, the best CoMSIA model was built based on the combination of SEHDA fields (in red).
Table 2 Statistical results of ten different combinations for CoMSIA models
| Combinations |
q2 |
ONC |
r2 |
SEE |
F |
rpred2 |
Field contribution (%) |
| S |
E |
H |
D |
A |
| CoMSIA |
| S + E |
0.666 |
7 |
0.936 |
0.203 |
48.159 |
0.496 |
0.606 |
0.394 |
— |
— |
— |
| S + D |
0.663 |
5 |
0.936 |
0.194 |
73.688 |
0.409 |
0.971 |
— |
— |
0.029 |
— |
| S + E + H |
0.671 |
8 |
0.983 |
0.108 |
156.330 |
0.638 |
0.205 |
0.304 |
0.492 |
— |
— |
| S + E + D |
0.668 |
9 |
0.994 |
0.092 |
87.329 |
0.576 |
0.832 |
0.150 |
— |
0.018 |
— |
| S + E + A |
0.685 |
8 |
0.956 |
0.172 |
59.929 |
0.507 |
0.544 |
0.364 |
— |
— |
0.092 |
| S + D + A |
0.667 |
4 |
0.936 |
0.192 |
94.518 |
0.531 |
0.834 |
— |
— |
0.046 |
0.120 |
| S + E + H + D |
0.659 |
8 |
0.985 |
0.101 |
178.371 |
0.658 |
0.193 |
0.290 |
0.488 |
0.029 |
— |
| S + E + H + A |
0.646 |
8 |
0.985 |
0.102 |
175.012 |
0.643 |
0.202 |
0.296 |
0.460 |
— |
0.042 |
| S + E + D + A |
0.700 |
8 |
0.955 |
0.175 |
58.090 |
0.531 |
0.536 |
0.358 |
— |
0.013 |
0.092 |
| S + E + H + D + A |
0.669 |
8 |
0.985 |
0.100 |
181.447 |
0.869 |
0.192 |
0.284 |
0.456 |
0.029 |
0.039 |
| Constraints |
>0.5 |
<10 |
>0.9 |
≪1 |
>100 |
>0.5 |
— |
— |
— |
— |
— |
Partial least squares (PLS) analysis and validation of the 3D-QSAR models
To attain more reliable 3D-QSAR models, partial least squares (PLS) regression analysis was carried out by using the leave-one-out (LOO) cross-validation, and the optimum number of components (ONC values) was determined.37 The final CoMFA and CoMSIA models of the h-DAAO inhibitors were in accordance with some statistical parameters. Actually, the predictive ability of the models was not completely evaluated with the cross-validated correlation coefficient (q2 > 0.5), the correlation coefficient (r2 > 0.9), ONC value (<10), the standard error of estimate (SEE ≪ 1) and probability value (F > 100).38 The external validation of both the training and test set is necessary for the reliability of the 3D-QSAR models. In this paper, the root-mean-square error (RMSE) and the mean error measure (MAE) values of the training set were calculated using eqn (1) and (2).39| |
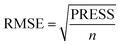 | (1) |
| |
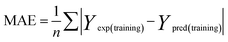 | (2) |
PRESS is the sum of the squared deviations between the actual and predicted biological data for each test set compound, and n is the number of compounds in the training set. The criteria for guaranteeing the accuracy of the 3D-QSAR models include MAE ≤ 0.1 × training set range and MAE + 3 × RMSE ≤ 0.2 × training set range.40 In order to validate the developed models, the external test set of 6 compounds with known activities was predicted, but not applied to the model generation. The predictive correlation coefficient (rpred2) values, in reference to eqn (3), were used for judging the quality of the 3D-QSAR model. Herein, SD is the sum of the squared deviations between the pIC50 values of the test set and the mean pIC50 value of the training set molecules.
| |
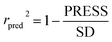 | (3) |
Some other external validation parameters R2, k, R02, Rm2 and t values are also necessary to evaluate the stability, reliability and predictability of the model. These parameters need to meet certain requirements (R2 > 0.6, Rm2 > 0.5, 0.85 ≤ k ≤ 1.15, t < 0.1) when the model has good external predictability. They were calculated by the following eqn (4)–(8).
| |
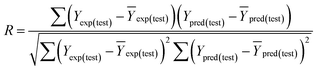 | (4) |
| |
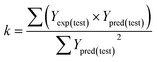 | (5) |
| |
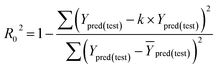 | (6) |
| |
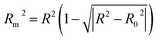 | (7) |
| |
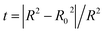 | (8) |
where
Yexp,
Ypred and
Ȳ are the experimental, predicted and mean activities of the compounds in the test set, respectively.
Molecular docking
Docking and scoring technologies contribute to predicting the binding mode of a bioactive ligand at the active site of the protein in the drug discovery process.41 Herein, the Surflex-Dock program was used to investigate the most appropriate conformation. The crystal structure of h-DAAO inhibitor was downloaded from the Protein Data Bank (PDB code: 3W4K). It was essential to do preparatory work on the protein before using the docking programs. Firstly, all water molecules were removed from the original 3W4K protein complex. The side chains and termini chains were repaired after analyzing the protein. The hydrogen atoms and the essential charges were then added to the protein and the protonation type and designated the atom types of the ligand were set. Afterwards, the ProtoMol file was generated by the ligand-based docking mode and each structure was semi-flexibly docked into the active pocket and yielded docking scores. The docking scoring function could be used for predicting the binding affinity between the ligand and the target protein. Finally, each compound could obtain ten different conformations. Taking into consideration the similarity of the orientation between the original ligand and the conformations, the conformation with highest docking score was chosen for the study of the binding mode and molecular dynamics simulation. Meanwhile, to check the accuracy and rationality of the docking method, the root-mean-square deviation (RMSD) value between the docked conformation and the crystal conformation was calculated.
Molecular dynamics (MD) simulation
MD simulations were carried out using the Amber 14.0 software. In the tleap module, the ff14SB force field and the typically general amber force field (gaff) were used for h-DAAO protein and ligands, respectively. The topology file of the ligand-protein structure was generated and the complex was simulated in a hexahedral TIP3P water box of size 78 Å × 78 Å × 78 Å. The distance between the border of the solvent box and protein was set to be larger than 8.5 nm. The total charge of the system was then neutralized by the addition of Na+. All MD simulations followed the procedures for minimization, heating, equilibration and production. Firstly, the energy minimizations of the entire system were divided into two steps. With a constraint force of 100 kcal mol−1 Å−2, the waters and Na+ were minimized by 500 steps of the steepest descent method and 500 steps of the conjugate gradient method. The whole system was then minimized by 5000 steps of the descent method, followed by 5000 steps of the conjugate gradient method. Secondly, the system temperature was heated from 0 to 300 K over 50 ps in the NVT ensemble. The systems were then equilibrated in the NPT ensemble at 300 K and 1 atm. A Langevin thermostat was used for temperature control and the SHAKE algorithm was applied to all bonds. Finally, a 10 ns production run was conducted. Trajectories were recorded for each 1 ps, and the last frame was chosen out of 1000 structures for the MD runs.
The stable molecule conformation obtained from MD simulations was used for the binding free energy calculations. In this process, the Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) method was used and the binding free energy (ΔGbind) was calculated using the following equation.
| ΔGbind = ΔGcomplex − ΔGprotein − ΔGligand ≈ ΔGgas + ΔGsol − TΔS |
| ΔGgas = ΔEELE + ΔEVDW; ΔGsol = ΔGGB + ΔGSA |
where Δ
Ggas is the interaction energy between the protein and ligand in the gas phase. It consists of Δ
EELE (electrostatic energy) and Δ
EVDW (van der Waals energy). Δ
Gsol is the sum of Δ
GGB (polar solvation energy) and Δ
GSA (non-polar solvation energy).
TΔ
S (conformation entropy) is ignored because of its heavy computational cost and weak influence. The above energy parameters were calculated by using the conformations extracted from the last 2 ns.
ADME and bioavailability prediction
In the drug discovery process, the assessments of bioavailability and pharmacokinetics become more important. The predictive models via the new SwissADME web tool42 are necessary and crucial for bioavailability and pharmacokinetics. In particular, when the candidate compounds are many, the computational modeling method is a good choice. In order to evaluate the bioavailability and pharmacokinetics of our newly designed h-DAAO inhibitors, the SwissADME web tool was adopted through the website http://www.swissadme.ch. Firstly, the 2D chemical structures of compounds were drawn in the molecular sketcher and then they were transferred to the SMILES list by clicking on the double-arrow button. Finally, the SMILES list was submitted, and the prediction for bioavailability and pharmacokinetics was calculated after clicking the “Run” button. This study can support the drug discovery efforts in computational chemistry. Specifically, the analysis of bioavailability involves lipophilicity, size, polarity, solubility, saturation and flexibility. These properties were well measured in terms of logP (octanol–water partition coefficient), MW (molecular weight), TPSA (topological polar surface area), logS, fraction C-sp3 and the number of rotatable bonds, respectively. In addition, the absorption, distribution, metabolism, and excretion (ADME) properties in vivo were predicted with the BOILED-Egg model,42 including human gastrointestinal absorption (HIA), blood–brain barrier (BBB) permeation, cytochrome P450-1A2 (CYP1A2) enzyme inhibition and skin permeation (logKp). Therefore, ADME and bioavailability predictions are very important for QSAR studies in the process of new drug design, and could be used to further screen out some designed compounds as potent new-type h-DAAO inhibitors.
Overall working process
In this work, we use CoMFA and CoMSIA methodologies to establish the appropriate models, and get the structure–activity relationships (SAR) information. On this basis, we further designed several new compounds and predicted their activities. Also, these compounds were analyzed in detail by molecular docking and molecular dynamics simulations, for directing the synthesis of higher bioactive h-DAAO inhibitors in the future. Simultaneously, the molecular computer-aided design procedure (MOLCAD) was carried out to further validate the 3D-QSAR model and the binding mode in the ligand–protein structure. Finally, three new designed compounds with higher efficiency were selected to predict the pharmacokinetics and bioavailability properties. Our work is presented in a flowchart in Fig. 3S.†
Results and discussions
3D-QSAR statistical results
Table 3 summarizes the statistical results of the 3D-QSAR models. The CoMFA model gave a q2 value of 0.613 with an ONC value of 6, r2 of 0.966, SEE of 0.144, and F-statistic value of 115.001. The contributions of the fields were 57.8% of the steric field and 42.2% of the electrostatic field descriptor. The CoMSIA model, obtaining a satisfactory q2 of 0.669 with an ONC value of 8, r2 of 0.985, SEE of 0.100, and F value of 181.447, was used for further study. The corresponding field contributions were 19.2%, 28.4%, 45.6%, 2.9% and 3.9% for steric, electrostatic, hydrophobic, hydrogen bond donor, and hydrogen bond acceptor fields, respectively. Comparatively, it was found that the steric, electrostatic and hydrophobic fields played important roles in the optimal CoMSIA model.
Table 3 The statistical results for the CoMFA and CoMSIA models
| Parameters |
CoMFA |
CoMSIA |
Constraints |
| MAE |
0.088 |
0.061 |
≤0.1 × training set range |
| RMSE |
0.127 |
0.085 |
MAE + 3 × RMSE ≤ 0.2 × training set range |
| Training set range |
3.398 |
3.398 |
— |
| q2 |
0.613 |
0.669 |
>0.5 |
| ONC |
6 |
8 |
— |
| r2 |
0.966 |
0.985 |
>0.9 |
| SEE |
0.144 |
0.100 |
≪1 |
| F |
115.001 |
181.447 |
>100 |
| rpred2 |
0.864 |
0.869 |
>0.5 |
| R2 |
0.894 |
0.900 |
>0.6 |
| k |
0.995 |
1.045 |
0.85 ≤ k ≤ 1.15 |
| R02 |
0.988 |
0.909 |
— |
| Rm2 |
0.565 |
0.812 |
>0.5 |
| t |
0.095 |
0.010 |
<0.1 |
|
| Field contribution (%) |
| Steric |
0.578 |
0.192 |
— |
| Electrostatic |
0.422 |
0.284 |
— |
| Hydrophobic |
— |
0.456 |
— |
| H-donor |
— |
0.029 |
— |
| H-acceptor |
— |
0.039 |
— |
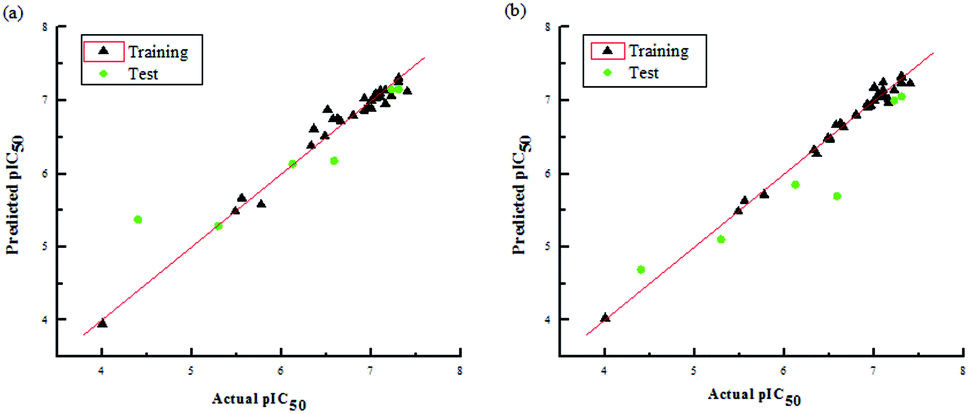
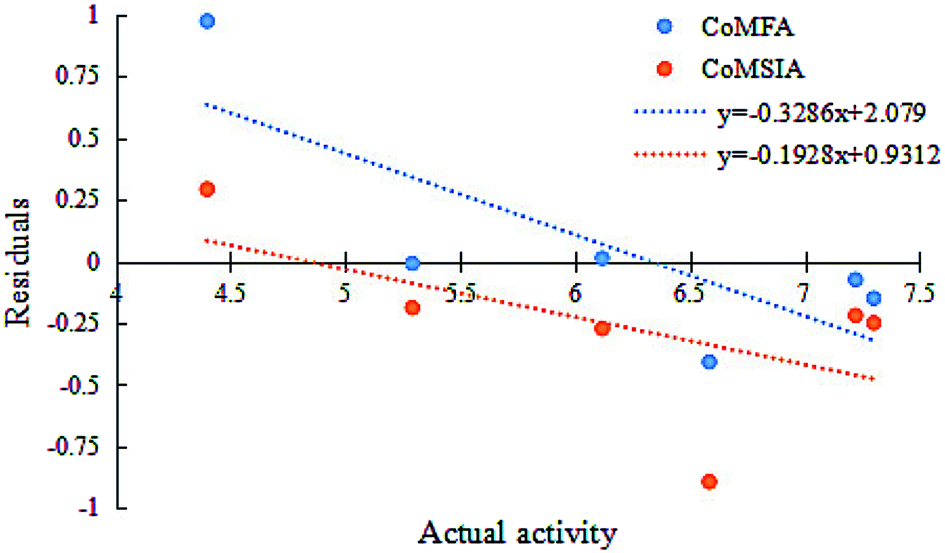
The two models had a statistically significant effect on the capability in predicting the activity. Fig. 3 indicates that the actual and predicted activities of the training and test set molecules had strong linear correlations. Furthermore, the high external predictability of the models was reflected in the external validation parameters. For the external validation of the training set, the CoMFA model gave a satisfactory MAE of 0.088, with RMSE value of 0.127, and the MAE and RMSE values of the CoMSIA model were 0.061 and 0.085, respectively. They both obey their corresponding constraints in Table 3. The rpred2 values of the CoMFA and CoMSIA model were 0.698 and 0.659, respectively, indicating the good external predictive capacity of the models. During the external validation process, the CoMFA model took good values of R2, k, Rm2 and t, at 0.894, 0.995, 0.565 and 0.095, respectively. For the optimal CoMSIA model, the external validation R2, k, Rm2 and t statistics were 0.900, 1.045, 0.812 and 0.010, respectively. In addition, the prediction errors of 3D-QSAR models in the form of a residual plot are clear at a glance in Fig. 4. It was found that the residual values of the test set were randomly distributed around zero; thus, the 3D-QSAR models had good predictability and reliability.
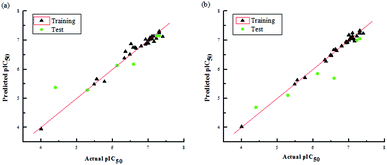 |
| | Fig. 3 Plots of actual pIC50 values against predicted pIC50 values for the data set in the optimal CoMFA (a) and CoMSIA (b) models. | |
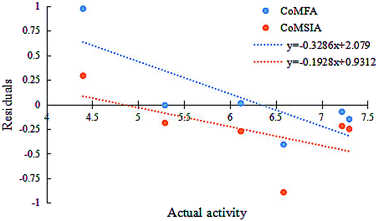 |
| | Fig. 4 Residuals vs. activity plots for the random distribution of prediction errors in CoMFA and CoMSIA models. | |
3D-QSAR contour map analysis
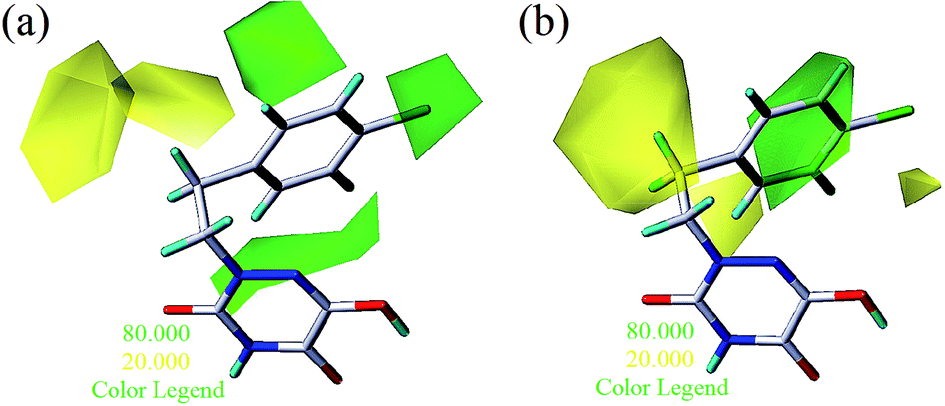
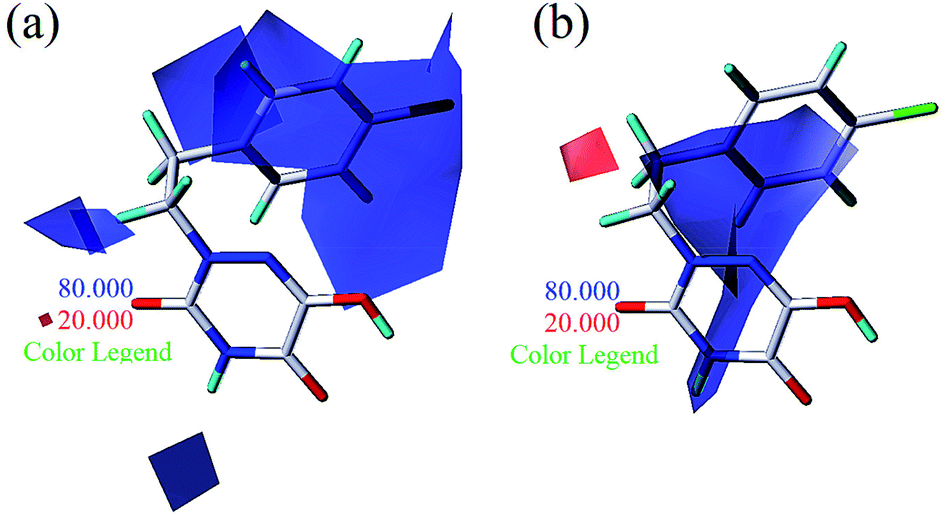
To facilitate understanding the effects of fields on activity in a structure-based manner, contour maps (Fig. 5–8) observed from CoMFA (a) and CoMSIA (b) were discussed by showing the regions in which the energy variations of the molecular fields were consistent with changes in bioactivity. The most active compound 13 was used as a reference structure to illustrate all contour maps of the optimal models. This offered insights into the key structural features for potent h-DAAO inhibitors. A default value of 80% contribution for favored regions was defined to visualize the contour maps, while the disfavored regions were 20%. For the steric contour maps, Fig. 5 offers important information on the spatial volume of groups substituted in different positions. The green contour maps mean that the bulky groups were beneficial for improving activity, while the yellow contour maps mean the bulky substituents were disfavored. It should be noted that the steric and electrostatic contour maps obtained from CoMFA were similar to these from CoMSIA. The CoMSIA model showed better predictability, so the contour maps of CoMSIA were used to analyze the biological data. A green contour map covering the R4 position indicated that a bulky group in this region generally got good biological activity. This was in good agreement with the experimental data. For example, 27 (R4 = Ph) > 18 (R4 = CF3) > 16 (R4 = CH3) > 24 (R4 = OH). There was a yellow contour map located in the position of X, indicating that compounds with bulky groups were not beneficial for inhibitory activity, as observed from 28 (X = CH3) < 29 (X = OH) < 31 (X = H).
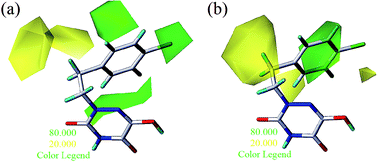 |
| | Fig. 5 Steric contours of CoMFA (a) and CoMSIA (b) based on compound 13. | |
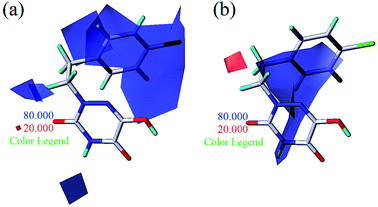 |
| | Fig. 6 Electrostatic contours of CoMFA (a) and CoMSIA (b) based on compound 13. | |
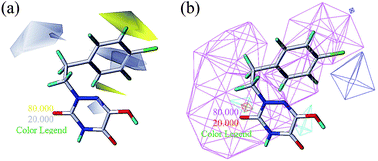 |
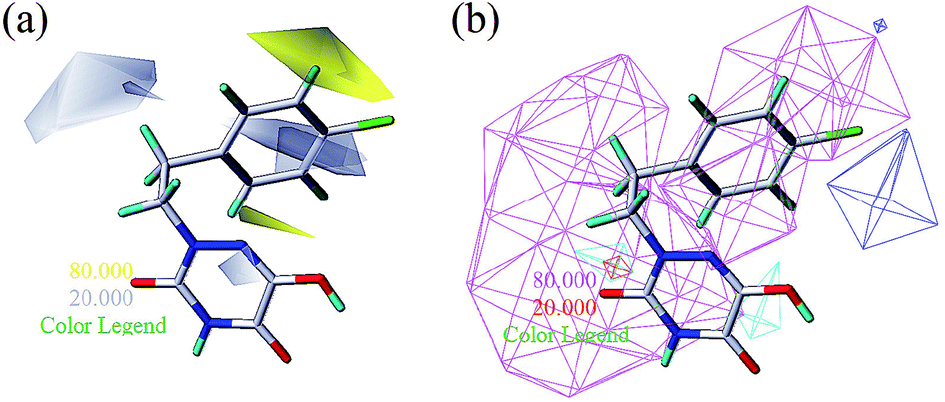
| | Fig. 7 Hydrophobic contours (a) and hydrogen bonding contours (b) of CoMSIA based on compound 13. Hydrogen bonding contours include H-bond donors and H-bond acceptors (displayed as lines). | |
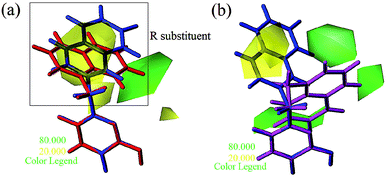 |
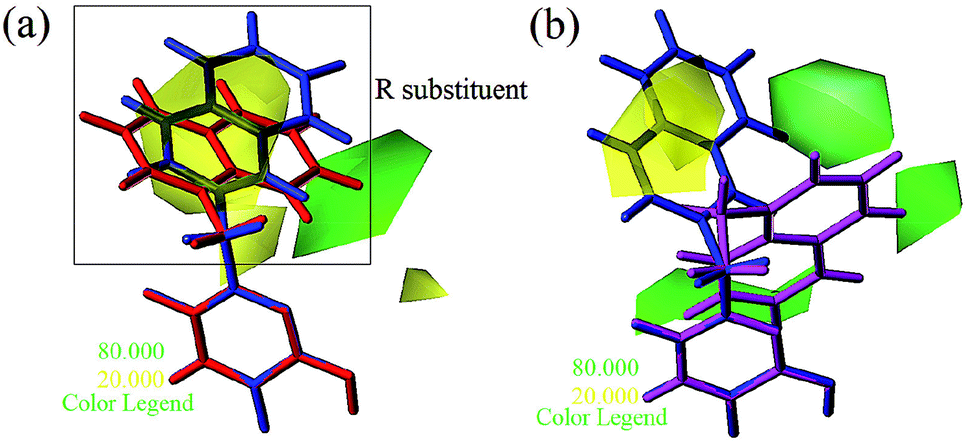
| | Fig. 8 Steric contours of CoMSIA (a) and CoMFA (b) models. (Blue: compound 4; red: compound 5; magenta: compound 6). | |
Fig. 6a and b show the electrostatic field contour maps in CoMFA and CoMSIA analysis, respectively. These contour maps are shown in red (electronegative groups were favorable) and blue (electropositive groups were favorable). A red contour map was nearest to the X position, indicating that having negative electrostatic substituents here was important for increasing the activity. The result also reflected the fact that compound 29 with X being a hydroxyl group obviously got better bioactivity than compound 28. One large blue contour appearing over the position of R3 illustrated that this region was suitable for improving the electropositivity. Therefore, the biological activity of compound 15 with a methyl substituent was significantly improved compared with compound 23. The substitutions of observed positions were also used to speed up the structural optimization.
From Table 3, the hydrophobic field (H) was considered to be the most important for the contributions in the developed CoMSIA model. It made a significant contribution to the activity, compared to the other four (S, E, D and A) fields. As shown in Fig. 7a, there was one large white (hydrophilic favorable) contour map around the R3 and X positions, suggesting that the introduction of hydrophilic moieties into these positions would be of benefit to biological activity. This was consistent with the actual data: 23 (R3 = OH) > 25 (R3 = OPh), 10 (R3 = H) > 17 (R3 = CF3), 29 (X = OH) > 28 (X = CH3). A small white contour map was in the proximity of the 1 N atom of the 1,2,4-triazine structure, indicating that the hydrophilic moieties had little effect on the inhibitory activity. Since the other small white contour maps were far from compound 13, the analysis of the map was ignored.
One medium-sized yellow (hydrophobic favorable) contour map was located at R5, indicating that the presence of higher hydrophobic groups at R5, may be more suitable. At the same time, this yellow contour map was close to R4, suggesting that hydrophobic groups could improve the inhibitory activity; e.g., 13 (R4 = Cl) > 10 (R4 = H) > 24 (R4 = OH). A small yellow contour map near R2 showed that the hydrophobic moieties were favorable. This agreed well with actual data: 14 (R2 = F) > 22 (R2 = OH). In Fig. 7b, the hydrogen bond donor and acceptor contour maps are displayed as lines. The purple color means that groups with a hydrogen bond donor were disfavored and the cyan color means that hydrogen bond donors were favored. On the contrary, the large magenta color contour indicated that the activity of compounds with a hydrogen bond acceptor, like compound 30 (Y = S), Test 5 (R3 = F), Test 6 (R4 = F), increased as compared with compound 10.
Apart from the compounds discussed above, two compounds 5 and 6 with bulky naphthalene moieties showed good inhibitory activities, even greater than the similar compound 4. As displayed in Fig. 8a, a yellow contour around the R substituent indicated that steric hindrance disfavored the activity. However, another green contour was located in the naphthalene moiety of compound 5. This is a possible reason why compound 5 (pIC50 = 7.301) was more potent than compound 4 (pIC50 = 6.658). From Fig. 8b, a yellow contour and three green contours appeared on the R groups of compounds 4 and 6, respectively. This meant that the bulky group of compound 6 would increase the activity. This result was supported by the activity order of 6 > 4.
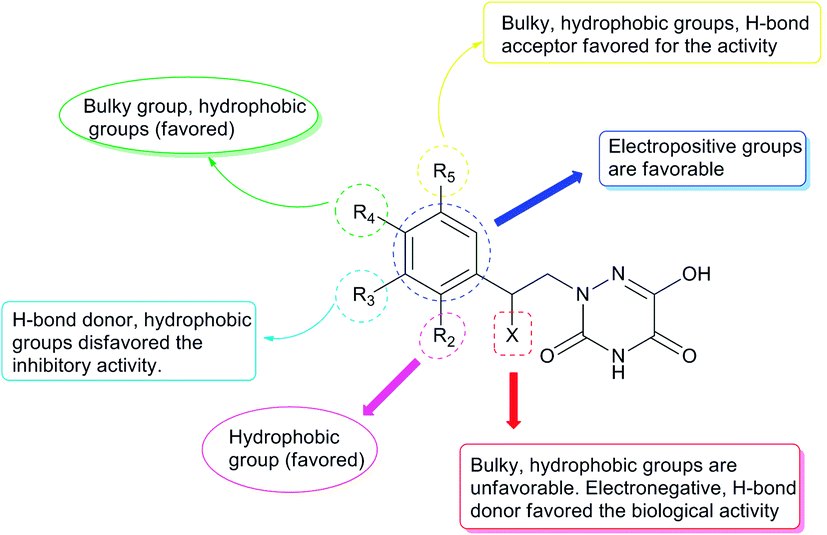
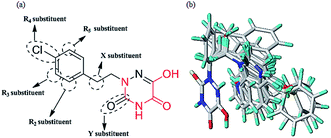
Design of new compounds with higher inhibitory activity
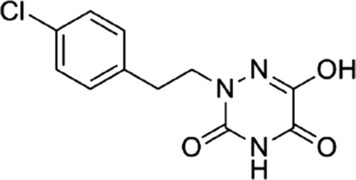
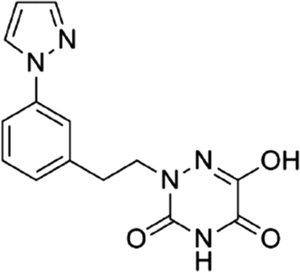
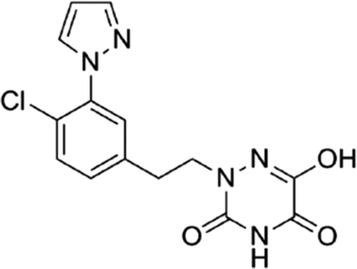
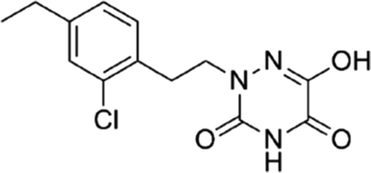
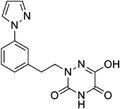
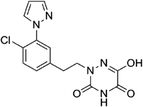
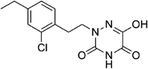
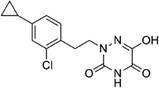
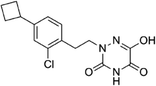
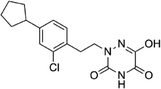
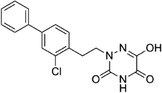
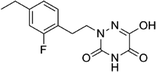
Based on the above comprehensive analysis of contour maps, the SAR information revealed by 3D-QSAR is illustrated in Fig. 9, which could guide the design of new compounds with high bioactivity. We designed a total of 9 compounds using compound 13 (which had the highest activity) as a reference molecule, and their solubility (logS) and lipophilicity (logP) were predicted using the SwissADME web tool42 in Table 4. The modified parts are mainly focused on three substituents (R2, R4 and R5). Since the bulky groups are favored for the activity at the R5 position, pyrazolyl was introduced in this position to yield compound D1. The previous contour maps suggested that some bulky substituents (ethyl, cyclopropyl, cyclobutyl, cyclopentyl and phenyl group) would improve the activity on the R4 position. Also, considering the advantages of hydrophobicity on the R2 position, the compounds D3, D4, D5, D6 and D7 were designed. Due to the fact that the introduction of fluorine can increase the liposolubility of drugs, compounds D8 and D9 were designed. Additionally, considering the advantageous conditions of R4 and R5 with a comprehensive understanding, compound D2 was obtained. Most of the compounds were designed according to steric and hydrophobic fields. Table 4 indicates that they had reasonable absorbency and solubility. In short, it was found that compounds D1, D3 and D8 showed higher inhibitory activity than other compounds.
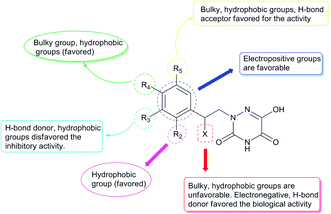 |
| | Fig. 9 Structure–activity relationship (SAR) information obtained from 3D-QSAR studies. | |
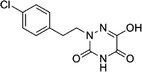
Table 4 Structures and predicted pIC50 activity, logP and logS values of newly designed h-DAAO inhibitors
| No. |
Structure |
Predicted (pIC50) |
logP |
logS |
Surflex-Dock (score) |
| CoMFA |
CoMSIA |
| 13 |
 |
7.118 |
7.228 |
1.55 |
−3.00 |
6.9737 |
| D1 |
 |
7.845 |
7.593 |
1.03 |
−2.80 |
8.4487 |
| D2 |
 |
7.513 |
7.689 |
1.53 |
−3.38 |
8.9393 |
| D3 |
 |
7.878 |
7.787 |
2.18 |
−3.57 |
8.2162 |
| D4 |
 |
7.851 |
7.720 |
2.34 |
−3.69 |
7.6383 |
| D5 |
 |
7.823 |
7.729 |
2.71 |
−4.10 |
7.9024 |
| D6 |
 |
7.813 |
7.751 |
2.96 |
−4.51 |
7.3262 |
| D7 |
 |
7.711 |
7.663 |
2.90 |
−4.50 |
9.0727 |
| D8 |
 |
7.723 |
7.820 |
1.99 |
−3.13 |
8.2472 |
| D9 |
 |
7.638 |
7.853 |
2.68 |
−4.06 |
9.3982 |
Research has shown that the introduction of halogen and alkyl groups into the aromatic ring should increase the efficacy of compounds theoretically, since these groups can respectively improve lipid solubility and chemical stability.43,44 Also, the introduction of naphthenic base can heighten the effectiveness of medications by enhancing both lipid solubility and chemical stability. The introduction of pyrazolyl can improve inhibitory activity by forming hydrogen-bonding interactions with receptors. This is consistent with computational modeling analysis, and the evidence strongly supports that D1 (R5 = pyrazolyl), D3 (R4 = ethyl), D4 (R4 = cyclopropyl) and D8 (R2 = F) have better biological activities than compound 13.
Practical application and evaluation of the 3D-QSAR model
Some h-DAAO inhibitors with different structures were reported by Tsukamoto et al.45 Among these were another four compounds (5y, 10b, 5z, 5m) based on the 6-hydroxy-1,2,4-triazine-3,5(2H,4H)-dione scaffold, which act as h-DAAO inhibitors, as shown in Table 5. The biological activity of compound 5y was higher than that of template compound 13. Furthermore, the activities of these four compounds could be explained well by the SAR information obtained from the 3D-QSAR model. The introduction of hydrophobic groups at the R4-position could improve the inhibitory activity as shown in Fig. 9; e.g., 5y (R4 = Cl) > 12 (R4 = H), 10b (R4 = F) > Test 5 (R4 = H). Similarly, the activity of compound 5z (R3 = Cl) was lower than that of compound Test 6 (R3 = H) due to the fact that the hydrophobic groups disfavored the inhibitory activity at the R3 position. Compound 5m exerted higher activity compared to compound 11 because the addition of Cl at the R4-position not only increased the steric field but also increased hydrophobicity, which contributed to the increasing activity and made all the sense in Fig. 9. We could therefore conclude that the SAR information obtained from the 3D-QSAR model could indicate the directions of the compound modification and save lots of manpower, material resources and research time.
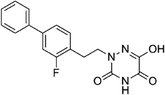
Table 5 Structures and actual pIC50 activity values of four new h-DAAO inhibitors, compared with their predicted activities by CoMFA and CoMSIA models
| No. |
Structure |
Actual pIC50 |
Predicted pIC50 |
| CoMFA |
CoMSIA |
| 5m |
 |
7.097 |
7.262 |
7.676 |
| 5y |
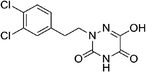 |
7.523 |
7.259 |
7.619 |
| 5z |
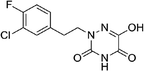 |
7.222 |
7.072 |
7.481 |
| 10b |
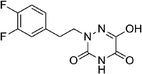 |
7.398 |
7.234 |
7.407 |
In this work, our 3D-QSAR model was used to predict the activities of these four compounds and the inhibitory activities are also listed in Table 5. The results showed that the predicted activity values were close to their experimental values, indicating that our model had good predictability, reliability and practical significance. However, only the activity of compound 5y was higher than that of the template compound 13 in these four compounds, and it is of great worth to study more efficient h-DAAO inhibitors with 1,2,4-triazine in silico. The model was also used to predict the newly designed compounds. On comparing Tables 4 and 5, it was found that the predicted activities of these newly designed compounds were much higher compared to compound 5y and compound 13. The findings suggested that the designed compounds had higher activities and could provide a reliable theoretical basis for the future synthesis of new potent h-DAAO inhibitors. These findings have profound guiding significance for the emergence and development of more efficient antipsychotic drugs.
Molecular docking analysis
Molecular docking is the most extensive program for identifying protein–ligand interactions. The program is effective in simulating the possible binding modes between small molecules and whole protein targets. In the evaluation of docking accuracy, the target ligand was re-docked into the crystal structure of the protein (PDB code: 3W4K). It has been reported that the root mean square deviation (RMSD) value should be less than 2.0.46 In this study, the corresponding RMSD and similarity of the re-docking result were 0.379 and 0.862, respectively, which indicated that the docking method was rational; the re-docking result is illustrated in Fig. 10. Although there was a small rotation angle, it was found that the re-docking structure and the original structure possessed the same binding site. It could also be seen that some key amino acids (Arg283, Gly313, Tyr224 and Tyr228) that interacted with the inhibitor at the binding site were consistent with existing reports. The compound was docked in the binding site via three hydrogen bonds and one π–π interaction. The key residues Arg283 and Tyr228 interacted with the inhibitor by hydrogen bonding. The hydrogen bond distances observed were 1.87 Å (Arg283–NH⋯OH), 1.97 Å (Arg283–NH⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ), and 2.40 Å (Tyr228–HO⋯HO). The π–π interaction was observed between Tyr224 and the triazine ring. It was concluded that the Surflex-Dock docking method and the re-docking results were reasonable and reliable, and Surflex-Dock was subsequently used for docking. Firstly, all compounds were docked into the binding pocket of the protein to explore the binding mode (Fig. 4S†), and hydrogen bond interactions were formed between the 1,2,4-triazine derivatives and several key residues, including Ala49, Leu50, His217, Tyr224, Tyr228, Arg283 and Gly313. The binding pocket of the protein is shown in Fig. 5S,† as well as the binding mode of compounds 4, 5 and 6. The geometry of the pocket looks like a “cave” and the three molecules were semi-flexibly docked onto the active pocket. Compound 5 had one residue more than compounds 4 and 6. Furthermore, the hydrogen bond interaction between molecule 4 and the protein was weaker compared to compounds 5 and 6, indicating that hydrogen bond interactions played an important role in the activity. It was found that the activity order was 5 > 6 > 4. These observations are in agreement with the analysis of the 3D-QSAR contour maps.
), and 2.40 Å (Tyr228–HO⋯HO). The π–π interaction was observed between Tyr224 and the triazine ring. It was concluded that the Surflex-Dock docking method and the re-docking results were reasonable and reliable, and Surflex-Dock was subsequently used for docking. Firstly, all compounds were docked into the binding pocket of the protein to explore the binding mode (Fig. 4S†), and hydrogen bond interactions were formed between the 1,2,4-triazine derivatives and several key residues, including Ala49, Leu50, His217, Tyr224, Tyr228, Arg283 and Gly313. The binding pocket of the protein is shown in Fig. 5S,† as well as the binding mode of compounds 4, 5 and 6. The geometry of the pocket looks like a “cave” and the three molecules were semi-flexibly docked onto the active pocket. Compound 5 had one residue more than compounds 4 and 6. Furthermore, the hydrogen bond interaction between molecule 4 and the protein was weaker compared to compounds 5 and 6, indicating that hydrogen bond interactions played an important role in the activity. It was found that the activity order was 5 > 6 > 4. These observations are in agreement with the analysis of the 3D-QSAR contour maps.
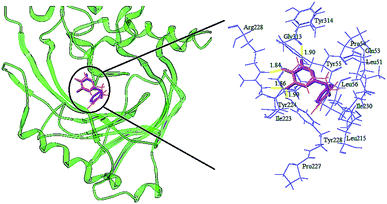 |
| | Fig. 10 Re-docking of the compound into the binding site of h-DAAO (3W4K). Hydrogen bonds are shown as yellow lines, with distance unit of Å (magenta: the re-docked ligand; red: the original ligand). | |
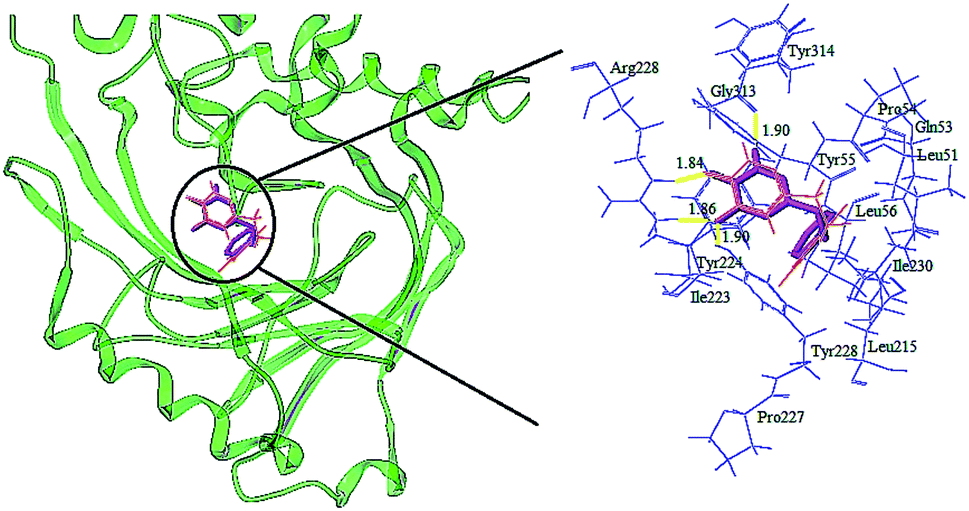
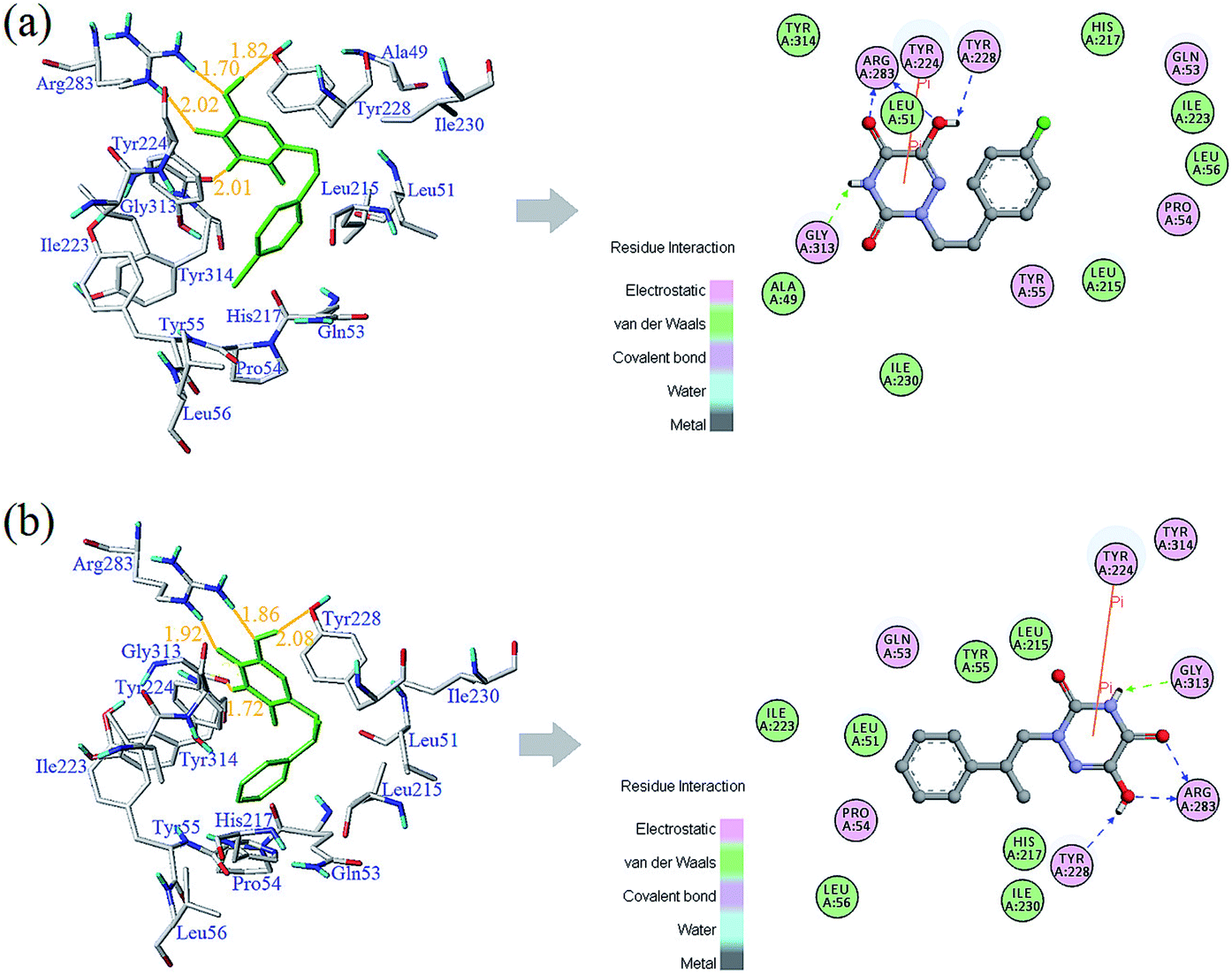
The detailed analyses of two molecules (13 and 28) are displayed in Fig. 11. Compound 13 was docked in the binding site via four H-bonds and one π–π interaction. The key residues Arg283, Tyr228 and Gly313 interacted with the inhibitor by H-bond. The H-bond distances were observed to be 1.70 Å (Arg283–NH⋯OH), 2.02 Å (Arg283–NH⋯O), 1.82 Å (Tyr228–HO⋯HO) and 2.01 Å (Gly313O⋯H–N). The 5-O group of the triazine ring formed one H-bond with the protein, consistent with the contour maps of CoMSIA as shown in Fig. 7b, where the 5-position group was found favorable as a H-bond acceptor. Also, two H-bonds of the 6-OH group matched the H-bond donor map of the 3D-QSAR model. The π–π interaction was observed between Tyr224 and the triazine ring. These two interactions were similar to the results in literature.25
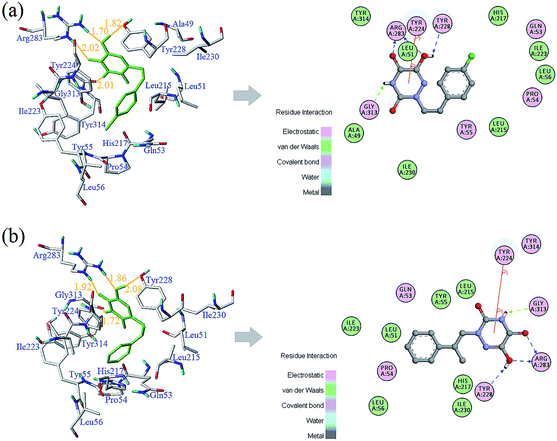 |
| | Fig. 11 Docking results and 2D maps of the selected compounds 13 (a) and 28 (b) in the binding site of the protein (PDB entry code: 3W4K). The inhibitor and the key residues are shown as stick models. Hydrogen bonds are described as yellow lines, with distance in Å units. | |
It was shown that the triazine ring was important for the activity of h-DAAO inhibitors, and agreed well with the high bioactivities of many triazine derivatives. In addition, electrostatic and van der Waals interactions were formed between the compound and several residues in the 2D maps obtained from Discovery Studio 4.5. The hydrophobic acting force had the greatest effects on bioactivity from the analysis results of CoMSIA and it was found that there were hydrophobic interactions with Leu51, His217, Gln53, Leu215, Arg283, Tyr224, Tyr228 and Gly313 residues located in the hydrophobic pocket. The binding mode of compound 13 indicated that eight residues Arg283, Tyr224, Tyr228, Leu51, His217, Gln53, Leu215 and Gly313 were necessary for interacting with h-DAAO inhibitors, and H-bonds and hydrophobic interactions were very important factors in improving the inhibitory activity.
The interactions between compound 28 and the active site are depicted in Fig. 11b. The ligand was docked into the binding site via four hydrogen bonds and one π–π interaction. The hydrogen bond distances observed were 1.86 Å (Arg283–NH⋯OH), 1.92 Å (Arg283–NH⋯O), 2.08 Å (Tyr228–HO⋯HO) and 1.72 Å (Gly313O⋯H–N). The π–π interactions were also observed between Tyr224 and the triazine ring. Some residues (such as Leu51, Leu56, Leu215, Ile223, Ile230, His217, Tyr55 and Pro54) formed van der Waals interactions with the target molecule. Some other residues, including Arg283, Gln53, Gly313, Tyr224 and Tyr228, formed electrostatic interactions. Compound 28 had the same hydrophobic residues as compound 13, but compound 13 had one more amino acid residue (Ala49) than compound 28. The residue Ala49 could interact with the compound via electrostatic and hydrophobic interactions, which affected the bioactivity of h-DAAO inhibitors from the field distributions in the CoMSIA model. This is the reason compound 13 had better activity than compound 28.
According to the docking results of all compounds, the key h-DAAO residues in the docking pockets of the protein (3W4K) were found to be Gly313, Arg283, Tyr224, Tyr228 and Leu51. To obtain new compounds with higher efficiency, the triazine structure has to be maintained. The substituents (type and position) on the benzene ring of the inhibitors can then be modified, aiming to form hydrophobic interactions with acid residues (His217, Leu215, Leu51) for stabilizing the ligand in the active site. In terms of increasing the activity of the h-DAAO inhibitors, it is vital to do modifications with some suitable substituents. Therefore, the binding mode was complementary to the CoMFA and CoMSIA models.
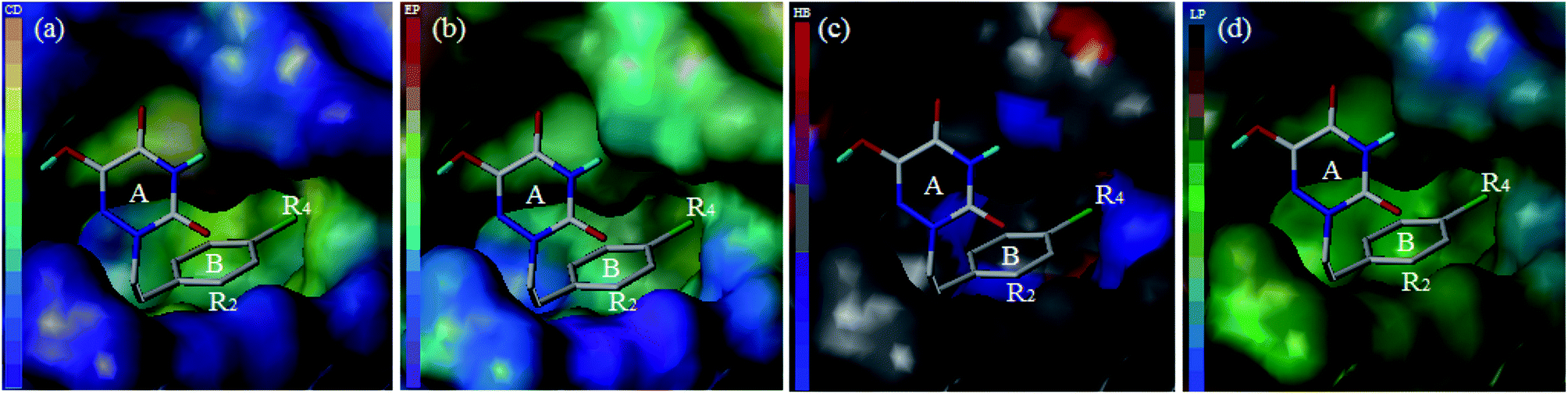
For the sake of further verification of the binding mode and 3D-QSAR models, the MOLCAD surface with compound 13 was determined (Fig. 12). MOLCAD computed and displayed four surface properties, namely, cavity depth (CD), electrostatic potential (EP), hydrogen bonding sites (HB) and lipophilic potential (LP). As shown in Fig. 12b, the color ramp on the left representing the surface of the EP ranges from red (positive) to purple (negative). The entire ring B, especially in the R2 position, was immersed in the purple area and without any red color on the surface, so the introduction of electropositive substituents into ring B was favored for the activity. In addition, the color ramp of HB is from red (H-bond donor) to blue (H-bond acceptor) in Fig. 12c. Therefore, the R4 substituent on ring B was oriented towards the red surface, indicating that it is advantageous to use hydrogen bond acceptor groups as R4 substituents. The color ramp also represents the hydrophobic degree of surface changes from hydrophobic (brown) to hydrophilic (blue). Thus, the area near the R4 substituent was light brown in Fig. 12d, suggesting that hydrophobic groups at the R4 position could increase the activity. All of the above conclusions are well in agreement with the SAR information obtained from 3D-QSAR studies, which also verified the correctness of the docking pocket. Finally, it can be concluded that the MOLCAD surface maps at the active site were highly consistent with the 3D-QSAR model in terms of hydrogen bonding, electrostatic and hydrophobic potentials.
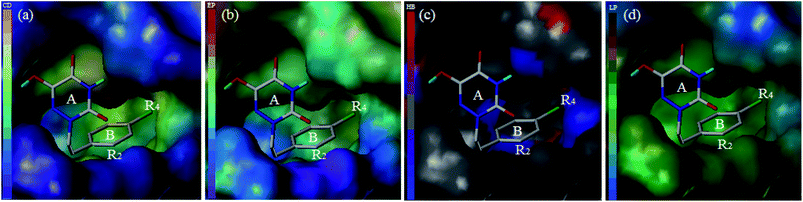 |
| | Fig. 12 MOLCAD surface displayed by cavity depth (a), electrostatic potential (b), hydrogen bonding sites (c) and lipophilic potential (d) for compound 13 at the active site of h-DAAO. | |
After the compounds with known activities were docked into protein h-DAAO, the docking with all designed molecules was studied, shown in Fig. 6S.† The docking results illustrated that most of the designed compounds (i.e., D1, D2, D3, D7, D8 and D9) had higher docking scores, and the orientation of the molecules was similar to the original ligand. Moreover, several residues, Tyr224, Gly313, Arg283, Ala49, Gly50 and Tyr228, interacted with the binding pocket, mainly through H-bonds with the triazine ring. This indicated that the H-bond interaction in the triazine region was beneficial for binding affinity and activity.
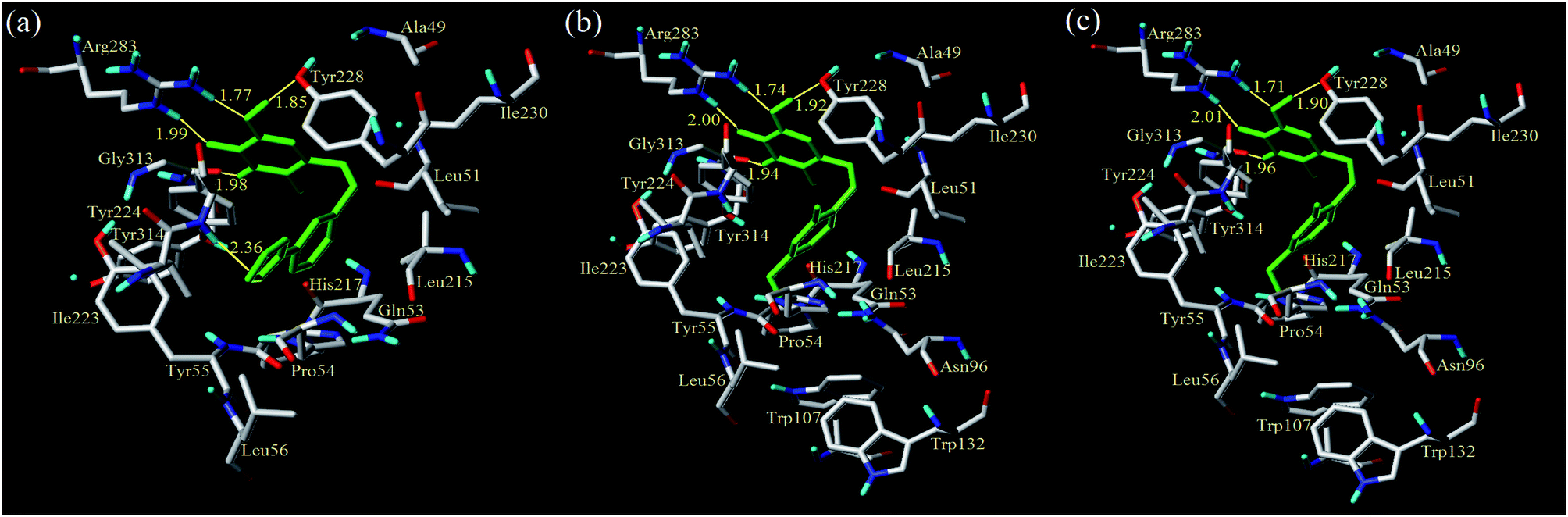
From Table 4, compounds D1, D3 and D8 had higher predicted pIC50 values and docking scores among the newly designed molecules, so they were further studied for in-depth analysis. As shown in Fig. 13, compound D1 docked into the cave-like pocket via five H-bonds and one π–π interaction. The 4,5,6-positions of the triazine ring formed four H-bonds with the amino acids Gly313, Arg283 and Tyr228, respectively. Another H-bond interaction was found between the pyrazolyl region and residue Tyr224 at distance of 2.36 Å. Additionally, the electrostatic and van der Waals interactions of D1 were similar to molecule 13. The strengthened H-bonds of D1 were helpful for the binding affinity, which obviously enhanced h-DAAO inhibitory activity. For compound D3, the H-bond distances were observed to be 1.74 Å (Arg283–NH⋯OH), 2.00 Å (Arg283–NH⋯O), 1.92 Å (Tyr228–HO⋯HO), and 1.94 Å (Gly313O⋯H–N), respectively. For compound D8, the H-bond distances were 1.71 Å (Arg283–NH⋯OH), 2.01 Å (Arg283–NH⋯O), 1.90 Å (Tyr228–HO⋯HO), and 1.96 Å (Gly313O⋯H–N), respectively. One π–π interaction was still observed between Tyr224 and the triazine ring. Some residues were used to form electrostatic interactions. Moreover, the other eight residues, such as Leu215, His217, Leu51, Tyr228, Arg283, Gly313, Tyr224 and Gln53, had the important hydrophobic interactions at the hydrophobic pocket. These interactions were the same as with compound 13. Some of the amino acids surrounding R2, R4 and R5 of the benzene ring were hydrophobic, declaring that the hydrophobic substituent at the R2, R4 and R5 positions could increase activity. Hydrophobic amino acids like Pro54, Tyr55, Leu51, Trp107, Ile223 and Ile230 were present in the proximity of R2, R4 and R5, which validated the hydrophobic contour maps of CoMSIA.
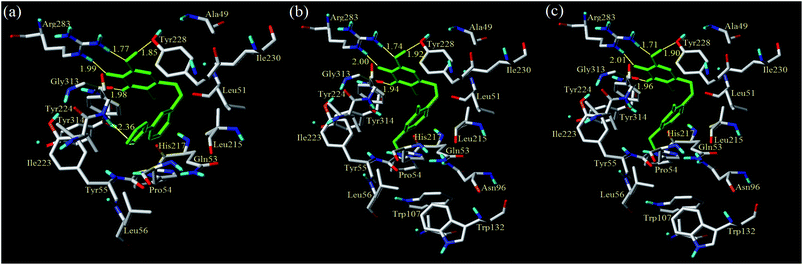 |
| | Fig. 13 Docking results of the designed compounds D1 (a), D3 (b) and D8 (c) in the binding sites of protein 3W4K (yellow lines: hydrogen bonds). | |
There are some significant differences between the newly designed compounds and the template compound 13. The number of key residues that interacted with the new compounds through van der Waals interactions to stabilize the ligand, like residues Trp107, Trp132 and Asn96, increased significantly. Furthermore, it was noted that the compounds D1, D3 and D8 had shorter hydrogen bond distances than 13, which would enhance the binding affinity between the inhibitors and protein. These findings accounted for the order of their inhibitory activities: compound D3 > 13 > 28. These were also consistent with the predicted activity in the 3D-QSAR model.
MD simulation analysis
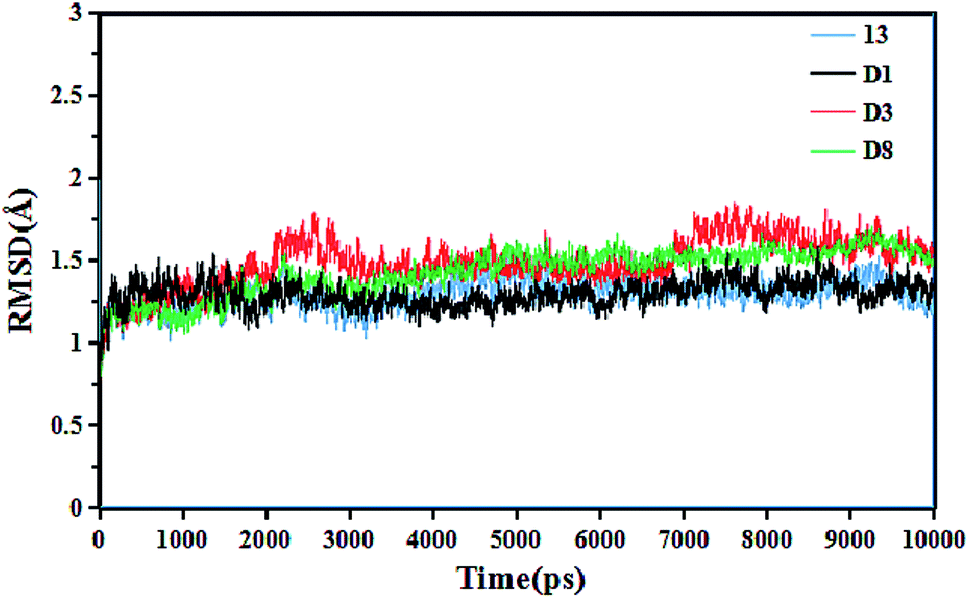
To gain insight into the dynamic interactions between the inhibitors and receptor protein (3W4K), MD simulations were run on our lab's server for four representative inhibitors 13, D1, D3 and D8. The system stability was determined in terms of the root-mean-square deviation (RMSD) of the complex backbone Cα atoms. The plots of the RMSD values versus the dynamic simulation time are illustrated in Fig. 14. It was found that all protein-ligand complexes were stable after 6 ns simulations, and the system had good convergence. In addition, the RMSD values fluctuated between 1 Å and 2 Å, indicating that the complexes were close to the native state. Moreover, the plots of total-energy and temperature versus time are shown in Fig. 7S.† This trend also indicated that the system had good stability. The superimposition of the original 13-3W4K complex and the equilibrium structure after 10 ns simulations are displayed in Fig. 8S.† Except for a small rotational angle, it can be recognized that the two structures are similar, and the original docked structure and the dynamics simulated structure adopted the same binding site of h-DAAO, which further determined the stability of the system and confirmed the reliability of the docking results.
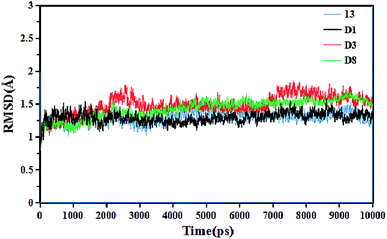 |
| | Fig. 14 Root-mean-square deviation (RMSD) of the ligand-3W4K complexes versus the dynamics simulation time. | |
The root mean square fluctuation (RMSF) value reflects the fluctuations in the protein amino acids residues. Fig. 9S† shows the relationship between the RMSF values of the complex backbone Cα atoms and the residue number in four dynamics simulated complexes. It can be seen that the inhibitor-protein complexes 13-3W4K, D1-3W4K, D3-3W4K and D8-3W4K had similar fluctuations, suggesting that the binding patterns of these inhibitors were similar. In Fig. 9S,† four residues Tyr224, Tyr228, Arg283 and Gly313 were labeled, which could form strong H-bonds interactions with inhibitors. Furthermore, the RMSF values of Tyr224, Tyr228, Arg283 and Gly313 were observed to be 0.3814 Å, 0.3149 Å, 0.3898 Å and 0.4055 Å, respectively. These amino acids residues with lower RMSF values showed rigid behaviours and good stability. Thus, compounds 13, D1, D3 and D8 had good binding affinities with h-DAAO.
Based on the MD simulations, MM/GBSA binding free energies for the four inhibitors of the last 2 ns trajectory were calculated in Table 6. It is generally recognized that the binding free energy values of inhibitors with higher activities are more negative. The binding free energies ΔGbind of compounds 13, D1, D3 and D8 were −24.6943 kcal mol−1, −32.0512 kcal mol−1, −28.7461 kcal mol−1 and −31.3676 kcal mol−1, respectively. This showed that ΔGbind values were in agreement with the predicted pIC50 values of the 3D-QSAR model; the corresponding pIC50 activity order is D1 (7.845) > D8 (7.820) > D3 (7.787) > 13 (7.398). It can also be seen that the greatest contributors to the whole binding free energy were van der Waals energy ΔEVDW, followed by electrostatic energy ΔEELE; therefore, ΔEVDW values were the key factors of ΔGbind. In addition, the ΔGGB values are positive, indicating that polar solvation energy is unfavourable for ΔGbind. In contrast, the ΔGSA values are negative, suggesting that non-polar solvation energy is favourable. The ΔGbind values of new compounds are more negative compared to 13, showing their greater inhibitory activities. From the structural viewpoint, one reason might be the introduction of F, Cl and pyrazolyl into the benzene ring, which could be confirmed by docking.
Table 6 Binding free energies of inhibitor-protein complexes and the different energy contributions
| No. |
ΔEELE (kcal mol−1) |
ΔEVDW (kcal mol−1) |
ΔGgas (kcal mol−1) |
ΔGGB (kcal mol−1) |
ΔGSA (kcal mol−1) |
ΔGsol (kcal mol−1) |
ΔGbind (kcal mol−1) |
pIC50 (predicted) |
Docking score |
| 13 |
−36.3352 |
−23.5881 |
−59.9233 |
39.8916 |
−4.6625 |
35.2291 |
−24.6943 |
7.398 |
6.9737 |
| D1 |
−40.2932 |
−26.6170 |
−66.9102 |
40.2745 |
−5.4155 |
34.8590 |
−32.0512 |
7.845 |
8.4487 |
| D3 |
−36.9914 |
−24.3826 |
−61.3740 |
37.4548 |
−4.8269 |
32.6279 |
−28.7461 |
7.787 |
8.2162 |
| D8 |
−40.2643 |
−26.3763 |
−66.6405 |
40.1342 |
−4.8613 |
35.2729 |
−31.3676 |
7.820 |
8.2472 |
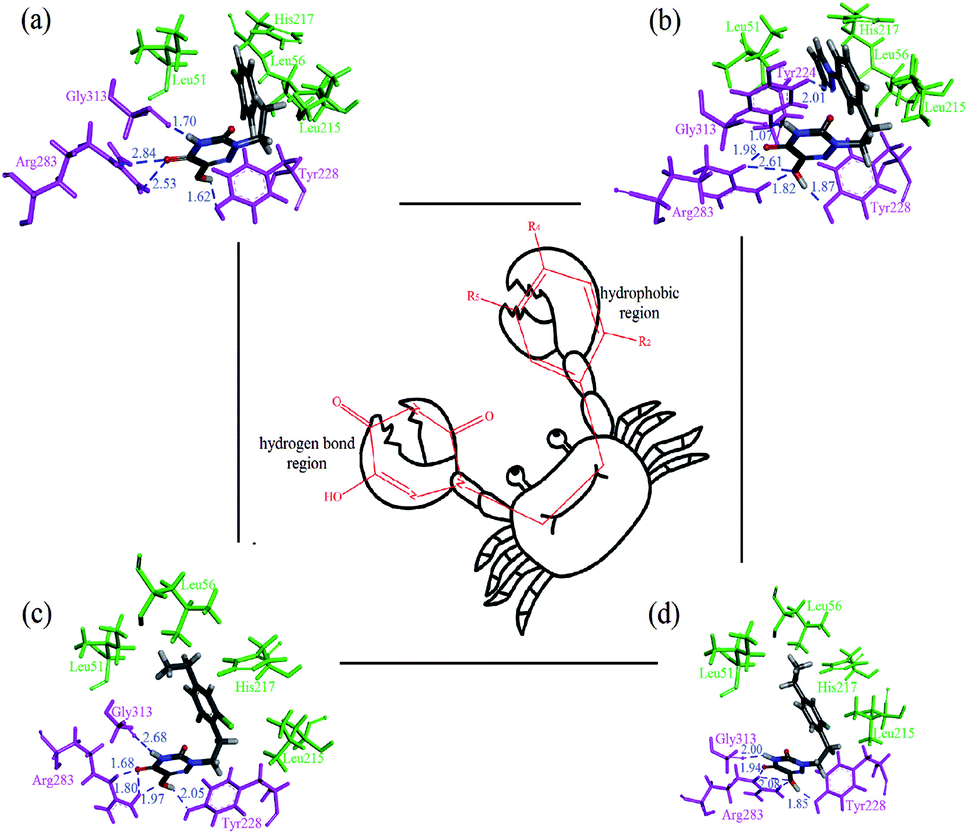
Subsequently, the dynamics stable complexes, 13-3W4K, D1-3W4K, D3-3W4K and D8-3W4K, were extracted for docking analysis. From the perspective of amino acids, the various interactions between triazine compounds and protein were explored. The designed molecules D1, D3 and D8 were perfectly docked onto the active site of the h-DAAO protein. The binding patterns were very similar to that of 13, and the triazine and benzene rings occupied the same position as the corresponding moieties of 13. The docking results of inhibitor-protein structures are displayed in Fig. 15. It can be seen that van der Waals and electrostatic interactions still existed after MD simulations, but hydrophobic and H-bond interactions became stronger. For the 13-3W4K complex, the carbonyl group at the 5-position of the triazine ring formed two H-bonds with NH of Arg283, The third H-bond was found between the NH at the 4-position of the triazine ring and the carbonyl of Gly313. Moreover, the OH group at the 6-position of the triazine ring formed H-bond interactions with Tyr228 at a distance of 1.62 Å. The stable H-bond interactions in the triazine ring region were considered to be a key factor for inhibitory activity, and the benzene ring was located in the hydrophobic region, which could be responsible for the good activity of compound 13. For inhibitors D1, D3 and D8, the 4,5,6-positions of the triazine ring formed five H-bonds with amino acids Gly313, Arg283 and Arg228 in the same way. The Cl of R2, ethyl of R4 and pyrazolyl of R5 interacted with the surrounding residues, making the benzene ring closer to the hydrophobic pocket, and the strong hydrophobic interaction favoured the binding stability of D1, D3 and D8. Therefore, compared with 13, the h-DAAO inhibitory activities of D1, D3 and D8 significantly improved. In short, the introduction of hydrophobic moieties into R2, R4 and R5 of 1,2,4-triazine derivatives is beneficial for inhibitory activity.
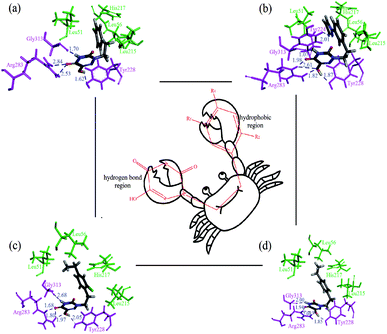 |
| | Fig. 15 Docking results of dynamics simulated ligand–3W4K complexes, and the ‘crab’ conformation of compounds at the binding site of the protein: (a) compound 13; (b) compound D1; (c) compound D3; (d) compound D8 (magenta: H-bond interaction; green: hydrophobic interaction). | |
The docking results were consistent with CoMFA and CoMSIA analyses. For example, the docking results showed that the ethyl groups of the benzene ring in D3 and D8 were helpful in forming stable hydrophobic interactions, which could enhance activity. As shown in Fig. 7a, a yellow contour map appeared on the R4 position of the benzene ring, suggesting that the introduction of hydrophobic groups at this position leads to higher activity. From Fig. 7b, the H-bond contour maps near the triazine ring indicate that strong H-bond interactions would improve the biological activity, and the docking results show that triazine rings formed strong H-bonds with the residues. It is also important to note that D1-3W4K not only formed H-bonds with the triazine region, but also formed H-bonds with the benzene region. Meanwhile, the docking scores of compounds D1 (8.4487), D3 (8.2162) and D8 (8.2472) are obviously higher compared to compound 13 (6.9737). This is inconsistent with the conclusion that D1, D3 and D8 have more negative binding free energies (stronger binding affinity) than molecule 13 in the MD simulations. These findings can also validate the accuracy of earlier SAR information obtained from 3D-QSAR. In conclusion, the newly designed compounds D1, D3 and D8 can be used as potential h-DAAO inhibitors.
Based on the results of these computational methods, we assumed the binding mode between inhibitor and protein to be a ‘crab’ conformation (Fig. 15). Through the comprehensive analysis of compounds 13, D1, D3 and D8, it was found that a C–C single bond formed the ‘mouth’, and the triazine ring and benzene ring served as the two big ‘pincers’, respectively. Firstly, the ‘mouth’ of the ‘crab’ was bound to the receptor via a C–C single bond and X substituent. Secondly, the left ‘pincer’ interacted with the protein mainly by hydrogen bonds and electrostatic interactions. Some hydrophobic residues (His217, Leu51, Leu56 and Leu215) interacted with the benzene ring, and we could consider the right ‘pincer’ as the important hydrophobic region. The above analysis shows that the results of CADD for h-DAAO inhibitors were valid. Therefore, we can draw the conclusion that the results of the 3D-QSAR, docking and dynamics simulations were reliable and verified each other.
ADME and bioavailability analysis
In the R&D process for new drugs, ADME and bioavailability analysis played significant roles in drug-likeness. The designed compounds D1, D3 and D8 with higher predicted activities and good stability were selected for ADME and bioavailability prediction. The ADME parameters calculated by the SwissADME web tool42 for the representative inhibitors are summarized in Table 7. The results showed that the logP values of the designed compounds D1, D3 and D8 were 1.03, 2.18 and 1.99, respectively, indicating that they have a reasonable absorbency. Meanwhile, D1 (logS = −2.80), D3 (logS = −3.57) and D8 (logS = −3.13) were considered to have good solubility in the body. The bioavailability radars of these inhibitors were analyzed intuitively in Fig. 10S.† The pink areas meant the optimum range of six properties, namely, lipophilicity, size, polarity, solubility, saturation and flexibility. It was found that compound 13 was outside the pink area, due to the inconformity of saturation. In contrast, the new inhibitors D1, D3 and D8 had superior bioavailability. In addition, the ADME descriptors were studied through a BOILED-Egg model (Fig. 11S†). Four compounds all exerted high HIA (in the white region) and the yolk (yellow region) represents the high probability of brain penetration. The new compounds D1, D3 and D8 were not brain penetrant, the same as compound 13. However, their logKp values were larger than that of compound 13, indicating that the skin permeability of the designed molecules is better. The “Yes” in Table 6 indicates that the compound has a greater probability of being the inhibitor of CYP1A2. Therefore, the newly designed compounds can be excreted via metabolic biotransformation by the inhibition of the cytochrome CYP1A2 enzyme. In short, bioavailability and pharmacokinetics predictions could improve the success rate of newly designed inhibitors. These are also beneficial for obtaining safer and more potent h-DAAO inhibitors for the treatment of schizophrenia.
Table 7 ADME and bioavailability predictions for newly designed compounds
| No. |
logP |
MW (g mol−1) |
TPSA (Å2) |
logS |
Fraction Csp3 |
Num. rotatable bonds |
HIA probability |
BBB |
CYP1A2 inhibition |
logKP (cm s−1) |
| 13 |
1.55 |
267.67 |
87.98 |
−3.00 |
0.18 |
3 |
High |
No |
Yes |
−6.57 |
| D1 |
1.03 |
299.28 |
87.98 |
−2.80 |
0.31 |
4 |
High |
No |
Yes |
−6.41 |
| D3 |
2.18 |
295.72 |
87.98 |
−3.57 |
0.31 |
4 |
High |
No |
Yes |
−6.17 |
| D8 |
1.99 |
279.27 |
87.98 |
−3.13 |
0.31 |
4 |
High |
No |
Yes |
−6.45 |
| Optimal range |
−0.7–5.0 |
150–500 |
20–130 |
≤6 |
≥0.25 |
≤9 |
— |
— |
— |
— |
Conclusion
The structure–activity relationship and binding interactions of 37 novel h-DAAO inhibitors were investigated theoretically and by means of CADD, including 3D-QSAR modelling, molecular docking and molecular dynamics simulation methods. A reliable 3D-QSAR model was established. The model exhibited good predictability and could be employed for predicting more new compounds. The contour maps of the 3D-QSAR revealed that hydrophobic, electrostatic and steric fields played an important role in designing the molecular structure, which was verified and supplemented by the MOLCAD molecular surface. The molecular docking and molecular dynamics studies with the ‘crab’ conformation revealed that the triazine ring and the substituents (type and position) of the benzene ring greatly affect the inhibitory activity. Meanwhile, the H-bonds that were formed by the 4,5,6-position of triazine with Gly313, Arg283 and Tyr228 were important for activity. The key hydrophobic residues, Leu51, His217, Gln53 and Leu215, were vital elements in the stability of the inhibitor at the binding site. Therefore, to obtain novel h-DAAO inhibitors with high efficiency, the triazine structure should be retained and the modified sections should mainly be in the R2 and R4 benzene ring positions with hydrophobic and bulky substituents, respectively. Moreover, to design compounds D3 and D8 with high inhibitory activity, chlorine or fluorine was introduced into the R2 position of the benzene ring, and ethyl was simultaneously set on the R4 position. D1 was obtained by the modification of pyrazolyl in the R5 position. The activities of all the newly designed compounds were superior to those of the published triazine compounds, 5y, 10b, 5z and 5m, which supports the reliability of the model and the accuracy of the design of the new compounds. These theories provide an understanding of the structural features of h-DAAO inhibitors and their binding interaction with protein, and therefore, the results could provide profound guidance and practical significance for the future design and experimental synthesis of novel h-DAAO inhibitors with high bioactivities. The results also supply great reference values for the emergence and development of new types of safe and effective antipsychotic drugs.
Conflicts of interest
The authors declare that there are no financial/commercial conflicts of interest.
Acknowledgements
We acknowledge the Shanghai Institute of Technology for support of this work.
References
- M. Möller, T. Swanepoel and B. H. Harvey, ACS Chem. Neurosci., 2015, 6, 987–1016 CrossRef PubMed.
- A. G. Awad and L. N. Voruganti, PharmacoEconomics, 2008, 26, 149–162 CrossRef PubMed.
- S. Grover, S. Chakrabarti, N. Hazari and A. Avasthi, Psychiatry Res., 2017, 249, 349–353 CrossRef PubMed.
- D. S. Berman and J. Davis-Berman, J. Exp. Educ., 2005, 28, 17–24 CrossRef.
- L. Orsolini, C. Tomasetti, A. Valchera, R. Vecchiotti, I. Matarazzo, F. Vellante, F. Iasevoli, E. F. Buonaguro, M. Fornaro, A. L. Fiengo, G. Martinotti, M. Mazza, G. Perna, A. Carano, A. De Bartolomeis, M. Di Giannantonio and D. De Berardis, Expert Opin. Drug Saf., 2016, 15, 1329–1347 CrossRef CAS PubMed.
- A. Abbott, Nature, 2010, 468, 158–159 CrossRef CAS PubMed.
- F. A. Mustafa, J. G. Burke, S. S. Abukmeil, J. J. Scanlon and M. Cox, Pharmacopsychiatry, 2015, 48, 11–14 CAS.
- I. J. Anwar, K. Miyata and A. Zsombok, J. Neurophysiol., 2016, 115, 1389–1398 CrossRef PubMed.
- M. Riedel, M. J. Schwarz, M. Strassnig, I. Spellmann, A. Müller-Arends, K. Weber, J. Zach, N. Müller and H. J. Möller, Eur. Arch. Psychiatry Clin. Neurosci., 2005, 255, 261–268 CrossRef PubMed.
- A. Rossi, F. Cañas, A. Fagiolini, I. Larmo, P. Levy, J. M. Montes, G. Papageorgiou, R. Sturlason, M. Zink and C. U. Correll, Postgrad. Med., 2011, 123, 135–159 CrossRef PubMed.
- A. Salmoiraghi and M. Odiyoor, J. Psychopharmacol., 2006, 20, 592–593 CrossRef PubMed.
- A. A. Shirzadi and S. N. Ghaemi, Harv. Rev. Psychiatry, 2006, 14, 152–164 CrossRef PubMed.
- E. A. Nunes, E. M. Mackenzie, D. Rossolatos, J. Perez-Parada, G. B. Baker and S. M. Dursun, Expert Rev. Neurother., 2012, 12, 801–812 CrossRef CAS PubMed.
- K. Wichapong, A. Nueangaudom, S. Pianwanit, F. Tanaka and S. Kokpol, Mol. Simul., 2014, 40, 1167–1189 CrossRef CAS.
- R. E. Williams and E. A. Lock, Toxicology, 2005, 207, 35–48 CrossRef CAS PubMed.
- K. Hashimoto, Y. Fujita, M. Horio, S. Kunitachi, M. Iyo, D. Ferraris and T. Tsukamoto, Biol. Psychiatry, 2009, 65, 1103–1106 CrossRef CAS PubMed.
- H. Mao, Y. Fujita, T. Ishima, M. Iyo, D. Ferraris, T. Tsukamoto and K. Hashimoto, Open Clin. Chem. J., 2009, 2, 16–21 CrossRef.
- T. Adage, A. C. Trillat, A. Quattropani, D. Perrin, L. Cavarec, J. Shaw, O. Guerassimenko, C. Giachetti, B. Gréco, I. Chumakov, S. Halazy, A. Roach and P. Zaratin, Eur. Neuropsychopharmacol., 2008, 18, 200–214 CrossRef CAS PubMed.
- S. M. Smith, J. M. Uslaner, L. Yao, C. M. Mullins, N. O. Surles, S. L. Huszar, C. H. McNaughton, D. M. Pascarella, M. Kandebo, R. M. Hinchliffe, T. Sparey, N. J. Brandon, B. Jones, S. Venkatraman, M. B. Young, N. Sachs, M. A. Jacobson and P. H. Hutson, J. Pharmacol. Exp. Ther., 2009, 328, 921–930 CrossRef CAS PubMed.
- N. Hin, B. Duvall, J. F. Berry, D. V. Ferraris, R. Rais, J. Alt, C. Rojas, B. S. Slusher and T. Tsukamoto, Bioorg. Med. Chem. Lett., 2016, 26, 2088–2091 CrossRef CAS PubMed.
- A. J. Duplantier, S. L. Becker, M. J. Bohanon, K. A. Borzilleri, B. A. Chrunyk, J. T. Downs, L. Y. Hu, A. El-Kattan, L. C. James, S. Liu, J. Lu, N. Maklad, M. N. Mansour, S. Mente, M. A. Piotrowski, S. M. Sakya, S. Sheehan, S. J. Steyn, C. A. Strick, V. A. Williams and L. Zhang, J. Med. Chem., 2009, 52, 3576–3585 CrossRef CAS PubMed.
- S. C. Zimmermann, R. Rais, J. Alt, C. Burzynski, B. S. Slusher and T. Tsukamoto, ACS Med. Chem. Lett., 2014, 5, 1251–1253 CrossRef CAS PubMed.
- J. F. Berry, D. V. Ferraris, B. Duvall, N. Hin, R. Rais, J. Alt, A. G. Thomas, C. Rojas, K. Hashimoto, B. S. Slusher and T. Tsukamoto, ACS Med. Chem. Lett., 2012, 3, 839–843 CrossRef CAS PubMed.
- T. Hondo, M. Warizaya, T. Niimi, I. Namatame, T. Yamaguchi, K. Nakanishi, T. Hamajima, K. Harada, H. Sakashita, Y. Matsumoto, M. Orita and M. Takeuchi, J. Med. Chem., 2013, 56, 3582–3592 CrossRef CAS PubMed.
- N. Hin, B. Duvall, D. Ferraris, J. Alt, A. G. Thomas, R. Rais, C. Rojas, Y. Wu, K. M. Wozniak, B. S. Slusher and T. Tsukamoto, J. Med. Chem., 2015, 58, 7258–7272 CrossRef CAS PubMed.
- S. P. Mandal, M. A. Garg, S. S. Sahetya, S. R. Nagendra, H. S. Sripad, M. M. Manjunath, S. M. Soni, R. N. Baig, S. V. Kumar and B. R. Prashantha Kumar, RSC Adv., 2016, 6, 58641–58653 RSC.
- Z. Qin, M. Wang and A. Yan, Bioorg. Med. Chem. Lett., 2017, 27, 2931–2938 CrossRef CAS PubMed.
- E. Cichero, C. Brullo, O. Bruno and P. Fossa, RSC Adv., 2016, 6, 61088–61108 RSC.
- S. Patel, B. Patel and H. Bhatt, J. Taiwan Inst. Chem. Eng., 2016, 59, 61–68 CrossRef CAS.
- J. Gasteiger and M. Marsili, Tetrahedron, 1980, 36, 3219–3228 CrossRef CAS.
- M. Clark, R. D. Cramer and N. V. Oppenbosch, J. Comput. Chem., 1989, 10, 982–1012 CrossRef CAS.
- R. D. Cramer, D. E. Patterson and J. D. Bunce, J. Am. Chem. Soc., 1988, 110, 5959–5967 CrossRef CAS PubMed.
- G. Klebe, U. Abraham and T. Mietzner, J. Med. Chem., 1994, 37, 4130–4146 CrossRef CAS PubMed.
- S. V. Jain, M. Ghate, K. S. Bhadoriya, S. B. Bari, A. Chaudhari and J. S. Borse, Org. Med. Chem. Lett., 2012, 2, 1–13 CrossRef PubMed.
- M. Böhm, J. Stürzebecher and G. Klebe, J. Med. Chem., 1999, 42, 458–477 CrossRef PubMed.
- U. Norinder, Perspect. Drug Discovery Des., 1998, 12, 25–39 CrossRef.
- E. Alciaturi, C. E. Scobar, E. Marcos, D. L. Cruz and C. Rincón, Rev. Tec. Fac. Ing., Univ. Zulia, 2003, 26, 197–204 Search PubMed.
- P. P. Roy, J. T. Leonard and K. Roy, Chemom. Intell. Lab. Syst., 2008, 90, 31–42 CrossRef CAS.
- T. Chai and R. R. Draxler, Geosci. Model Dev., 2014, 7, 1247–1250 CrossRef.
- K. Roy, P. Ambure and R. B. Aher, Chemom. Intell. Lab. Syst., 2017, 162, 44–54 CrossRef CAS.
- A. R. Leach, B. K. Shoichet and C. E. Peishoff, J. Med. Chem., 2006, 49, 5851–5855 CrossRef CAS PubMed.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717 CrossRef PubMed.
- M. Remko, Comput. Theor. Chem., 2009, 897, 73–82 CAS.
- R. B. Silverman, The Organic Chemistry of Drug Design and Drug Action, Elsevier, Amsterdam, 2nd edn, 2004 Search PubMed.
- T. Tsukamoto, B. S. Slusher, D. V. Ferraris, C. Rojas, N. Hin B. Duvall, US Pat. 9505753B2, 2016.
- W. Shen and Y. H. Lu, Bangladesh J. Pharmacol., 2013, 8, 156–170 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra00094h |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*