
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Substituted L-tryptophan-L-phenyllactic acid conjugates produced by an endophytic fungus Aspergillus aculeatus using an OSMAC approach†
Hao Wanga,
Peter M. Eze b,
Simon-Patrick Höfertc,
Christoph Janiakc,
Rudolf Hartmannd,
Festus B. C. Okoyee,
Charles O. Esimoneb,
Raha S. Orfalif,
Haofu Daig,
Zhen Liu*a and
Peter Proksch*a
b,
Simon-Patrick Höfertc,
Christoph Janiakc,
Rudolf Hartmannd,
Festus B. C. Okoyee,
Charles O. Esimoneb,
Raha S. Orfalif,
Haofu Daig,
Zhen Liu*a and
Peter Proksch*a
aInstitute of Pharmaceutical Biology and Biotechnology, Heinrich-Heine-University Düsseldorf, Universitätsstrasse 1, 40225 Düsseldorf, Germany. E-mail: zhenfeizi0@sina.com; proksch@uni-duesseldorf.de
bDepartment of Pharmaceutical Microbiology and Biotechnology, Faculty of Pharmaceutical Sciences, Nnamdi Azikiwe University, Awka, Nigeria
cInstitute of Inorganic and Structural Chemistry, Heinrich-Heine-University Düsseldorf, Universitätsstrasse 1, 40225 Düsseldorf, Germany
dInstitute of Complex Systems: Structural Biochemistry, Forschungszentrum Juelich, Wilhelm-Johnen-Straße, 52428 Juelich, Germany
eDepartment of Pharmaceutical and Medicinal Chemistry, Faculty of Pharmaceutical Sciences, Nnamdi Azikiwe University, Awka, Nigeria
fDepartment of Pharmacognosy, Faculty of Pharmacy, King Saud University, Riyadh, Saudi Arabia
gKey Laboratory of Biology and Genetic Resources of Tropical Crops, Ministry of Agriculture, Institute of Tropical Bioscience and Biotechnology, Chinese Academy of Tropical Agricultural Sciences, Haikou 571101, China
First published on 19th February 2018
Abstract
The endophytic fungus Aspergillus aculeatus isolated from leaves of the papaya plant Carica papaya was fermented on solid rice medium, yielding a new L-tryptophan-L-phenyllactic acid conjugate (1) and thirteen known compounds (11, 14–25). In addition, an OSMAC approach was employed by adding eight different sodium or ammonium salts to the rice medium. Addition of 3.5% NaNO3 caused a significant change of the metabolite pattern of the fungus as indicated by HPLC analysis. Subsequent isolation yielded several new substituted L-tryptophan-L-phenyllactic acid conjugates (1–10) in addition to three known compounds (11–13), among which compounds 2–10, 12–13 were not detected in the rice control culture. All structures were unambiguously elucidated by one and two dimensional NMR spectroscopy and by mass spectrometry. The absolute configuration of the new compounds was determined by Marfey's reaction and X-ray single crystal diffraction. Compounds 19–22 showed cytotoxicity against the L5178Y mouse lymphoma cell line with IC50 values of 3.4, 1.4, 7.3 and 23.7 μM, respectively.
Introduction
Endophytic fungi thrive widely in different healthy tissues of living plants, and have significant influence on the growth of their hosts.1,2 Endophytes also comprise a large reservoir of structurally diverse secondary metabolites including alkaloids, steroids, terpenoids, xanthones, peptides and quinones, which exhibit a variety of biological activities including anticancer, antibacterial, antifungal, anti-inflammatory and antidepressant activity.2,3 For example, 14-membered macrolides isolated from the endophytic fungus Pestalotiopsis microspora showed significant cytotoxicity against the murine lymphoma cell line L5178Y while fusaric acid derivatives produced by the fungal endophyte Fusarium oxysporum showed significant phytotoxicity to leaves of barley.4,5 However, the high rediscovery rate of known compounds from fungi is a severe obstacle for the search for new drug leads.6 One of the approaches to increase the diversity of metabolites from fungi involves the application of the OSMAC (One Strain Many Compounds) method which is based on systematic variations of the cultivation parameters (media type and composition, pH value, temperature etc.).7–9 For instance, the fermentation of the fungus Gymnascella dankaliensis on solid rice medium following addition of 3.5% NaBr led to the isolation of ten brominated tyrosine-derived alkaloids, which showed cytotoxicity against the L5178Y mouse lymphoma cell line.10,11 In addition, cultivation of Fusarium tricinctum on fruit and vegetable juice-supplemented solid rice media that led to the production of the new fusarielins K and L.12The fungus Aspergillus aculeatus has been reported to produce several bioactive secondary metabolites, such as secalonic acids D and F, aculeatusquinone B and D, aculeacins A–G and aspergillusol A.13–17 In this study, A. aculeatus was isolated from leaves of Carica papaya collected in Awka in Nigeria. The fungus was fermented on solid rice medium, yielding a new L-tryptophan-L-phenyllactic acid conjugate (1) together with thirteen known compounds including N-[(2S)-2-hydroxy-1-oxo-3-phenylpropyl]-L-tryptophan methyl ester (11),18 okaramine A (14),19 oxaline (15),20 emindole SB (16),21 16-keto-aspergillimide (17),22 JBIR 75 (18),23 secalonic acids D and F (19 and 20),13 asperdichrome (21),24 RF 3192C (22),25 pannorin (23),26 altechromone A (24),27 and variecolactone (25).28 Due to the pronounced chemical diversity of this fungus we decided to subject A. aculeatus to an OSMAC approach by adding either 3.5% NaCl, 3.5% NaBr, 3.5% NaI, 1% NaF, 3.5% NaNO3, 3.5% NH4Cl, 3.5% (NH4)2SO4 or 3.5% NH4OAc to the rice medium. The selection of these salts for the OSMAC study was based on previous experiments with other fungi that had indicated the usefulness of these chemical stimuli for the accumulation of cryptic metabolites.10,11,29 The fungus did not grow on rice medium containing 1% NaF or 3.5% NH4OAc. Addition of 3.5% NaNO3, however, caused a significant change of the metabolite pattern as indicated by HPLC analysis. Addition of the remaining salts had no influence. Subsequent workup of the extract resulting from addition of 3.5% NaNO3 yielded several new substituted L-tryptophan-L-phenyllactic acid conjugates (1–10) in addition to three known analogues N-[(2S)-2-hydroxy-1-oxo-3-phenylpropyl]-L-tryptophan methyl ester (11), N-[(2S)-2-hydroxy-1-oxo-3-phenylpropyl]-L-tryptophan (12) and acu-dioxomorpholine (13).18 Compounds 2–10, 12–13 were not detected in the rice control culture. On the other hand, from all compounds isolated from the rice control culture, only oxaline (15), JBIR 75 (18), secalonic acid F (20) and RF 3192C (22) could be detected in the fungal culture after addition of 3.5% NaNO3. In this study we report the structure elucidation and the biological activities of the isolated compounds.
Results and discussion
Compound 1 had the molecular formula C22H24O4N2 as established by HRESIMS exhibiting 12 degrees of unsaturation. The 13C NMR spectrum of 1 (Table 1) included 22 signals, corresponding to two methyls, two methylenes, two alphatic methines and ten aromatic methines as well as six quaternary carbons. The 1H NMR spectrum of 1 (Table 2) showed the presence of a monosubstituted benzene ring and an indole moiety as indicated by signals between δH 6 and 8, two –CH2–CH– units and two methyl groups (δH 3.70 and 3.62). These data were similar to those of the co-isolated known compound N-[(2S)-2-hydroxy-1-oxo-3-phenylpropyl]-L-tryptophan methyl ester (11).18 However, the detection of an additional methyl substituent at δC 32.7 and δH 3.70 and the HMBC correlation from the protons of this methyl group to C-10 (δC 138.5) and C-12 (δC 129.0) indicated its attachment to N-11. Detailed analysis of the 2D NMR spectra of 1 (Fig. 2) revealed that its remaining substructure was identical to that of 11. The absolute configuration of 1 was determined by X-ray single crystal analysis as 2S, 2′S (Fig. 3), being identical to that of 11. Thus, the structure of 1, for which the trial name aculeatine A is proposed, was elucidated as shown in Fig. 1.| No. | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| 1 | 173.4, C | 174.8, C | 173.9, C | 173.8, C | 173.5, C |
| 2 | 53.7, CH | 53.7, CH | 55.3, CH | 54.3, CH | 53.6, CH |
| 3 | 28.4, CH2 | 28.4, CH2 | 29.2, CH2 | 28.0, CH2 | 28.5, CH2 |
| 4 | 109.2, C | 109.5, C | 107.5, C | 106.0, C | 109.4, C |
| 5 | 129.1, C | 129.4, C | 130.3, C | 129.1, C | 129.4, C |
| 6 | 119.6, CH | 119.8, CH | 119.2, CH | 119.0, CH | 119.6, CH |
| 7 | 119.9, CH | 119.9, CH | 120.1, CH | 120.0, CH | 120.0, CH |
| 8 | 122.6, CH | 122.5, CH | 122.7, CH | 122.0, CH | 122.5, CH |
| 9 | 110.2, CH | 110.1, CH | 109.7, CH | 109.7, CH | 110.6, CH |
| 10 | 138.5, C | 138.5, C | 139.0, C | 138.4, C | 137.8, C |
| 12 | 129.0, CH | 129.0, CH | 142.2, C | 138.8, C | 127.6, CH |
| 13 | 32.7, CH3 | 32.7, CH3 | 33.8, CH3 | 30.0, CH3 | |
| 14 | 41.9, C | 24.8, CH2 | 44.8, CH2 | ||
| 15 | 149.1, CH | 122.4, CH | 121.5, CH | ||
| 16 | 112.8, CH2 | 133.9, C | 137.1, C | ||
| 17 | 30.0, CH3 | 25.8, CH3 | 25.8, CH3 | ||
| 18 | 29.9, CH3 | 18.1, CH3 | 18.1, CH3 | ||
| 1′ | 175.8, C | 175.8, C | 176.0, C | 176.1, C | 175.9, C |
| 2′ | 73.4, CH | 73.6, CH | 73.7, CH | 73.7, CH | 73.5, CH |
| 3′ | 41.4, CH2 | 41.5, CH2 | 41.2, CH2 | 41.4, CH2 | 41.6, CH2 |
| 4′ | 138.8, C | 139.0, C | 139.1, C | 138.9, C | 138.9, C |
| 5′, 9′ | 130.9, CH | 130.9, CH | 130.6, CH | 130.6, CH | 130.9, CH |
| 6′, 8′ | 129.1, CH | 129.1, CH | 129.0, CH | 129.0, CH | 129.1, CH |
| 7′ | 127.5, CH | 127.5, CH | 127.3, CH | 127.4, CH | 127.5, CH |
| OMe | 52.8, CH3 | 52.6, CH3 | 52.8, CH3 | 52.8, CH3 |
| No. | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| 2 | 4.74, dd (6.2, 5.4) | 4.71, dd (6.2, 5.2) | 4.69, dd (8.5, 7.3) | 4.65, dd (7.2, 6.6) | 4.74, dd (6.1, 5.4) |
| 3 | 3.21, dd (14.6, 6.2), 3.07, dd (14.6, 5.4) | 3.24, dd (14.7, 6.2), 3.14, dd (14.7, 5.2) | 3.45, dd (14.9, 7.3), 3.26, dd (14.9, 8.5) | 3.15, dd (14.7, 6.6), 3.10, dd (14.7, 7.2) | 3.22, dd (14.7, 6.1), 3.05, dd (14.7, 5.4) |
| 6 | 7.41, d (7.9) | 7.49, d (7.9) | 7.50, d (7.8) | 7.40, d (7.8) | 7.40, d (7.9) |
| 7 | 7.03, dd (7.9, 7.2) | 7.02, dd (7.9, 7.2) | 7.07, dd (7.8, 7.2) | 7.02, dd (7.8, 7.1) | 7.02, dd (7.9, 7.1) |
| 8 | 7.15, dd (8.2, 7.2) | 7.14, dd (8.2, 7.2) | 7.14, dd (8.1, 7.2) | 7.10, dd (8.2, 7.1) | 7.13, dd (8.2, 7.1) |
| 9 | 7.29, d (8.2) | 7.29, d (8.2) | 7.25, d (8.1) | 7.26, d (8.2) | 7.29, d (8.2) |
| 12 | 6.61, s | 6.66, s | 6.71, s | ||
| 13 | 3.70, s | 3.70, s | 3.72, s | 3.60, s | |
| 14 | 3.48, dd (16.6, 6.6), 3.44, dd (16.6, 6.6) | 4.66, d (6.9) | |||
| 15 | 6.23, dd (17.5, 10.6) | 5.11, br t (6.6) | 5.32, br t (6.9) | ||
| 16 | 5.11, d (10.6), 4.98, d (17.5) | ||||
| 17 | 1.66, s | 1.74, s | 1.74, s | ||
| 18 | 1.66, s | 1.82, s | 1.83, s | ||
| 2′ | 4.24, dd (7.1, 4.0) | 4.22, dd (7.4, 3.8) | 4.10, dd (8.1, 3.7) | 4.17, dd (7.8, 3.8) | 4.22, dd (7.3, 3.9) |
| 3′ | 2.97, dd (13.9, 4.0), 2.78, dd (13.9, 7.1) | 2.96, dd (13.9, 3.8), 2.73, dd (13.9, 7.4) | 2.64, dd (13.9, 3.7), 2.24, dd (13.9, 8.1) | 2.85, dd (13.9, 3.8), 2.56, dd (13.9, 7.8) | 2.96, dd (13.9, 3.9), 2.76, dd (13.9, 7.3) |
| 5′, 9′ | 7.20, d (7.0) | 7.19, t (7.1) | 7.05, d (7.1) | 7.11, d (7.2) | 7.20, d (7.2) |
| 6′, 8′ | 7.24, t (7.0) | 7.22, d (7.1) | 7.15, t (7.1) | 7.16, t (7.2) | 7.23, t (7.2) |
| 7′ | 7.17, t (7.0) | 7.17, t (7.1) | 7.12, t (7.1) | 7.12, t (7.2) | 7.17, t (7.2) |
| OMe | 3.62, s | 3.49, s | 3.60, s | 3.63, s |
 | ||
| Fig. 1 Structures of compounds 1–25 isolated from A. aculeatus. | ||
Compound 2 possessed the molecular formula C21H22O4N2 as determined by HRESIMS, which indicated the loss of a methyl group compared to 1. This was confirmed by the absence of the 1H and 13C signals of the methoxy group attached to C-1 in the NMR spectra of 2 (Tables 1 and 2). The structure of 2 was confirmed by detailed analysis of the 2D NMR spectra. Its absolute configuration was determined as 2S, 2′S by X-ray single crystal analysis (Fig. 3).
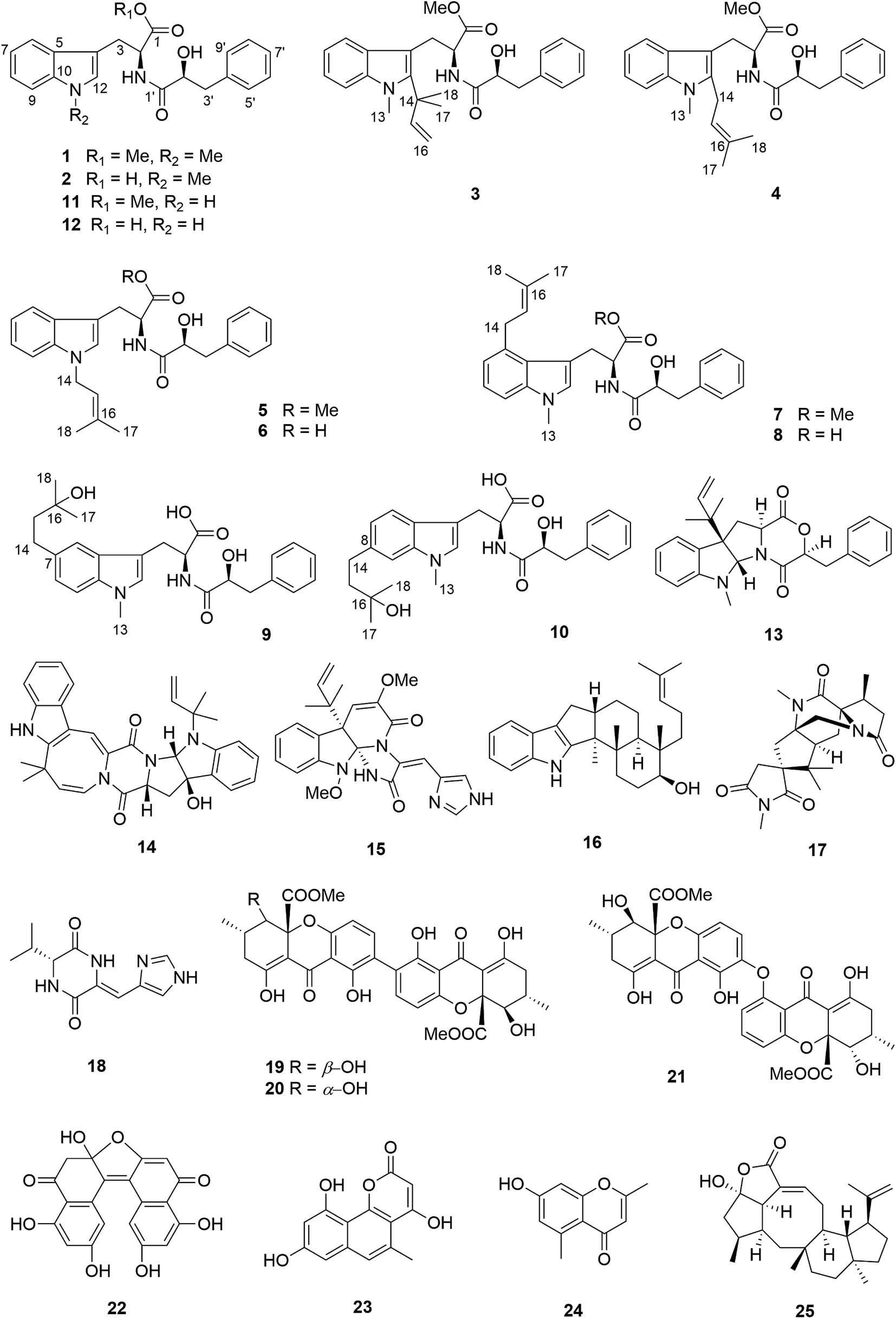
The molecular formula of compound 3 was determined as C27H32O4N2 by HRESIMS. The 13C NMR spectrum of 3 exhibited five additional carbons compared to 1. The 1H NMR spectrum of 3 was similar to that of compound 1 except for the observation of two singlet methyls at δH 1.66 (Me-17 and 18) and signals of a terminal double bond at δH 6.23 (H-15), 5.11 and 4.98 (H2-16) as well as the absence of the singlet aromatic proton at C-12. The COSY correlations between H-15 and H2-16 together with the HMBC correlations from Me-17 and Me-18 to C-12 (δC 142.2), C-14 (δC 41.9) and C-15 (δC 149.1) and from Me-13 (δH 3.72) to C-12 and C-10 (δC 139.0) indicated the attachment of a 1,1-dimethylprop-2-en-1-yl side chain at C-12. Detailed analysis of the 2D NMR spectra of 3 (Fig. 2) revealed that its remaining substructure was identical to that of 1.
 | ||
| Fig. 2 COSY and key HMBC correlations of compounds 1, 3 and 4. | ||
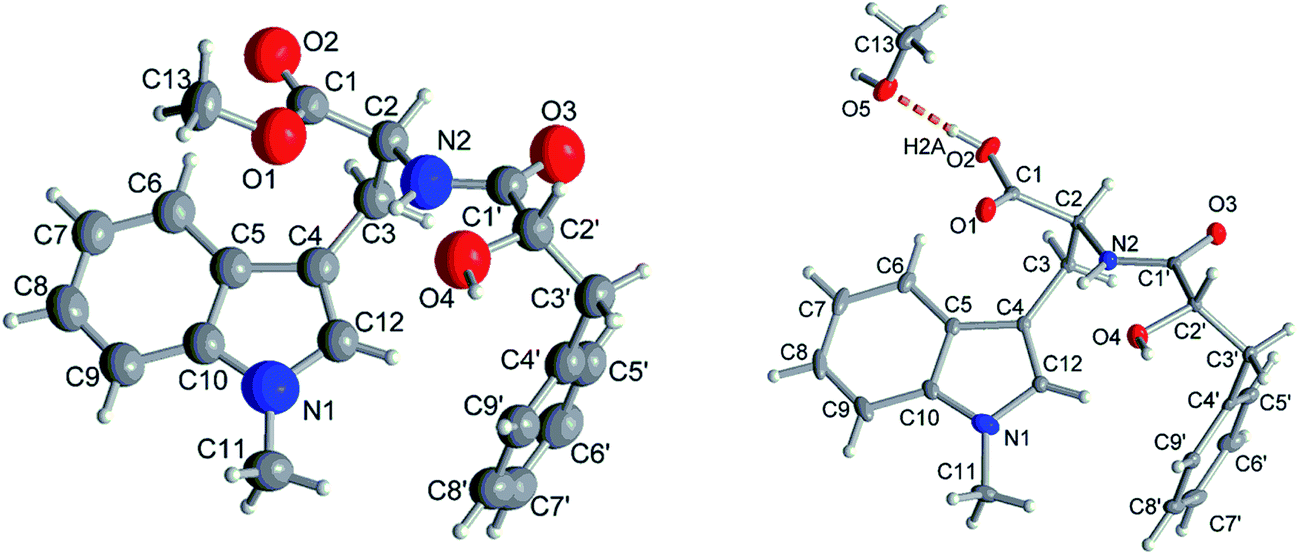 | ||
| Fig. 3 Molecular structures of 1 and 2 from single-crystal X-ray diffractometry (50% thermal ellipsoids, H-atoms with arbitrary radii). The structure drawing of 2 shows also the hydrogen bond (orange dashed line) to the methanol crystal solvent molecule. | ||
Compound 4 had the same molecular formula as 3. The 1H and 13C NMR data were also similar to those of 3. The appearance of a broad triplet proton (δH 5.11, H-15) and one methylene group (δH 3.48 and 3.44, H2-14) together with two olefinic methyl groups (δH 1.74 and 1.82, Me-17 and 18) in the 1H NMR spectrum of 4 suggested the existence of a 3-methyl-but-2-en-1-yl group at C-12, which was confirmed by the COSY correlations between H-15 and H2-14 together with the HMBC correlations from Me-17 and 18 to C-15 (δC 122.4) and C-16 (δC 133.9), and from H2-14 to C-4 (δC 106.0) and C-12 (δC 138.8) (Fig. 2).
Compound 5 was isolated as white powder. Its molecular formula was established as C26H30O4N2 by HRESIMS. The 1H and 13C NMR spectra of 5 were similar to those of 4 except for the appearance of a singlet methine at δH 6.70 (H-12) and the absence of the methyl group attached to N-11. The COSY correlations between H-15 (δH 5.32) and H2-14 (δH 4.66) along with the HMBC correlations from H-14 to C-10 (δC 137.8) and C-12 (δC 127.6), and from both Me-17 (δH 1.74) and Me-18 (δH 1.83) to C-15 (δC 121.5) and C-16 (δC 137.1) indicated the presence of a 3-methyl-but-2-en-1-yl residue and its attachment to N-11 of compound 5 (Fig. 4).
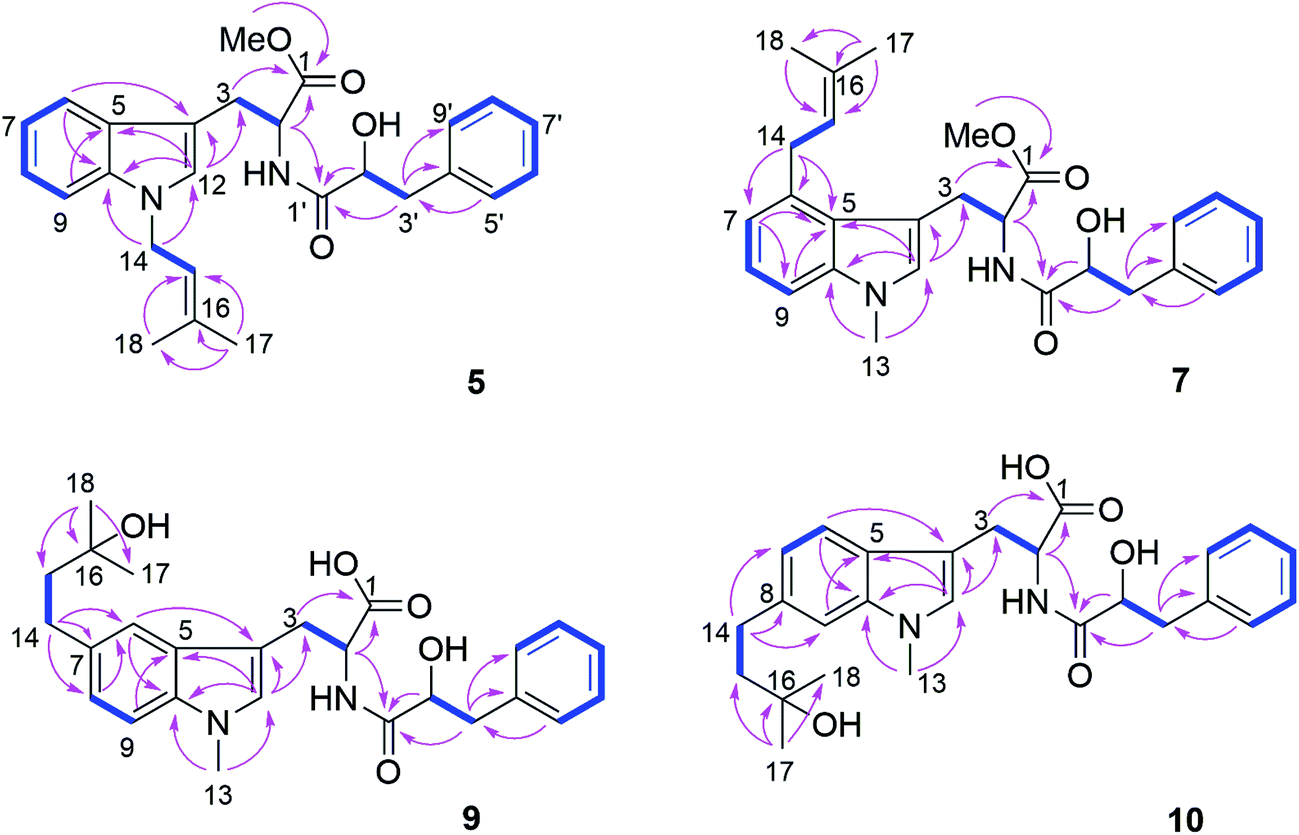 | ||
| Fig. 4 COSY and key HMBC correlations of compounds 5, 7, 9 and 10. | ||
The molecular formula of 6 was determined as C25H28O4N2, indicating the lack of a methyl group compared to 5, which was confirmed by the absence of signals of the methoxy group at C-1 compared to 5 (Tables 3 and 4). The remaining substructure of 6 was unambiguously elucidated to be identical to that of 5 by detailed analysis of 2D NMR spectra.
| No. | 6a | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|
| a Measured at 175 MHz. | |||||
| 1 | 176.4, C | 173.7, C | 175.0, C | 175.8, C | 175.6, C |
| 2 | 54.3, CH | 54.7, CH | 54.4, CH | 54.3, CH | 54.1, CH |
| 3 | 28.5, CH2 | 30.3, CH2 | 30.4, CH2 | 28.3, CH2 | 28.4, CH2 |
| 4 | 110.0, C | 110.1, C | 110.4, C | 109.4, C | 109.7, C |
| 5 | 129.6, C | 126.8, C | 127.0, C | 129.8, C | 127.7, C |
| 6 | 119.8, CH | 135.4, C | 135.5, C | 118.9, CH | 119.8, CH |
| 7 | 119.5, CH | 120.7, CH | 120.7, CH | 134.4, C | 121.0, CH |
| 8 | 122.0, CH | 122.7, CH | 122.6, CH | 123.5, CH | 137.7, C |
| 9 | 110.1, CH | 108.5, CH | 108.4, CH | 110.0, CH | 109.4, CH |
| 10 | 137.4, C | 139.3, C | 139.3, C | 137.1, C | 138.8, C |
| 12 | 127.3, CH | 129.4, CH | 129.3, CH | 129.1, CH | 128.5, CH |
| 13 | 32.9, CH3 | 32.9, CH3 | 32.7, CH3 | 32.7, CH3 | |
| 14 | 44.5, CH2 | 33.3, CH2 | 33.4, CH2 | 32.0, CH2 | 32.2, CH2 |
| 15 | 121.4, CH | 125.5, CH | 125.5, CH | 47.8, CH2 | 47.8, CH2 |
| 16 | 136.5, C | 132.9, C | 132.9, C | 71.4, C | 71.4, C |
| 17 | 25.5, CH3 | 25.9, CH3 | 25.9, CH3 | 29.3, CH3 | 29.3, CH3 |
| 18 | 17.8, CH3 | 18.2, CH3 | 18.3, CH3 | 29.2, CH3 | 29.2, CH3 |
| 1′ | 175.4, C | 176.1, C | 176.1, C | 175.9, C | 175.8, C |
| 2′ | 73.6, CH | 73.8, CH | 73.8, CH | 73.8, CH | 73.6, CH |
| 3′ | 41.3, CH2 | 41.7, CH2 | 41.7, CH2 | 41.6, CH2 | 41.4, CH2 |
| 4′ | 138.9, C | 138.9, C | 139.0, C | 139.1, C | 139.1, C |
| 5′, 9′ | 130.5, CH | 130.6, CH | 130.6, CH | 130.8, CH | 130.8, CH |
| 6′, 8′ | 128.8, CH | 129.1, CH | 129.1, CH | 129.1, CH | 129.1, CH |
| 7′ | 127.1, CH | 127.4, CH | 127.4, CH | 127.4, CH | 127.4, CH |
| OCH3 | 52.7, CH3 | ||||
| No. | 6a | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|
| a Measured at 700 MHz. | |||||
| 2 | 4.66, m | 4.71, dd (8.7, 5.6) | 4.70, dd (8.9, 4.8) | 4.67, m | 4.67, m |
| 3 | 3.23, dd (14.5, 6.1), 3.17, dd (14.5, 4.7) | 3.38, dd (15.0, 5.6), 3.18, dd (15.0, 8.7) | 3.45, dd (15.1, 4.8), 3.20, dd (15.1, 8.9) | 3.21, dd (14.8, 5.8), 3.16, dd (14.8, 4.7) | 3.20, dd (14.8, 5.9), 3.13, dd (14.8, 4.7) |
| 6 | 7.53, d (7.8) | 7.36, s | 7.40, d (8.0) | ||
| 7 | 7.01, dd (7.8, 7.1) | 6.82, d (7.2) | 6.82, d (7.0) | 6.91, d (8.0) | |
| 8 | 7.11, dd (8.3, 7.1) | 7.06, dd (8.6, 7.2) | 7.06, dd (8.3, 7.0) | 7.02, d (8.3) | |
| 9 | 7.27, d (8.3) | 7.16, d (8.6) | 7.15, d (8.3) | 7.20, d (8.3) | 7.11, s |
| 12 | 6.80, s | 6.82, s | 6.84, s | 6.68, s | 6.63, s |
| 13 | 3.70, s | 3.69, s | 3.68, s | 3.68, s | |
| 14 | 4.65, m | 3.73, m | 3.77, dd (16.3, 6.8), 3.73, dd (16.3, 6.8) | 2.77, m | 2.78, m |
| 15 | 5.32, br t (6.8) | 5.31, br t (6.6) | 5.32, br t (6.8) | 1.80, m | 1.79, m |
| 17 | 1.72, s | 1.76, s | 1.76, s | 1.24, s | 1.26, s |
| 18 | 1.83, s | 1.77, s | 1.77, s | 1.25, s | 1.26, s |
| 2′ | 4.16, dd (8.0, 3.5) | 4.19, dd (7.8, 3.9) | 4.18, dd (8.0, 3.8) | 4.19, dd (7.8, 3.6) | 4.19, dd (7.6, 3.7) |
| 3′ | 2.93, dd (13.9, 3.5), 2.63, dd (13.9, 8.0) | 2.88, dd (13.9, 3.9), 2.62, dd (13.9, 7.8) | 2.87, dd (13.9, 3.8), 2.58, dd (13.9, 8.0) | 2.95, dd (13.9, 3.6), 2.65, dd (13.9, 7.8) | 2.93, dd (13.9, 3.7), 2.66, dd (13.9, 7.6) |
| 5′, 9′ | 7.16, d (7.2) | 7.11, d (6.9) | 7.11, d (6.9) | 7.17, d (7.2) | 7.17, d (7.2) |
| 6′, 8′ | 7.20, t (7.2) | 7.14, t (6.9) | 7.12, t (6.9) | 7.20, t (7.2) | 7.22, t (7.2) |
| 7′ | 7.15, t (7.2) | 7.10, t (6.9) | 7.10, t (6.9) | 7.15, t (7.2) | 7.16, t (7.2) |
| OCH3 | 3.66, s | ||||
Compound 7 possessed the molecular formula C27H32O4N2 as deduced from the HRESIMS data. The 1H and 13C NMR spectra of 7 were similar to those of 4, revealing the presence of one monosubstituted benzene ring, an indole moiety and a 3-methyl-but-2-en-1-yl side chain. However, the appearance of an aromatic proton signal at δH 6.82 (H-7), which was split to a doublet, suggested that the 3-methyl-but-2-en1-yl group was substituted on the benzene ring of the tryptophan moiety. The HMBC correlation from H2-14 (δH 3.73) to C-5 (δC 126.8), C-6 (δC 135.4) and C-7 (δC 120.7), and from H-7 to C-5, C-9 (δC 108.5) and C-14 (δC 33.3) indicated the 3-methyl-but-2-enyl group to be located at C-6. The structure of 7 was determined by analysis of the 2D NMR spectra as shown in Fig. 4.
Compound 8 possessed the molecular formula C26H30O4N2 as established by HRESIMS, thus lacking a methyl group compared to 7. The 1H and 13C NMR spectra were almost identical to those of 7 except for the absence of signals of the methoxy group at C-1, which was further confirmed by detailed analysis of the 2D NMR data of 8.
Compound 9 had the molecular formula C26H32O5N2 as determined by HRESIMS, corresponding to 12 degrees of unsaturation. The 1H NMR spectra of 9 (Table 4) revealed the presence of a 1,2,4-trisubstituted benzene ring and a monosubstituted benzene ring, suggesting a substituent at C-7 or C-8 of the indole moiety. A 3-hydroxy-3-methylbutyl moiety was established based on the correlation between H2-14 (δH 2.77) and H2-15 (δH 1.80) together with the HMBC correlations from Me-17 (δH 1.24) and Me-18 (δH 1.25) to C-16 (δC 71.4) and C-15 (δC 47.8). In addition, the HMBC correlation from H2-14 to C-6 (δC 118.9), C-7 (δC 134.4) and C-8 (δC 123.5), from H-6 (δH 7.36) to C-4 (δC 109.4), C-8, C-10 (δC 137.1) and C-14 (δC 32.0), and from H-8 (δH 7.02) to C-6, C-10 and C-14 indicated the 3-hydroxy-3-methylbutyl moiety to be attached at C-7 (Fig. 4).
Compound 10 shared the same molecular formula as 9. The 1H NMR data of 10 resembled those of compound 9 except for the chemical shifts of the aromatic protons of the indole moiety. The HMBC correlation from H2-14 (δH 2.78) to C-7 (δC 121.0), C-8 (δC 137.7) and C-9 (δC 109.4), from H-7 (δH 6.91) to C-5 (δC 127.7), C-9 and C-14 (δC 32.3), and from H-9 (δH 7.11) to C-5, C-7 and C-14 confirmed that the 3-hydroxy-3-methylbutyl moiety was located at the C-8 position. The remaining substructure of 10 was the same as that of 9 as confirmed by detailed analysis of 2D NMR spectra of 10 (Fig. 4).
Compounds 3–10 share the same core structure with compounds 1 and 2, for which the absolute configuration had been assigned through X-ray analysis. Hence it is concluded on biogenetic terms that the absolute configuration of the former compounds is also 2S, 2′S. Compounds 11 and 12 were determined to have 2S absolute configuration by Marfey's reaction. Based on the close biogenetic similarity, the absolute configuration at C-2′ of the latter two compounds is assumed to be S.
A plausible biosynthetic pathway of the substituted L-tryptophan-L-phenyllactic acid conjugates obtained from A. aculeatus is proposed to start from L-tryptophan and phenylpyruvate. The important intermediate 12 is suggested to be formed by a condensation reaction between L-tryptophan and L-phenyllactic acid. The indole metabolites isolated in this study could be produced by further methylation of 12 and prenylation of the indole nucleus of tryptophan as shown in Fig. 5.
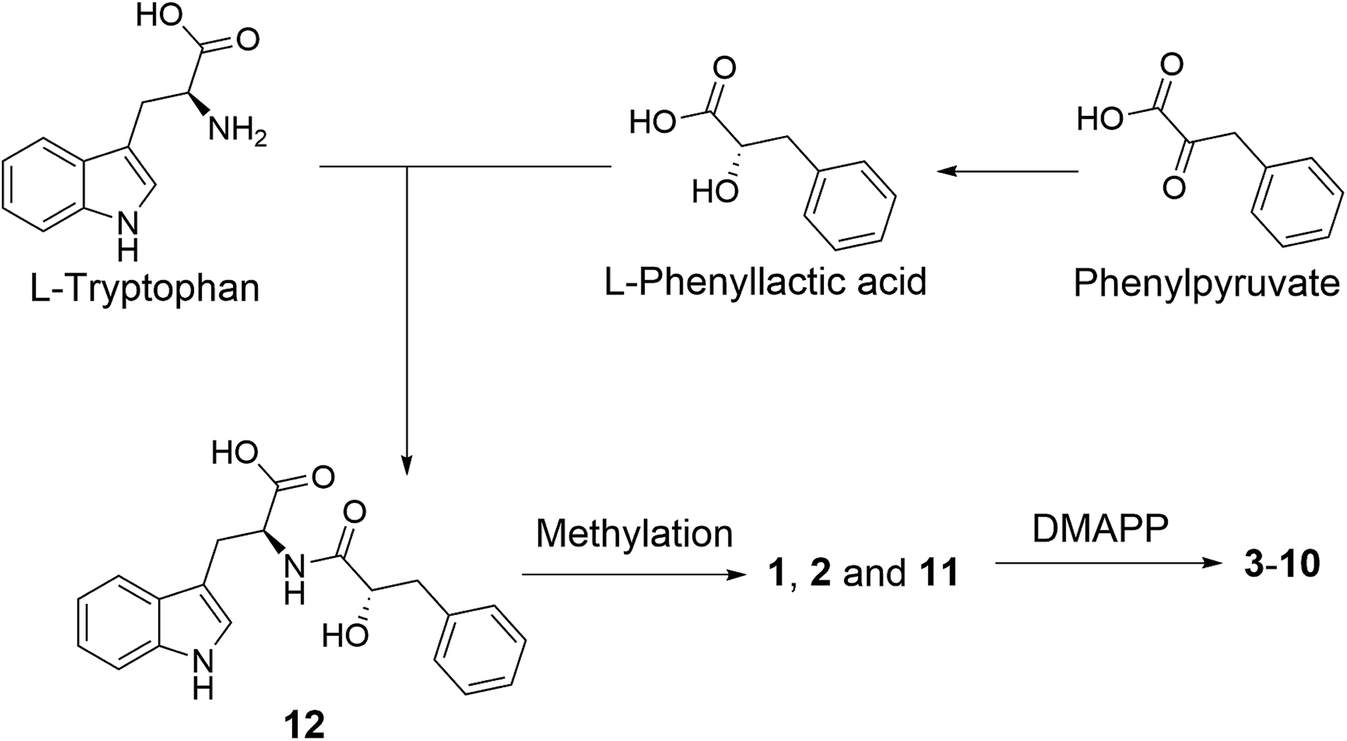 | ||
| Fig. 5 Plausible biosynthesis of substituted L-tryptophan-L-phenyllactic acid conjugates from A. aculeatus. | ||
All isolated compounds (1–25) were evaluated for their cytotoxicity against the L5178Y mouse lymphoma cell line. Secalonic acids D and F (19 and 20), asperdichrome (21) and RF 3192C (22) showed cytotoxicity with IC50 values of 3.4, 1.4, 7.3 and 23.7 μM, respectively, whereas the remaining compounds proved to be inactive when assayed at a dose of 10 μg mL−1.
The results of the OSMAC experiments indicated that substituted L-tryptophan-L-phenyllactic acid conjugates were induced by nitrate but not by sodium in the medium. In the absence of nitrogen sources favoured by the fungus including ammonium and glutamine, fungi are able to use secondary nitrogen sources such as nitrate, purines, urea, amines and amides etc., which is commonly known as nitrogen metabolite repression (NMR).30 In fungi, the activity of nitrogen regulators for derepression of NMR genes, which also affect secondary metabolites formation, is regulated by the intracellular nitrogen status and extracellular nitrogen availability.31,32 For example, the main GATA transcriptional regulator of nitrogen metabolism AreA accumulates in the nucleus in Fusarium graminearum with nitrate as sole nitrogen source, which is required for activation of the nitrate assimilation system including the nitrate reductase genes.33 Thus, the biosynthesis of fungal secondary metabolites can be affected by the quality and quantity of the nitrogen sources. For example, 67% of secondary metabolites silent gene clusters of Fusarium fujikuroi were expressed based on the modification of nitrogen sources.34 Furthermore, the natural product beauvericin was accumulated by Fusarium oxysporum by utilizing nitrate as sole nitrogen source.35 In this study, the production of substituted L-tryptophan-L-phenyllactic acid conjugates was stimulated by the activation of the nitrate assimilation system in A. aculeatus due to the presence of sodium nitrate in medium.
The substituted L-tryptophan-L-phenyllactic acid conjugates identified in this study showed no cytotoxic or antibacterial activity. However, the two known compounds (11 and 12) were claimed as plant growth regulators in a patent and showed pronounced rooting promoting effect.36 It may hence be hypothesized that a high concentration of nitrate in host plants, as simulated in this study by addition of sodium nitrate to solid rice medium, may induce the production of plant growth stimulating indole metabolites of the endophytic fungus A. aculeatus.37 This could lead to an increased growth and production of biomass by the host plant. In return, the fungus could receive nutrients, water, minerals and nitrogen from its host. Further studies will be necessary to evaluate this hypothesis.
Experimental section
General procedures
A Jasco P-2000 polarimeter was used to measure the optical rotation. 1D and 2D NMR spectra were recorded on Bruker Avance DMX 600 or 700 NMR spectrometers. Chemical shifts were referenced to the solvent residual peaks. Mass spectra were recorded with a LC-MS HP1100 Agilent Finnigan LCQ Deca XP Thermoquest and HRESIMS were measured with a UHR-QTOF maXis 4G (Bruker Daltonics) mass spectrometer. HPLC analysis was performed on a Dionex 3000 RS system coupled with an Ultimate 3000 pump and a photodiode array detector (DAD 300RS). The analytical column (125 × 4 mm) was prefilled with Eurosphere-10 C18 (Knauer, Germany), and the following gradient solvent system was used: 0 min (10% MeOH), 5 min (10% MeOH), 35 min (100% MeOH), and 45 min (100% MeOH). Semi-preparative HPLC was performed using a Merck Hitachi HPLC System (UV detector L-7400; pump L-7100; Eurosphere-100 C18, 300 × 8 mm, Knauer) with MeOH–H2O as mobile phase and a flow rate of 5.0 mL min−1. Column chromatography was carried out using Merck MN silica gel 60 M (0.04–0.063 mm). TLC plates with silica gel F254 (Merck) were used to monitor and collect fractions under detection at 254 and 366 nm. Distilled and spectral-grade solvents were used for column chromatography and spectroscopic measurements, respectively.Fungal material and cultivation
A. aculeatus was isolated from leaves of Carica papaya collected in Awka in Nigeria and was identified by DNA amplification, sequencing of ITS region and by comparing with GenBank data (GeneBank accession no. KX137846) following standard procedures.38The fungal strain was grown on solid rice medium (100 g rice and 100 mL distilled water autoclaved) in ten Erlenmeyer flasks (1 L each) at 22 °C under static conditions for 14 days. The OSMAC experiments were performed on rice medium containing either 3.5% NaCl, 3.5% NaBr, 3.5% NaI, 1% NaF, 3.5% NaNO3, 3.5% NH4Cl, 3.5% (NH4)2SO4 or 3.5% NH4OAc under static conditions until they reached their stationary phase of growth (16 days except for rice media spiked with 1% NaF or 3.5% NH4OAc where the fungus failed to grow).
Extraction and isolation
Fungal cultures were extracted with EtOAc followed by evaporation under reduced pressure. Initial purification of the EtOAc extract (12.5 g) of the fungal culture fermented on rice was performed by partitioning between n-hexane and 90% aqueous MeOH. The 90% aqueous MeOH phase (9.2 g) was centrifuged and then fractionated by vacuum liquid chromatography on reversed-phase silica gel using a gradient elution of H2O–MeOH (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :90 – 0:100) to give 10 fractions (Fr.1 to Fr.10).
:90 – 0:100) to give 10 fractions (Fr.1 to Fr.10).
Fr.2 (152 mg) was chromatographed on a Sephadex LH-20 column with MeOH followed by further purification using semi-preparative HPLC to give 17 (1.5 mg), 18 (7.0 mg) and 24 (0.5 mg). Compound 19 (30.2 mg) was obtained from Fr.3 (835 mg) by recrystallization. Part of Fr.3 was purified by semi-preparative HPLC to give 20 (20.0 mg). Fr.4 (208 mg) was subjected to a Sephadex LH-20 column with MeOH to give two subfractions (Fr.4-1 and Fr.4-2). Fr.4-1 was further purified by a silica gel column with DCM/MeOH as mobile phase to give 15 (3.8 mg). Compound 1 (13.1 mg) was obtained by recrystallization from Fr.4-2. Fr.5 (160 mg) was separated by a Sephadex LH-20 column followed by purification using semi-preparative HPLC to yield 14 (0.7 mg) and 21 (8.2 mg). Compound 11 (3.2 mg), 22 (1.2 mg) and 25 (2.4 mg) were obtained from Fr.6 (54 mg) by semi-preparative HPLC. Fr.7 (101 mg) was subjected to a Sephadex LH-20 column with MeOH as mobile phase and then purified by semi-preparative HPLC to give 23 (2.0 mg) and 16 (0.8 mg).
The fungal cultures from the OSMAC experiments that were grown on rice medium containing different salts were extracted with EtOAc (2 × 500 mL) followed by solvent evaporation under reduced pressure. The obtained crude extracts were analyzed by HPLC. The EtOAc extracts (28.6 g) of fungal cultures that had been grown on rice medium (30 flasks) after adding 3.5% NaNO3 were dissolved in MeOH and then subjected to vacuum filtering. The obtained MeOH solution was evaporated under reduced pressure and fractionated by vacuum liquid chromatography on silica gel using a gradient elution of n-hexane–EtOAc to give 24 fractions (Fr.N1 to Fr.N24). Fr.N19 (221 mg) was separated by a Sephadex LH-20 column with MeOH as mobile phase followed by semi-preparative HPLC to give 3 (3.5 mg) and 4 (15.0 mg). Fr.N22 (465 mg) was separated by a Sephadex LH-20 column with MeOH as mobile phase to give 3 fractions. Fr.N22-2 (54 mg) was further purified by semi-preparative HPLC to give 1 (10.1 mg), 2 (10.4 mg), 11 (3.5 mg), 12 (3.4 mg) and 13 (2.5 mg). Fr.N22-3 (380 mg) was further purified by a silica gel column with DCM–MeOH as mobile phase followed by semi-preparative HPLC to give 5 (2.4 mg), 6 (0.9 mg), 7 (4.8 mg), 8 (4.7 mg), 9 (1.2 mg) and 10 (1.2 mg).
Aculeatine A (1), white needle crystals; [α]20D +23 (c 0.25, MeOH); UV (MeOH): λmax 221, 288 nm; HRESIMS m/z 381.1810 [M + H]+ (calcd 381.1809 for C22H25O4N2); 1H and 13C NMR data see Tables 1 and 2.
Aculeatine B (2), colorless needle crystals; [α]20D +3 (c 0.40, MeOH); UV (MeOH): λmax 227, 288 nm; HRESIMS m/z 367.1653 [M + H]+ (calcd 367.1652 for C21H23O4N2); 1H and 13C NMR data see Tables 1 and 2.
Aculeatine C (3), white amorphous powder; [α]20D −54 (c 0.70, MeOH); UV (MeOH): λmax 227, 288 nm; HRESIMS m/z 449.2439 [M + H]+ (calcd 449.2435 for C27H33O4N2); 1H and 13C NMR data see Tables 1 and 2.
Aculeatine D (4), white amorphous solid; [α]20D −47 (c 1.7, MeOH); UV (MeOH): λmax 233, 286 nm; HRESIMS m/z 449.2435 [M + H]+ (calcd 449.2435 for C27H33O4N2); 1H and 13C NMR data see Tables 1 and 2.
Aculeatine E (5), white amorphous powder; [α]20D −2 (c 0.48, MeOH); UV (MeOH): λmax 228, 287 nm; HRESIMS m/z 435.2281 [M + H]+ (calcd 435.2278 for C26H31O4N2); 1H and 13C NMR data see Tables 1 and 2.
Aculeatine F (6), white amorphous solid; [α]20D −11 (c 0.20, MeOH); UV (MeOH): λmax 228, 288 nm; HRESIMS m/z 421.2122 [M + H]+ (calcd 421.2122 for C25H29O4N2); 1H and 13C NMR data see Tables 3 and 4.
Aculeatine G (7), white amorphous powder; [α]20D −24 (c 0.20, MeOH); UV (MeOH): λmax 225, 291 nm; HRESIMS m/z 449.2435 [M + H]+ (calcd 435.2435 for C27H33O4N2) and m/z 471.2252 [M + Na]+ (calcd 471.2254 for C27H32O4N2Na); 1H and 13C NMR data see Tables 3 and 4.
Aculeatine H (8), white amorphous powder; [α]20D −41 (c 0.94, MeOH); UV (MeOH): λmax 226, 291 nm; HRESIMS m/z 435.2282 [M + H]+ (calcd 435.2278 for C26H31O4N2) and m/z 457.2099 [M + Na]+ (calcd 457.2098 for C26H30O4N2Na); 1H and 13C NMR data see Tables 3 and 4.
Aculeatine I (9), white amorphous solid; [α]20D −35 (c 0.24, MeOH); UV (MeOH): λmax 225, 292 nm; HRESIMS m/z 453.2383 [M + H]+ (calcd 453.2384 for C26H33O5N2); 1H and 13C NMR data see Tables 3 and 4.
Aculeatine J (10), light yellow amorphous solid; [α]20D −19 (c 0.24, MeOH); UV (MeOH): λmax 226, 287 nm; HRESIMS m/z 453.2380 [M + H]+ (calcd 453.2384 for C26H33O5N2) and m/z 475.2201 [M + Na]+ (calcd 475.2203 for C26H32O5N2Na); 1H and 13C NMR data see Tables 3 and 4.
X-ray crystallographic analysis of compounds 1 and 2
The absolute structures of compound 1 and 2 were determined using anomalous dispersion from Cu-Kα radiation, resulting in Flack parameters of −0.14(10) (1) and 0.00(3) (2) using Parsons quotient method.43 Due to a rather high Flack parameter for 1, its absolute structure was determined using likelihood methods.44 DIAMOND was used for the drawing of all graphics.45 PLATON for Windows was used for the analyses of hydrogen bonds and CH–π interactions.46 The structural data for 2 has been deposited in the Cambridge Crystallographic Data Center (CCDC no. 1589955). Crystals of 1 did not diffract beyond θ = 44.9° (cf. desired 67.7°) for Cu-Kα radiation, resulting in only 1538 total (1484 observed with I > 2σ(I)) reflections versus 263 parameters for anisotropic refinement. Therefore the cif did not meet the requirements for publication and refinement data of compound 1 is only given in the ESI.†
751, N = 1538 (Rint = 0.051), R1 = 0.022, wR2 = 0.053, S = 1.12, Flack parameter = −0.14(10), Hooft parameter = −0.09(8), probability for correct absolute structure P2 = 1.000.796, N = 3416 (Rint = 0.027), R1 = 0.025, wR2 = 0.067, S = 1.09, Flack parameter = 0.00(3), Hooft parameter = 0.00(3), probability for correct absolute structure P2 = 1.000.Marfey's reaction for compounds 11 and 12
Compounds 11 and 12 (0.5 mg) were hydrolyzed with 2 mL 6 M HCl containing 0.4% β-mercaptoethanol at 110 °C for 24 h. The hydrolysate was evaporated to dryness and treated separately with 4 M NaOH at room temperature for 4 h. 4 M HCl was used to adjust the pH to 4. The above resulting solutions were evaporated until complete elimination of HCl and then resuspended in 50 μL H2O. To 25 μL of each resulting solutions was added 50 μL FDAA (1% 1-fluoro-2-4-dinitrophenyl-5-L-alanine amide in acetone) and 10 μL NaHCO3. The reaction tubes were covered with an aluminum paper and heated over a hot plate at 40 °C for 1 h. After cooling to room temperature, 5 μL of 2 M HCl was added and then evaporated to dryness. The residue was dissolved in 500 μL MeOH. L-Tryptophan and D-tryptophan were treated separately with FDAA in the same manner. The analysis of FDAA derivatives were carried out using HPLC and LC-MS by comparison of the retention time and molecular weight.Cytotoxicity assay
Cytotoxicity was tested against the L5178Y mouse lymphoma cell line (European Collection of Authenticated Cell Cultures, Catalogue no. 87111908) using the MTT method as described before.11 Kahalalide F was used as positive control with a IC50 value of 4.3 μM.Conflicts of interest
There are no conflicts to declare.Acknowledgements
H. W. wishes to thank the China Scholarship Council, the Ministry of Education of China, for a doctoral scholarship. Financial support by the DFG (GRK 2158) and by the Manchot Foundation to P. P. is gratefully acknowledged. P. P. and R. S. O. acknowledge support by the International Scientific Partnership Program of King Saud University (ISPP# 0065).Notes and references
- M. Jia, L. Chen, H. L. Xin, C. J. Zheng, K. Rahman, T. Han and L. P. Qin, Front. Microbiol., 2016, 7, 906–919 CrossRef PubMed.
- J. J. Kellogg and H. A. Raja, Phytochem. Rev., 2016, 16, 1–23 CrossRef.
- H. Nisa, A. N. Kamili, I. A. Nawchoo, S. Shafi, N. Shameem and S. A. Bandh, Microb. Pathog., 2015, 82, 50–59 CrossRef CAS PubMed.
- S. Liu, H. Dai, G. Makhloufi, C. Heering, C. Janiak, R. Hartmann, A. Mándi, T. Kurtán, W. E. G. Müller, W. Lin, Z. Liu and P. Proksch, J. Nat. Prod., 2016, 79, 2332–2340 CrossRef CAS PubMed.
- S. Liu, H. Dai, R. S. Orfali, W. Lin, Z. Liu and P. Proksch, J. Agric. Food Chem., 2016, 64, 3127–3132 CrossRef CAS PubMed.
- K. L. Kurita, E. Glassey and R. G. Linington, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 11999–12004 CrossRef CAS PubMed.
- H. B. Bode, B. Bethe, R. Höfs and A. Zeeck, ChemBioChem, 2002, 3, 619–627 CrossRef CAS PubMed.
- L. H. Meng, X. M. Li, Y. Liu, G. M. Xu and B. G. Wang, RSC Adv., 2017, 7, 55026–55033 RSC.
- S. S. Gao, X. M. Li, K. Williams, P. Proksch, N. Y. Ji and B. G. Wang, J. Nat. Prod., 2016, 79, 2066–2074 CrossRef CAS PubMed.
- L. Hammerschmidt, A. H. Aly, M. Abdel-Aziz, W. E. G. Müller, W. Lin, G. Daletos and P. Proksch, Bioorg. Med. Chem., 2015, 23, 712–719 CrossRef CAS PubMed.
- H. Wang, H. Dai, C. Heering, C. Janiak, W. Lin, R. S. Orfali, W. E. G. Müller, Z. Liu and P. Proksch, RSC Adv., 2016, 6, 81685–81693 RSC.
- C. F. Hemphill, P. Sureechatchaiyan, M. U. Kassack, R. S. Orfali, W. Lin, G. Deletos and P. Proksch, J. Antibiot., 2017, 70, 726–732 CrossRef CAS PubMed.
- R. Andersen, G. Büchi, B. Kobbe and A. L. Demain, J. Org. Chem., 1977, 42, 352–353 CrossRef CAS PubMed.
- K. Mizuno, A. Yagi, S. Satoi, M. Takada and M. Hayashi, J. Antibiot., 1977, 30, 297–302 CrossRef CAS PubMed.
- M. Miyata, J. Kitamura and H. Miyata, Arch. Microbiol., 1980, 127, 11–16 CrossRef CAS PubMed.
- S. Satoi, A. Yagi, K. Asano, K. Mizuno and T. Watanabe, J. Antibiot., 1977, 30, 303–307 CrossRef CAS PubMed.
- N. Ingavat, J. Dobereiner, S. Wiyakrutta, C. Mahidol, S. Ruchirawat and P. Kittakoo, J. Nat. Prod., 2009, 72, 2049–2052 CrossRef CAS PubMed.
- Z. G. Khalil, X. Huang, R. Raju, A. M. Piggott and R. J. Capon, J. Org. Chem., 2014, 79, 8700–8705 CrossRef CAS PubMed.
- H. Hayashi, K. Furutsuka and Y. Shiono, J. Nat. Prod., 1999, 62, 315–317 CrossRef CAS PubMed.
- D. W. Nagel, K. G. R. Pachler, P. S. Steyn, R. Vleggaar and P. L. Wessels, Tetrahedron, 1976, 32, 2625–2631 CrossRef CAS.
- K. Nozawa, S. Nakajima, K. Kawai and S. Udagawa, J. Chem. Soc., Perkin Trans. 1, 1988, 9, 2607–2610 RSC.
- R. M. Banks, S. E. Blanchflower, J. R. Everett, B. R. Manger and C. Reading, J. Antibiot., 1997, 50, 840–846 CrossRef CAS PubMed.
- M. Takagi, K. Motohashi and K. Shin-Ya, J. Antibiot., 2010, 63, 393–395 CrossRef CAS PubMed.
- H. Yamazaki, K. Ukai and M. Namikoshi, Tetrahedron Lett., 2016, 57, 732–735 CrossRef CAS.
- S. V. Jain, K. S. Bhadoriya and S. B. Bari, Med. Chem. Res., 2012, 21, 1665–1676 CrossRef CAS.
- H. Ogawa, K. Hasumi, K. Sakai, S. Murakawa and A. Endo, J. Antibiot., 1991, 44, 762–767 CrossRef CAS PubMed.
- P. Königs, B. Rinker, L. Maus, M. Nieger, J. Rheinheimer and S. R. Waldvogel, J. Nat. Prod., 2010, 73, 2064–2066 CrossRef PubMed.
- H. Takahashi, T. Hosoe, K. Nozawa and K. Kawai, J. Nat. Prod., 1999, 62, 1712–1713 CrossRef CAS.
- H. Wang, B. O. Umeokoli, P. Eze, C. Heering, C. Janiak, W. E. G. Müller, R. S. Orfali, R. Hartmann, H. Dai, W. Lin, Z. Liu and P. Proksch, Tetrahedron Lett., 2017, 58, 1702–1705 CrossRef CAS.
- G. A. Marzluf, Microbiol. Mol. Biol. Rev., 1997, 61, 17–32 CAS.
- M. X. Caddick, M. G. Jones, J. M. van Tonder, H. le Cordier, F. Narendja, J. Strauss and I. Y. Morozov, Mol. Microbiol., 2006, 62, 509–519 CrossRef CAS PubMed.
- B. Tudzynski, Front. Microbiol., 2014, 5, 656 Search PubMed.
- K. Min, Y. Shin, H. Son, J. Lee, J. C. Kim, G. J. Choi and Y. W. Lee, FEMS Microbiol. Lett., 2012, 334, 66–73 CrossRef CAS PubMed.
- P. Wiemann and B. Tudzynski, Fusarium: Genomics, Molecular and Cellular Biology, 2013, pp. 111–142 Search PubMed.
- M. S. López-Berges, K. Schäfer, C. Hera and A. di Pietro, Fungal Genet. Biol., 2014, 62, 78–84 CrossRef PubMed.
- Y. Maki, H. Soejima and T. Sugiyama, JP2014080406A, 2014.
- S. Siebrecht, K. Herdel, U. Schurr and R. Tischner, Planta, 2003, 217, 783–793 CrossRef CAS PubMed.
- J. Kjer, A. Debbab, A. H. Aly and P. Proksch, Nat. Protoc., 2010, 5, 479–490 CrossRef CAS PubMed.
- Apex2, Data Collection Program for the CCD Area-Detector System; SAINT, Data Reduction and Frame Integration Program for the CCD Area-Detector System: Bruker Analytical X-ray Systems, Madison, WI, USA, 1997–2012 Search PubMed.
- G. M. Sheldrick, SADABS: Area-detector absorption correction, University of Goettingen, Germany, 1996 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- (a) H. D. Flack and G. Bernardinelli, Acta Crystallogr., Sect. A: Found. Crystallogr., 1999, 55, 908–915 CrossRef; (b) H. Flack, Acta Crystallogr., Sect. A: Found. Crystallogr., 1983, 39, 876–881 CrossRef.
- R. W. W. Hooft, L. H. Straver and A. L. Spek, J. Appl. Crystallogr., 2008, 41, 96–103 CrossRef CAS PubMed.
- K. Brandenburg, Diamond (Version 4.4), Crystal and Molecular Structure Visualization, Bonn, Germany, 2009–2017 Search PubMed.
- (a) A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed; (b) A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS; (c) A. L. Spek, PLATON – A Multipurpose Crystallographic Tool, Utrecht University, Utrecht, The Netherlands, 2008 Search PubMed; (d) L. J. Farrugia, Windows Implementation, Version 40608, University of Glasgow, Scotland, 2008 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: UV, HRESIMS, 1D and 2D NMR data of all the new compounds 1–10 and results of X-ray analysis of compounds 1 and 2. CCDC 1589955. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra00200b |
| This journal is © The Royal Society of Chemistry 2018 |