
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Poly-histidine grafting leading to fishbone-like architectures†
Vincenzo Razzano§ a,
Marco Paolino§a,
Annalisa Reale§a,
Germano Giuliani§a,
Alessandro Donati§a,
Gianluca Giorgi§a,
Roberto Artusi§b,
Gianfranco Caselli§b,
Michela Visintin§b,
Francesco Makovec§b,
Salvatore Battiato§c,
Filippo Samperi§c,
Francesca Villafiorita-Monteleone‡§
d,
Chiara Botta§d and
Andrea Cappelli§*a
a,
Marco Paolino§a,
Annalisa Reale§a,
Germano Giuliani§a,
Alessandro Donati§a,
Gianluca Giorgi§a,
Roberto Artusi§b,
Gianfranco Caselli§b,
Michela Visintin§b,
Francesco Makovec§b,
Salvatore Battiato§c,
Filippo Samperi§c,
Francesca Villafiorita-Monteleone‡§
d,
Chiara Botta§d and
Andrea Cappelli§*a
aDipartimento di Biotecnologie, Chimica e Farmacia, European Research Centre for Drug Discovery and Development, Università di Siena, Via A. Moro 2, 53100 Siena, Italy. E-mail: andrea.cappelli@unisi.it; vincy_box@hotmail.it; paomar@oneonline.it; reale5@student.unisi.it; germix67@hotmail.com; alessandro.donati@unisi.it; gianluca.giorgi@unisi.it; Tel: +39 577 234320
bRottapharm Biotech S.p.A., Via Valosa di Sopra 9, 20900 Monza, Italy. E-mail: Roberto.Artusi@rottapharmbiotech.com; Gianfranco.Caselli@rottapharmbiotech.com; Michela.Visintin@rottapharmbiotech.com; Francesco.Makovec@rottapharmbiotech.com
cIstituto per i Polimeri, Compositi e Biomateriali (IPCB), U.O.S. di Catania, CNR, Via Gaifami 18, 95126 Catania, Italy. E-mail: salvatore.battiato@cnr.it; fsamperi@unict.it
dIstituto per lo Studio delle Macromolecole (CNR), Via A. Corti 12, 20133 Milano, Italy. E-mail: villafiorita@ismac.cnr.it; chiara.botta@ismac.cnr.it
First published on 26th February 2018
Abstract
A small series of Morita–Baylis–Hillman adduct (MBHA) derivatives was synthesized and made to react with imidazole, N-acetylhistidine, and N-acetylhexahistidine as models of poly-histidine derivatives. Intriguingly, the reaction of MBHA derivatives 1a and b with imidazole in acetonitrile–phosphate buffered saline (PBS) gave the imidazolium salt biadducts 3a and b as the main reaction products. These results were confirmed by experiments performed with N-acetylhistidine and 1b and suggested the possible occurrence of these structures in the products of poly-histidine labeling with MBHA derivatives 1a and b. These compounds were then transformed into the corresponding water-soluble derivatives 1c–e by introducing oligo(ethylene glycol) chains and their reactivity was evaluated in preliminary experiments with imidazole and then with N-acetylhexahistidine in PBS. The structure of polymeric materials Ac-His-6-MBHA-1d and Ac-His-6-MBHA-1e obtained using ten-fold excesses of compounds 1d and e was investigated using mass spectrometry, NMR spectroscopy, and photophysical studies, which suggested the presence of biadduct residues in both polymeric materials. These results provide the basis for the preparation of fishbone-like polymer brushes, the characterization of their properties, and the exploration of their potential applications in different fields of science such as in vivo fluorogenic labeling, fluorescence microscopy, protein PEGylation, up to the production of smart materials and biosensors.
Introduction
Histidine is an amino acid of crucial importance in many life processes. The imidazole residue confers peculiar characteristics to this amino acid and its derivatives.1 In fact, thanks to the imidazole pKa (about 6.0), histidine plays an important role as a buffer within the cellular environment (usually neutral) due to annular nitrogen protonation.2,3 Moreover, histidine is capable of complexing metal ions and is thus involved in many catalytic sites.4 Not uncommon, histidine is used in the synthesis of aminoacidic polymers. Poly-histidine (poly-His) showing different molecular weight was synthesized by controlled ring opening polymerization of L-histidine-N-carboxyanhydride.2,5 These polymers have been studied as such or used as functional side chain of other polymers (such as poly-lysine) or as fragments in block copolymers with polyethylene glycol (PEG) and acrylic polymers.6–8 Most histidine polymers have in common a high biocompatibility, a good fusogenic activity, a buffering capacity, and a peculiar proton-sponge effect. By virtue of these properties, they are often used as non-toxic gene delivery agents and investigated as component of drug delivery systems for intracellular cytotoxic drugs.6,7,9 In fact, the presence of imidazole residues ensures, at physiological pH, the presence of positive charges on the surface of the polymers that promotes adhesion to cell membranes and internalization by endocytosis of complexes polymer/pDNA (poliplex) or polymer/drug. In addition, the proton-sponge effect inactivates lysosomal enzymes and the fusogenic activity allows the endosome escape.10,11However, the systemic administration of the poly-histidine derivatives (and of cationic polymers in general) may show some complications. In fact, the cationic surface charge of this type of vectors promotes many nonspecific interactions with cellular components of blood, of the endothelium of blood vessels and plasma protein, dramatically reducing the plasma half-life and biodistribution. To overcome this problem, it has been developed the “shielding strategy”, which consists in protecting the residual surface charge with a steric barrier.12 This strategy is feasible by means of the conjugation to poly-histidines of neutral hydrophilic polymers such as the polyethylene glycol (PEG), which confers an increase in the solubility, a reduced interaction with serum proteins (e.g. albumin), improved biocompatibility, and a prolonged blood circulation.13
Furthermore, the insertion of poly-histidine tags in recombinant proteins represents a standard methodology in isolation and purification of recombinant proteins. Furthermore, their presence in the protein sequence provides an interesting opportunity for the selective covalent functionalization of these engineered proteins.14–18
Although alkylation of histidine residues in poly-histidine derivatives could be a difficult task owing to the low reactivity of imidazole rings in conventional alkylation reactions, a successful example of N-acetylhexahistidine (Ac-His-6) alkylation with Morita–Baylis–Hillman adduct (MBHA) derivatives was accomplished by using transition metal cations interacting with a nitrilotriacetate ligand linked to the leaving group of MBHA derivative.19
In order to explore the synthesis, the structure, and the properties of graft copolymers of poly-histidine with fluorescent molecules, a small series of MBHA derivatives20 were designed and synthesized to be evaluated in the reaction with Ac-His-6 as models of poly-histidine species (Fig. 1).
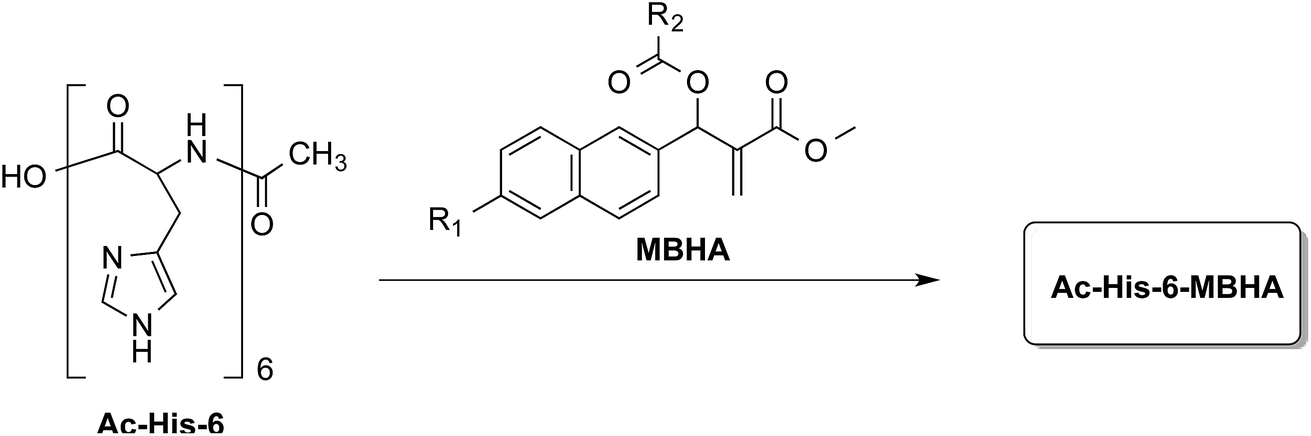 | ||
| Fig. 1 Grafting N-acetylhexahistidine (Ac-His-6) with Morita–Baylis–Hillman adduct (MBHA) derivatives. | ||
Thus in the work described herein, a small series of MBHA derivatives were designed, synthesized, and made to react with imidazole in order to evaluate their reactivity and the photophysical properties of the corresponding imidazole derivatives. The most promising compounds were then evaluated in their reaction with Ac-His-6 and the structural and photophysical features of the corresponding graft copolymers were analyzed in details.
Results and discussion
Synthesis
The synthetic works started with the preparation of MBHA derivatives 1a,b from 6-hydroxy-2-naphthaldehyde (4) or 6-bromo-2-naphthaldehyde (7), respectively (Schemes 1 and 2).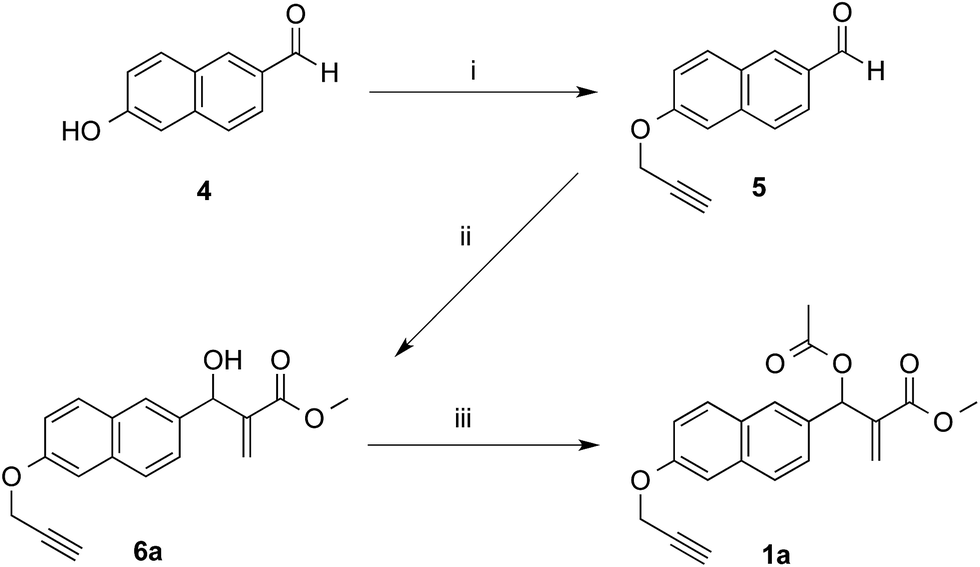 | ||
| Scheme 1 Synthesis of MBHA derivative 1a. Reagents: (i) propargyl bromide, K2CO3, acetonitrile; (ii) methyl acrylate, DABCO, CH3OH, THF; (iii) CH3COCl, TEA, CH2Cl2. | ||
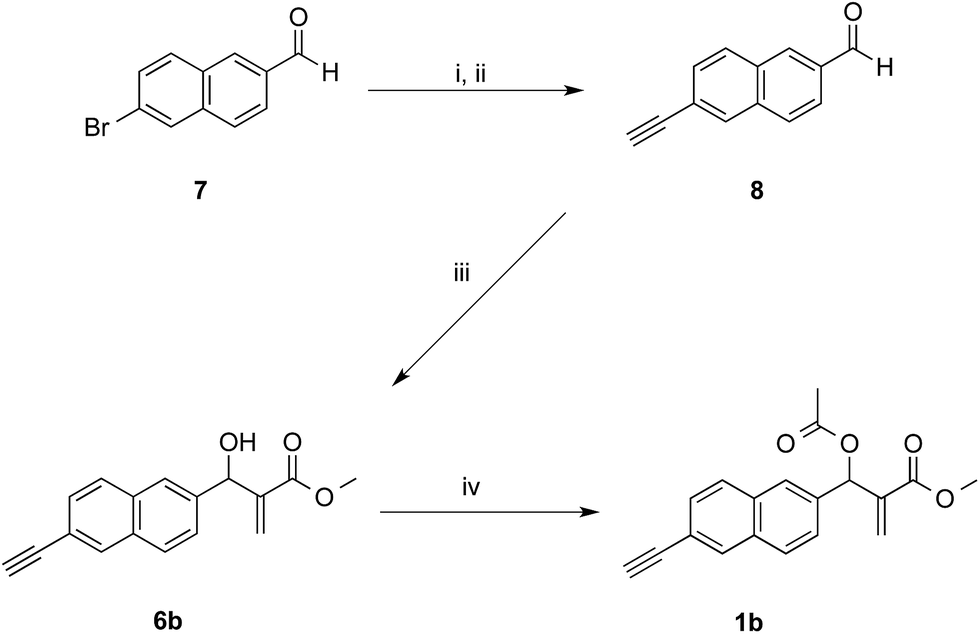 | ||
| Scheme 2 Synthesis of MBHA derivative 1b. Reagents: (i) ethynyltrimethylsilane, Pd(PPh3)2Cl2, CuI, TEA, THF; (ii) K2CO3, MeOH; (iii) DABCO, methyl acrylate, MeOH, THF; (iv) CH3COCl, TEA, CH2Cl2. | ||
As expected, the reaction of naphthalene MBHA derivative 1a with 1.5 equivalents of imidazole in THF–water (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) under reflux afforded imidazole derivative 2a in 78% yield. Similar yields (i.e. 80%) were obtained when 1a was reacted with 6 equivalents of imidazole in acetonitrile–phosphate buffered saline (PBS) as the reaction medium at room temperature for 24 h. On the other hand, when 1.5 equivalents of imidazole were used in the same reaction medium, along the expected imidazole derivative 2a, imidazolium salt 3a was isolated as the main reaction product (Scheme 3).
:1) under reflux afforded imidazole derivative 2a in 78% yield. Similar yields (i.e. 80%) were obtained when 1a was reacted with 6 equivalents of imidazole in acetonitrile–phosphate buffered saline (PBS) as the reaction medium at room temperature for 24 h. On the other hand, when 1.5 equivalents of imidazole were used in the same reaction medium, along the expected imidazole derivative 2a, imidazolium salt 3a was isolated as the main reaction product (Scheme 3).
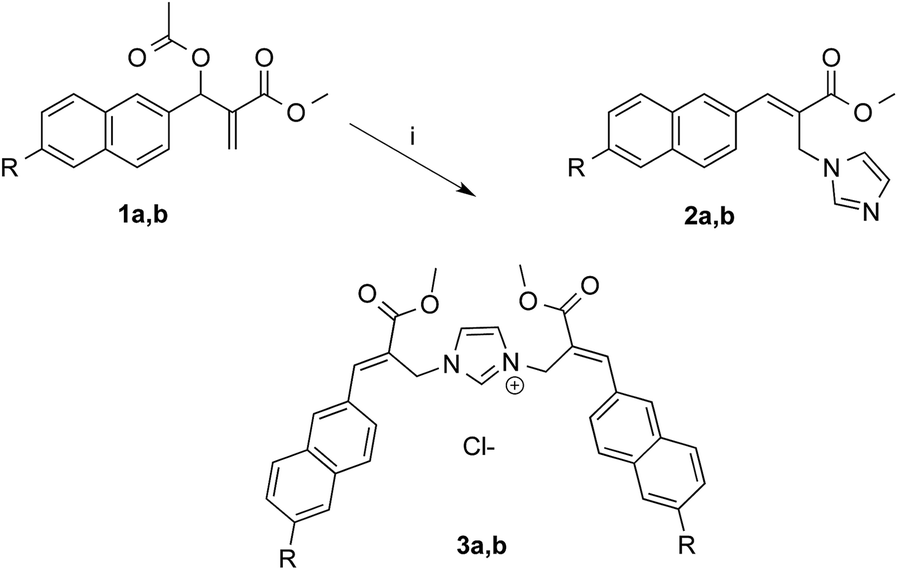 | ||
| Scheme 3 Reaction of MBHA derivatives 1a,b with imidazole. Reagents: (i) imidazole, CH3CN, PBS. Substituents: R = –OCH2CCH (1a, 2a, 3a); R = –CCH (1b, 2b, 3b). | ||
The analysis of the crystallographic structure of imidazolium salt 3a (Fig. 2) suggested that aromatic specific (i.e. π-stacking) interactions play an important role in stabilizing the solid state of this imidazole biadduct.
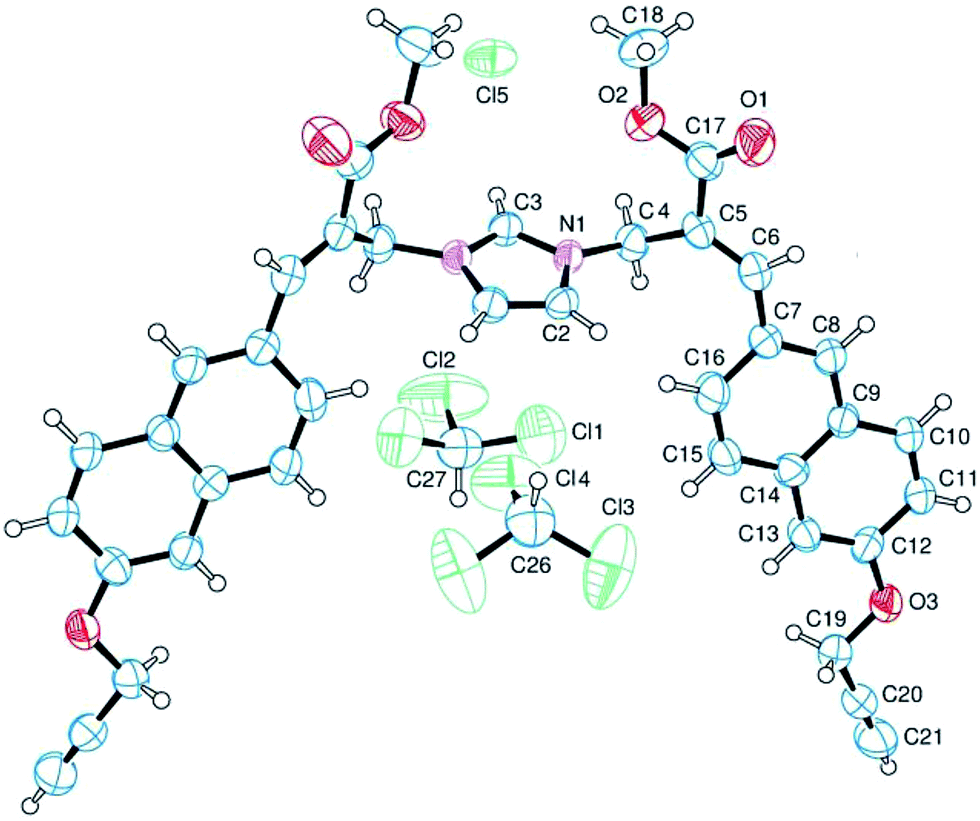 | ||
| Fig. 2 Structure of imidazolium salt 3a obtained by crystallography. The ellipsoids enclose 50% probability. | ||
These results were confirmed with MBHA derivative 1b. In fact, imidazole derivative 2b was obtained in 92% yield by using 6 equivalents of imidazole in acetonitrile–PBS at room temperature for 24 h, whereas imidazolium salt 3b was isolated in 62% yield when 1.5 equivalents of imidazole were used in the same reaction conditions.
On the other hand, the reaction of 1b with 1.5 equivalents of N-acetylhistidine21 in acetonitrile–PBS at room temperature was rather slow and required heating to be carried out in a reasonable time. Thus, 1b reacted with 1.5 equivalents of N-acetylhistidine in acetonitrile–PBS at the refluxing temperature to give biadduct 3c as the main product (i.e. yields around 80%) along with small amounts (i.e. yields around 5%) of 2c (Scheme 4).
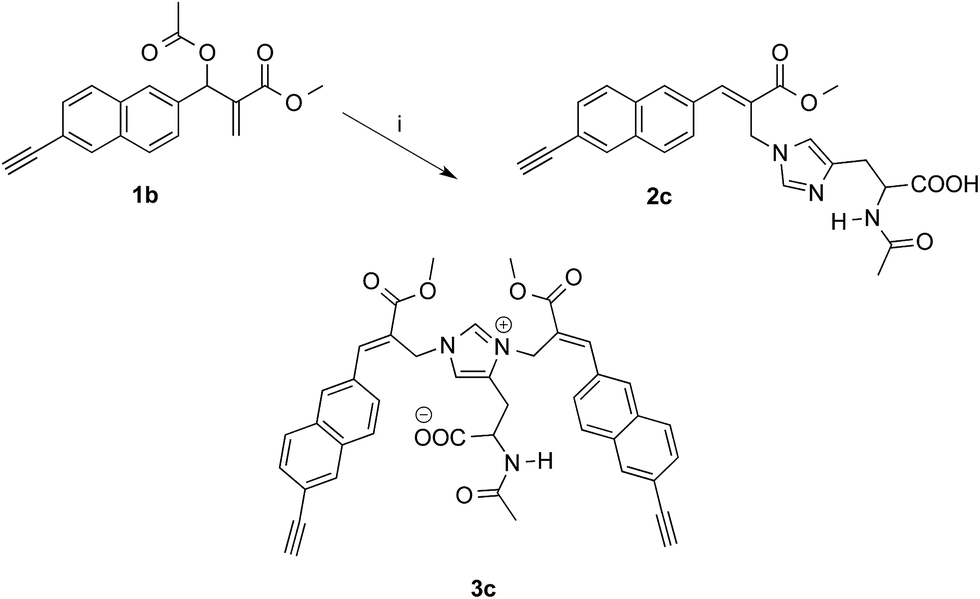 | ||
| Scheme 4 Reaction of MBHA derivative 1b with N-acetylhistidine. Reagents: (i) N-acetylhistidine, CH3CN, PBS. | ||
In the aim of evaluating the reactivity of MBHA derivatives 1a,b in the absence of organic solvents, the structures of these compounds were manipulated by introducing solubilizing groups in the form of oligo(ethylene glycol) (OEG) chains capable of solubilizing the corresponding water-soluble derivatives in PBS.
The first approach to the water-soluble derivatives of compounds 1a,b was based on the use of a solubilizing leaving group containing the undeca(ethylene glycol) side chain as in compounds 1c,d (Scheme 5). These latter MBHA derivatives were synthesized by acylation of 6a,b with commercially available O-(2-carboxyethyl)-O′-methyl-undeca(ethylene glycol) via the corresponding acyl chloride.
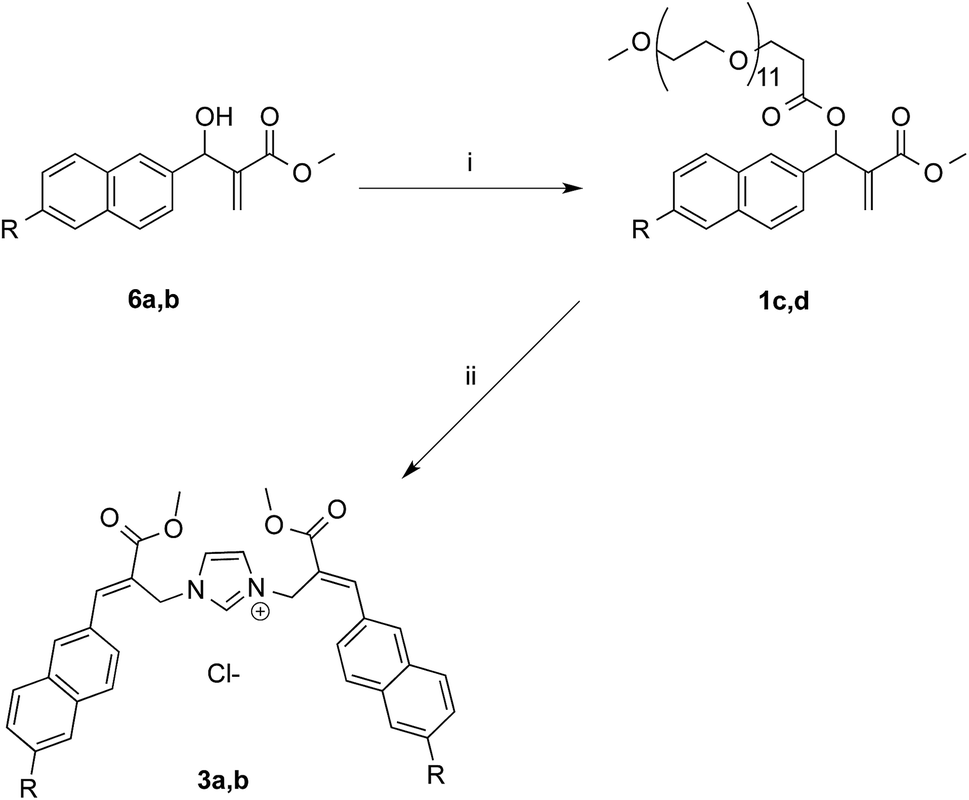 | ||
| Scheme 5 Synthesis of water-soluble MBHA derivatives 1c,d and their reactivity towards imidazole. Reagents: (i) CH3O(CH2CH2O)11CH2CH2COCl, TEA, CH2Cl2; (ii) imidazole, PBS. Substituents: R = OCH2CCH (1c, 3a, 6a); CCH (1d, 3b, 6b). | ||
The reactivity of PEGylated derivatives 1c,d was firstly tested with imidazole in PBS at room temperature. The experiments performed with 1.5 equivalents of imidazole showed the almost exclusive formation of imidazolium salts 3a,b and a slightly higher reactivity of acetylene derivative 1d with respect to propargyloxy derivative 1c.
Owing to the apparently higher reactivity of water soluble 1d, this compound was selected to be used in the preliminary coupling experiments with Ac-His-6 (Scheme 6).
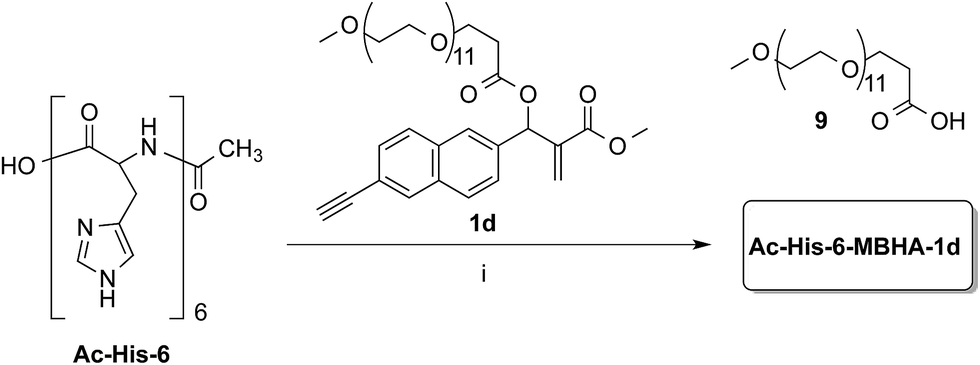 | ||
| Scheme 6 Coupling of water-soluble MBHA derivative 1d with Ac-His-6. Reagents: (i) PBS, D2O. | ||
The experiments were performed in vials containing a PBS medium prepared with deuterium oxide and solid PBS (from Aldrich) in order to follow the reaction trend by NMR spectroscopy by integrating the signals attributed to the methylene group in position 2 of the O-(2-carboxyethyl)-O′-methyl-undeca(ethylene glycol) 9, which was produced by the concerted addition–elimination process. A ten-fold excess of compound 1d was used in the aim of saturating all the possible reactive sites in Ac-His-6 molecules. Initially, the reaction mixtures showed the appearance of clear solutions from which a white precipitate separated in the time course suggesting that an insoluble material was formed. NMR analysis of the reaction mixture at regular time intervals showed that after stirring at room temperature for 24 h Ac-His-6 was almost completely converted, whereas the conversion of 1d was only partial (around 30% of the initial amount). After 48 h at the same temperature, Ac-His-6 was completely converted and the conversion of 1d reached ca. 50%. After one week of stirring at room temperature the conversion of 1d reached 70% that became ca. 85% after three weeks. The polymeric material Ac-His-6-MBHA-1d was extracted with chloroform, purified by flash chromatography, and characterized by mass spectrometry (MS) and NMR spectroscopy techniques.
The MS studies began with the characterization of the starting Ac-His-622 sample by MALDI-TOF and ESI-TOF in positive-ion mode, and the corresponding spectra are displayed in Fig. 3.
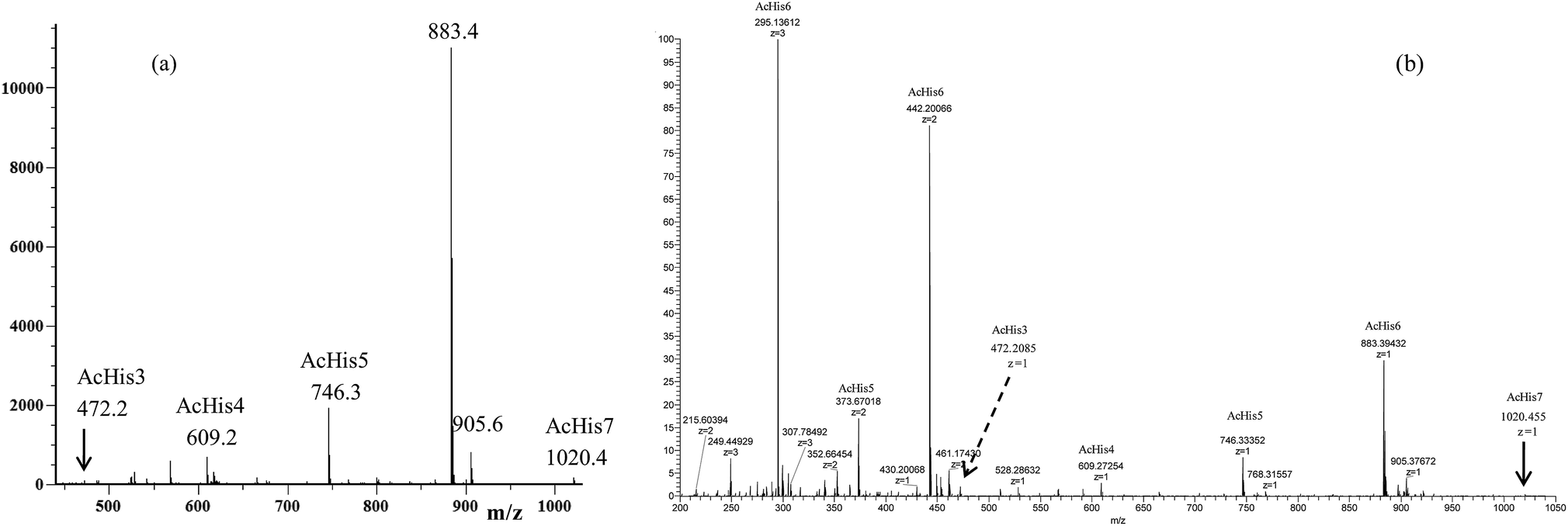 | ||
| Fig. 3 (a) MALDI-TOF mass spectrum (positive-ion mode) and (b) ESI mass spectrum (positive-ion mode) of the starting Ac-His-6 sample. | ||
The MALDI-TOF mass spectrum shown in Fig. 3a was recorded using α-cyano-4-hydroxycinnamic acid as a matrix, and very similar mass spectra were also obtained by using 2-(4-hydroxyphenylazo)benzoic acid or 2,5-dihydroxybenzoic acid matrices. The mass spectra confirmed that the starting Ac-His-6 sample was constituted of a high percentage (i.e. 80%) of N-acetylhexahistidine (m/z 883.4). However, both mass spectra revealed also the presence of N-acetylheptahistidine (Ac-His-7, m/z 1020.4), N-acetylpentahistidine (Ac-His-5, m/z 746.3), N-acetyltetrahistidine (Ac-His-4, m/z 609.2), and N-acetyltrihistidine (Ac-His-3, m/z 472.2) molecules. On the base of the relative intensity of the corresponding peaks present in the MALDI mass spectrum, we calculated the molar composition of the starting Ac-His-6 sample (i.e. Ac-His-7: 1.5%; Ac-His-6: 80%; Ac-His-5: 13%; Ac-His-4: 4.5%; Ac-His-7: 1%). The ESI mass spectrum (Fig. 3b) confirmed the composition of the starting Ac-His-6 sample.
MALDI-TOF-MS measurements were then performed on Ac-His-6-MBHA-1d material and the results (Fig. 4) were compared with those obtained with the starting Ac-His-6 sample.
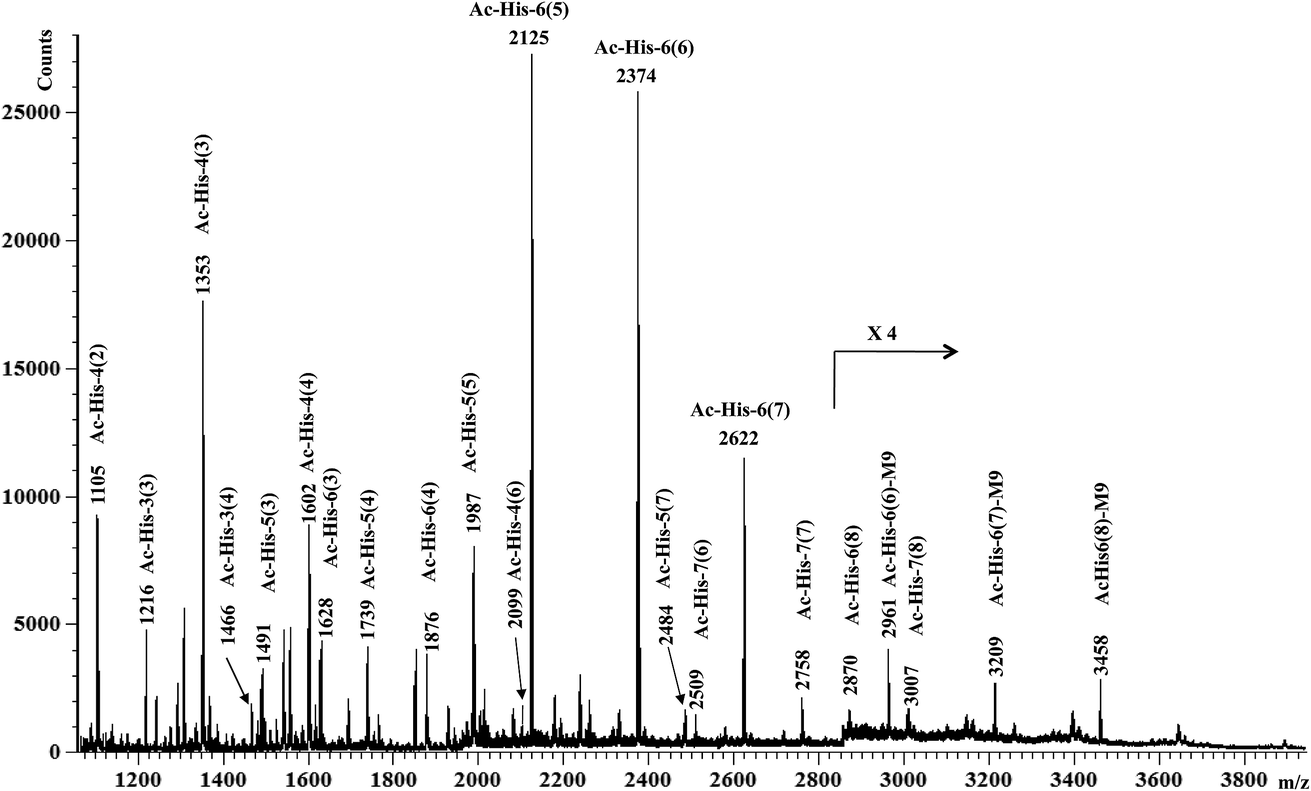 | ||
| Fig. 4 MALDI-TOF mass spectrum (positive-ion mode) of Ac-His-6-MBHA-1d obtained by reaction of Ac-His-6 with a ten-fold excess of 1d. In each assignment, the value in parentheses is the number of naphthalene substituents for each Ac-His molecule. M9 denotes the peaks assigned to the highly substituted species bearing compound 9 as a counterion of the imidazolium salt moieties. | ||
This comparison suggested the existence of a considerable variability in the grafting degree (i.e. the number of the imidazole residues of each Ac-His-6 molecule functionalized by means of the concerted addition–elimination processes with naphthalene substituents). In particular, the peaks at m/z 2125, 2374, 2622, 2870 were assigned to Ac-His-6 molecules bearing 5, 6, 7, and 8 naphthalene substituents, respectively. Furthermore, the peaks at m/z 2622 and 2870 indicated, unequivocally, that at two naphthalene substituents were linked to the same imidazole ring belonging to one His unit of Ac-His-6 molecule, producing imidazolium salt (biadduct) moieties. The formation of biadduct residues was also confirmed by the mass peaks at m/z 2099 and 2484 corresponding to the Ac-His-4 (6) and Ac-His-5 (7), respectively. The mass peaks at m/z 2961, 3209, and 3461 were attributed to the interaction of imidazolium moieties with the anion of compound 9 (Scheme 6), which acted as a counterion and was referred as M9 in the labels reported in Fig. 4. The presence of compound 9 was also confirmed by electrospray ionization mass spectrometry (ESI-MS) analysis in negative mode, since the mass spectrum of Ac-His-6-MBHA-1d sample (Fig. ESI-1, see ESI†) showed an intense peak at 587.3 Da corresponding to molecular mass of the anion.
Interestingly, the most represented macromolecular species appeared to show a grafting degree in the range of 5–7.
NMR spectroscopic studies confirmed the polymeric nature of Ac-His-6-MBHA-1d. In fact, the 1H NMR spectrum (Fig. 5) showed the presence of very broad signals, which were analyzed in comparison with the spectrum of biadduct 3c, which was taken into consideration as a model of the monomeric unity potentially represented into Ac-His-6-MBHA-1d structure.
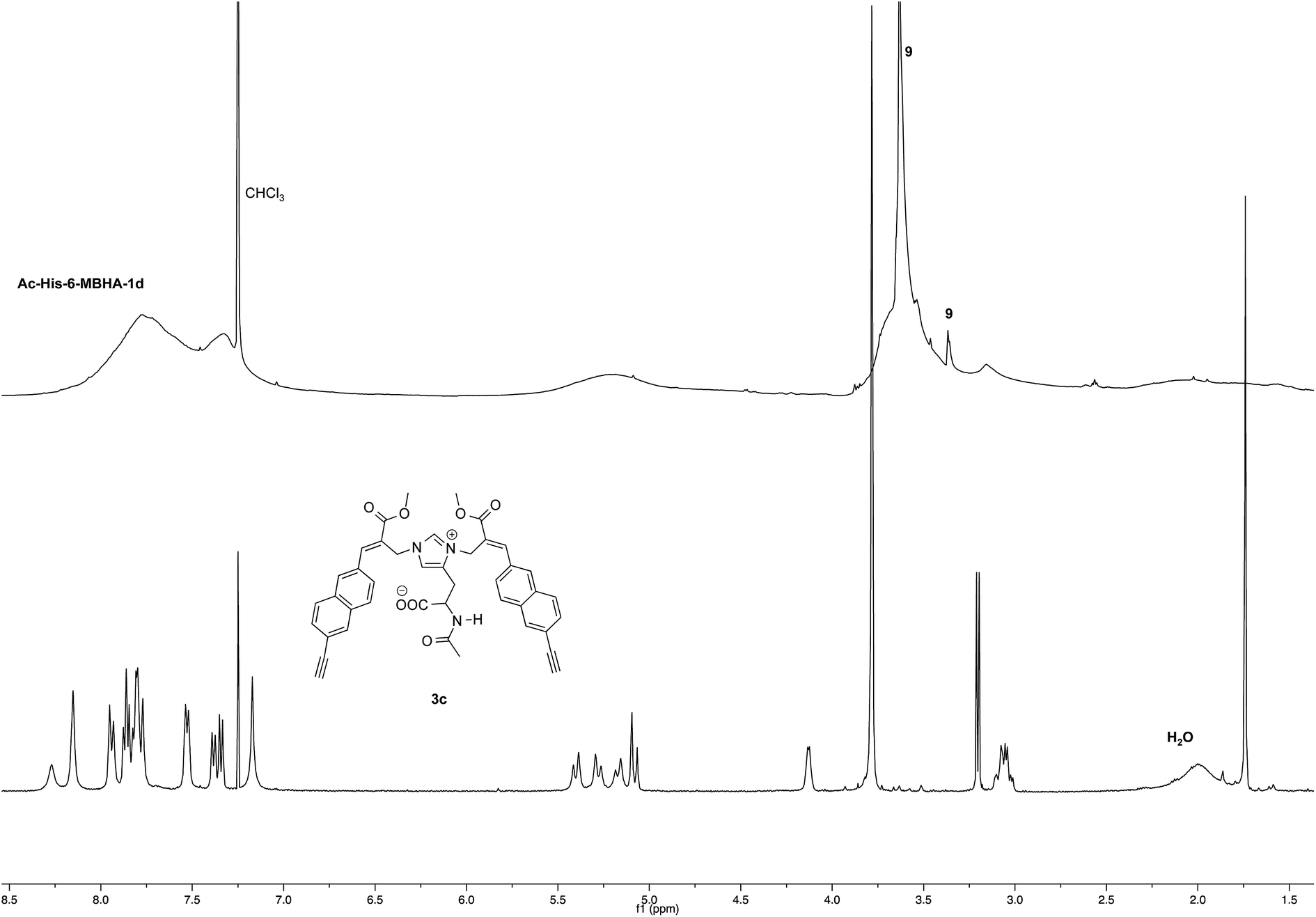 | ||
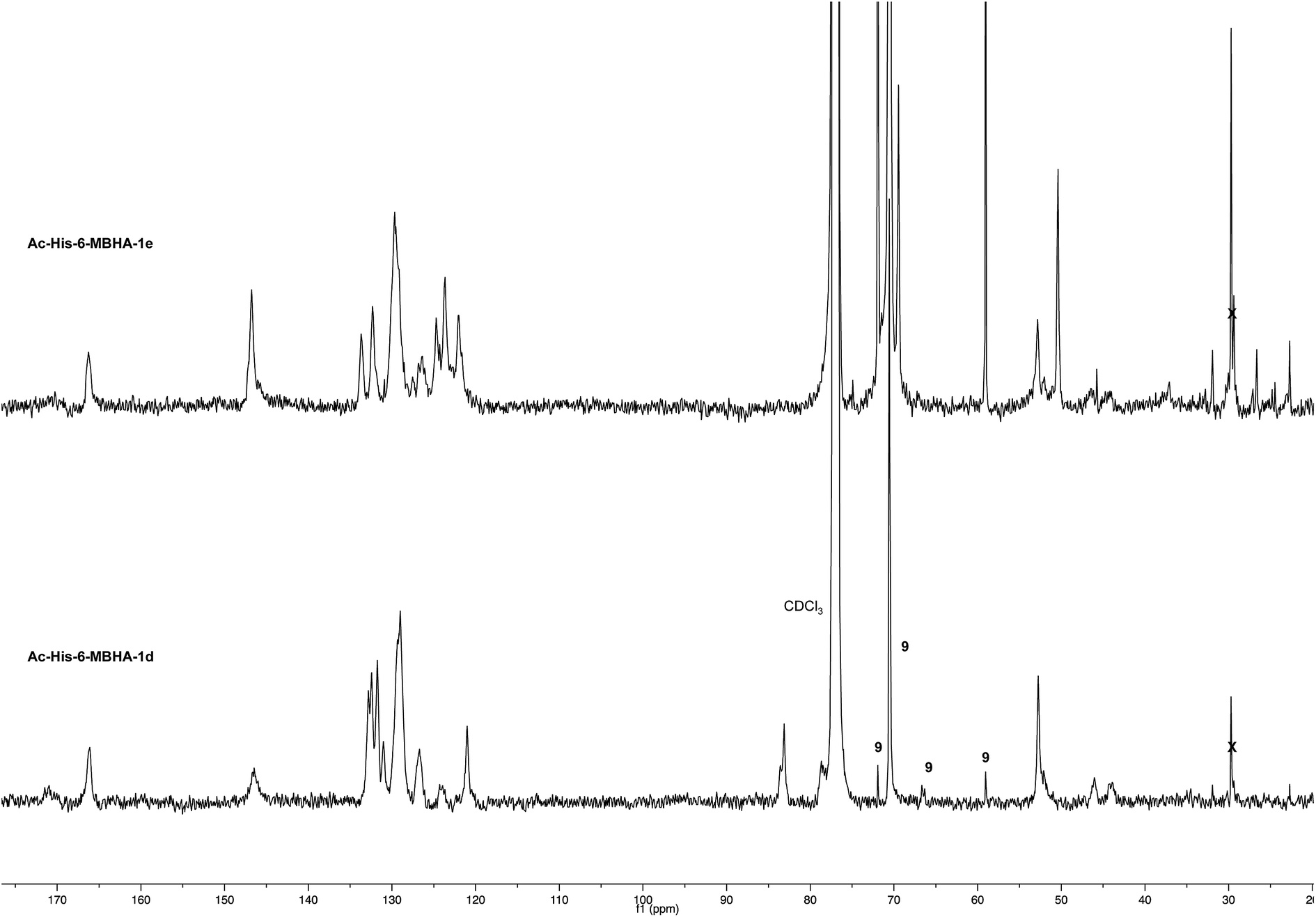
| Fig. 5 1H NMR spectrum (500 MHz, CDCl3) of Ac-His-6-MBHA-1d obtained by reaction of Ac-His-6 with a ten-fold excess of 1d compared with that of biadduct 3c. The peaks marked by 9 were attributed to compound 9, which was produced in the concerted addition–elimination process and remained as a counterion of the imidazolium salt moieties. | ||
Very interestingly, the comparison of the 1H NMR spectrum of the polymeric material with that of biadduct 3c confirmed the presence of a considerable amount of biadduct residues. In fact, a close correspondence appeared to exist between the sharp signals of 3c spectrum and the broad signals in the spectrum of polymeric material of Ac-His-6-MBHA-1d. This apparent correspondence was even more evident in the comparison of 13C NMR spectra shown in Fig. 6.
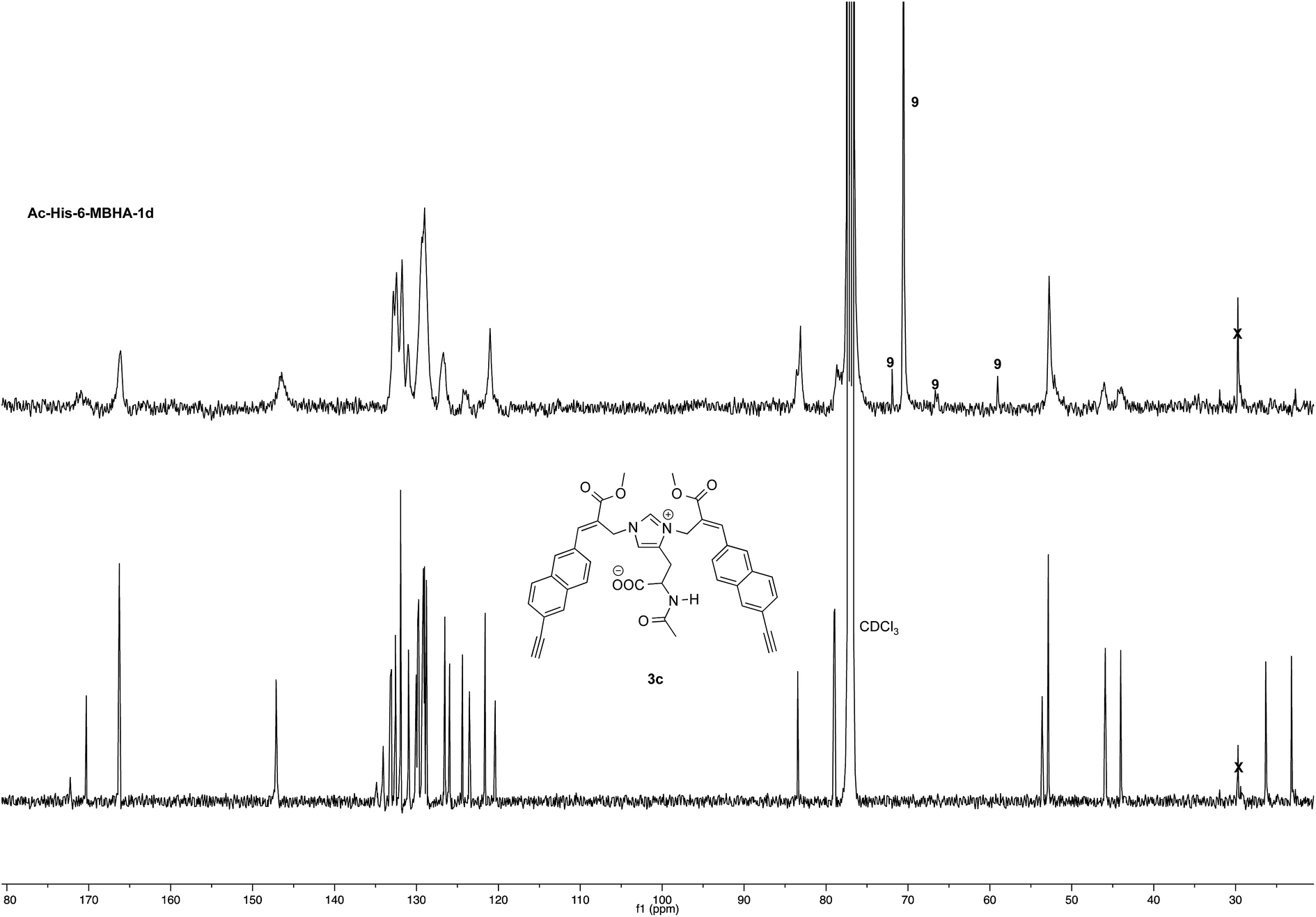 | ||
| Fig. 6 13C NMR spectrum (125, CDCl3) of Ac-His-6-MBHA-1d obtained by reaction of Ac-His-6 with a ten-fold excess of 1d compared with that of biadduct 3c. The peaks marked by 9 were attributed to compound 9, which was produced in the concerted addition–elimination process and remained as a counterion of the imidazolium salt moieties. | ||
As expected, the higher resolution of the 13C NMR spectrum of Ac-His-6-MBHA-1d allowed a more profitable signal-to-signal comparison to be made. In particular in the spectrum of biadduct 3c, the signals at 44.0 and 45.9 ppm attributed to the methylene bridges directly bound to the imidazole moieties of histidine appeared in Ac-His-6-MBHA-1d spectrum as two slightly broader peaks showing very similar chemical shift values (i.e. 44.1 and 46.0 ppm). This correspondence strongly supported the large preponderance of biadduct residues with respect to monoadduct ones. More in general, the close correspondence between the 13C NMR spectrum of Ac-His-6-MBHA-1d with the corresponding one of biadduct 3c confirmed this latter compound as the model of the main monomeric unit of Ac-His-6-MBHA-1d.
In the aim of investigate on the role of OEG side chain in the reactivity of these MBHA derivatives, the position of the solubilizing moiety was shifted from the leaving group in the close proximity of the reactive center to the naphthalene ring as in compound 1e (Scheme 7). This structural modification was assumed to have a considerable impact in the reactivity of MBHA derivative 1e. Moreover, the solubilizing group remained covalently attached to resulting adduct after the concerted addition–elimination process.
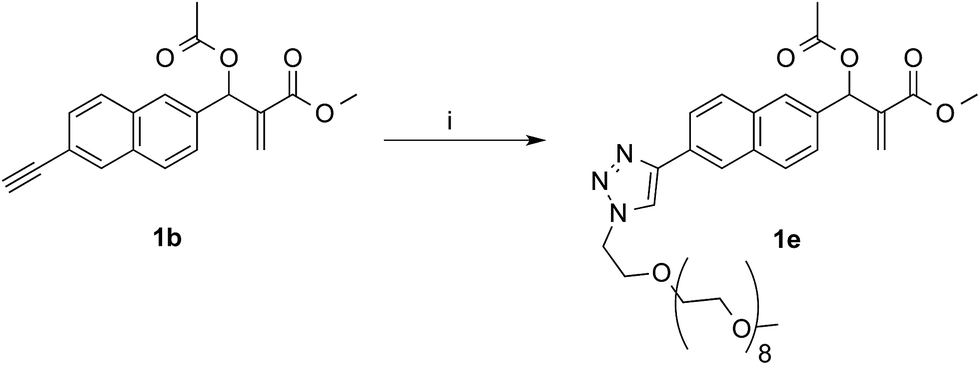 | ||
| Scheme 7 Synthesis of water-soluble MBHA derivative 1e. Reagents: (i) CH3(OCH2CH2)9N3, CuSO4, sodium ascorbate, tert-butanol, PBS. | ||
Compound 1b was reacted with O-(2-azidoethyl)-O′-methyl-octa(ethylene glycol) in the conditions of copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) in tert-butanol–PBS to afford triazole derivative 1e, which was found to be sufficiently soluble and stable in PBS to be used in the coupling experiments with Ac-His-6 (Scheme 8).
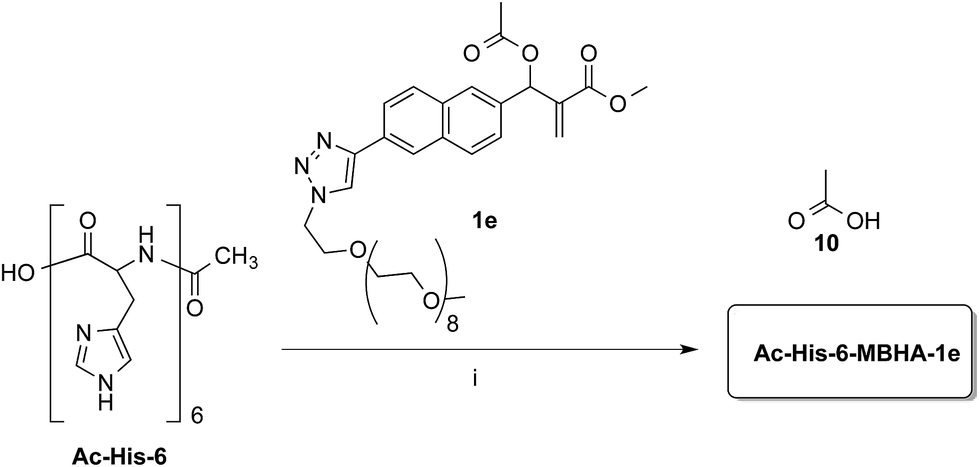 | ||
| Scheme 8 Coupling of water-soluble MBHA derivative 1e with Ac-His-6. Reagents: (i) PBS, D2O. | ||
Experiments with a ten-fold excess of compound 1e were carried out in order to saturate all the possible reactive sites in Ac-His-6 molecules with PEGylated fluorogens and the reaction progress was monitored by NMR spectroscopy.
After 24 h at room temperature, the 1H NMR experiments (Fig. 7) demonstrated that 1e was partially (about 40%) converted, while Ac-His-6 was completely converted into a soluble polymeric material.
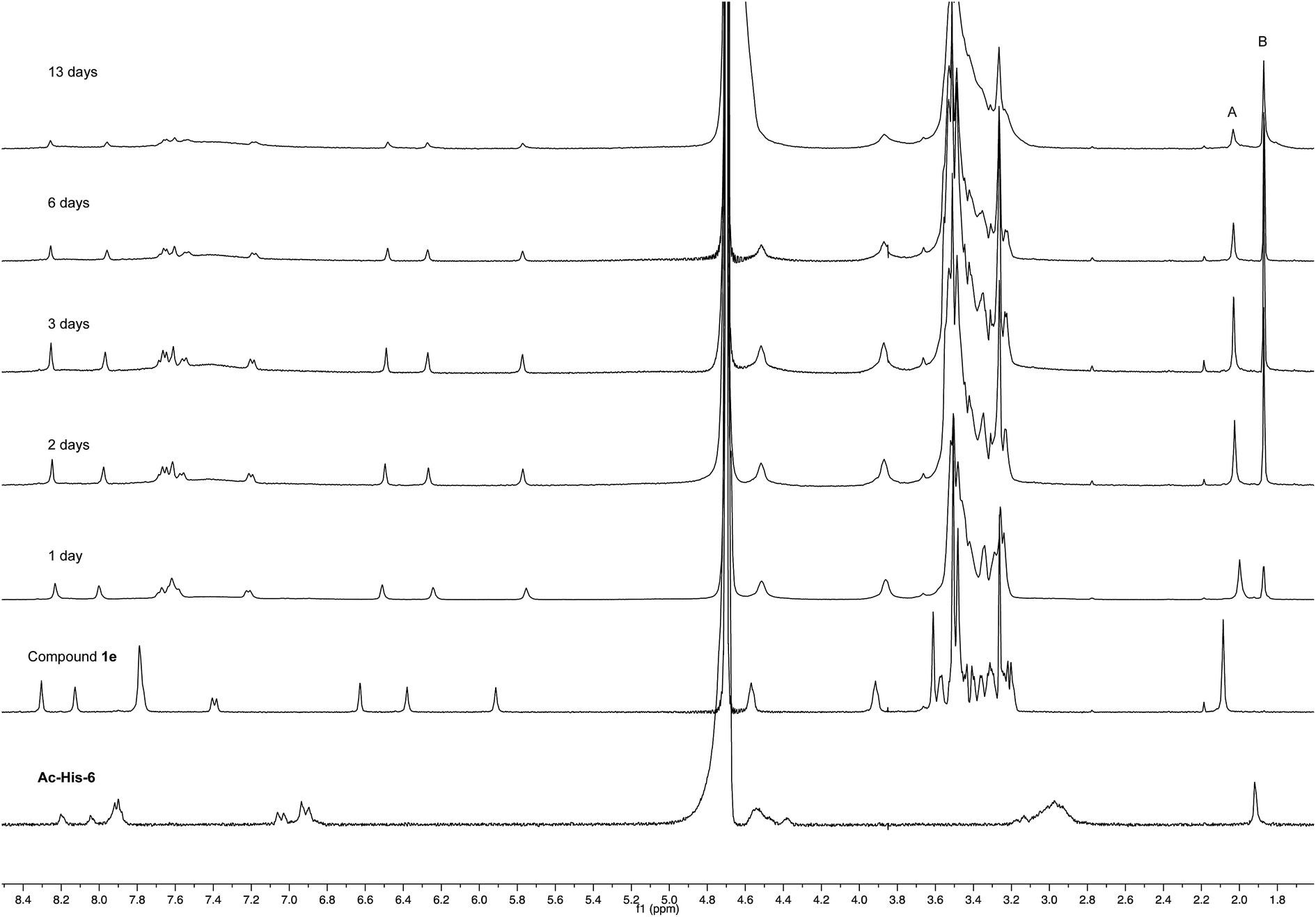 | ||
| Fig. 7 1H NMR spectra (400 MHz, PBS-D2O) of the mixtures obtained by reaction of Ac-His-6 with a ten-fold excess of compound 1e. The peaks marked with A (attributed to the acetyl moiety of 1e) and B (attributed to acetate 10 produced by the concerted addition–elimination process) were used to follow the reaction progress. | ||
After 48 h (two days) at room temperature, the conversion reached about 60% suggesting that each histidine residue could be attacked by 1e. After one-two weeks, the reaction reached a plateau characterized by around 70% conversion of 1e. These results suggested that when the solubilizing OEG chain was shifted from the leaving group in the close proximity of the reactive center (as in 1d) to the naphthalene tail (as in 1e) the reactivity increased, but the persistence of the OEG side chain attached to Ac-His-6 backbone produced by the reaction of 1e decreased the number of the sites available for functionalization. In fact, Ac-His-6 appeared to tolerate a mean of about 6–7 electrophilic attack events by compound 1e, as confirmed by the mass spectrum of polymeric material Ac-His-6-MBHA-1e (Fig. 8).
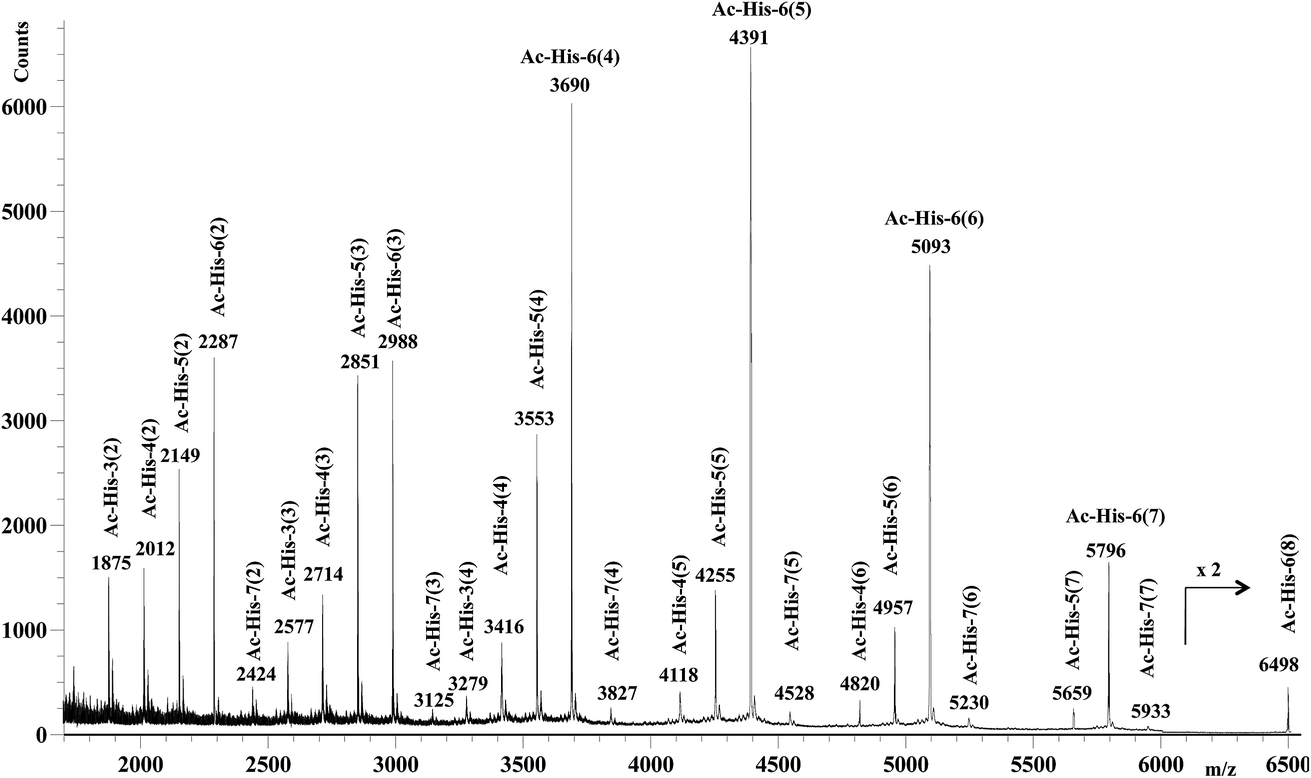 | ||
| Fig. 8 MALDI-TOF mass spectrum (positive-ion mode) of the polymeric material Ac-His-6-MBHA-1e obtained by reaction of Ac-His-6 with a ten-fold excess of compound 1e. The polymerization degree of the corresponding Ac-His molecule is reported for each monoisotopic mass peak along with the grafting degree (i.e. the number of naphthalene substituents for each Ac-His molecule) noted in parentheses. | ||
Fig. 8 shows the mass spectrum recorded in positive ion mode using 2,5-dihydroxybenzoic acid as a matrix, but very similar mass spectra were also obtained using 2-(4-hydroxyphenylazo)benzoic acid or α-cyano-4-hydroxycinnamic acid as the matrix. Similarly to the results obtained with Ac-His-6-MBHA-1d, the peaks at m/z 4820 [Ac-His-4 (6)], 5659 [Ac-His-5 (7)], and 6498 [Ac-His-6 (8)] supported the formation of biadducts (imidazolium moieties) since two naphthalene substituents derived from reagent 1e (Scheme 8) were indubitably linked to the same imidazole ring belonging to the relevant Ac-His molecule.
The comparison of 13C NMR spectrum of Ac-His-6-MBHA-1e with the corresponding one obtained with Ac-His-6-MBHA-1d (Fig. 9) confirmed a similar reactivity in spite of the difference in the position of the solubilizing moiety. In fact, the diagnostic peaks attributed to the methylene bridges directly bound to the imidazole moieties of Ac-His-6 backbone showed roughly the same chemical shift values in Ac-His-6-MBHA-1e spectrum (44.5 and 46.6) as Ac-His-6-MBHA-1d spectrum (44.1 and 46.0 ppm). Thus, the NMR spectroscopic studies supported the preponderance of biadducts in Ac-His-6-MBHA-1e as well as in Ac-His-6-MBHA-1d.
 | ||
| Fig. 9 13C NMR spectrum (CDCl3) of Ac-His-6-MBHA-1e obtained by reaction of Ac-His-6 with a ten-fold excess of 1e compared with that of Ac-His-6-MBHA-1d obtained by reaction of Ac-His-6 with a ten-fold excess of 1d. The peaks marked by 9 were attributed to compound 9, which was produced in the concerted addition–elimination process and remained as a counterion of the imidazolium salt moieties. | ||
Thus, if the first electrophilic attack was assumed to occur randomly, the subsequent attacks could be governed by the propensity to the formation of biadducts featuring the characteristics of an imidazolium salt, which could be stabilized by the amphoteric properties of the vicinal imidazole or by the presence of suitable anions. This mechanism led to conceive the existence, within a heterogeneous polymeric population deriving from the randomness of the first attack, of molecular entities showing a more organized structure characterized by the presence of biadduct sequences as in a fishbone-like architecture (Fig. 10). However, the presence of this architecture in longer poly-histidine derivatives could be affected by their conformational preferences and their liability to form coil-like structures, which may hamper the formation and/or decrease the stability of biadduct residues. This could be particularly relevant in compound 1e because the additional steric bulk of the OEG side chain.
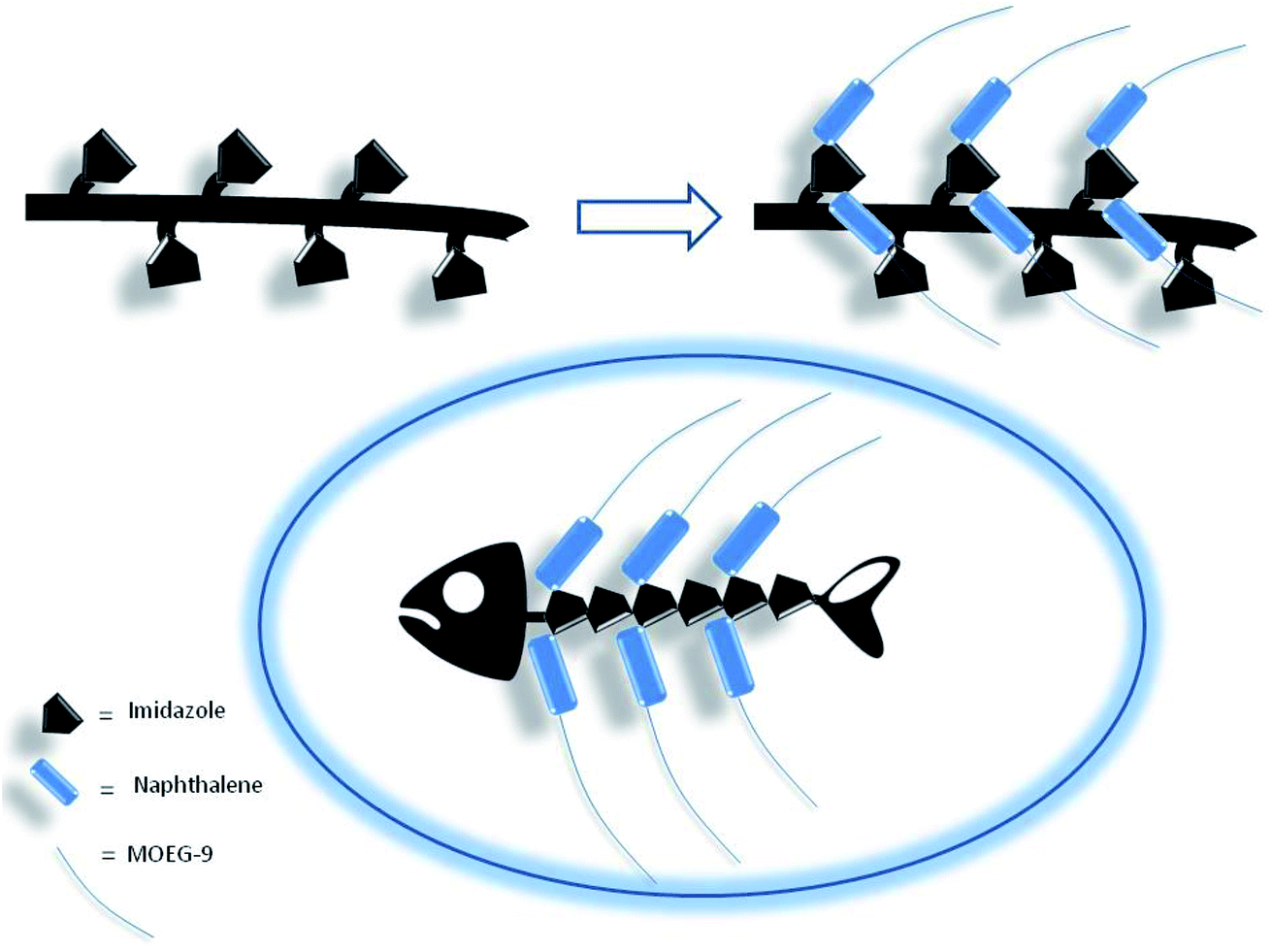 | ||
| Fig. 10 Fishbone-like architecture assumed for some organized sequences of poly-histidine derivatives grafted with naphthalene derivatives. | ||
These results may have intriguing implications in the application of MBHA in different fields of covalent poly-histidine modification/conjugation such as in vivo fluorogenic labeling, fluorescence microscopy, and protein PEGylation up to the production of smart materials and biosensors, and pave the way for the preparation of fishbone-like polymer brushes and the exploration of their properties.
Photophysical properties
The UV-visible absorption and emission features of imidazole derivatives 2a–d were characterized both in solution (i.e. methanol as the solvent) and in the solid state and compared in Table 1 with those of biadducts 3a–c and polymeric materials Ac-His-6-MBHA-1d and Ac-His-6-MBHA-1e.
|
|||||
|---|---|---|---|---|---|
| Compd | Solutiona | Solidb | |||
| λab (nm) | λemc (nm) | PLQY (%) | λem (nm)d | PLQY (%) | |
| a Methanol.b Powders.c λexc = 320 nm.d λexc = 340 nm.e Compound 2d was synthesized as a model compound from 2b as described in the Experimental section.f λexc = 460 nm. | |||||
| 2a | 266, 320 | 423 | 0.7 | 428 | 15 |
| 2b | 263, 315 | 404 | 4.4 | 454 | 15 |
| 2c | 264, 318 | 406 | 0.3 | 438 | 11 |
| 2de | 260, 318 | 430 | 1.1 | 465 | 23 |
| 3a | 270, 325 | 437 | 0.8 | 465 | 12 |
| 3b | 260, 318 | 420 | 1.1 | 490 | 10 |
| 3c | 265, 320 | 420 | 1.0 | 520 | 12 |
| Ac-His-6-MBHA-1d | 265, 319 | 425 | 1.5 | 570f | 3 |
| Ac-His-6-MBHA-1e | 260, 329 | 470 | 0.2 | 570f | 7 |
As shown in Table 1 all the naphthalene-based push–pull structures in solution display absorption bands at about 320 nm and 260 nm, with a quite weak photoluminescence (PL). A significant increase in PL quantum yield (QY) of the solution was obtained by transforming the propargyloxy group of 2a into the acetylene one of 2b. Interestingly, for all the compounds the low PLQY displayed by the solutions increased in the solid state with a parallel red shift, as it generally occurs in a typical AIE phenomenon.23 (see Fig. 11–14).
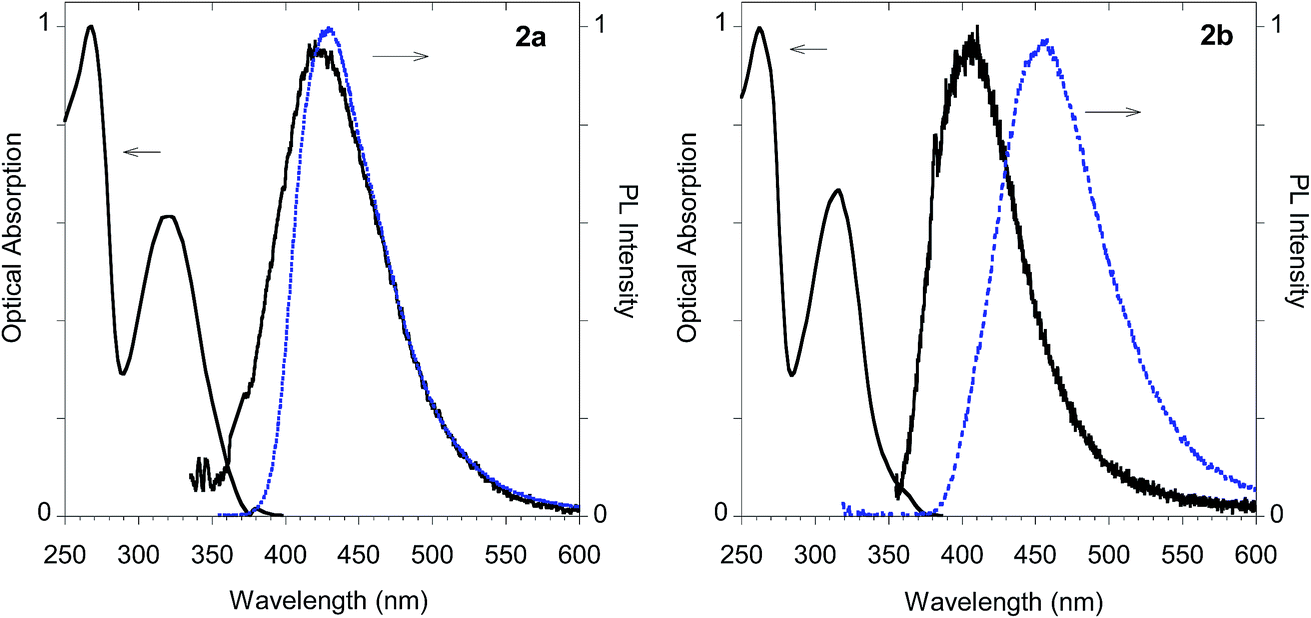 | ||
| Fig. 11 Optical properties of imidazole derivative 2a (left) and 2b (right). Normalized absorption and emission spectra obtained in methanol (black solid line) and emission spectrum of the powder (dashed blue line). | ||
The highest PLQY value of 23% in the solid state is obtained by transformation of the acetylene moiety of 2b into the triazole moiety of the model compound 2d. In fact, powders of the triazole derivative 2d showed a sharp emission switch on in the solid state, with bright blue emissive powders, as shown in Fig. 12.
 | ||
| Fig. 12 Optical properties of imidazole derivative 2d. Left panel: normalized absorption and emission spectra obtained in methanol (black solid line) and emission spectrum of the powder (dashed blue line). Right panel: fluorescence microscopy image (290 μm width) obtained with solid 2d. | ||
The imidazolium salts 3a and 3b conserved the interesting emission features and the AIE behavior shown by parent compounds 2a and 2b (Fig. 13).
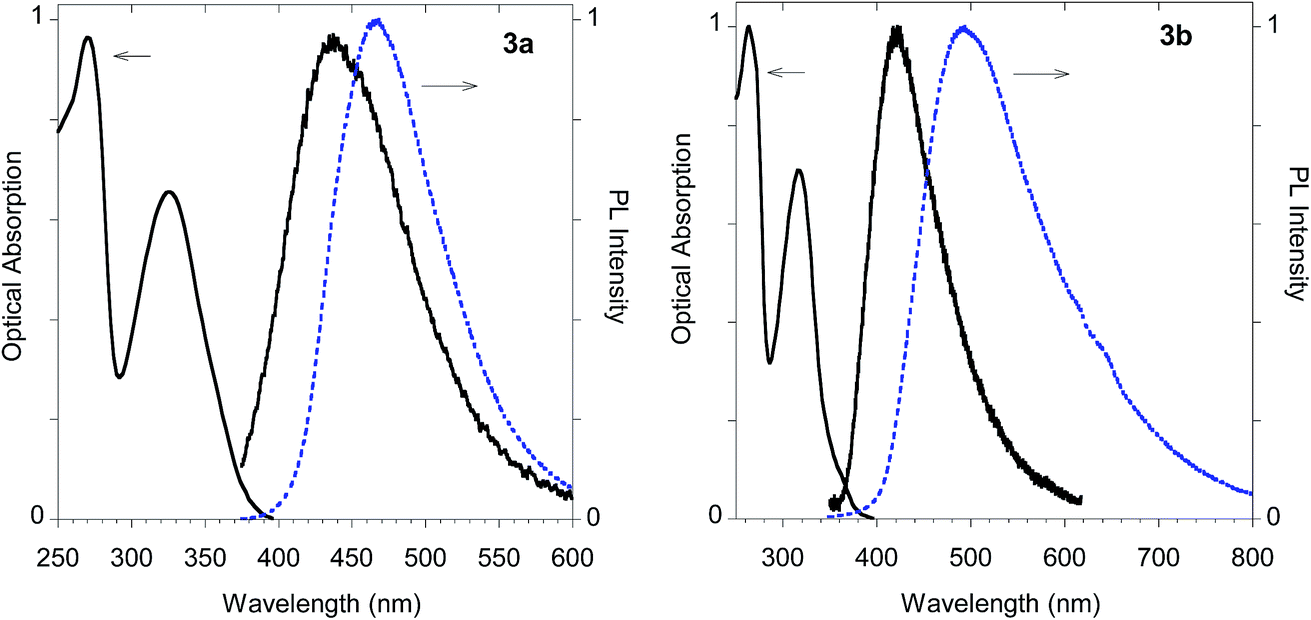 | ||
| Fig. 13 Optical features of imidazolium salts 3a and 3b. Normalized absorption and emission spectra obtained in methanol (black solid line) and emission spectrum of the powder (dashed blue line). | ||
On the other hand, significant discrepancies were observed in the couple of histidine derivatives 2c and 3c (Fig. 14).
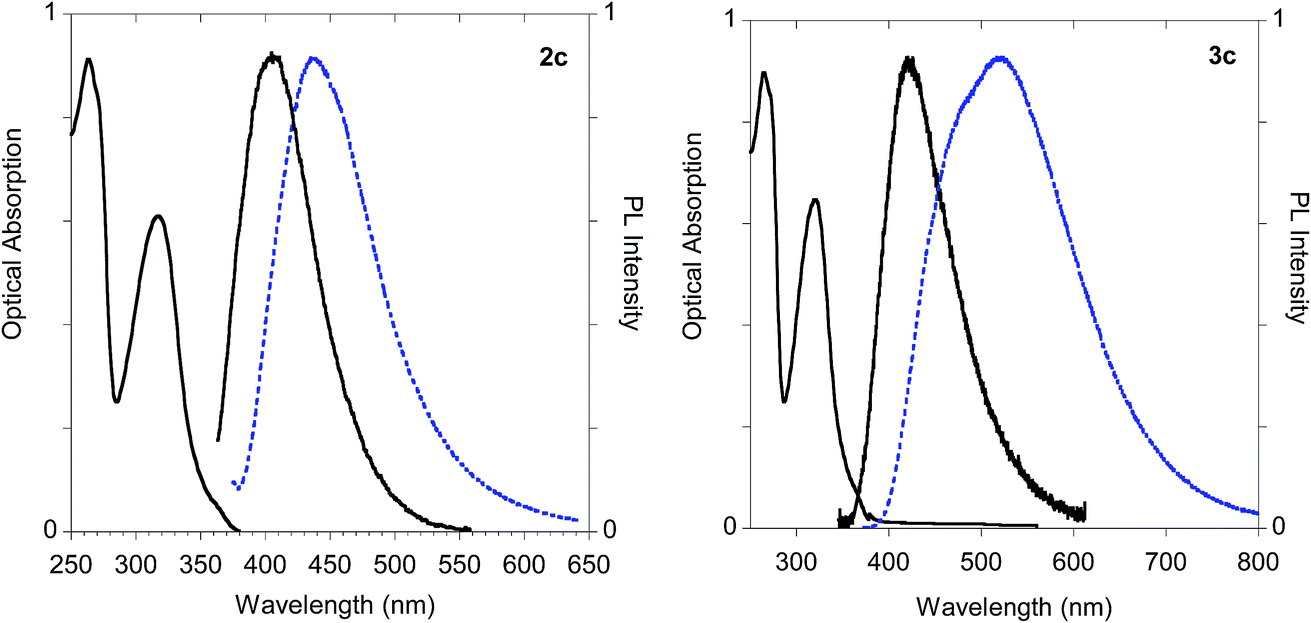 | ||
| Fig. 14 Optical features of histidine derivatives 2c and 3c. Normalized absorption and emission spectra obtained in methanol (black solid line) and emission spectrum of the powder (dashed blue line). | ||
In fact, monoadduct 2c showed a particularly low PLQY value (i.e. 0.3%) in solution and a moderate (i.e. 32 nm) red shift in the solid-state emission, whereas the corresponding biadduct 3c showed emission features in the solution aligned with those obtained with the other compounds and a peculiar (i.e. 100 nm) red shift in the solid state emission up to 520 nm. Interestingly, a similar red shift was observed in the polymeric materials Ac-His-6-MBHA-1d and Ac-His-6-MBHA-1e (Fig. 15), the latter showed excitation dependent emissions, suggesting the existence of different emitting species (see ESI†). These results confirm the assumption on the presence of biadduct sequences in the polymeric materials Ac-His-6-MBHA-1d and Ac-His-6-MBHA-1e as in a fishbone-like architecture.
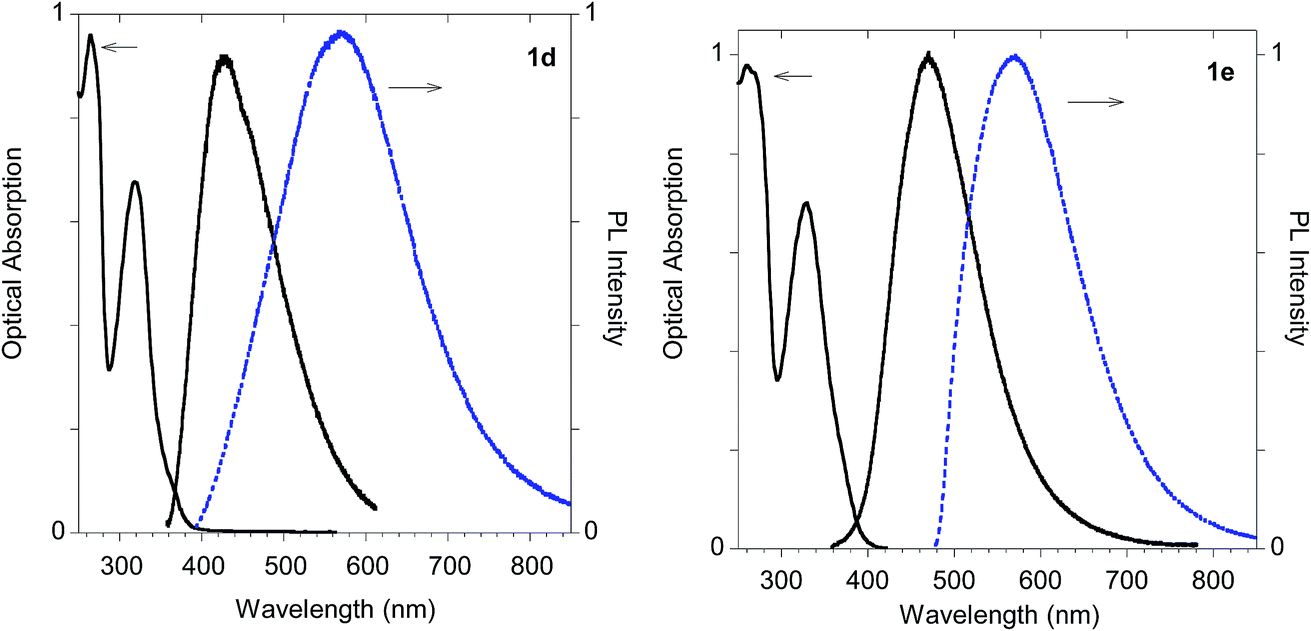 | ||
| Fig. 15 Optical features of polymeric materials Ac-His-6-MBHA-1d and Ac-His-6-MBHA-1e. Normalized absorption and emission spectra obtained in methanol (black solid line) and emission spectrum of the powder (dashed blue line). | ||
Conclusions
In order to explore the synthesis, the structure, and the properties of graft copolymers of poly-histidine with fluorescent molecules, a small series of Morita–Baylis–Hillman adduct (MBHA) derivatives was synthesized, and made to react with imidazole in order to evaluate their reactivity and the photophysical properties of the corresponding imidazole derivatives. Intriguingly, the reaction of MBHA derivatives 1a,b with 1.5 equivalents of imidazole in acetonitrile–phosphate buffered saline (PBS) as the reaction medium gave, along the expected mono-adducts 2a,b, the biadducts featuring the characteristics of imidazolium salt derivatives 3a,b as the main reaction products. These results were confirmed by experiments performed with N-acetylhistidine and 1b and suggest the possible occurrence of these structures in the products of poly-histidine labeling with MBHA derivatives 1a,b. These compounds were then transformed into the corresponding water-soluble derivatives 1c–e by introducing solubilizing groups in the form of oligo(ethylene glycol) chains in the leaving group in the close proximity of the reactive center (as in compounds 1c,d) or attached to the naphthalene moiety, far from the reactive center as in compound 1e. The solubility of these PEGylated MBHA derivatives in water allowed their reactivity to be evaluated in PBS, and, after some preliminary experiments with imidazole, compounds 1d,e were coupled with Ac-His-6. The coupling experiments were performed with ten-fold overloads of compounds 1d,e in the aim of saturating all the possible reactive sites of Ac-His-6 molecules in the polymeric materials Ac-His-6-MBHA-1d and Ac-His-6-MBHA-1e. The monitorization of the reaction progress by NMR spectroscopy suggested a higher reactivity of 1e with respect to the 1d probably caused by the steric bulk of the solubilizing OEG moiety near the reactive center of 1d. However, the most reactive 1e was observed to produce a lower grafting degree with respect to 1d, probably caused by the persistence of the solubilizing OEG chains attached to Ac-His-6 backbone of Ac-His-6-MBHA-1e. The structure of the polymeric materials Ac-His-6-MBHA-1d and Ac-His-6-MBHA-1e was investigated by mass spectrometry and NMR spectroscopy studies, which suggested the presence of biadduct residues in both the polymeric materials. These results pave the way for the preparation of fishbone-like polymer brushes, the characterization of their properties, and the exploration of their potential applications in different fields of the science such as in vivo fluorogenic labeling, fluorescence microscopy, protein PEGylation, up to the production of smart materials and biosensors. For example, the PEGylation of engineered proteins bearing terminal polyhistidine sequences with compound 1e could lead to the formation of PEG umbrella at the desired sequence end potentially capable of solubilizing, protecting, and detecting the engineered protein.The analysis of the photophysical features showed that a significant emission was observed in all the naphthalene derivatives 2a–d, and imidazolium salts 3a–c, all featuring the aggregation-induced emission (AIE) phenomenon, with photoluminescence quantum yields in the solid state sensitively higher than the corresponding values measured in solution. Interestingly, biadduct 3c showed a peculiar (i.e. 100 nm) red shift in the solid state emission up to 520 nm. Since a similar red shift was observed in the polymeric materials Ac-His-6-MBHA-1d and Ac-His-6-MBHA-1e, these results appeared to confirm the assumption about the presence of biadduct sequences in the polymeric materials as in a fishbone-like architecture.
Experimental section
Chemistry
All chemicals used were of reagent grade. Yields refer to purified products and are not optimized. Melting points were determined in open capillaries on a Gallenkamp apparatus and are uncorrected. Merck silica gel 60 (230–400 mesh) was used for column chromatography. Merck TLC plates, silica gel 60 F254 were used for TLC. NMR spectra were recorded by means of either a DRX 400 AVANCE or a Bruker DRX 500 AVANCE spectrometer in the indicated solvents (TMS as internal standard); the values of the chemical shifts (δ) are expressed in ppm and the coupling constants (J) in Hz. Mass spectra were recorded on an Agilent 1100 LC/MSD.:1) as the eluent to obtain compound 5 (0.13 g, yield 76%) as a white solid melting at 107–108 °C. 1H NMR (500 MHz, CDCl3): 2.58 (t, J = 2.4, 1H), 4.85 (d, J = 2.4, 2H), 7.26–7.30 (m, 2H), 7.83 (d, J = 8.5, 1H), 7.91–7.94 (m, 2H), 8.27 (s, 1H), 10.10 (s, 1H). 13C NMR (125 MHz, CDCl3): 55.9, 76.2, 77.9, 107.7, 120.0, 123.7, 128.0, 128.3, 131.3, 132.7, 134.2, 137.9, 158.0, 192.0. MS (ESI) m/z: [M + Na]+ calcd for C14H10O2Na 233.1; found 233.1.:1) containing Pd(PPh3)2Cl2 (21 mg, 0.0299 mmol) and CuI (6.0 mg, 0.0315 mmol) was added ethynyltrimethylsilane (90 μL, 0.64 mmol). After stirring overnight at room temperature, the reaction mixture was concentrated under reduced pressure. The residue was dissolved in dichloromethane and washed with water. The organic layer was dried over sodium sulfate and concentrated under reduced pressure. The residue was purified by flash chromatography with petroleum ether–ethyl acetate (9:1) as the eluent to afford 6-[(trimethylsilyl)ethynyl]-2-naphthaldehyde (88 mg, yield 82%), which was promptly used in the subsequent step. 1H NMR (400 MHz, CDCl3): 0.28 (s, 9H), 7.59 (dd, J = 8.4, 1.6, 1H), 7.83–7.98 (m, 3H), 8.03 (s, 1H), 8.29 (s, 1H), 10.14 (s, 1H).A mixture of 6-[(trimethylsilyl)ethynyl]-2-naphthaldehyde (80 mg, 0.317 mmol) and potassium carbonate (4.0 mg, 0.0289 mmol) in methanol (5.0 mL) was stirred overnight at room temperature and then concentrated under reduced pressure. The resulting residue was partitioned between dichloromethane and water. The organic layer was dried over sodium sulfate and concentrated under reduced pressure. Purification of the residue by flash chromatography with petroleum ether–ethyl acetate (9:1) as the eluent gave compound 8 (56 mg, yield 98%) as a white solid. 1H NMR (500 MHz, CDCl3): 3.24 (s, 1H), 7.62 (dd, J = 8.5, 1.65, 1H), 7.89 (d, J = 8.5, 1H), 7.94–7.98 (m, 2H), 8.06 (s, 1H), 8.31 (s, 1H), 10.15 (s, 1H). 13C NMR (125 MHz, CDCl3): 79.3, 83.3, 122.8, 123.7, 128.9, 129.6, 129.8, 132.2, 134.0, 134.7, 135.8, 192.0. MS (ESI) m/z: [M + Na]+ calcd for C13H8ONa 203.1; found 203.1.
:2) as the eluent to obtain compound 6a (0.26 g, yield 37%) as a colorless oil, which crystallized on standing (mp 77–78 °C). 1H NMR (400 MHz, CDCl3): 2.54 (t, J = 2.4, 1H), 3.03 (d, J = 5.6, 1H), 3.71 (s, 3H), 4.80 (d, J = 2.4, 2H), 5.71 (d, J = 5.5, 1H), 5.87 (s, 1H), 6.36 (s, 1H), 7.18 (dd, J = 8.9, 2.6, 1H), 7.22 (d, J = 2.2, 1H), 7.44 (dd, J = 8.7, 1.7, 1H), 7.71–7.80 (m, 3H). 13C NMR (125 MHz, CDCl3): 52.0, 55.9, 73.4, 75.7, 78.4, 107.3, 119.0, 125.2, 125.4, 126.3, 127.3, 129.1, 129.8, 133.9, 136.8, 141.9, 155.7, 166.8. MS (ESI) m/z: [M + Na]+ calcd for C18H16O4Na 319.1; found 319.0.:2:1) as the eluent to obtain compound 6b (0.49 g, yield 61%) as a pale yellow oil. 1H NMR (500 MHz, CDCl3): 3.14 (s, 1H), 3.18 (br s, 1H), 3.72 (s, 3H), 5.71 (s, 1H), 5.86 (t, J = 1.1, 1H), 6.38 (s, 1H), 7.48 (dd, J = 8.5, 1.7, 1H), 7.51 (dd, J = 8.4, 1.5, 1H), 7.77 (d, J = 8.4, 2H), 7.83 (s, 1H), 7.99 (s, 1H). 13C NMR (125 MHz, CDCl3): 52.1, 73.3, 77.6, 83.9, 119.6, 125.4, 126.7, 128.1, 128.2, 128.9, 132.0, 132.4, 132.9, 139.8, 141.6, 166.8. MS (ESI) m/z: [M + Na]+ calcd for C17H14O3Na 289.1; found 289.0.:2) as the eluent to afford the corresponding Morita–Baylis–Hillman acetate (1a,b).:1) containing imidazole (1.5 equivalents) was heated under reflux overnight. After cooling to room temperature, the reaction mixture was diluted with brine and extracted with dichloromethane. The organic layer was dried over sodium sulfate and concentrated under reduced pressure. Purification of the residue by flash chromatography with ethyl acetate as the eluent gave the corresponding imidazole derivative (2a,b).Compound 2a was also prepared by means of procedure B as it follows. A mixture of 1a (50 mg, 0.148 mmol) in acetonitrile (5.0 mL) and water (6.0 mL) containing phosphate buffered saline (PBS, pH 7.4) powder (Aldrich, 60 mg) and imidazole (60 mg, 0.89 mmol) was stirred at room temperature for 24 h, then diluted with brine, and extracted with chloroform. The organic layer was dried over sodium sulfate and concentrated under reduced pressure. The residue was purified by flash chromatography with ethyl acetate as the eluent to afford compound 2a (41 mg, yield 80%).
Compound 2b was also prepared by means of procedure B as it follows. A mixture of 1b (50 mg, 0.162 mmol) in acetonitrile (5.0 mL) and water (6.0 mL) containing phosphate buffered saline (PBS, pH 7.4) powder (Aldrich, 60 mg) and imidazole (66 mg, 0.97 mmol) was stirred at room temperature for 24 h, then diluted with brine, and extracted with chloroform. The organic layer was dried over sodium sulfate and concentrated under reduced pressure. The residue was purified by flash chromatography with ethyl acetate as the eluent to afford compound 2b (47 mg, yield 92%).
:1) to obtain compound 3a as a white solid (23 mg, yield 47%). An analytical sample of compound 3a was prepared by recrystallization from chloroform–methanol (5:1) by slow evaporation to obtain X-ray quality colorless plates (mp 190 °C dec). 1H NMR (400 MHz, CDCl3): 2.55 (t, J = 2.4, 2H), 3.82 (s, 6H), 4.79 (d, J = 2.4, 4H), 5.58 (s, 4H), 7.00 (s, 2H), 7.18–7.23 (m, 4H), 7.46–7.51 (m, 2H), 7.82–7.93 (m, 6H), 8.20 (s, 2H), 10.86 (s, 1H). 13C NMR (125 MHz, CDCl3): 46.2, 52.9, 55.9, 76.1, 78.0, 107.3, 120.1, 121.0, 122.8, 126.5, 128.2, 128.4, 128.8, 130.2, 130.7, 135.1, 139.3, 147.5, 157.1, 166.7. MS (ESI) m/z: [M]+ calcd for C39H33N2O6 625.2; found 625.2.:1) to obtain compound 3b as an off-white solid (30 mg, yield 62%). An analytical sample of compound 3b was prepared by recrystallization from chloroform–methanol (5:1) by slow evaporation to a pale yellow microcrystalline powder (mp 135 °C dec). 1H NMR (400 MHz, CDCl3): 3.20 (s, 2H), 3.83 (s, 6H), 5.55 (s, 4H), 6.96 (s, 2H), 7.48 (d, J = 8.2, 2H), 7.53 (d, J = 8.3, 2H), 7.86 (d, J = 8.4, 2H), 7.95–7.99 (m, 6H), 8.20 (s, 2H), 10.87 (s, 1H). 13C NMR (125 MHz, CDCl3): 46.1, 53.0, 78.8, 83.5, 121.1, 121.5, 124.3, 126.5, 129.0, 129.1, 129.5, 129.8, 131.0, 131.9, 132.6, 133.1, 139.4, 146.8, 166.3. MS (ESI) m/z: [M]+ calcd for C37H29N2O4 565.2; found 565.2.:1) as the eluent gave compound 2d (78 mg, yield 98%) as a pale yellow oil, which crystallized on standing. 1H NMR (500 MHz, CDCl3): 3.32 (s, 3H), 3.51–3.53 (m, 2H), 3.61–3.68 (m, 6H), 3.84 (s, 3H), 3.93 (t, J = 5.0, 2H), 4.63 (t, J = 4.9, 2H), 5.06 (s, 2H), 6.93 (s, 1H), 7.06 (s, 1H), 7.42 (dd, J = 8.5, 1.5, 1H), 7.54 (s, 1H), 7.76 (s, 1H), 7.87 (d, J = 8.6, 1H), 7.93 (d, J = 8.5, 1H), 7.99 (dd, J = 8.5, 1.6, 1H), 8.16 (s, 1H), 8.18 (s, 1H), 8.38 (s, 1H). 13C NMR (125 MHz, CDCl3): 43.2, 50.5, 52.6, 59.0, 69.5, 70.5, 71.9, 118.8, 121.7, 124.0, 125.1, 126.3, 126.9, 129.1, 129.4, 129.9, 131.4, 132.6, 133.7, 137.1, 145.0, 147.2, 167.1. MS (ESI) m/z: [M + H]+ calcd for C27H32N5O5 506.2; found 506.2.:1) as the eluent to afford compound 3c as an off-white solid (90 mg, yield 80%, mp 140 °C dec). 1H NMR (500 MHz, CDCl3): 1.74 (s, 3H), 3.01–3.11 (m, 2H), 3.20 (s, 1H), 3.21 (s, 1H), 3.78 (s, 6H), 4.10–4.15 (m, 1H), 5.08 (d, J = 14.6, 1H), 5.17 (d, J = 14.6, 1H), 5.28 (d, J = 15.0, 1H), 5.40 (d, J = 15.0, 1H), 7.17 (s, 2H), 7.34 (d, J = 8.8, 1H), 7.38 (d, J = 8.5, 1H), 7.53 (d, J = 8.2, 2H), 7.77–7.88 (m, 6H), 7.93 (s, 1H), 7.95 (s, 1H), 8.15 (s, 2H), 8.27 (s, 1H). 13C NMR (125 MHz, CDCl3): 23.2, 26.3, 44.0, 45.9, 52.9, 53.6, 79.0, 83.5, 120.4, 121.6, 123.5, 124.4, 126.0, 126.5, 128.8, 129.0, 129.1, 129.2, 129.7, 129.9, 130.0, 130.9, 131.0, 131.9, 132.5, 132.6, 133.0, 133.2, 134.1, 134.9, 147.1, 166.3, 170.3, 172.3. MS (ESI) m/z: [M + H]+ calcd for C42H36N3O7 694.3; found 693.7.Compound 2c was obtained from the above flash chromatography purification (as a more polar fraction) by using dichloromethane–methanol (8:2) as the eluent (8.0 mg, yield 5.5%). An analytical sample of compound 2c was prepared by recrystallization from methanol by slow evaporation to obtain rosettes of white needles (mp 180 °C dec). 1H NMR (500 MHz, DMSO-d6): 1.71 (s, 3H), 2.64–2.69 (m, 1H), 2.82–2.92 (m, 1H), 3.75 (s, 3H), 4.12–4.23 (m, 1H), 4.37 (s, 1H), 4.98 (s, 2H), 6.74 (s, 1H), 7.47 (s, 1H), 7.53–7.67 (m, 3H), 7.91–8.02 (m, 3H), 8.06 (s, 1H), 8.14 (s, 1H). 13C NMR (125 MHz, DMSO-d6): 23.2, 31.3, 43.1, 52.9, 54.0, 82.6, 84.0, 116.2, 120.9, 127.6, 127.8, 128.8, 129.5, 129.7, 131.9, 132.6, 132.7, 132.9, 137.0, 139.4, 143.7, 167.1, 169.0, 176.3. MS (ESI) m/z: [M + H]+ calcd for C25H24N5O5 446.2; found 445.8.
:5) as the eluent afforded compound 1c (245 mg, yield 67%) as a pale yellow oil. 1H NMR (400 MHz, CDCl3): 2.54 (t, J = 2.3 Hz, 1H), 2.58–2.70 (m, 2H), 3.37 (s, 3H), 3.52–3.78 (m, 49H), 4.79 (d, J = 2.3 Hz, 2H), 5.96 (s, 1H), 6.41 (s, 1H), 6.82 (s, 1H), 7.17 (dd, J = 8.8, 2.4, 1H), 7.20 (d, J = 2.0, 1H), 7.44 (d, J = 8.4, 1H), 7.69–7.76 (m, 3H). 13C NMR (125 MHz, CDCl3): 35.3, 52.0, 55.8, 59.1, 66.6, 70.4, 70.6, 71.9, 73.4, 75.8, 78.3, 107.3, 119.1, 125.8, 126.0, 127.1, 127.3, 128.9, 129.9, 133.2, 134.2, 139.4, 155.9, 165.4, 170.1. MS (ESI) m/z: [M + Na]+ calcd for C44H66NaO17 889.4; found 889.6.:1) to obtain compound 3a as a white solid (3.5 mg, yield 92%).:5) as the eluent to afford compound 1d (204 mg, yield 57%) as a colorless oil. 1H NMR (500 MHz, CDCl3): 2.66 (m, 2H), 3.15 (s, 1H), 3.37 (s, 3H), 3.53–3.66 (m, 44H), 3.69 (s, 3H), 3.75 (t, J = 6.3, 2H), 5.98 (s, 1H), 6.44 (s, 1H), 6.83 (s, 1H), 7.47–7.52 (m, 2H), 7.76 (d, J = 8.4, 2H), 7.82 (s, 1H), 7.98 (s, 1H). 13C NMR (125 MHz, CDCl3): 35.3, 52.1, 59.1, 66.6, 70.4, 70.6, 71.9, 73.3, 77.8, 83.8, 119.9, 126.0, 126.2, 127.0, 128.2, 128.3, 129.0, 132.0, 132.6, 132.7, 136.3, 139.2, 165.3, 170.1. MS (ESI) m/z: [M + Na]+ calcd for C43H64NaO16 859.4; found 858.8.:1) to obtain compound 3b as an off-white solid (14 mg, yield 78%).:2.5:2.5) for 3 h. Also the Nim-trityl group was removed during the release from the resins. The resin was removed by filtration, and the crude peptide was purified by precipitation in cold anhydrous diethyl ether from a solution containing the minimum amount of methanol to give Ac-His-6 as a white powder. 1H NMR (400 MHz, D2O – PBS): Fig. 16. MS: Fig. 3.
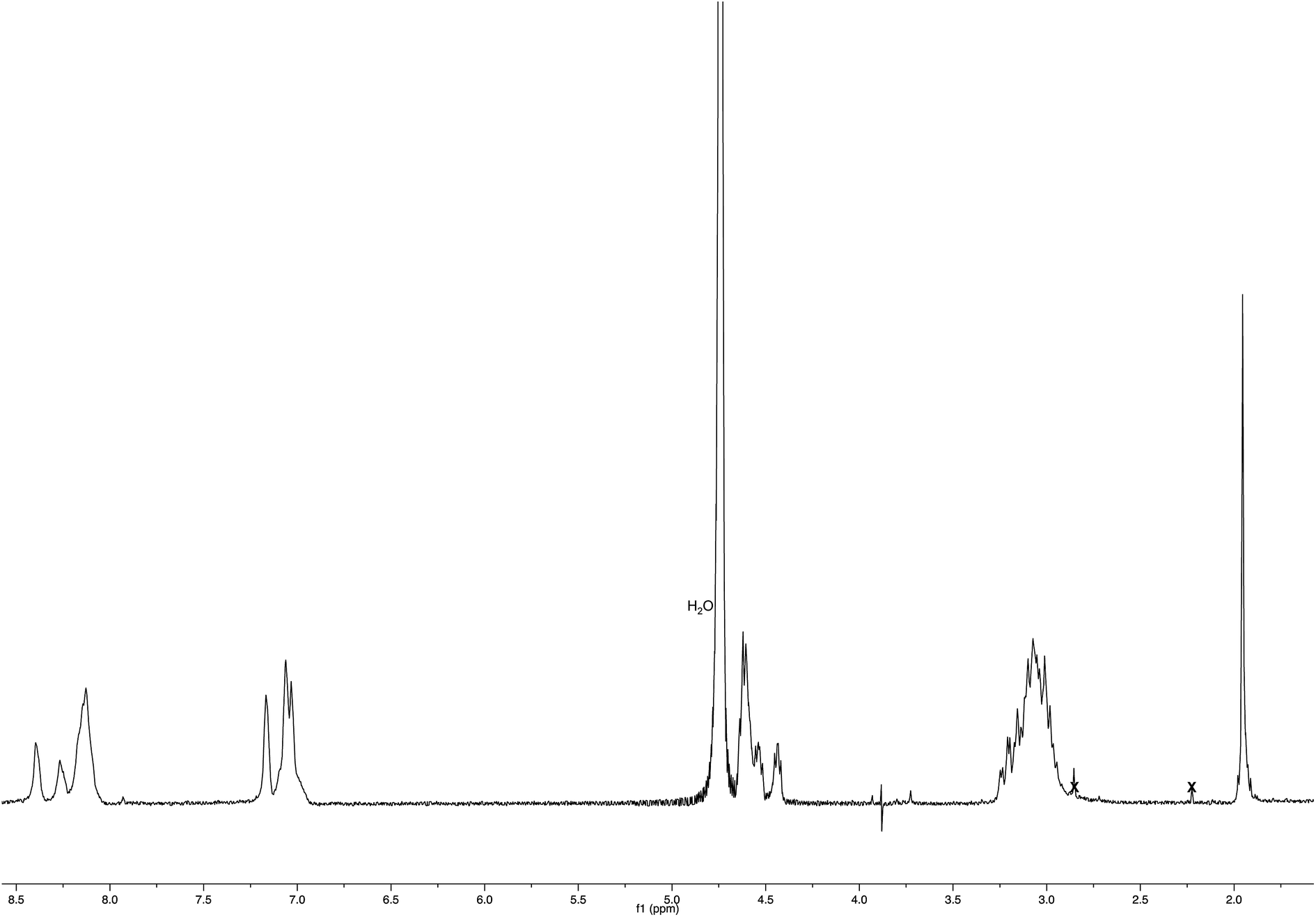 | ||
| Fig. 16 1H NMR spectrum (400 MHz, D2O – PBS) of Ac-His-6. | ||
:2) to obtain polymeric material Ac-His-6-MBHA-1d as a brown glassy solid (10.5 mg). 1H NMR (500 MHz, CDCl3): Fig. 5. 13C NMR (125 MHz, CDCl3): Fig. 6. MS: Fig. 4 and ESI-1.†:1) as the eluent to afford compound 1e (120 mg, yield 72%) as a pale yellow oil.1H NMR (500 MHz, CDCl3): 2.13 (s, 3H), 3.36 (s, 3H), 3.50–3.54 (m, 2H), 3.58–3.64 (m, 30H), 3.70 (s, 3H), 3.92–3.94 (m, 2H), 4.60–4.63 (m, 2H), 5.94 (s, 1H), 6.45 (s, 1H), 6.84 (s, 1H), 7.47 (dd, J = 8.5, 1.6, 1H), 7.83–7.88 (m, 3H), 7.94 (dd, J = 8.5, 1.4, 1H), 8.12 (s, 1H), 8.34 (s, 1H). 13C NMR (125 MHz, CDCl3): 21.2, 50.5, 52.1, 59.1, 69.5, 70.5, 71.9, 73.2, 121.5, 124.0, 124.4, 125.8, 126.0, 127.0, 128.6, 128.7, 128.8, 132.7, 133.4, 135.3, 139.5, 147.5, 165.4, 169.5. MS (ESI) m/z: [M + Na]+ calcd for C38H55N3NaO13 784.4; found 784.3.
:2) to obtain polymeric material Ac-His-6-MBHA-1e as a brown gummy solid (25 mg). 1H NMR (400 MHz, CDCl3): Fig. 17. 13C NMR (125 MHz, CDCl3): Fig. 9. MS: Fig. 8.
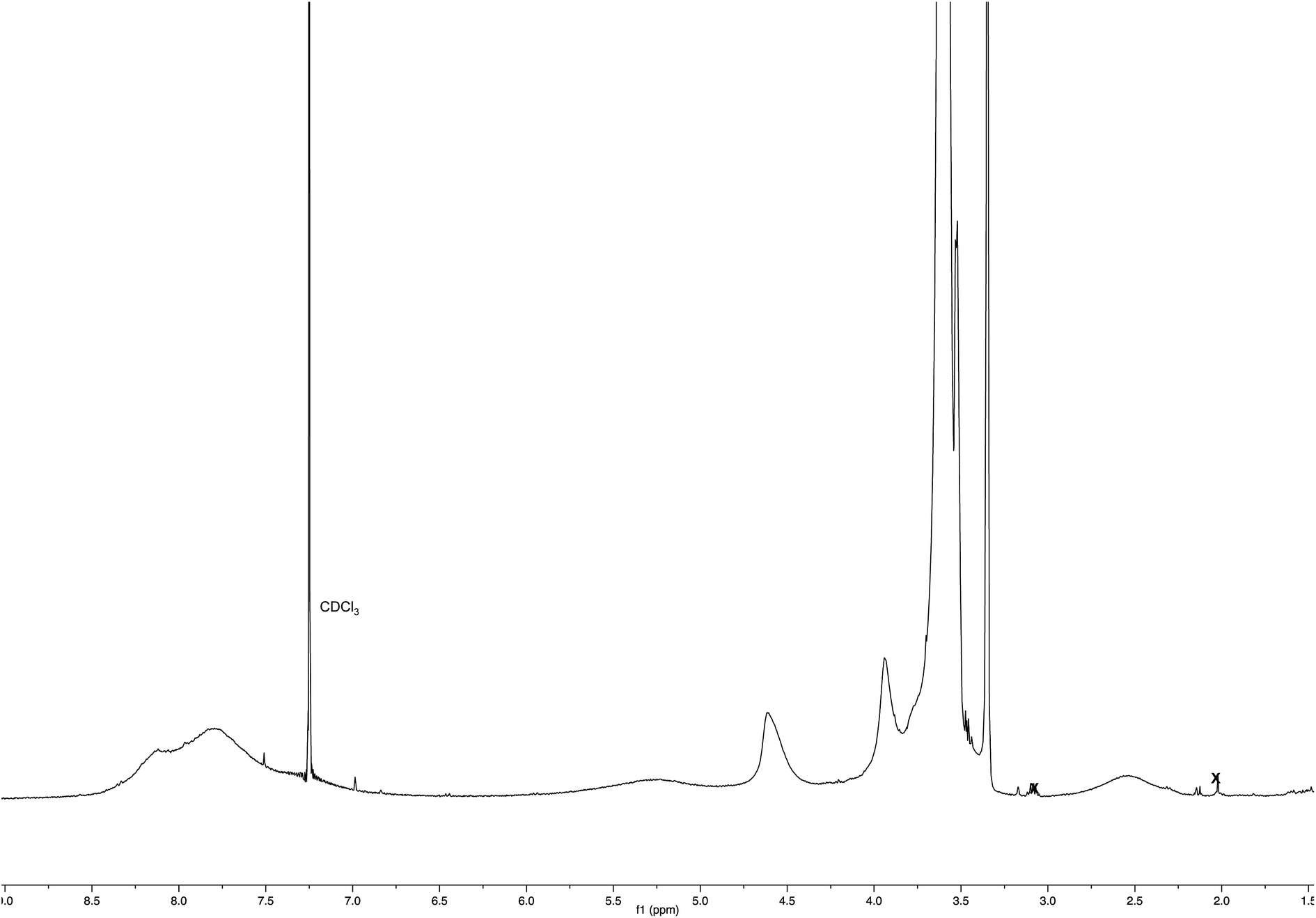 | ||
| Fig. 17 1H NMR spectrum (CDCl3) of Ac-His-6-MBHA-1e obtained by reaction of Ac-His-6 with a ten-fold excess of 1e. | ||
X-ray crystallography
A single crystal of compound 3a was submitted to X-ray data collection on an Oxford-Diffraction Xcalibur Sapphire 3 diffractometer with a graphite monochromated Mo-Kα radiation (λ = 0.71073 Å) at 293 K. The structure was solved by direct methods implemented in SHELXS-97 program.24 The refinement was carried out by full-matrix anisotropic least-squares on F2 for all reflections for non-H atoms by means of the SHELXL-97 program.25 Crystallographic data (excluding structure factors) have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 1504772.†Mass spectrometry measurements
MALDI-TOF mass spectra were recorded both in reflectron and linear mode by means of a 4800 Proteomic Analyzer (Applied Biosystems) MALDI-TOF/TOF instrument equipped with a Nd:YAG laser at a wavelength of 355 nm with <500 ps pulse and 200 Hz firing rate. The accelerating voltage was 20 kV. External calibration was performed using an Applied Biosystems calibration mixture consisting of polypeptides with different molar mass values. The irradiance was maintained slightly above the threshold, to obtain in reflectron mode a mass resolution of about 4000–6000 FWHM (isotopic resolution was observed in the mass range from m/z 300 up to m/z 5000). Mass accuracy was about 50–100 ppm. All measurements were performed in positive ion mode. Approximately 1500 laser shots were accumulated for each mass spectrum in reflectron mode, and 250 laser shots in linear one too. For the analysis of all the samples, several matrices such as 2-(4-hydroxyphenylazo)benzoic acid, 2,5-dihydroxybenzoic acid, and α-cyano-4-hydroxycinnamic acid were used. The best spectra were obtained using α-cyano-4-hydroxycinnamic and 2,5-dihydroxybenzoic acid, matrices, and operating in reflectron mode.ESI mass spectra were acquired by a Thermo Scientific Exactive Plus Orbitrap MS (Thermo Fisher Scientific, San Jose, CA), using a heated electrospray ionization (HESI II) interface. Mass spectra were recorded operating both in positive and negative ion mode in the m/z range 200–4000 at a resolving power of 25000 (full-width-at-half-maximum, at m/z 300, RFWHM), resulting in a scan rate of >1.5 scans per sec when using automatic gain control target of 1.0 × 106 and a C-trap injection time of 100 ms. under the following conditions in positive mode: capillary temperature 300 °C, heater temperature 300 °C, nebulizer gas (nitrogen) with a flow rate of 20 arbitrary units; auxiliary gas flow rate of 3 arbitrary units; source voltage 3.5 kV; capillary voltage 50 V; tube lens voltage 150 V, skimmer voltage 23 V. The same negative values of source voltage, capillary voltage, tube lens voltage and skimmer voltage, were used in negative ion mode. 1 μL of each sample was injected with autosampler into the mass spectrometer, using methanol/acetonitrile (50:50, v/v) as solvent at a flow rate of 100 μL min−1. The Orbitrap MS system was tuned and calibrated in: (a) positive modes, by infusion of solutions of a standard mixture of caffeine (MM 194.1 Da), MRFA peptide (MM 423.6 Da) and ultramark-mixture 1621; (b) negative ion mode by infusion of a solution of a standard mixture of sodium dodecyl sulfate (MM 265.3), sodium taurocholate (MM 514.5) and ultramark-mixture 1621. Data acquisition and analysis were performed using of the Xcalibur software.
Optical properties and photoluminescence
UV-vis absorption spectra are obtained with a Perkin Elmer Lambda 900 spectrometer. PL spectra are obtained with a SPEX 270 M monochromator equipped with a N2 cooled charge-coupled device exciting with a monochromated 450 W Xe lamp. The spectra are corrected for the instrument response. PL QY values of solutions were obtained by using quinine sulfate as the reference, with an experimental error of about 5% for values below 0.1%. PL QY of solid powders were measured with a home-made integrating sphere, with an experimental error of 10% and a sensitivity of about 0.1%, according to the procedure reported elsewhere.26Conflicts of interest
The authors declare no competing financial interest.References
- E. A. Barnard and W. D. Stein, The Roles of Imidazole in Biological Systems, Advances in Enzymology and Related Areas of Molecular Biology, ed, F. F. Nord, Wiley, 2006, vol. 20; pp. 51–110 Search PubMed.
- E. S. Shin, H. J. Lee, K. Na and Y. H. Bae, J. Controlled Release, 2003, 90, 363–374 CrossRef.
- E. S. Lee, K. Na and Y. H. Bae, Nano Lett., 2005, 5, 325–329 CrossRef CAS PubMed.
- R. J. Sundberd and R. B. Martin, Chem. Rev., 1974, 74, 471–517 CrossRef.
- D. Mavrogiorgis, P. Bilalis, A. Karatzas, D. Skoulas, G. Fotinogiannopoulou and H. Iatrou, Polym. Chem., 2014, 5, 6256–6278 RSC.
- H. S. Hwang, J. Hu, K. Na and Y. H. Bae, Biomacromolecules, 2014, 15, 3577–3586 CrossRef CAS PubMed.
- X. Zhang, D. Chen, S. Ba, J. Zhu, J. Zhang, W. Hong, X. Zhao, H. Hu and M. Qiao, Biomacromolecules, 2014, 15, 4032–4045 CrossRef CAS PubMed.
- M. Casolaro, Y. Ito, T. Ishii, S. Bottari, F. Samperi and R. Mendichi, Polym. Lett., 2008, 2, 165–183 CrossRef CAS.
- R. P. Johnson, S. Uthaman, J. V. John, H. R. Lee, S. J. Lee, H. Park, I.-K. Park, H. Suh and I. Kim, ACS Appl. Mater. Interfaces, 2015, 7, 21770–21779 CAS.
- M. L. Read, S. Singh, Z. Ahmed, M. Stevenson, S. S. Briggs, D. Oupicky, L. B. Barrett, R. Spice, M. Kendall, M. Berry, J. A. Preece, A. Logan and L. W. Seymour, Nucleic Acids Res., 2005, 33, e86 CrossRef PubMed.
- J. M. Benns, J. S. Choi, R. I. Mahato, J. S. Park and S. W. Kim, Bioconjugate Chem., 2000, 11, 637–645 CrossRef CAS PubMed.
- Y. Huang, Y. Jiang, H. Wang, J. Wang, M. C. Shin, Y. Byun, H. He, Y. Liang and V. C. Yang, Adv. Drug Delivery Rev., 2013, 65, 1299–1315 CrossRef CAS PubMed.
- D. Putnam, A. N. Zelikin, V. A. Izumrudov and R. Langer, Biomaterials, 2003, 24, 4425–4433 CrossRef CAS PubMed.
- X. Chen and Y. W. Wu, Org. Biomol. Chem., 2016, 14, 5417–5439 CAS.
- S. Uchinomiya, A. Ojida and I. Hamachi, Inorg. Chem., 2014, 53, 1816–1823 CrossRef CAS PubMed.
- A. N. Kapanidis, Y. W. Ebright and R. H. Ebright, J. Am. Chem. Soc., 2001, 123, 12123–12125 CrossRef CAS PubMed.
- Y. T. Lai, Y. Y. Chang, L. Hu, A. Chao, Z. Y. Du, J. A. Tanner, M. L. Chye, C. Qian, K. M. Ng, H. Li and H. Sun, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 2948–2953 CrossRef CAS PubMed.
- Z. Wang, X. Ding, S. Li, J. Shi and Y. Li, RSC Adv., 2014, 4, 7235–7245 RSC.
- X. X. Liu and A. Melman, Chem. Commun., 2013, 49, 9042–9044 RSC.
- V. Razzano, M. Paolino, A. Reale, G. Giuliani, R. Artusi, G. Caselli, M. Visintin, F. Makovec, A. Donati, F. Villafiorita-Monteleone, C. Botta and A. Cappelli, ACS Omega, 2017, 2, 5453–5459 CrossRef CAS.
- Y. Zheng, C. Duanmu and Y. Gao, Org. Lett., 2006, 8, 3215–3217 CrossRef CAS PubMed.
- M. R. Mehlenbacher, F. Bou-Abdallah, X. X. Liu and A. Melman, Inorg. Chim. Acta, 2015, 437, 152–158 CrossRef CAS.
- E. Cariati, V. Lanzeni, E. Tordin, R. Ugo, C. Botta, A. Giacometti Schieroni, A. Sironi and D. Pasini, Phys. Chem. Chem. Phys., 2011, 13, 18005–18014 RSC.
- G. M. Sheldrick, SHELXS-97, Rel. 97–2, A Program for Automatic Solution of Crystal Structures, Göttingen University, 1997 Search PubMed.
- G. M. Sheldrick, SHELXL-97, Rel. 97–2, A Program for Crystal Structure Refinement, Göttingen University, 1997 Search PubMed.
- J. Moreau, U. Giovanella, J.-P. Bombenger, W. Porzio, V. Vohra, L. Spadacini, G. Di Silvestro, L. Barba, G. Arrighetti, S. Destri, M. Pasini, M. Saba, F. Quochi, A. Mura, G. Bongiovanni, M. Fiorini, M. Uslenghi and C. Botta, ChemPhysChem, 2009, 10, 647–653 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: ESI mass spectrum of Ac-His-6-MBHA-1d material; 1H and 13C NMR of compounds 1a–e, 2a–d, 3a–c, 5, 6a,b, and 8; photoluminescence of the polymeric materials obtained by exciting at different wavelengths. CCDC 1504772. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra00315g |
| ‡ Present address: Photosynthesis Research Unit, Institute of Biophysics (IBF-CNR), via Celoria 26, 20133 Milano, Italy. |
| § The manuscript was written through contributions of all authors. |
| This journal is © The Royal Society of Chemistry 2018 |