
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Michellamines A6 and A7, and further mono- and dimeric naphthylisoquinoline alkaloids from a Congolese Ancistrocladus liana and their antiausterity activities against pancreatic cancer cells†
Blaise Kimbadi Lombe ab,
Doris Feineisa,
Virima Mudogob,
Reto Bruncd,
Suresh Awale*e and
Gerhard Bringmann*a
ab,
Doris Feineisa,
Virima Mudogob,
Reto Bruncd,
Suresh Awale*e and
Gerhard Bringmann*a
aInstitute of Organic Chemistry, University of Würzburg, Am Hubland, D-97074 Würzburg, Germany. E-mail: bringman@chemie.uni-wuerzburg.de; Fax: +49 931 31 84755
bFaculté des Sciences, Université de Kinshasa, B.P. 202, Kinshasa XI, Democratic Republic of the Congo
cSwiss Tropical and Public Health Institute, Socinstrasse 57, CH-4002 Basel, Switzerland
dUniversity of Basel, Petersplatz 1, CH-4003 Basel, Switzerland
eDivision of Natural Drug Discovery, Institute of Natural Medicine, University of Toyoma, 2630 Sugitani, Toyama 930-0194, Japan. E-mail: suresh@inm.u-toyama.ac.jp
First published on 31st January 2018
Abstract
Michellamines A6 (1) and A7 (2) are the first dimers of 5,8′-coupled naphthylisoquinoline alkaloids with cis-configured stereocenters in both tetrahydroisoquinoline subunits. They were isolated from the leaves of a recently discovered, yet unidentified Congolese Ancistrocladus liana that shares some morphological characteristics with Ancistrocladus likoko. Two further new dimeric analogs, michellamines B4 (3) and B5 (4), were obtained, along with two previously likewise unknown monomers, ancistrobonsolines A1 (5) and A2 (6), which, besides one single known other example, are the only naphthyldihydroisoquinolines with an M-configured biaryl axis and R-configuration at C-3. Moreover, five compounds earlier reported from other Ancistrocladus species were identified, ancistroealaine C (7), korupensamines A (8a) and B (8b), and michellamines A2 (9) and E (10). Their complete structural elucidation succeeded due to the fruitful interplay of spectroscopic, chemical, and chiroptical methods. Chemotaxonomically, the stereostructures of the metabolites clearly delineate this Congolese Ancistrocladus liana from all known related species, showing that it might be a new taxon. Ancistrobonsolines A1 (5) and A2 (6) exhibited strong preferential cytotoxicities against human PANC-1 pancreatic cancer cells under nutrient-deprived conditions, without displaying toxicity in normal, nutrient-rich medium. Against cervical HeLa cancer cells, the dimeric alkaloids michellamines A6 (1) and E (10) displayed the highest cytotoxic activities, comparable to that of the standard agent, 5-fluorouracil. Furthermore, ancistrobonsolines A1 (5) and A2 (6) showed weak-to-moderate antiprotozoal activities.
Introduction
Plants belonging to the genus Ancistrocladus Wall. (Ancistrocladaceae) continue to receive much attention, mainly because they produce a class of structurally, biosynthetically, and pharmacologically unusual compounds, the naphthylisoquinoline alkaloids.1 The genus Ancistrocladus comprises 18 botanically accepted species of tropical lianas,2–4 of which four occur in the Democratic Republic of the Congo (DRC), namely, A. ealaensis J. Léonard,5 A. congolensis J. Léonard,5 A. likoko J. Léonard,5 and A. ileboensis G. Heubl, V. Mudogo & G. Bringmann.4 The number of Congolese Ancistrocladus species may, however, be actually much higher, as there have not yet been thorough and specific botanical campaigns covering the entire Congolese forest, which is so vast and so rich in biodiversity. This assumption is also supported by recent phylogenetic investigations on Ancistrocladus samples collected at different sites in DRC,6 which hinted at the presence of a potentially new taxon from the region near the village Bonsolerive, close to the town Mbandaka, in the northwestern part of DRC.Most recently, likewise close to the village Bonsolerive, we have discovered another Ancistrocladus liana. Particularly striking were its leaves, which were much larger (about 57 cm long and 12 cm wide) than those of any other Ancistrocladus plant species found nearby. This liana had hooked inflorescences, which, together with the aforementioned large size of the leaves, were reminiscent of A. likoko, the only known Congolese species with similar – yet different – morphological characteristics.2,5 Comparative LC-UV-MS profiling of the leaf alkaloid pattern of this liana and that of an authentic sample of A. likoko, however, revealed substantial phytochemical differences between the two samples. This metabolic divergence and the fact that A. likoko had so far not been found to occur in that sampling area indicated that the liana might belong to a new, as yet undescribed plant species or subspecies. The isolation and structural elucidation of its alkaloids thus became a rewarding task, not only for the chemotaxonomic characterization of this liana and its possible delineation from other Ancistrocladus species, but also for their biological evaluation.
Herein we report on the isolation and structural elucidation of eleven mono- and dimeric naphthylisoquinoline alkaloids from the leaves of this yet unidentified Ancistrocladus liana (Fig. 1). The isolated metabolites comprise six previously unknown compounds: michellamines A6 (1), A7 (2), B4 (3), and B5 (4), ancistrobonsoline A1 (5) and its 6-O-methyl derivative, ancistrobonsoline A2 (6), and five analogs described from earlier work on other Ancistrocladus plants: ancistroealaine C (7),7 korupensamines A (8a) and B (8b),8 and michellamines A2 (9)9 and E (10).10 Furthermore, we discuss the chemotaxonomic position of this liana relative to other Ancistrocladus species. The cytotoxic activities of the isolated compounds against cervical HeLa cancer cells and their antiausterity potencies against PANC-1 human pancreatic cancer cells are also described, as well as the antiprotozoal properties of the new monomeric compounds.
 | ||
| Fig. 1 Metabolites isolated from an as yet unidentified Congolese Ancistrocladus liana, among them six new compounds: michellamines A6 (1), A7 (2), B4 (3), and B5 (4), ancistrobonsolines A1 (5) and A2 (6), and five previously known ones: ancistroealaine C (7), korupensamines A (8a) and B (8b), and michellamines A2 (9) and E (10). Yellow ellipses on 1 and 2 highlight the combination of two 1,3-cis-configurations, never observed in any other related dimer; for reason of comparison, see the structure of the well-known michellamine A (11) with its 1,3-trans-configurations (underlaid in gray) in both molecular halves – this compound is not produced by the investigated Congolese liana. | ||
Results and discussion
Isolation and structural elucidation of metabolites
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), followed by concentration under reduced pressure, provided a crude residue, which was then submitted to a cation-exchange column to remove undesired, non-basic metabolites. The resulting alkaloid-enriched fraction was partitioned between water and dichloromethane, and monitored by HPLC (Fig. 2).
:1), followed by concentration under reduced pressure, provided a crude residue, which was then submitted to a cation-exchange column to remove undesired, non-basic metabolites. The resulting alkaloid-enriched fraction was partitioned between water and dichloromethane, and monitored by HPLC (Fig. 2).
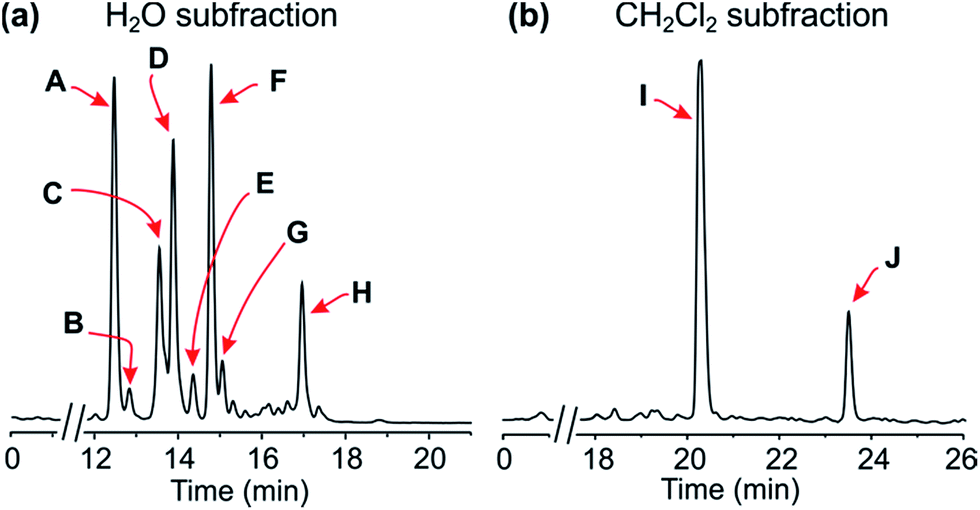 | ||
| Fig. 2 HPLC profiles of the alkaloid-enriched subfractions from the leaves of the plant. | ||
Resolution of the water-phase subfraction on a preparative HPLC column permitted isolation of nine pure constituents, obtained as amorphous solids. The fastest-eluting compound (Peak A, Fig. 2a) was found to be ancistroealaine C (7), earlier known from Ancistrocladus ealaensis.7 The second alkaloid (Peak B) was korupensamine A (8a), while peak C contained two alkaloids overlapping each other, korupensamine B (8b) and michellamine A2 (9), and peak G was michellamine E (10). These four metabolites (8a/b–10) had previously been isolated from other Ancistrocladus species, yet only from Central Africa: The korupensamines A (8a) and B (8b), first known from the Cameroonian plant A. korupensis8 as well as michellamine E (10),10 had both also been reported from A. likoko,11,12 where they occur as major metabolites. Korupensamine A (8a) is likewise the main naphthylisoquinoline alkaloid in A. congolensis,9,13 in which michellamine A2 (9) was discovered9 before its more recent detection as a minor constituent of A. likoko.12 The other four remaining compounds from the aqueous subfraction (Peaks D, E, F, and H, Fig. 2a), as well as those from the dichloromethane subfraction (Peaks I and J, Fig. 2b), were as yet unknown.
The 1H NMR spectrum (Table 1) exhibited four aromatic protons. Two of them displayed a meta-coupling, typical of protons at C-1′ and C-3′,7–10 while the other two appeared as singlets, which suggested the presence of a symmetric dimer of 5,6′-, 5,8′-, 7,6′-, or 7,8′-coupled naphthylisoquinoline monomers. The aliphatic region, with two three-proton doublets (Me-1 and Me-3), signals for two aryl substituents (a methyl group and a methoxy function), a quartet, a multiplet, and two doublet of doublets, was, however, closely similar to that of the likewise isolated known ancistroealaine C (7), so that a 5,8′-coupled molecular framework was expected. In agreement with this assumption, one of the two aromatic singlets was assigned as H-7, from its HMBC correlations with C-6, C-8, and C-9 (Fig. 3a). This showed that the other, remaining singlet had to be at C-7′ and, hence, clearly excluded any biaryl linkage involving C-7.
| Position | 1b | 5 | 6 | |||
|---|---|---|---|---|---|---|
| δH | δC | δH | δC | δH | δC | |
| a 1H and 13C NMR data were recorded in methanol-d4 (δ in ppm).b These data are identical for the second molecular half of the C2-symmetric dimer 1. | ||||||
| 1 | 4.64, q (6.5) | 52.4 | 175.8 | 176.3 | ||
| 3 | 3.27, m | 51.0 | 3.70, m | 49.5 | 3.71, m | 49.6 |
| 4eq. | 2.64, dd (3.3, 17.3) | 33.3 | 2.41, dd (5.8, 16.9) | 33.6 | 2.42, dd (5.8, 16.9) | 33.4 |
| 4ax | 2.27, dd (12.0, 17.3) | 33.3 | 2.48, dd (11.3, 16.9) | 33.6 | 2.49, dd (11.3, 16.9) | 33.4 |
| 5 | 119.4 | 122.5 | 123.6 | |||
| 6 | 156.8 | 167.8 | 168.7 | |||
| 7 | 6.47, s | 102.9 | 6.67, s | 99.3 | 6.83, s | 95.9 |
| 8 | 156.5 | 165.8 | 166.4 | |||
| 9 | 113.0 | 108.8 | 109.2 | |||
| 10 | 135.2 | 142.9 | 141.6 | |||
| 1′ | 6.84, pt (0.9) | 119.3 | 6.72, pt (1.1) | 118.7 | 6.64, pt (1.1) | 118.6 |
| 2′ | 137.7 | 138.2 | 138.2 | |||
| 3′ | 6.86, pd (1.3) | 108.1 | 6.83, pd (0.9) | 107.9 | 6.82, pd (0.9) | 107.8 |
| 4′ | 158.2 | 158.2 | 158.1 | |||
| 5′ | 152.4 | 156.4 | 156.2 | |||
| 6′ | 120.4 | 6.80, d (7.9) | 110.4 | 6.78, d (7.9) | 110.3 | |
| 7′ | 7.29, s | 134.9 | 7.05, d (7.9) | 131.7 | 7.00, d (7.9) | 131.4 |
| 8′ | 124.3 | 123.1 | 123.3 | |||
| 9′ | 137.1 | 136.7 | 136.6 | |||
| 10′ | 115.3 | 115.0 | 114.8 | |||
| 1-Me | 1.82, d (6.5) | 20.0 | 2.79, pd (1.5) | 24.8 | 2.82, pd (1.5) | 24.9 |
| 3-Me | 1.26, d (6.5) | 18.9 | 1.26, d (6.6) | 18.2 | 1.27, d (6.6) | 18.1 |
| 2′-Me | 2.37, s | 22.3 | 2.34, pd (0.6) | 22.3 | 2.32, pd (0.8) | 22.3 |
| 6-OMe | 3.83, s | 57.2 | ||||
| 8-OMe | 4.04, s | 56.8 | 4.15, s | 57.0 | ||
| 4′-OMe | 4.10, s | 57.1 | 4.10, s | 57.0 | 4.10, s | 57.0 |
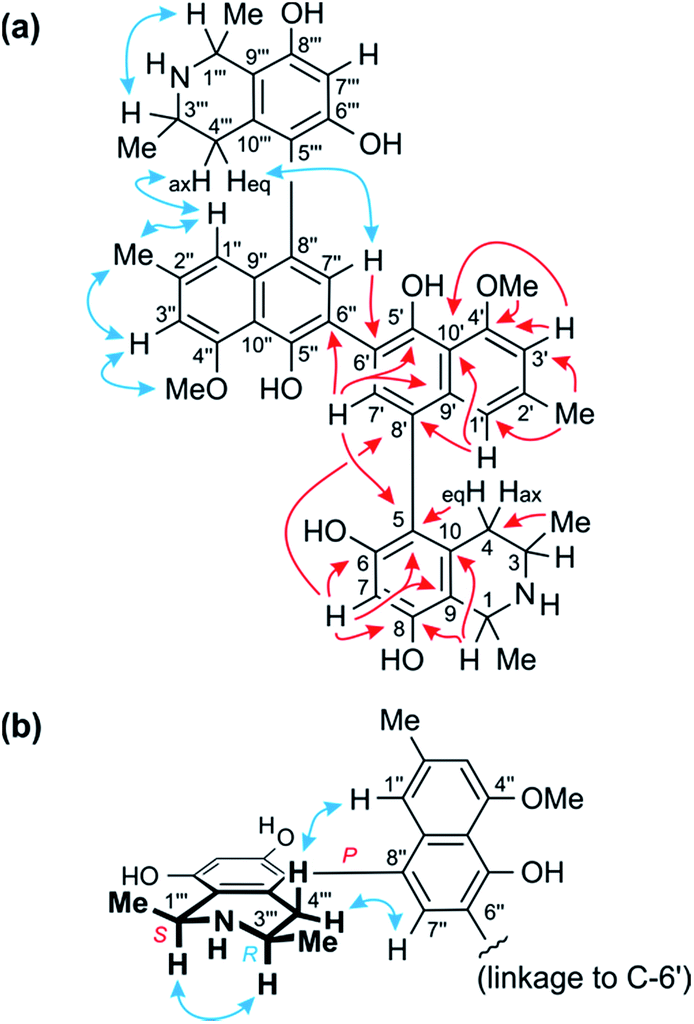 | ||
| Fig. 3 (a) Key HMBC (red single arrows) and ROESY (blue double arrows) interactions evidencing the constitution of the homodimer 1. (b) ROESY interactions indicating the relative and – given the results of oxidative degradation – absolute configurations of the two identical portions (=ancistroealaine C, 7) of 1. | ||
The two remaining possible coupling types (5,6′ or 5,8′) were further discriminated by joint correlative HMBC signals from H-7 and H-1′ to C-8′ (Fig. 3a), which excluded a 5,6′-coupling, where such interactions would not have been observed. Thus, the naphthalene and isoquinoline portions were linked to each other by a 5,8′-axis, and the two monomeric naphthylisoquinoline halves were therefore connected via C-6′. These couplings, in particular the 6′,6′′-linkage between the naphthalene portions, were further corroborated by the chemical shifts of C-6′ (δC 120.4) and of the surrounding nuclei, C-5′ (δC 152.4) and C-7′ (δC 134.9), which were near-identical to those of other dimers with similar molecular frameworks (e.g., michellamine A2 (9):9 δC 120.3 for C-6′, 152.3 for C-5′, and 134.7 for C-7′). The location of the methoxy group at C-4′ was deduced from NOE interactions of its protons with H-3′, in conjunction with joint HMBC correlations from both the O-methyl protons and H-3′ to C-4′ (Fig. 3a).
Based on NOESY interactions of the proton at C-3 and the one at C-1 (Fig. 3a), the relative configuration at the stereocenters C-1 and C-3 was determined to be cis. Ruthenium-mediated oxidative degradation14 of this alkaloid led to the R-enantiomer of 3-aminobutyric acid (derived from C-3), as monitored by GC-MSD analysis of the Mosher derivatives. This revealed C-3 to be R-configured and, given the aforementioned relative cis-configuration, C-1 had to be S-configured.
At the naphthalene-isoquinoline biaryl axes, within the 5,8′-coupled monomeric halves, the stereo-array was determined by NOESY interactions of the proton at C-7′ with the equatorial proton at C-4 (δH = 2.64 ppm; J = 17.3, 3.3 Hz) and between the axial proton (δH = 2.27 ppm; J = 17.3, 12.0 Hz) and the aromatic proton at C-1′. This, in conjunction with the absolute R-configuration at C-3, as depicted in Fig. 3b, indicated P-configuration at the outer axes, revealing these portions to be structurally identical to the co-occurring known ancistroealaine C (7). Hence, the metabolite was found to be the 6′-homocoupling product of 7, with the absolute stereostructure 1 presented in Fig. 1. This was further confirmed by the similarity of the ECD (electronic circular dichroism) spectrum of 1 with that of the co-occurring, likewise P,P-configured michellamine A2 (9). Owing to this combination of two P-configured outer biaryl axes, the new compound was included in the ‘series A’ of michellamines – which so far comprised michellamines A–A5 – and was thus named michellamine A6. Its 1,3-cis-configurations in both tetrahydroisoquinoline portions, as highlighted in Fig. 1, is a unique structural feature that had not been found in any other similarly coupled naphthylisoquinoline dimers.
| Position | 2 | 3 | 4 | |||
|---|---|---|---|---|---|---|
| δH | δC | δH | δC | δH | δC | |
| a The NMR data were recorded in methanol-d4 (δ in ppm).b This signal was overlapped by that of 2′-Me, but could be deduced to be dd, from the respective 2D NMR cross peaks.c These signals overlapped each other. | ||||||
| 1/1′′′ | 4.64, q (6.6)/4.64, q (6.6) | 62.6/52.5 | 4.74, q (6.7)/4.64, q (6.5) | 49.5/52.4 | —/4.64, q (6.6) | 175.8/52.4 |
| 3/3′′′ | 3.23, m/3.29, m | 60.6/51.0 | 3.66, m/3.29, m | 45.4/50.9 | 3.72, m/3.29, m | 49.4/50.9 |
| 4eq./4′′′eq. | 2.36b, dd/2.65, dd (4.3, 17.8) | 34.5/33.4 | 2.37b, dd/2.61, dd (3.3, 17.4) | 34.1/33.3 | 2.52, ddc/2.59, ddc | 33.6/33.3 |
| 4ax/4′′′ax | 2.69, dd (11.4; 18.0)/2.28, dd (12.2, 17.8) | 2.53, dd (11.7; 18.1)/2.26, dd (12.0, 17.4) | 2.61, ddc/2.25, dd (11.9, 17.2) | |||
| 5/5′′′ | 119.1/119.4 | 119.1/119.2 | 122.3/119.2 | |||
| 6/6′′′ | 157.0/156.8 | 157.0/156.8 | 167.8/156.8 | |||
| 7/7′′′ | 6.47, s/6.47, s | 102.6/102.9 | 6.46, s/6.48, s | 102.2/102.9 | 6.69, s/6.48, s | 99.4/102.9 |
| 8/8′′′ | 155.7/156.5 | 155.7/156.5 | 165.8/156.5 | |||
| 9/9′′′ | 113.4/113.0 | 113.4/113.0 | 108.8/113.0 | |||
| 10/10′′′ | 135.2/135.2 | 133.2/135.2 | 143.0/135.2 | |||
| 1′/1′′ | 6.78, s/6.85, s | 119.5/119.4 | 6.84, s/6.84, s | 119.4/119.3 | 6.77, pt (1.2)/6.84, pt (1.2) | 118.9/119.4 |
| 2′/2′′ | 137.6/137.7 | 137.8/137.8 | 138.2/137.8 | |||
| 3′/3′′ | 6.87, s/6.86, s | 108.2/108.1 | 6.87, s/6.87, s | 108.3/108.1 | 6.88, d (1.2)/6.86, d (1.4) | 108.3/108.1 |
| 4′/4′′′ | 158.3/158.2 | 158.2/158.2 | 158.2/158.3 | |||
| 5′/5′′ | 152.5/152.4 | 152.4/152.4 | 152.5/152.9 | |||
| 6′/6′′ | 120.4/120.4 | 120.3/120.3 | 120.0/120.4 | |||
| 7′/7′′ | 7.33, s/7.29, s | 135.7/135.0 | 7.27, s/7.30, s | 135.3/134.9 | 7.30, s/7.30, s | 135.2/134.9 |
| 8′/8′′ | 124.0/124.3 | 124.3/124.3 | 122.6/124.3 | |||
| 9′/9′′ | 136.5/137.1 | 136.6/137.2 | 136.1/137.2 | |||
| 10′/10′′ | 115.2/115.3 | 115.4/115.3 | 115.2/115.3 | |||
| 1-Me/1′′′-Me | 1.77, d (6.7)/1.82, d (6.6) | 19.6/20.0 | 1.70, d (6.7)/1.82, d (6.5) | 18.5/20.0 | 2.77, d (1.4)/1.82, d (6.6) | 24.9/20.0 |
| 3-Me/3′′′-Me | 1.33, d (6.6)/1.27, d (6.5) | 18.2/18.9 | 1.28, d (6.3)/1.24, d (6.4) | 19.4/18.9 | 1.28, d (6.7)/1.24, d (6.5) | 18.2/18.9 |
| 2′-Me/2′′-Me | 2.36, s/2.37, s | 22.3/22.3 | 2.37, s/2.37, s | 22.3/22.3 | 2.37, s/2.37, s | 22.3/22.3 |
| 8-OMe | 4.03, s | 56.8 | ||||
| 4′-OMe/4′′-OMe | 4.12, s/4.10, s | 57.1/57.1 | 4.11, s/4.11, s | 57.2/57.2 | 4.11, s/4.10, s | 57.1/57.2 |
| N-Me | 3.04, s | 41.4 | ||||
Further 1D and 2D NMR data revealed that methyl group to be attached to the nitrogen atom in the ‘southeastern’ portion. Key evidence of the exact location of this N-methyl group were the HMBC correlations of its protons with both, C-1 and C-3 (Fig. 4a) and the signals of these methine carbons, which appeared deshielded (62.6 ppm for C-1 and 60.6 for C-3) as compared to those of the N-demethylated congeners – like, for example, in the case of 1 (Tables 1 and 2). This assignment was further supported by the ROESY series {Me-1 ↔ N–Me ↔ Me-3}, which, in addition, revealed a 1,3-diaxial relationship of H-1 and H-3 and, thus, a relative cis-configuration at the stereocenters C-1 and C-3 (Fig. 4a). For the stereocenters in the other, ‘northwestern’ part of 2, a relative cis-configuration was observed, too, again evidenced by the ROESY interactions of H-1′′′ with H-3′′′, as in 1 (Fig. 3b).
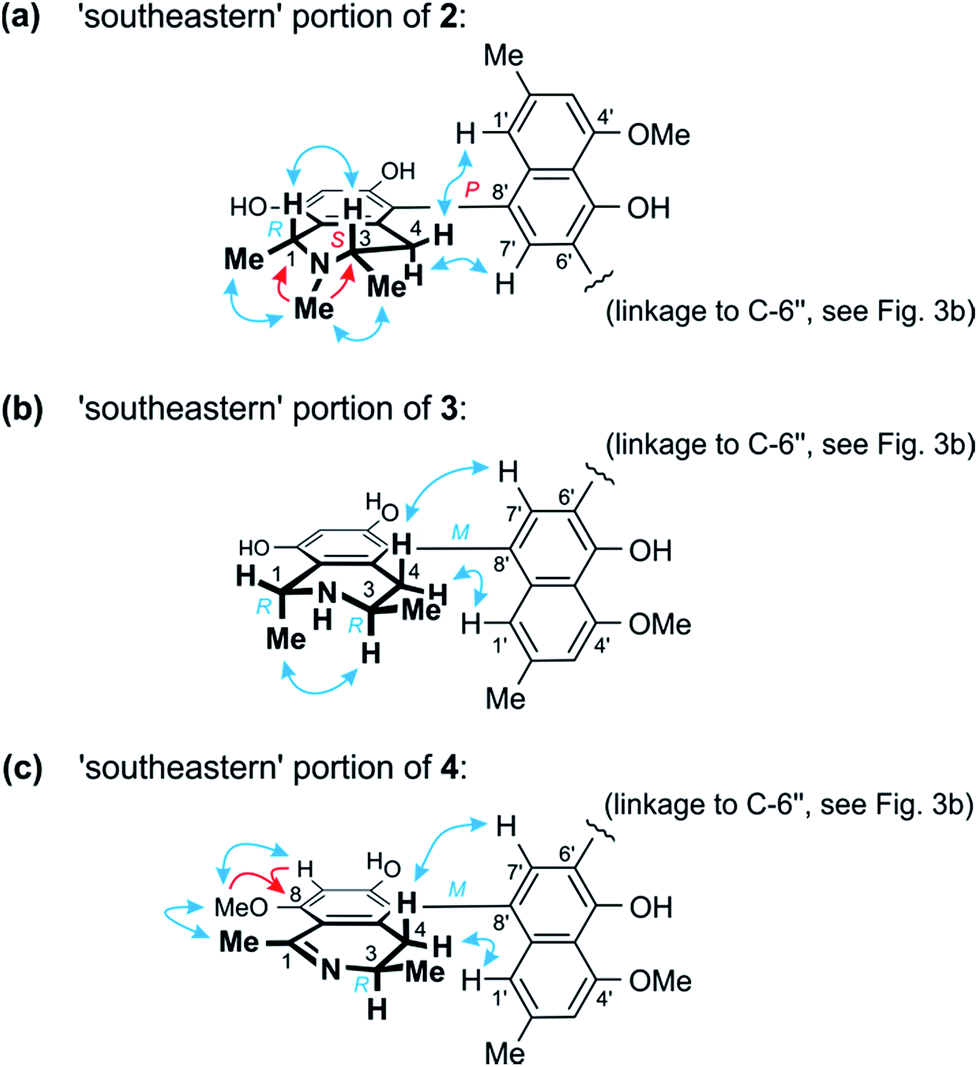 | ||
| Fig. 4 (a) Selected HMBC (red single arrows) and ROESY (blue double arrows) interactions within the ‘southeastern’ portion of 2 establishing its constitution and – together with the results of the oxidative degradation – absolute configuration (=korupensamine D, Fig. S1†). (b) ROESY correlations evidencing the relative and – based on the results of the oxidative degradation – absolute configurations in the southeastern part (=korupensamine B, 8a) of 3. (c) Key HMBC and ROESY for the ‘southeastern’ portion of 4 that, together with the results of the oxidative degradation, establish the absolute stereostructure of this portion (=the new compound ancistrobonsoline A1, 5). | ||
The ruthenium-mediated oxidative degradation of 2 yielded both, R- and S-configured 3-aminobutyric acids, showing that this alkaloid was either 3R,3′′′S- or 3S,3′′′R-configured. The simultaneous formation of the N-methylated analog of 3-aminobutyric acid only as its S-enantiomer, however, clearly indicated that the S-configured stereocenter was located next to the N-methyl group, i.e. at C-3. Thus, this compound was found to be 3S,3′′′R-configured, which, given the relative cis-configurations in both halves as mentioned above, showed that C-1 and C-1′′′ were R- and S-configured, respectively.
From ROESY interactions between H-4ax and H-7′ and between H-4eq. and H-1′ (Fig. 4a), and based on the above-assigned S-configuration at C-3, the configuration at the southeastern axis was determined to be P, thus revealing this portion to correspond to korupensamine D, a known naphthylisoquinoline alkaloid from A. korupensis (for its structure, see Fig. S1†).8 At the other, northwestern axis of 2, the ROESY interactions were complementary: H-4′′′ax with H-1′′ and H-4′′′eq. with H-7′′, which, together with the opposite R-configuration at C-3′′′ established above, indicated that axis to be P-configured, too, as in 1 (Fig. 3b). This metabolite thus had to be a cross-coupling product of ancistroealaine C (7) and korupensamine D (Fig. S1†),8 and possessed the absolute structure 2 presented in Fig. 1. It was named michellamine A7, due to its P,P-configured outer biaryl axes, as in 1. It is the second dimer of 5,8′-coupled naphthylisoquinolines with the unusual combination of two 1,3-cis-configured halves.
The 1H NMR spectrum of 1 matched the half-set of signals of 3 nearly perfectly (ΔδH ≤ 0.02, see Tables 1 and 2), which suggested that the two compounds had one molecular portion in common. Further analysis of the 1D and 2D NMR data of 3 established the constitution of 3 to be identical to that of 1, that is, two 5,8′-coupled naphthylisoquinoline halves connected to each other via C-6′ (Fig. 3a).
In one portion, the northwestern part of 3, the relative configuration at the stereocenters C-1′′′ versus C-3′′′ was deduced to be cis from ROE interactions of H-1′′′ and H-3′′′, as also (even twice) in 1 (Fig. 3b). In the southeastern part of 3, however, the stereocenters were trans-configured, as evidenced from ROESY interactions between H-3 and Me-1 (Fig. 4b). The ruthenium-mediated oxidative degradation of 3 yielded 3-aminobutyric acid as its R-enantiomer exclusively, which demonstrated that both, C-3 and C-3′′′ were R-configured. This, together with the aforementioned cis- and trans-arrays, established the absolute configurations at C-1′′′ and C-1 to be S and R, respectively.
From ROESY interactions of H-4′′′ax with H-1′′ and of H-4′′′eq. with H-7′′ and in view of the absolute R-configuration at C-3′′′, the absolute axial configuration in the northwestern half was assigned to be P, revealing this portion to be identical to 7, as in 1 (Fig. 3b). In the other, southeastern part, the respective protons interacted in a complementary way: H-4ax with H-7′ and H-4eq. with H-1′, as shown in Fig. 4b. These correlations, together with the known R-configuration at C-3 (see above), evidenced the southeastern biaryl axis to be M-configured, thus showing the corresponding moiety to have the same structure as the likewise isolated korupensamine B (8b) (Fig. 4b). Therefore, compound 3 had to be the 6′,6′′-cross-coupling product of ancistroealaine C (7) and korupensamine B (8b), with the absolute stereostructure given in Fig. 1. It was named michellamine B4, in continuation of the series of michellamines with opposite configurations at the two outer biaryl axes (type B).
This assumption was corroborated by 1D and 2D NMR data, which led to a molecular architecture similar to that of 3, but with one additional methyl group attached to the oxygen function at C-8 and a double bond between C-1 and the adjacent nitrogen atom in the southeastern portion (Fig. 4c). Key NMR features of the location of the methoxy function at C-8 were the joint HMBC interactions from H-7 and OMe-8 to C-8 and the ROESY series {H-7 ↔ OMe-8 ↔ Me-1} (Fig. 4c).
The oxidative degradation procedure delivered only (R)-3-aminobutyric acid, showing that both, C-3 and C-3′′′ were R-configured. In the northwestern portion, ROESY interactions at the stereocenters C-1′′′ versus C-3′′′ and across the naphthalene–isoquinoline linkage were the same as in 1 (Fig. 3b), so that C-1 was S-configured and the biaryl axis had the P-configuration. In the southeastern portion, the ROESY interaction between H-4ax and H-7′ indicated these two spin systems to be on the same side of the molecule, as depicted in Fig. 4c, which evidenced an M-configuration at the biaryl axis. Therefore, this metabolite possessed the absolute stereostructure 4, as shown in Fig. 1. It was named michellamine B5, due to its stereochemical similarity with michellamine B4 (3). Within a list of now 16 known natural michellamine-type dimers, it is only the third example that possesses a dihydroisoquinoline ring system; the other two analogs with such a structural peculiarity are michellamines F10 and A4.9
The ROESY interactions across the biaryl axis of 5 were all similar to those in the southeastern portion of 4 (see Fig. 4c), as were also the results of the oxidative degradation. This monomeric alkaloid thus possessed the absolute stereostructure 5, as presented in Fig. 1. It was, consequently, the as yet undescribed enantiomer of the known alkaloid 6,5′-O,O-didemethylancistroealaine A.15 In agreement with their opposite absolute configurations, their ECD spectra were fully mirror-image like (Fig. 5). Instead of the rational, but long name ent-6,5′-O,O-didemethylancistroealaine A, the new compound was named ancistrobonsoline A1, after the Congolese village Bonsolerive, where the plant had been collected.
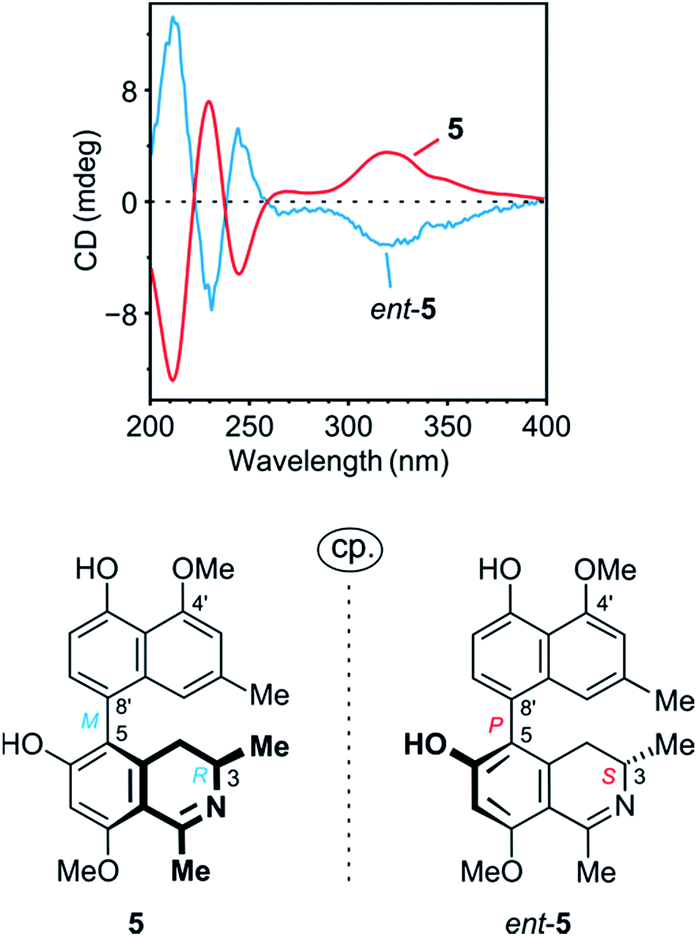 | ||
| Fig. 5 Comparison of the ECD spectrum of 5 with that of its previously known natural enantiomer, ent-5 (=6,5′-O,O-didemethylancistroealaine A),15 further confirming the absolute stereostructure of 5. | ||
The absolute configuration at the stereocenter C-3 of 6 was determined to be R by oxidative degradation, which, as in the case of 5, provided (R)-3-aminobutyric acid. Across the biaryl axis the specific relationships between the diastereotopic protons at C-4 and the aromatic protons H-1′ and H-7′ were the same as in 5 (Fig. 4c): H-4ax (‘up’) interacted with H-7′ and H-4eq. (‘down’) with H-1′, which was consistent with an M-configuration at the 5,8′-axis. This absolute stereochemical assignment was further corroborated by the close similarity of the ECD spectrum of 6 with that of 5. Therefore, the new alkaloid had to possess the absolute stereostructure 6, as presented in Fig. 1, and was, thus, the 6-O-methylated derivative of 5. It was named ancistrobonsoline A2. With their R-configuration at the stereocenter and M at the biaryl axis, ancistrobonsolines A1 (5) and A2 (6) belong to a very small subgroup of naphthyldihydroisoquinolines displaying such stereochemical features, with previously only one single representative, ancistrolikokine F (Fig. S1†).12
Chemotaxonomic and biosynthetic significance of the isolated metabolites
The molecular architectures of the alkaloids thus isolated from this as yet unidentified Ancistrocladus taxon are remarkable in many respects. Firstly, it is striking that all of these compounds belong to the same 5,8′-coupling type and, where dimers are concerned, they are all based on a central 6′,6′′-axis. Such a specific phenol-oxidative coupling – and, thus strict enzymic assistance – have so far been observed only in A. likoko.11,12,16These findings thus reveal a close phylogenetic relationship between the two taxa (in addition to their aforementioned morphological similarities), but delineate one from the other by the fact that the naphthalene moieties of the metabolites produced by the plant from the proximity of Bonsolerive are, all of them, O-methylated only at C-4′ (and C-4′′, in the case of dimers), not at C-5′ (nor at C-5′′). And their configuration at C-3 (or/and C-3′′′, for dimeric compounds) is always R, except for the N-methylated isoquinoline portions, where an exclusive S-configuration is observed (see Fig. 1). Such a regioselective O-methylation in the naphthalene subunits and a stereospecificity at C-3 (and/or C-3′′′) are not known from A. likoko (or from any other related plant). Likewise demarcating these two taxa is the fact that A. likoko is a virtually exclusive producer of monomeric alkaloids (out of 21 metabolites, only two dimers have so far been isolated from this species),12 whereas the plant investigated here is a rich source of dimeric compounds.
Moreover, the remarkable occurrence of dimers with a symmetric 6′,6′′-coupling at the central axis chemotaxonomically delineates this taxon from the other – likewise still unidentified – liana that occurs in the same area.6 The latter is known to produce a drastically different metabolic pattern, including dimers with unsymmetric, 6′,1′′-coupled central axes,17 with additional oxygen bridges18 and/or acetal linkages,21 but so far no symmetric, 6′,6′′-coupled ones, not even in traces.
Secondly, it is particularly noteworthy that this liana produces the 1,3-trans-configured korupensamine A (8a) only in small quantities (Peak B, Fig. 2a) and shows no hints at the presence of its dimer, michellamine A (11), not even in traces.20 This is exceptional, since all of the plants known to produce michellamine-type alkaloids, viz. A. korupensis,10,20 A. congolesis,9 and A. likoko,12 always contain michellamine A (11) as one of the main dimeric compounds. Moreover, 11 is always accompanied by its monomer, korupensamine A (8a), likewise occurring as a major metabolite in those plants.9,10,20 For this reason, it has even been suggested9 that other (minor) dimeric analogs differing from michellamine A (11) by the configurations at the stereocenters may originate from the preformed ‘parent’ dimer michellamine A (11), i.e. by modification after the dimerization step. In the case of the metabolites of the plant investigated here, however, one of the main constituents (Peak D, Fig. 2a), michellamine A6 (1), is twofold 1,3-cis-configured and can be regarded as a bis-epimer of michellamine A (11) at C-1 and C-1′′′. Since 1 is accompanied by an unusually large quantity of its – likewise cis-configured – monomer (Peak A, Fig. 2a), ancistroealaine C (=1-epi-korupensamine A) (7), it seems likely that 1 (as also 2) originates from the correspondingly preformed cis-monomers.
Biological activities of the isolated metabolites
| Compound | IC50 (μM) | |||||
|---|---|---|---|---|---|---|
| P. falciparum | T. brucei rhodesiense STIB900 | T. cruzi Tulahuen C2C4 Lac Z | L. donovani axenic amastigotes MHOM/ET/67/L82 | Cytotoxicity (L6 cells) | ||
| K1 | NF54 | |||||
| a Values reported earlier (see, ref. 15).b Chloroquine.c Melarsoprol.d Benznidazole.e Miltefosine.f Podophyllotoxin. | ||||||
| 5 | 2.7 | 2.1 | 44.4 | 109.3 | 87.9 | 114.9 |
| ent-5a | 5.4 | n. d | 24.8 | 16.3 | 43.4 | >230 |
| 6 | 1.8 | 2.4 | 12.6 | 80.1 | 246.6 | 52.8 |
| Standard | 0.31b | 0.01b | 0.04c | 5.69d | 1.08e | 0.01f |
| Compound | HeLa (IC50 in μM) | PANC-1a (PC50 in μM) |
|---|---|---|
| a Concentration at which 50% of the PANC-1 pancreatic cancer cells were killed preferentially in nutrient-deprived medium (NDM).b 5-Fluorouracil.c Arctigenin. | ||
| 1 | 14.8 | 54.2 |
| 2 | 20.6 | 24.3 |
| 3 | 46.3 | 50.3 |
| 4 | 29.8 | 60.2 |
| 5 | 14.3 | 7.5 |
| 6 | 21.5 | 12.1 |
| 7 | 30.5 | >100 |
| 8a | 48.3 | >100 |
| 8b | 37.8 | 94.9 |
| 9 | 32.1 | 19.3 |
| 10 | 8.8 | 18.9 |
| Standard | 13.9b | 0.8c |
Michellamine E (10) was, therefore, investigated for its effects on cell morphology and apoptosis using two distinct staining methods, the Hoechst 33342 staining and the ethidium bromide–acridine orange (EB–AO) double staining assay. In the Hoechst 33342 staining, the dye penetrates through the cell membrane and intercalates with DNA and emits blue fluorescence.28 As shown in Fig. 6a, untreated HeLa cells (the control) displayed regular cell morphology with the intact nuclei. However, treatment with 12.5 μM of 10 induced nuclear fragmentation, suggestive of cells undergoing apoptosis, as indicated by fragmented nuclei exemplarily shown by white arrows in Fig. 6a. The EB–AO assay, on the other hand, allows visualizing the cellular morphology as well as the distinction between the live and dead cells. Acridine orange (AO) is a cell-membrane permeable dye emitting bright-green fluorescence in live cells, and ethidium bromide (EB) penetrates only the membrane of dying or dead cells staining them red.29 As shown in Fig. 6b, untreated HeLa cells (the control) displayed intact, regular cell morphology with exclusive bright green fluorescence in AO–EB staining. Treatment of the tumor cells with 12.5 μM of 10, however, disrupted the cellular integrity leading to rounding of the cell membrane, membrane rupture, and disintegration of cellular contents resulting in an increased population of red EB fluorescence of dead cells (Fig. 6b).
 | ||
| Fig. 6 Morphological changes of HeLa human cervical cancer cells induced by 12.5 μM of michellamine E (10) in comparison to untreated control: (a) Hoechst 33342 nuclei staining and (b) acridine orange (AO) – ethidium bromide (EB) staining. | ||
The effects of ancistrobonsolines A1 (5) and A2 (6), as representatives of the compounds showing potent antiausterity activities, on the cell morphology and apoptosis were further investigated by the ethidium bromide (EB) – acridine orange (AO) double-staining fluorescence assay as described above. As shown in Fig. 7(A and A′), the untreated PANC-1 cells, serving as the control, emitted the typical bright-green color, characteristic of living cells, exhibiting an intact cellular morphology. This, however, changed significantly when the cells were treated with 12.5 μM of 5 and 6, as revealed by the yellow fluorescence resulting from the overlapping of light emitted by AO (green) and EB (red) (Fig. 7B′ and D′). With 25 μM, both compounds induced a dramatic morphological alteration and disintegration of cellular organelles, leading to total cell death as illustrated by the virtually exclusive red stain in Fig. 7(C′ and E′). All these results suggest that naphthylisoquinoline alkaloids are promising lead structures for anticancer drug development.
 | ||
| Fig. 7 Morphological changes of PANC-1 human pancreatic cancer cells induced by the new compounds ancistrobonsolines A1 (5) and A2 (6) in comparison to the untreated ones (control). | ||
Experimental section
General experimental procedures
UV/Vis spectra were measured with a Shimadzu UV-1800 spectrophotometer, IR spectra with a Jasco FT-IR-4600 type A spectrometer, and optical rotations with a Jasco P-1020 polarimeter. ECD spectra were obtained on a J-715 spectropolarimeter (Jasco) at room temperature, using a standard cell (0.02 cm) and spectrophotometric-grade methanol, and are reported in Δε values (cm2 mol−1) at the given wavelength λ (nm). GC-MSD analyses were performed on a GCMS-QP 2010SE (Shimadzu). 1D and 2D NMR spectra were recorded on Bruker Avance III HD 400 (400 MHz) and 600 (600 MHz) instruments in deuterated methanol. Chemical shifts (δ) are reported in parts per million (ppm) with the 1H and 13C signals of the solvent (1H, δ = 3.31 ppm; 13C, δ = 49.15 ppm) as the internal reference. HRESIMS spectra were obtained on a microTOF-focus and micrOTOF-Q III mass spectrometers (Bruker). Preparative HPLC separation was performed on a Jasco HPLC system (PU-2087, UV-2077, LC-NetII/ADC), using a SymmetryPrep C18 column (Waters, 19 × 300 mm, 7 μm) with the UV absorption wavelengths set at 232, 254, and 310 nm. Organic solvents were analytical grade or distilled prior to use.Plant material
Ancistrocladus plant material, morphologically related to those of A. likoko, were collected in August 2015 (Global Positioning System coordinates: 00°06.572S, 018°20.146E, 315 ± 22 m) by one of us (B. K. L.) in the vicinity of the village Bonsolerive, which is located at about 20 km southeast of the town of Mbandaka in the Democratic Republic of the Congo. A voucher specimen (No. 105) has been deposited at the Herbarium Bringmann, Institute of Organic Chemistry, University of Würzburg.Extraction and isolation
Air-dried leaves (200 g) were ground and repeatedly extracted by maceration with a neutral mixture of MeOH – CH2Cl2 (1:1, 2 L) with mechanical shaking (160 RPM). After three cycles of 24 h each, the filtrates were evaporated to dryness and the marc was extracted with an acidified (HCl) mixture of MeOH – CH2Cl2 (1:1, pH = 2–3, 2 L), again with mechanical shaking, for 48 h. The acidified extract was neutralized with NaOH and mixed with the neutral one, after the similarity between the two extracts was established by analytical HPLC. The resulting total crude residue was submitted to a cation-exchange column (Amberlyst-15 from Fluka, Ø 3 cm) to remove undesired, non-basic metabolites.21 The obtained alkaloid-enriched fraction was partitioned between water and dichloromethane, giving, after evaporation, 800 mg of an aqueous subfraction and 30 mg of dichloromethane subfraction. The polar subfraction was dissolved in methanol and resolved by preparative HPLC, using the following gradient: A/B: 0 min 10% B; 25 min 25% B; 30 min 28% B; 31 min 100% B; 34 min 100% B; 35 min 10% B, in which A = 90% H2O + 0.05% trifluoroacetic acid and B = 90% MeCN + 0.05% trifluoroacetic acid. From this subfraction, eight metabolites were obtained, including the alkaloids 1–3 and 6–10. Resolution of the lipophilic subfraction by preparative HPLC yielded compounds 4 and 5.
Ruthenium-mediated oxidative degradation
Following a procedure described earlier,14 ca. 0.8 mg of the compounds 1–10 were submitted to a ruthenium(III)-catalyzed periodate oxidation, followed by derivatization of the resulting amino acids with MeOH/HCl and (R)-α-methoxy-α-trifluoromethylphenylacetyl chloride [(R)-MTPA-Cl, prepared from (S)-MTPA] and analyzed by GC-MSD.Antiprotozoal activities
The assessment of the antiprotozoal properties of the compounds 5 and 6 against Plasmodium falciparum (NF54 and K1), Trypanosoma cruzi (Tulahuen C2C4 with the Lac Z gene incorporated) amastigotes in mouse macrophages, Trypanosoma brucei rhodesiense (STIB900) bloodstream stages, and Leishmania donovani (MHOM/ET/67/L82) axenic amastigotes and the testing of the cytotoxicity against mammalian host cells (rat skeletal myoblast L6 cells) were done in vitro as previously described.33Cytotoxicity assay
The cytotoxicity assays against HeLa cell line (RCB0007, Tsukuba, Japan) were carried out as described previously.24 In brief, exponentially growing cells were harvested and plated in 96-well plates (2 × 103 per well) in DMEM and allowed to attach for 24 h in the humidified CO2 incubator at 37 °C. The cells were then washed with phosphate-buffered saline (PBS) followed by the addition of serially diluted test samples in DMEM. For each concentration, three replications were performed. After 72 h of incubation, the cells were washed twice with PBS, and 100 μL of DMEM containing 10% WST-8 cell counting kit (Dojindo Molecular Technologies, Inc., Rockville, MD, USA) solution were added. After 3 h of incubation, the absorbance at 450 nm was measured on an EnSpire Multimode plate reader (PerkinElmer, Inc., Waltham, MA, USA). Cell viability was calculated from the mean values from three wells using the following equation:| Cell viability (%) = [(Abstest sample − Absblank)/(Abscontrol − Absblank)] × 100% |
Antiausterity assay
The human pancreatic cancer PANC-1 (RBRC-RCB2095, Tsukuba, Japan) cell line was purchased from the Riken BRC cell bank and maintained in the standard DMEM with 10% FBS supplement under a humidified atmosphere of 5% CO2 in the incubator at 37 °C. For the antiausterity evaluation, exponentially growing cells were seeded in 96-well plates (1.5 × 104 per well) in DMEM and incubated for 24 h for the cell attachment. After this incubation time, the cells were washed twice with PBS, the medium was changed to serially diluted test samples in both nutrient-rich medium (DMEM) and nutrient-deprived medium (NDM) with a control and a blank in each test plate. The composition of the NDM was as follows: 0.1 mg L−1 Fe(NO3)3 (9H2O), 265 mg L−1 CaCl2 (2H2O), 400 mg L−1 KCl, 200 mg L−1 MgSO4 (7H2O), 6400 mg L−1 NaCl, 700 mg L−1 NaHCO3, 125 mg L−1 NaH2PO4, 15 mg L−1 phenol red, 25 mM L−1 HEPES buffer (pH 7.4), and MEM vitamin solution (Life Technologies, Inc., Rockville, MD, USA); the final pH was adjusted to 7.4 with 10% aqueous NaHCO3. After 24 h of incubation with the respective test compound in DMEM and NDM, the cells were washed twice with PBS and replaced by 100 μL of DMEM containing 10% WST-8 cell counting kit solution. After 3 h of incubation, cell viability was measured and calculated as described above.Morphological assessment of cancer cells
For studies on morphological changes, HeLa and PANC-1 cells were seeded in 24-well plates (1 × 105) and incubated in a humidified CO2 incubator for 24 h for the cell attachment. The cells were then washed twice with PBS and treated with vehicle control or test compounds in DMEM for HeLa cells; and vehicle control or test compounds in NDM for PANC-1 cells, and incubated for 24 h. For nuclei staining, two drops of the NucBlue® ready probe (Hoechst 33342) was directly added to the cells in full media and was incubated for further 25 min at the end of the experiment. For cellular morphology, 10 μL of EB/AO reagent (dye mixture: 100 μg mL−1 AO and 100 μg mL−1 EB in PBS) was added to each test well and further incubated for 10 min. The cellular images were captured in the fluorescent and phase contrast modes, using an EVOS FL digital microscope (20× objectives).Conclusions
In summary, this paper describes the discovery of the first twofold 1,3-cis-configured michellamine-type dimers, 1 and 2, and of four other mono- and dimeric naphthylisoquinoline alkaloids, 3–6, in the leaves of an as yet unidentified Congolese Ancistrocladus liana related to A. likoko. These new compounds, obtained along with five previously described analogs, 7–10, chemotaxonomically delineate this Congolese plant from all known related taxa, thus suggesting that it is probably a new species. Further taxonomic investigations, including DNA analysis, are planned. Some of the isolated metabolites have shown attractive cytotoxicities against the HeLa cell line and very good antiausterity activities against PANC-1 human pancreatic cancer cells. The weak-to-moderate antiprotozoal properties of the new monomeric metabolites 5 and 6 highlight the consequence of the stereochemical features on the biological activities and provide valuable information for the ongoing SAR investigations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (project Br 699/14-2; SFB 630 “Agents against Infectious Diseases” to G. B.) and by grants from the Deutscher Akademischer Austauchdienst (DAAD) and the Excellence Scholarship Program BEBUC (htpp://www.foerderverein-uni-kinshasa.de) to B. K. L. The assessment of cytotoxicities and antiausterity activities was supported by a grant from the Japanese Society for the Promotion of Science (Japan, Kakenhi, 16K08319) to S. A. We gratefully acknowledge technical assistance from Dr M. Grüne and Mrs P. Altenberger (NMR), Dr M. Büchner and Mrs J. Adelmann (MS), and Mrs M. Michel (oxidative degradation), all from the University of Würzburg.Notes and references
- For reviews on naphthylisoquinoline alkaloids, see: (a) G. Bringmann and F. Pokorny, in The Alkaloids, ed. G. A. Cordell, Academic Press Inc, New York, 1995, vol. 46, pp. 127–271 Search PubMed; (b) S. R. M. Ibrahim and G. A. Mohamed, Fitoterapia, 2015, 106, 194–225 CrossRef CAS PubMed.
- C. M. Taylor, R. E. Gereau and G. M. Walters, Ann. Mo. Bot. Gard., 2005, 92, 360–399 Search PubMed.
- H. Rischer, G. Heubl, H. Meimberg, M. Dreyer, H. A. Hadi and G. Bringmann, Blumea, 2005, 50, 357–365 CrossRef.
- G. Heubl, F. Turini, V. Mudogo, I. Kajahn and G. Bringmann, Plant Ecology and Evolution, 2010, 143, 63–69 CrossRef.
- J. Léonard, Bull. Soc. R. Bot. Belg., 1949, 82, 27–40 Search PubMed.
- F. G. Turini, C. Steinert, G. Heubl, G. Bringmann, B. K. Lombe, V. Mudogo and H. Meimberg, Taxon, 2014, 63, 329–341 CrossRef.
- D. T. Tshitenge, PhD thesis, Julius-Maximilians-Universität Würzburg, 2017.
- Y. F. Hallock, K. P. Manfredi, J. W. Blunt, J. H. Cardellina II, M. Schäffer, K. P. Gulden, G. Bringmann, A. Y. Lee, J. Clardy, G. François and M. R. Boyd, J. Org. Chem., 1994, 59, 6349–6355 CrossRef CAS.
- G. Bringmann, C. Steinert, D. Feineis, V. Mudogo, J. Betzin and C. Scheller, Phytochemistry, 2016, 128, 71–81 CrossRef CAS PubMed.
- Y. F. Hallock, K. P. Manfredi, J. R. Dai, J. H. Cardellina II, R. J. Gulakowski, J. B. McMahon, M. Schäffer, M. Stahl, K. P. Gulden, G. Bringmann, G. François and M. R. Boyd, J. Nat. Prod., 1997, 60, 677–683 CrossRef CAS PubMed.
- G. Bringmann, C. Günther, W. Saeb, J. Mies, A. Wickramasinghe, V. Mudogo and R. Brun, J. Nat. Prod., 2000, 63, 1333–1337 CrossRef CAS.
- S. Fayez, D. Feineis, V. Mudogo, S. Awale and G. Bringmann, RSC Adv., 2017, 7, 53740–53751 RSC.
- G. Bringmann, K. Messer, R. Brun and V. Mudogo, J. Nat. Prod., 2002, 65, 1096–1101 CrossRef CAS.
- G. Bringmann, R. God and M. Schäffer, Phytochemistry, 1996, 43, 1393–1403 CrossRef CAS.
- G. Bringmann, J. Spuziak, J. H. Faber, T. Gulder, I. Kajahn, M. Dreyer, G. Heubl, R. Brun and V. Mudogo, Phytochemistry, 2008, 69, 1065–1075 CrossRef CAS PubMed.
- G. Bringmann, W. Saeb, M. Rückert, J. Mies, M. Michel, V. Mudogo and R. Brun, Phytochemistry, 2003, 62, 631–636 CrossRef CAS PubMed.
- G. Bringmann, B. K. Lombe, C. Steinert, K. Ndjoko Ioset, R. Brun, F. Turini, G. Heubl and V. Mudogo, Org. Lett., 2013, 15, 2590–2594 CrossRef CAS PubMed.
- B. K. Lombe, T. Bruhn, D. Feineis, V. Mudogo, R. Brun and G. Bringmann, Org. Lett., 2017, 19, 1342–1345 CrossRef CAS PubMed.
- B. K. Lombe, T. Bruhn, D. Feineis, V. Mudogo, R. Brun and G. Bringmann, Org. Lett., 2017, 19, 6740–6743 CrossRef CAS PubMed.
- M. R. Boyd, Y. F. Hallock, J. H. Cardellina II, K. P. Manfredi, J. W. Blunt, J. B. McMahon, R. W. Buckheit Jr, G. Bringmann, M. Schäffer, G. M. Cragg, D. W. Thomas and J. G. Jato, J. Med. Chem., 1994, 37, 1740–1745 CrossRef CAS PubMed.
- G. Bringmann, G. Zhang, T. Büttner, G. Bauckmann, T. Kupfer, H. Braunschweig, R. Brun and V. Mudogo, Chem.–Eur. J., 2013, 19, 916–923 CrossRef CAS PubMed.
- E. Izumi, T. Ueda-Nakamura, B. P. D. Filho, V. F. V. Júnior and C. V. Nakamura, Nat. Prod. Rep., 2011, 28, 809–823 RSC.
- M. M. Salem and K. A. Werbovetz, Curr. Med. Chem., 2006, 13, 2571–2598 CrossRef CAS PubMed.
- J. Li, R. Seupel, T. Bruhn, D. Feineis, M. Kaiser, R. Brun, V. Mudogo, S. Awale and G. Bringmann, J. Nat. Prod., 2017, 80, 2807–2817 CrossRef CAS PubMed.
- J. Li, R. Seupel, D. Feineis, V. Mudogo, M. Kaiser, R. Brun, D. Brunnert, M. Chatterjee, E.-J. Seo, T. Efferth and G. Bringmann, J. Nat. Prod., 2017, 80, 443–458 CrossRef CAS PubMed.
- G. Bringmann, R. Seupel, D. Feineis, G. Zhang, M. Xu, J. Wu, M. Kaiser, R. Brun, E.-J. Seo and T. Efferth, Fitoterapia, 2016, 115, 1–8 CrossRef CAS PubMed.
- D. B. Longley, D. P. Harkin and P. G. Johnston, Nat. Rev. Cancer, 2003, 3, 330–338 CrossRef CAS PubMed.
- G. Van Zant and C. G. Fry, Cytometry, 1983, 4, 40–46 CrossRef CAS PubMed.
- K. Liu, P.-C. Liu, R. Liu and X. Wu, Med. Sci. Monit. Basic Res., 2015, 21, 15–20 CrossRef PubMed.
- K. Izuishi, K. Kato, T. Ogura, T. Kinoshita and H. Esumi, Cancer Res., 2000, 60, 6201–6207 CAS.
- H. Esumi, K. Izuishi, K. Kato, K. Hashimoto, Y. Kurashima, A. Kishimoto, T. Ogura and T. Ozawa, J. Biol. Chem., 2002, 277, 32791–32798 CrossRef CAS PubMed.
- S. Awale, E. M. Nakashima, S. K. Kalauni, Y. Tezuka, Y. Kurashima, J. Lu, H. Esumi and S. Kadota, Bioorg. Med. Chem. Lett., 2006, 16, 581–583 CrossRef CAS PubMed.
- I. Orhan, B. Sener, M. Kaiser, R. Brun and D. Tasdemir, Mar. Drugs, 2010, 8, 47–58 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: MS, NMR, CD, and IR spectra, and the results of the oxidative degradation. See DOI: 10.1039/c8ra00363g |
| This journal is © The Royal Society of Chemistry 2018 |