
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
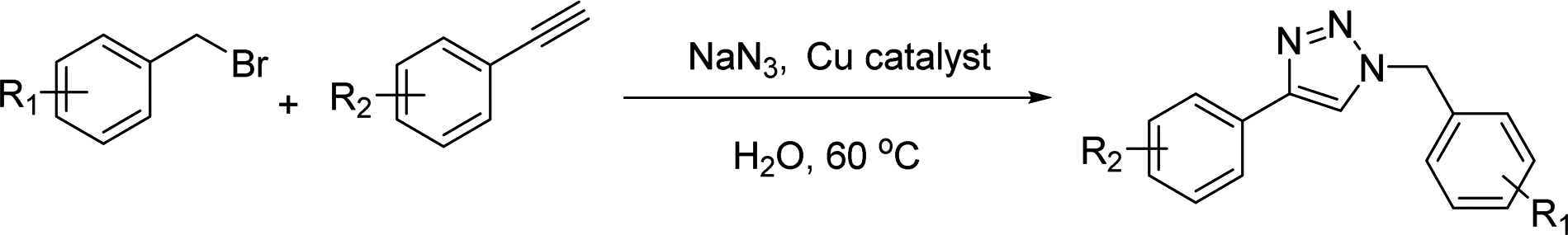
Azide–alkyne cycloaddition reactions in water via recyclable heterogeneous Cu catalysts: reverse phase silica gel and thermoresponsive hydrogels†
Minkyung Lim‡
a,
Heejin Lee‡a,
Minseok Kangb,
Woncheol Yoo b and
Hakjune Rhee*ab
b and
Hakjune Rhee*ab
aDepartment of Bionanotechnology, Hanyang University, 55 Hanyangdaehak-ro, Sangnok-gu, Ansan, Gyeonggi-do 15588, South Korea.
bDepartment of Applied Chemistry, Hanyang University, 55 Hanyangdaehak-ro, Sangnok-gu, Ansan, Gyeonggi-do 15588, South Korea. E-mail: hrhee@hanyang.ac.kr
First published on 6th February 2018
Abstract
Functionalized reverse phase silica gel and thermoresponsive hydrogels were synthesized as heterogeneous catalysts supports. Cu(I) and Cu(II) catalysts immobilized onto two types of supports were prepared and characterized. The copper catalyzed azide–alkyne cycloaddition was performed in water via a one-pot reaction and yielded good results. These catalysts are air stable and reusable over multiple uses.
1. Introduction
Triazole is a very useful motif within the fields of medicinal chemistry,1–4 carbohydrate chemistry,5–7 and materials science.8–12 Triazole has also been studied as a strong ligand for metal coordination with wide applicability.13–16 The thermal Huisgen 1,3-dipolar cycloaddition of organoazides and alkynes is a classic method for the synthesis of triazole.17,18 However, this method is slow even at high temperatures, producing a mixture of 1,4- and 1,5-disubstituted triazoles. Numerous synthetic methods using metal catalysts have been reported;19–29 copper has been shown to especially accelerate the reaction. The copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) reaction initiated by Sharpless30 and Meldal31 is considered to be a powerful pathway with a high regioselectivity and yield. However, copper catalyzed methods also possess drawbacks due to limited catalyst recyclability, the use of organic solvents, and the use of relatively expensive copper complexes that are difficult to remove.32–35 Therefore, the development of copper immobilized heterogeneous catalysts has attracted attention due to its reusability and ease of catalyst separation.36–41 A variety of solid supports have been applied including zeolites,42 charcoal,43,44 silica,45,46 and polysaccharides.47 Additionally, the use of water as a solvent is a promising approach with regard to green chemistry.37,38,48–52Recently, we developed two types of solid supports. One is an aminopropyl-functionalized reverse phase silica gel, which is end capped with a hydrophobic alkyl group. The other one is a thermoresponsive poly(N-isopropylacrylamide-co-4-vinylpyridine) (pNIPAM-VP) exhibiting hydrophilicity and hydrophobicity depending on temperature. These properties make it possible to use water as a solvent for organic reactions. These solid supports were applied to the syntheses of Pd, Au immobilized heterogeneous catalysts and organic reactions such as hydrogenations,53 Suzuki–Miyaura couplings,54,55 Heck–Mizoroki couplings,56 Sonogashira couplings,56 Tsuji–Trost reactions,57 and A-3 coupling reactions58 that were performed in water. Herein, we wish to demonstrate that our supports can be extended to other metal catalyzed organic reaction applications. In this study, Cu(I) and Cu(II) catalysts immobilized onto two support types were synthesized and characterized. The copper catalyzed azide–alkyne cycloaddition was performed in water.
2. Results and discussion
2.1. Copper catalysts immobilized onto reverse phase silica gel
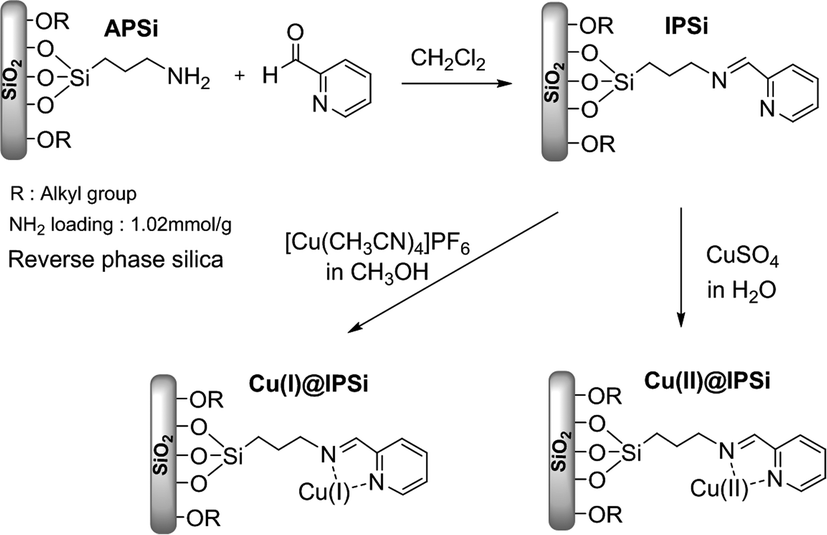 | ||
| Scheme 1 Synthesis of Cu(I)@IPSi and Cu(II)@IPSi. | ||
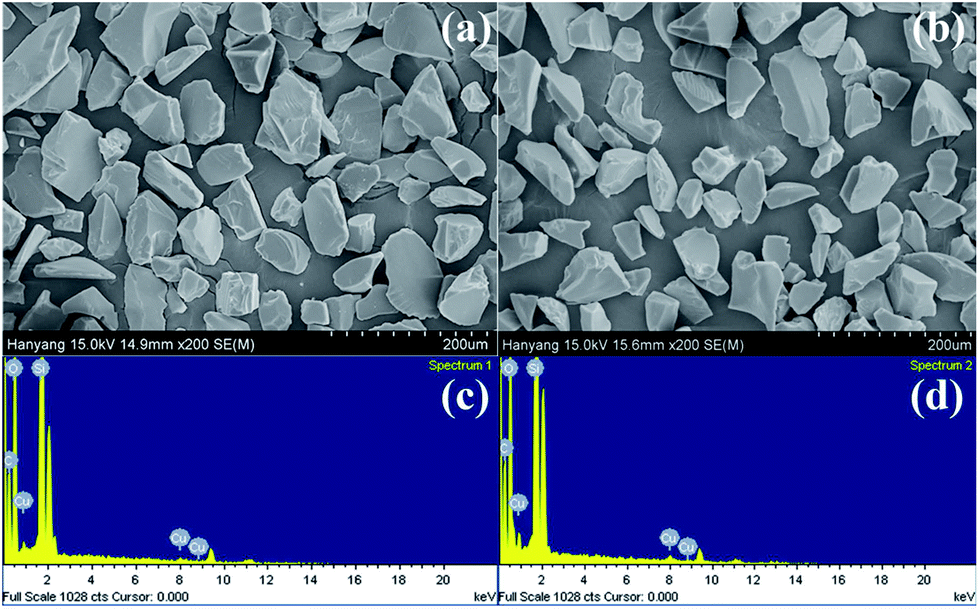 | ||
| Fig. 1 Scanning electron microscope (SEM) image and energy dispersive X-ray analysis (EDXA) of Cu(I)@IPSi (a, c) and Cu(II)@IPSi (b, d). | ||
The reverse phase silica gel possessed an end capped alkyl group yielding hydrophobicity. We anticipated that transport of the hydrophobic substrate towards the hydrophobic surface would enhanced reactivity by bringing the substrate and catalyst in close proximity.47 This property would also enable the azide–alkyne cycloaddition reaction in water (Fig. 2).
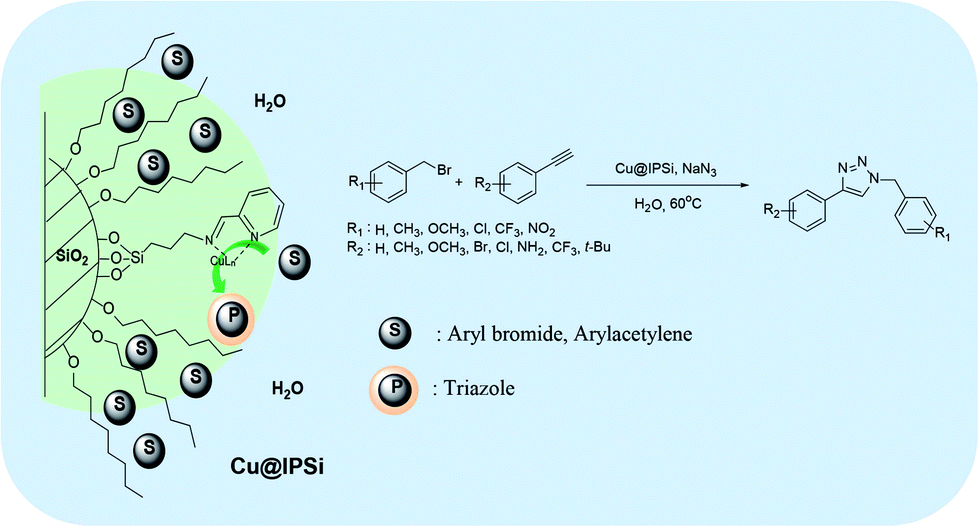 | ||
| Fig. 2 Catalytic behavior of the reverse phase silica gel in water. | ||
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Mol% | Temp. (°C) | Time (h) | Yieldb (%) | TONc (mol mol−1) | TOFd (h−1) |
| a Reaction conditions: benzyl bromide (1.0 mmol), phenyl acetylene (1.0 mmol), sodium azide (1.0 mmol), H2O (2 mL).b Isolated yield.c TON (turnover number): reactant moles converted/catalyst moles.d TOF (turnover frequency): TON/reaction time. | |||||||
| 1 | Cu(I)@IPSi | 5 | r.t. | 24 | 94 | 18.8 | 0.78 |
| 2 | Cu(I)@IPSi | 5 | 60 | 2 | 92 | 18.4 | 9.2 |
| 3 | Cu(I)@IPSi | 5 | 80 | 2 | 89 | 17.8 | 8.9 |
| 4 | Cu(I)@IPSi | 2.5 | 60 | 2 | 92 | 36.8 | 18.4 |
| 5 | Cu(I)@APSi | 2.5 | 60 | 2 | 84 | 35.2 | 17.6 |
| 6 | Cu(II)@IPSi | 5 | r.t. | 24 | 94 | 18.8 | 0.78 |
| 7 | Cu(II)@IPSi | 5 | 60 | 2 | 93 | 18.6 | 9.3 |
| 8 | Cu(II)@IPSi | 5 | 80 | 2 | 87 | 17.4 | 8.7 |
| 9 | Cu(II)@IPSi | 2.5 | 60 | 2 | 87 | 34.8 | 17.4 |
| 10 | Cu(II)@APSi | 5 | 60 | 2 | 78 | 15.6 | 7.8 |
We considered various substrates for further investigation as can be seen in Table 2. The azide–alkyne cycloaddition was performed under the established reaction conditions of 60 °C in water (Table 1, 4 & 7). Both catalysts were effective and Cu(I)@IPSi was more reactive than Cu(II)@IPSi. The reaction proceeded smoothly in water, an environmentally friendly solvent. All reactants, aryl bromides and arylacetylenes regardless of attached electron withdrawing and electron donating substituents, yielded good results (74%-quant. yield).
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Product | Cu(I)@IPSia | Cu(II)@IPSib | Entry | Product | Cu(I)@IPSia | Cu(II)@IPSib | ||||
| Time (h) | Isolated yield (%) | Time (h) | Isolated yield (%) | Time (h) | Isolated yield (%) | Time (h) | Isolated yield (%) | ||||
| a Reaction conditions: aryl bromide (1.0 mmol), aryl acetylene (1.0 mmol), sodium azide (1.0 mmol), Cu(I)@IPSi (2.5 mol% of Cu), H2O (2 mL).b Cu(II)@IPSi (5.0 mol% of Cu).c 4-Aminobenzyl azide was prepared by the reaction of 4-aminobenzyl bromide with sodium azide for 2 hours; phenyl acetylene was subsequently added to the reaction mixture.d Benzylbromide (1.2 mmol). | |||||||||||
| 1 |  |
2 | 92 | 2 | 93 | 10d |  |
6 | 80 | 6 | 83 |
| 2 |  |
2 | 93 | 2 | 90 | 11 |  |
4 | 90 | 4 | 90 |
| 3 | 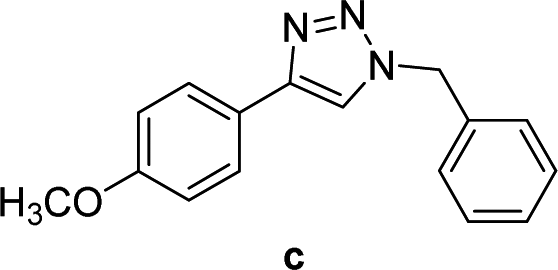 |
2 | Quant. | 2 | 88 | 12 | 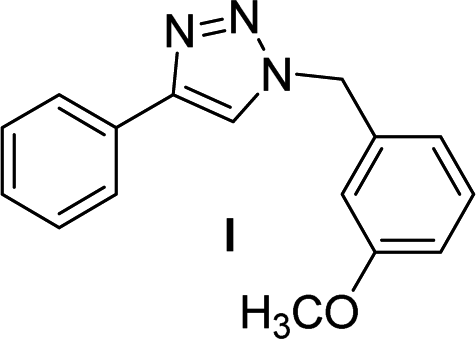 |
6 | 96 | 6 | 96 |
| 4 | 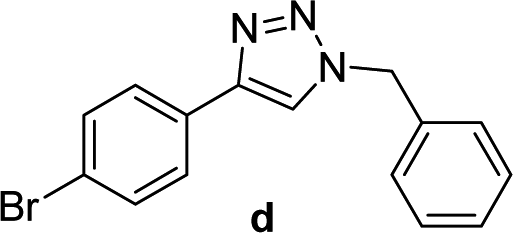 |
2 | 97 | 2 | 91 | 13d | 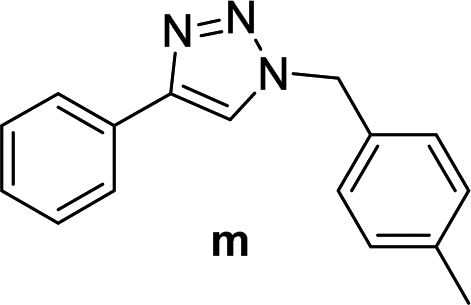 |
4 | 84 | 4 | 84 |
| 5c |  |
4 | 76 | 4 | 74 | 14 |  |
4 | 91 | 6 | 91 |
| 6 |  |
2 | 90 | 2 | 90 | 15 |  |
6 | 90 | 6 | 93 |
| 7 |  |
2 | 96 | 2 | 88 | 16 |  |
6 | 95 | 6 | 94 |
| 8 |  |
2 | 96 | 2 | 86 | 17 |  |
6 | 97 | 6 | 86 |
| 9 | 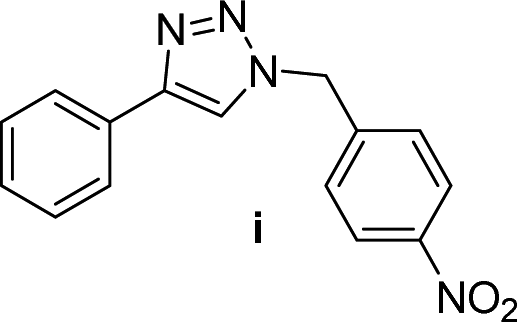 |
6 | 94 | 6 | 94 | ||||||
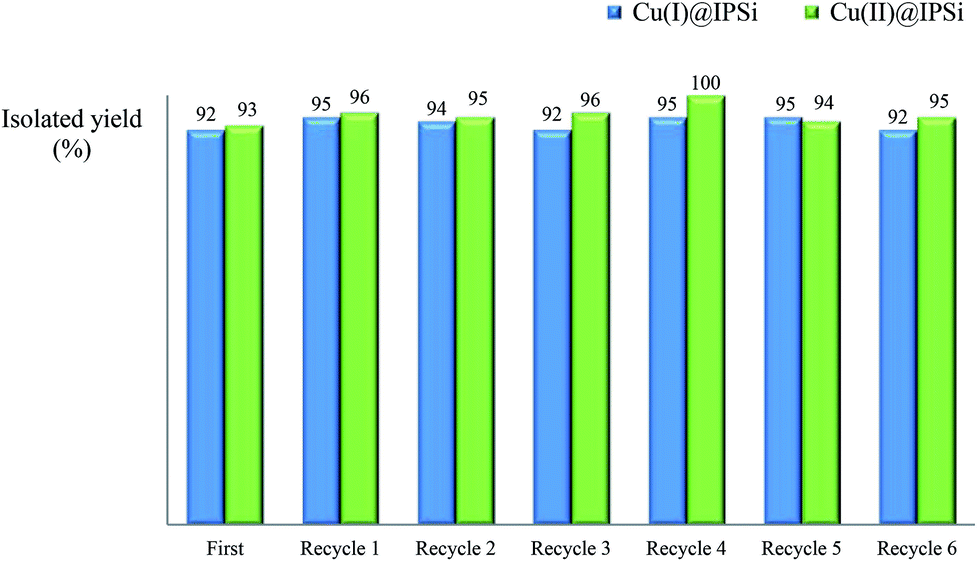 | ||
| Fig. 3 Recycling test of Cu(I)@IPSi and Cu(II)@IPSi. | ||
Fortunately, the ICP data showed no significant copper loss from both catalysts. XPS analysis was performed to confirm the oxidation state of freshly prepared catalyst and recovered catalyst (more details, see ESI†).
2.2. Copper catalyst immobilized on thermoresponsive hydrogels
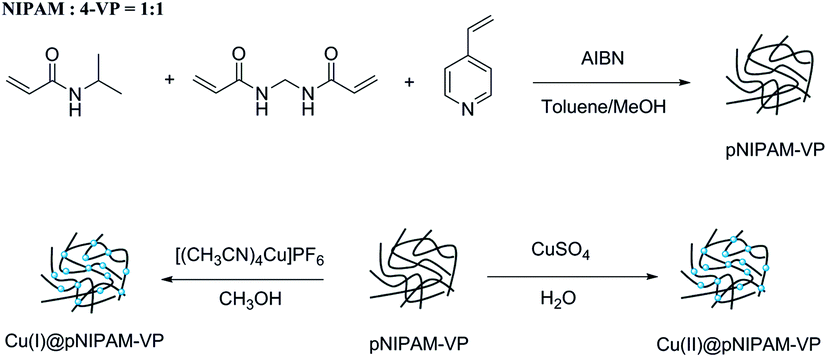 | ||
| Scheme 2 Synthesis of Cu(I)@pNIPAM-VP and Cu(II)@pNIPAM-VP. | ||
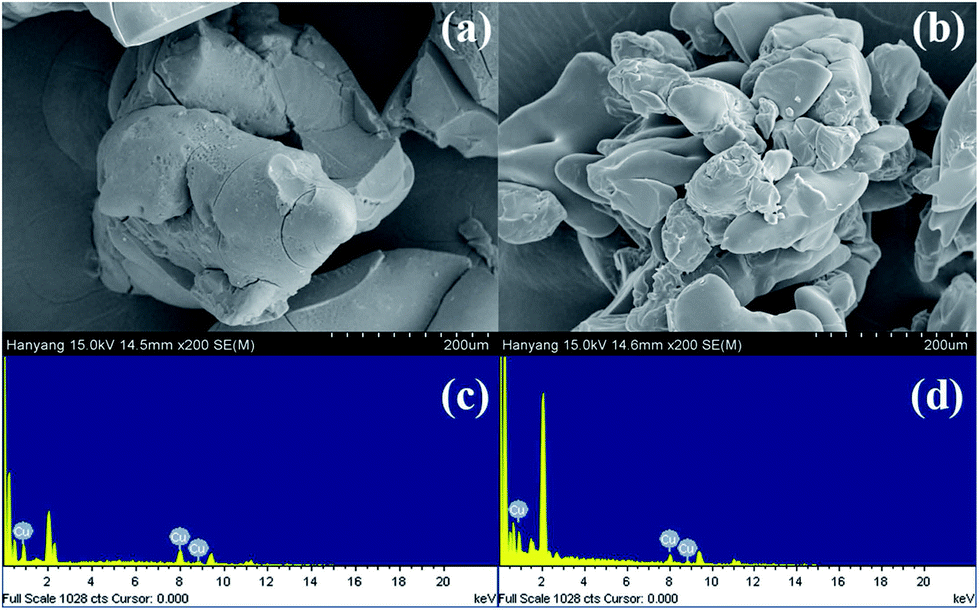 | ||
| Fig. 4 Scanning electron microscope (SEM) images and energy dispersive X-ray analysis (EDXA) of Cu(I)@pNIPAM-VP (a, c) and Cu(II)@pNIPAM-VP (b, d). | ||
The loading values of copper were determined via ICP and the results revealed loading levels of 0.374 mmol g−1 for Cu(I)@pNIPAM-VP and 0.166 mmol g−1 for Cu(II)@pNIPAM-VP. pNIPAM-VP supports featured a special low critical solution temperature (LCST) that imparted additional control to accelerate the reaction. According to the differential scanning calorimetry (DSC) data, the LCST of the synthesized polymer was 47 °C, as can be seen in Fig. 5. At temperatures higher than 47 °C, hydrogen bonds between the polymer and solvent molecules become weak and the polymer coils around itself, shrinking in size as intramolecular hydrogen bonding becomes significant.59,60 This mechanism was proposed for the coil-to-globule transition of polymer coils that took place in hydrophilic solvents (Fig. 5).
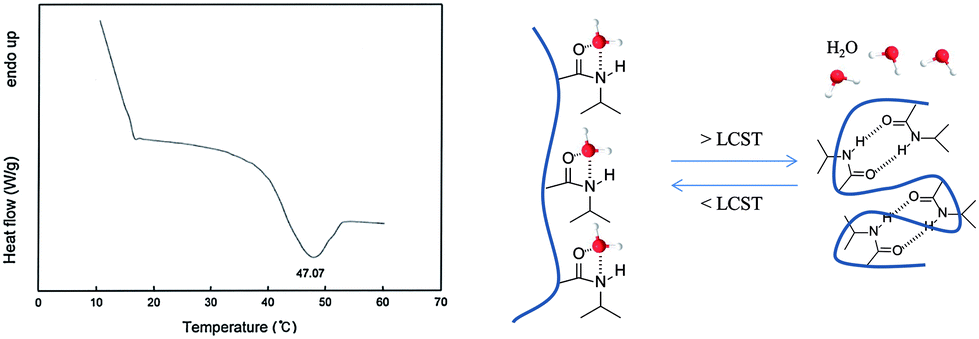 | ||
| Fig. 5 Differential scanning calorimetry curve for pNIPAM-VP. | ||
2.3. Azide–alkyne cycloaddition
One-pot azide–alkyne cycloaddition reactions were performed to explore the catalytic activity of the prepared catalysts. Initially, we chose benzyl bromide and phenylacetylene as substrates and the reaction was performed at 60 °C above the LCST (47 °C). The reaction was attempted by varying the quantity of catalyst and the TON and TOF values of the corresponding reaction were compared as can be seen in Table 3. Cu(II)@pNIPAM-VP (1 mol%) was able to effectively perform the reaction (91%). In case of Cu(I)@pNIPAM-VP (5 mol%), the product yield was 94% (Table 3, entries 1 & 4).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Mol% | Time (h) | Yieldb (%) | TONc (mol mol−1) | TOFd (h−1) |
| a Reaction conditions: benzyl bromide (1.0 mmol), phenyl acetylene (1.0 mmol), sodium azide (1.0 mmol), H2O (2 mL).b Isolated yield.c TON (turnover number): reactant moles converted/catalyst moles.d TOF (turnover frequency): TON/reaction time. | ||||||
| 1 | Cu(I)@pNIPAM-VP | 5 | 2 | 94 | 18.8 | 9.4 |
| 2 | Cu(I)@pNIPAM-VP | 2 | 2 | 89 | 44.5 | 22.3 |
| 3 | Cu(I)@pNIPAM-VP | 1 | 2 | 62 | 62 | 31 |
| 4 | Cu(II)@pNIPAM-VP | 1 | 2 | 91 | 91 | 45.5 |
The azide–alkyne cycloaddition was performed using various substrates with electron withdrawing and electron donating substituents (Table 4). Reactions with both catalysts proceeded in water and Cu(II)@pNIPAM-VP was more reactive than Cu(I)@pNIPAM-VP.
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Product | Cu(I)@pNIPAM-VPa | Cu(II)@pNIPAM-VPb | Entry | Product | Cu(I)@pNIPAM-VPa | Cu(II)@pNIPAM-VPb | ||||
| Time (h) | Isolated yield (%) | Time (h) | Isolated yield (%) | Time (h) | Isolated yield (%) | Time (h) | Isolated yield (%) | ||||
| a Reaction conditions: aryl bromide (1.0 mmol), aryl acetylene (1.0 mmol), sodium azide (1.0 mmol), Cu(I)@pNIPAM-VP (5 mol% of Cu), H2O (2 mL).b Cu(II)@pNIPAM-VP (1.0 mol% of Cu).c 4-Aminobenzyl azide was prepared by the reaction of 4-aminobenzyl bromide with sodium azide for 2 hours; phenyl acetylene was subsequently added to the reaction mixture.d Benzylbromide (1.2 mmol). | |||||||||||
| 1 |  |
2 | 94 | 2 | 91 | 10 |  |
6 | 88 | 8 | 75 |
| 2 |  |
3 | 92 | 2 | 93 | 11d |  |
4 | 62 | 4 | 88 |
| 3 |  |
2 | 92 | 2 | 92 | 12 |  |
6 | 89 | 6 | 88 |
| 4 |  |
4 | 93 | 4 | 92 | 13d |  |
6 | 70 | 6 | 73 |
| 5c |  |
6 | 75 | 6 | 71 | 14 | 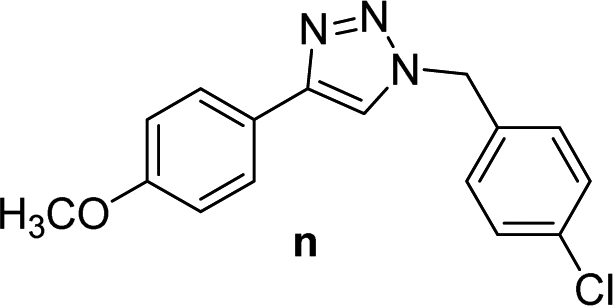 |
6 | 92 | 6 | 90 |
| 6 | 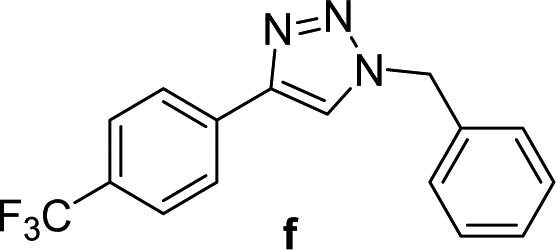 |
4 | 75 | 4 | 90 | 15 | 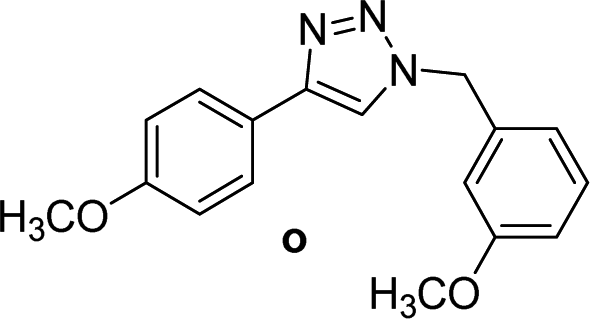 |
6 | 90 | 6 | 92 |
| 7 | 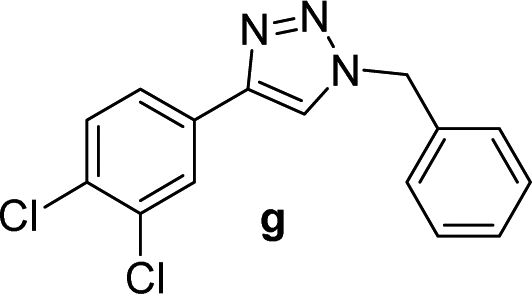 |
2 | 89 | 2 | 90 | 16 |  |
6 | 91 | 6 | 93 |
| 8 |  |
4 | 84 | 4 | 90 | 17 |  |
6 | 92 | 6 | 90 |
| 9d |  |
8 | 72 | 8 | 74 | ||||||
2.4. Recycling test
Recycling tests were performed using recovered catalysts under the established reaction conditions of 60 °C in water (Table 3, entries 1 & 4). As can be seen in Fig. 6, the Cu(I)@pNIPAM-VP catalyst was reusable several times, exhibiting little to no change to the product yield (90–94%). After the reaction, catalysts were collected by hot filtration and the filtrate was checked via ICP to confirm the leaching of copper. Fortunately, ICP data revealed no significant copper losses from the Cu(I)@pNIPAM-VP catalyst. On the other hand, in case of Cu(II)@pNIPAM-VP, the yield decreased remarkably after being recycled three times. XPS analysis was also performed to confirm the oxidation state of freshly prepared catalyst and recovered catalyst (ESI).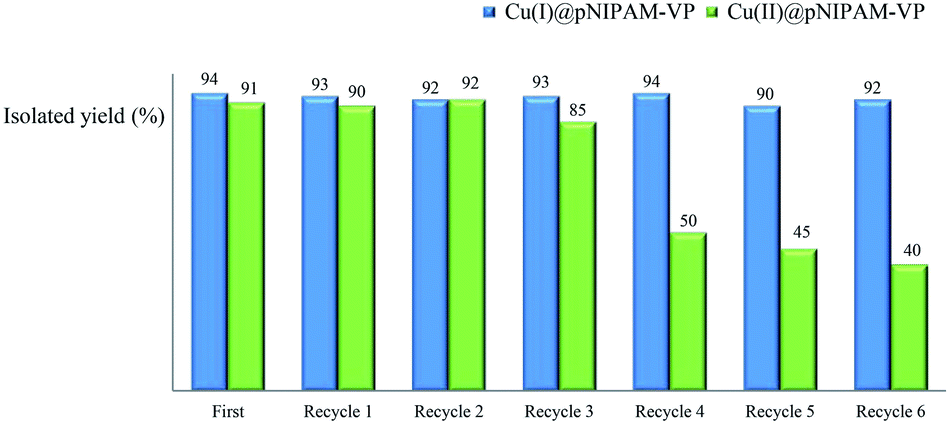 | ||
| Fig. 6 Recycling test of Cu(I)@pNIPAM-VP and Cu(II)@pNIPAM-VP. | ||
3. Experimental
3.1. Material and characterization
All chemicals were purchased from commercial sources (Sigma Aldrich, TCI and Alfa Aesar) and were used without further purification unless specifically mentioned. 2-Pyridinecarboxaldehyde and 4-vinylpyridine (4-VP) were distilled under reduced pressure prior to use. The silica gel support used to prepare the catalysts was commercially available 9% functionalized 3-aminopropyl silica gel with 1 mmol NH2 per gram (particle size 40–63 μm; pore size 60 Å; surface area 550 m2 g−1). The copper catalyst loading on the silica support and the azide–alkyne cycloaddition reaction were performed using a shaker (Eyela, Mixer CM-1000) at 12 × 103 rpm. Scanning electron microscopy (SEM) and energy dispersive X-ray analysis (EDXA) were performed on a HRTEM JEOL electron microscope at an acceleration voltage of 300 kV. The copper catalyst loading values were estimated via inductively coupled plasma (ICP) analysis with a JY Ultima2C. ICP analysis was also used to check for Cu leaching after the recycle test. X-ray photoelectron spectroscopy (XPS) analysis was performed on a VG SCIENTA R3000 to show the electronic state of copper. 1H and 13C NMR spectra were obtained in CDCl3 with a Bruker NMR at 400 MHz for 1H and at 100 MHz for 13C with TMS as an internal standard. For solid-state NMR experiments, 1D 13C spectra were measured via a cross polarization (CP) pulse sequence (contact time: 2 ms) on a Bruker 400 MHz NMR spectrometer equipped with a 4 mm magic angle spinning (MAS) probe (Bruker Biospin, Billerica, MA) operating at a 10 kHz spinning rate. Low temperature nitrogen adsorption–desorption isotherms were measured at −196 °C on an absorption volumetric analyzer BEL MINI manufactured by BEL, Inc. (Japan). Specific surface areas were determined using the Brunnauer–Emmet–Teller (BET) method from nitrogen adsorption isotherms of gas adsorbed at the relative pressure P/P0 = 0.99. Infrared spectra were recorded on a Bruker Alpha FT-IR spectrometer. Melting points were determined with a Sanyo Gallenkamp melting point apparatus. Analytical thin layer chromatography (TLC) was performed with E. Merck 60 F254 aluminum-backed silica gel plates (0.2 mm) containing a fluorescent indicator.3.2. Synthesis of Cu(I)@IPSi and Cu(II)@IPSi
3-Aminopropyl functionalized silica gel (3 mmol NH2 unit, 1.0 equiv., 3 g) was added to a jacketed vial containing a solution of 2-pyridinecarboxaldehyde (1.1 equiv., 0.34 mL) in 15 mL of distilled CH2Cl2. After 3 hours of vigorous stirring, the reaction mixture was filtered, washed with CH2Cl2, and dried under vacuum at 40 °C. To coordinate Cu(I) with silica, imine functionalized silica gel (IPSi, 1.0 equiv., 1.1 g) and [Cu(CH3CN)4]PF6 (1.1 equiv., 0.21 g) were added to 25 mL of methanol. The reaction mixture was shaken at room temperature for 12 hours, followed by filtration, washing with methanol, and drying under vacuum at 40 °C. The loading value of Cu(I)@IPSi was 0.498 mmol g−1. For the preparation of Cu(II)@IPSi, imine functionalized silica gel (1.0 equiv., 1.1 g) was added to a jacketed vial containing CuSO4·5H2O (1.1 equiv., 0.27 g) dissolved in 25 mL of H2O. The mixture was stirred for 12 hours at room temperature, followed by filtration, washing with H2O, and drying under vacuum at 40 °C. The loading value of Cu(II)@IPSi was 0.319 mmol g−1.3.3. Synthesis of Cu(I)@pNIPAM-VP and Cu(II)@pNIPAM-VP
Poly(N-isopropylacrylamide-co-4-vinylpyridine) (pNIPAM-VP) hydrogels were prepared via free radical solution polymerization using N-isopropylacrylamide (NIPAM), N,N′-methylenebis-acrylamide (MBAAm), and 4-vinylpyridine (4-VP) according to a previously reported procedure.55 NIPAM (10.1 mmol, 1.173 g) dissolved in 2 mL of methanol, MBAAm (1.28 mmol, 0.2 g) dissolved in 2 mL of methanol, and 4-VP (10.1 mmol, 1.06 g) dissolved in 16 mL of toluene were placed in a 100 mL flask. The polymerization was initiated by adding 2 mol% AIBN and heating the mixture at 80 °C for 4 h. A white solid polymer was filtered off, washed with methanol and diethyl ether, and dried under vacuum at 40 °C for 24 h. To coordinate Cu(I) with the polymer, pNIPAM-VP (1.0 g) and [Cu(CH3CN)4]PF6 (0.192 g) were added to 20 mL of methanol. The reaction mixture was shaken at room temperature for 12 hours, followed by filtration, washing with methanol, and drying under vacuum at 40 °C. The loading value of Cu(I)@pNIPAM-VP was 0.374 mmol g−1. For the preparation of Cu(II)@pNIPAM-VP, polymer (1.0 g) was added to a jacketed vial containing CuSO4·5H2O (0.13 g) dissolved in 20 mL of H2O. The mixture was stirred for 12 hours at room temperature, followed by filtration, washing with H2O, and drying under vacuum at 40 °C. The loading value of Cu(II)@pNIPAM-VP was 0.166 mmol g−1.3.4. General procedure for triazole synthesis
For a typical triazole synthesis experiment, benzylbromide (1.0 mmol), sodium azide (1.0 mmol), phenylacetylene (1.0 mmol), and catalysts were added to a 20 mL of jacketed vial containing 2 mL of H2O. After the addition of the Cu(I)@IPSi catalyst (2.5 mol%), Cu(II)@IPSi catalyst (5.0 mol%), Cu(I)@pNIPAM-VP catalyst (5.0 mol%), or Cu(II)@pNIPAM-VP catalyst (1.0 mol%), the reaction mixture was stirred at 60 °C. The reaction was monitored via thin layer chromatography (TLC). After completion, the reaction mixture was cooled and 6 mL of CH2Cl2 was added, followed by filtration, and washing with CH2Cl2 and H2O. The reaction mixture was filtered through a pad of celite and dried in vacuo. The product was purified by silica gel column chromatography with hexane/ethyl acetate and analyzed by 1H and 13C NMR spectroscopy.Compounds a,61 b,62 c,63 d,64 e,65 f,66 h,67 i,67 j,66 k,42 l,68 m,69 n,50 and o![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 70 are known compounds.
70 are known compounds.
4. Conclusions
Two types of solid supports were synthesized for the preparation of heterogeneous catalysts. One was aminopropyl-functionalized reverse phase silica gel, which possessed an end capped hydrophobic alkyl group. The other was a thermoresponsive poly(N-isopropylacrylamide-co-4-vinylpyridine) (pNIPAM-VP), which exhibited hydrophilicity and hydrophobicity according to temperature. These catalyst properties enabled one-pot azide–alkyne cycloaddition reactions in water. A series of 1,4-disubstituted-1,2,3-triazoles were synthesized with good results and the catalysts could be reused multiple times.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education (2015R1D1A1A09058536) and the Korean Ministry of Education through the BK21-Plus Project of the Hanyang University Graduate Program.References
- E. Bonandi, M. S. Christodoulou, G. Fumagalli, D. Perdicchia, G. Rastelli and D. Passarella, Drug Discovery Today, 2017, 22, 1572–1581 CrossRef CAS PubMed.
- D. Dheer, V. Singh and R. Shankar, Bioorg. Chem., 2017, 71, 30–54 CrossRef CAS PubMed.
- R. S. Keri, S. A. Patil, S. Budagumpi and B. M. Nagaraja, Chem. Biol. Drug Des., 2015, 86, 410–423 CAS.
- S. G. Agalave, S. R. Maujan and V. S. Pore, Chem.–Asian J., 2011, 6, 2696–2718 CrossRef CAS PubMed.
- X. P. He, Y. L. Zeng, Y. Zang, J. Li, R. A. Field and G. R. Chen, Carbohydr. Res., 2016, 429, 1–22 CrossRef CAS PubMed.
- V. K. Tiwari, B. B. Mishra, K. B. Mishra, N. Mishra, A. S. Singh and X. Chen, Chem. Rev., 2016, 116, 3086–3240 CrossRef CAS PubMed.
- D. Kushwaha, P. Dwivedi, S. K. Kuanar and V. K. Tiwari, Curr. Org. Synth., 2013, 10, 90–135 CrossRef CAS.
- P. Gaur, S. Dev, S. Kumar, M. Kumar, A. A. Vargeese, P. Soni, P. F. Siril and S. Ghosh, ACS Omega, 2017, 2, 8227–8233 CrossRef CAS.
- A. Sinopoli, F. A. Black, C. J. Wood, E. A. Gibson and P. I. Elliott, Dalton Trans., 2017, 46, 1520–1530 RSC.
- J. Huo, Z. Hu, D. Chen, S. Luo, Z. Wang, Y. Gao, M. Zhang and H. Chen, ACS Omega, 2017, 2, 5557–5564 CrossRef CAS.
- G. Delaittre, N. K. Guimard and C. Barner-Kowollik, Acc. Chem. Res., 2015, 48, 1296–1307 CrossRef CAS PubMed.
- D. Fournier, R. Hoogenboom and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 1369–1380 RSC.
- R. A. Vasdev, D. Preston and J. D. Crowley, Dalton Trans., 2017, 46, 2402–2414 RSC.
- Q. V. C. van Hilst, N. R. Lagesse, D. Preston and J. D. Crowley, Dalton Trans., 2018, 47, 997–1002 RSC.
- P. A. Scattergood and P. I. P. Elliott, Dalton Trans., 2017, 46, 16343–16356 RSC.
- I. Oura, K. Shimizu, K. Ogata and S. Fukuzawa, Org. Lett., 2010, 12, 1752–1755 CrossRef CAS PubMed.
- R. Huisgen, G. Szeimies and L. Möbius, Chem. Ber., 1967, 100, 2494–2507 CrossRef CAS.
- R. Huisgen, Angew. Chem., Int. Ed., 1963, 2, 565–598 CrossRef.
- C. P. Mencio, D. R. Garud, Y. Doi, Y. Bi, H. Vankayalapati, M. Koketsu and B. Kuberan, Bioorg. Med. Chem. Lett., 2017, 27, 5027–5030 CrossRef CAS PubMed.
- Z. Chen, Z. Liu, G. Cao, H. Li and H. Ren, Adv. Synth. Catal., 2017, 359, 202–224 CrossRef CAS.
- P. Destito, J. R. Couceiro, H. Faustino, F. Lopez and J. L. Mascarenas, Angew. Chem., 2017, 56, 10766–10770 CrossRef CAS PubMed.
- V. Castro, H. Rodriguez and F. Albericio, ACS Comb. Sci., 2016, 18, 1–14 CrossRef CAS PubMed.
- E. Haldon, M. C. Nicasio and P. J. Perez, Org. Biomol. Chem., 2015, 13, 9528–9550 CAS.
- M. Boominathan, N. Pugazhenthiran, M. Nagaraj, S. Muthusubramanian, S. Murugesan and N. Bhuvanesh, ACS Sustainable Chem. Eng., 2013, 1, 1405–1411 CrossRef CAS.
- M.-Y. Chen, T. Song, Z.-J. Zheng, Z. Xu, Y.-M. Cui and L.-W. Xu, RSC Adv., 2016, 6, 58698–58708 RSC.
- Z. J. Zheng, D. Wang, Z. Xu and L.-W. Xu, Beilstein J. Org. Chem., 2015, 11, 2557–2576 CrossRef CAS PubMed.
- T. Song, L. Li, W. Zhou, Z. J. Zheng, Y. Deng, Z. Xu and L.-W. Xu, Chemistry, 2015, 21, 554–558 CrossRef CAS PubMed.
- C.-Y. Wang, J.-F. Zou, Z.-J. Zheng, Z.-J. Huang, L. Li and L.-W. Xu, RSC Adv., 2014, 4, 54256–54262 RSC.
- Z. J. Zheng, F. Ye, L. S. Zheng, K. F. Yang, G. Q. Lai and L.-W. Xu, Chemistry, 2012, 18, 14094–14099 CrossRef CAS PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef.
- D. Wang, N. Li, M. Zhao, W. Shi, C. Ma and B. Chen, Green Chem., 2010, 12, 2120–2123 RSC.
- J. Kalisiak, K. B. Sharpless and V. V. Fokin, Org. Lett., 2008, 10, 3171–3174 CrossRef CAS PubMed.
- L.-H. Lu, J.-H. Wu and C.-H. Yang, J. Chin. Chem. Soc., 2008, 55, 414–417 CrossRef CAS.
- G. Molteni, C. L. Bianchi, G. Marinoni, N. Santo and A. Ponti, New J. Chem., 2006, 30, 1137–1139 RSC.
- S. Chassaing, V. Bénéteau and P. Pale, Catal. Sci. Technol., 2016, 6, 923–957 CAS.
- W. Yu, L. Jiang, C. Shen, W. Xu and P. Zhang, Catal. Commun., 2016, 79, 11–16 CrossRef CAS.
- A. Pourjavadi and Z. Habibi, RSC Adv., 2015, 5, 99498–99501 RSC.
- A. Pourjavadi, N. Safaie, S. H. Hosseini and C. Bennett, Appl. Organomet. Chem., 2015, 29, 601–607 CrossRef CAS.
- J. Albadi, Acta Chim. Slov., 2015, 617–624 CrossRef CAS.
- B. S. P. Anil Kumar, K. Harsha Vardhan Reddy, K. Karnakar, G. Satish and Y. V. D. Nageswar, Tetrahedron Lett., 2015, 56, 1968–1972 CrossRef CAS.
- S. Chassaing, A. Sani Souna Sido, A. Alix, M. Kumarraja, P. Pale and J. Sommer, Chemistry, 2008, 14, 6713–6721 CrossRef CAS PubMed.
- F. Alonso, Y. Moglie, G. Radivoy and M. Yus, Org. Biomol. Chem., 2011, 9, 6385–6395 CAS.
- F. Alonso, Y. Moglie, G. Radivoy and M. Yus, Adv. Synth. Catal., 2010, 352, 3208–3214 CrossRef CAS.
- J. M. Collinson, J. D. Wilton-Ely and S. Diez-Gonzalez, Chem. Commun., 2013, 49, 11358–11360 RSC.
- R. Hosseinzadeh, H. Sepehrian and F. Shahrokhi, Heteroat. Chem., 2012, 23, 415–421 CrossRef CAS.
- A. Kumar, S. Aerry, A. Saxena, A. de and S. Mozumdar, Green Chem., 2012, 14, 1298–1301 RSC.
- A. Banan, A. Bayat and H. Valizadeh, Appl. Organomet. Chem., 2017, 31, e3604 CrossRef.
- K. Lal and P. Rani, Arkivoc, 2016, 2016, 307–341 CrossRef.
- S. S. E. Ghodsinia, B. Akhlaghinia and R. Jahanshahi, RSC Adv., 2016, 6, 63613–63623 RSC.
- Y. Wang, J. Liu and C. Xia, Adv. Synth. Catal., 2011, 353, 1534–1542 CrossRef CAS.
- M. K. Barman, A. K. Sinha and S. Nembenna, Green Chem., 2016, 18, 2534–2541 RSC.
- M. Lim, K. A. De Castro, S. Oh, K. Lee, Y.-W. Chang, H. Kim and H. Rhee, Appl. Organomet. Chem., 2011, 25, 1–8 CrossRef CAS.
- S. Shabbir, S. Lee, M. Lim, H. Lee, H. Ko, Y. Lee and H. Rhee, J. Organomet. Chem., 2017, 846, 296–304 CrossRef CAS.
- M. C. Hong, M. C. Choi, Y. W. Chang, Y. Lee, J. Kim and H. Rhee, Adv. Synth. Catal., 2012, 354, 1257–1263 CrossRef CAS.
- Y. Lee, M. C. Hong, H. Ahn, J. Yu and H. Rhee, J. Organomet. Chem., 2014, 769, 80–93 CrossRef CAS.
- Y. Lee, S. Shabbir, S. Lee, H. Ahn and H. Rhee, Green Chem., 2015, 17, 3579–3583 RSC.
- S. Shabbir, Y. Lee and H. Rhee, J. Catal., 2015, 322, 104–108 CrossRef CAS.
- J. Cheng, G. Shan and P. Pan, Ind. Eng. Chem. Res., 2017, 56, 1223–1232 CrossRef CAS.
- Y. Hertle and T. Hellweg, J. Mater. Chem. B, 2013, 1, 5874–5885 RSC.
- H. Sharghi, R. Khalifeh and M. M. Doroodmand, Adv. Synth. Catal., 2009, 351, 207–218 CrossRef CAS.
- Q. Yang, C. Kuang, X. Cheng and Y. Yang, Synthesis, 2011, 2011, 2907–2912 CrossRef.
- K. Kamata, Y. Nakagawa, K. Yamaguchi and N. Mizuno, J. Am. Chem. Soc., 2008, 130, 15304–15310 CrossRef CAS PubMed.
- F. Wang, H. Fu, Y. Jiang and Y. Zhao, Green Chem., 2008, 10, 452–456 RSC.
- Z. Novák, K. Lőrincz and P. Kele, Synthesis, 2009, 2009, 3527–3532 CrossRef.
- K. Asano and S. Matsubara, Org. Lett., 2010, 12, 4988–4991 CrossRef CAS PubMed.
- Z. Jia, K. Wang, T. Li, B. Tan and Y. Gu, Catal. Sci. Technol., 2016, 6, 4345–4355 CAS.
- L. Rinaldi, K. Martina, F. Baricco, L. Rotolo and G. Cravotto, Molecules, 2015, 20, 2837–2849 CrossRef PubMed.
- C. Shao, X. Wang, Q. Zhang, S. Luo, J. Zhao and Y. Hu, J. Org. Chem., 2011, 76, 6832–6836 CrossRef CAS PubMed.
- D. Linares, O. Bottzeck, O. Pereira, A. Praud-Tabaries and Y. Blache, Bioorg. Med. Chem. Lett., 2011, 21, 6751–6755 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra00306h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |