
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRole of ceria in the improvement of NO removal of lanthanum-based perovskite-type catalysts
Xiaochen Li and
Hongwei Gao *
*
Key Laboratory of Plant Resources and Chemistry in Arid Regions, Xinjiang Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Urumqi 830011, China. E-mail: gaohongw369@ms.xjb.ac.cn; Fax: +86-991-3858319; Tel: +86-991-3858319
First published on 27th March 2018
Abstract
Lanthanum-based perovskite-type oxides represented by LaBO3 (B = Co, Fe, Mn) have been thought to present strong limitations for practical application although they are active for catalytic removal of NO. Cerium (Ce) substitution has been extensively studied to modify the properties of perovskites. It is noted that a new phase of ceria (CeO2) can be separated from perovskites when the doping ratio exceeds the solution limit (x > S). This review outlines the relationship between the existence of CeO2 phase and catalytic activity. CeO2 dispersing on the lattice surface or small particles are beneficial for catalytic activity, but larger particles are adverse. Ce-doped LaBO3 perovskites exhibiting the best activity must contain additional CeO2 phases. In addition, CeO2-supported LaBO3 perovskite catalysts are discussed.
1. Introduction
The major pollutants discharged from vehicle engines to the environment are particulate matter (PM), carbon dioxide, hydrocarbons and nitrogen oxides (NOx). Among these contaminants, NOx is paid increasing attention, owing to the fact that it is not only harmful to human health but also responsible for acid rain and photochemical smog.1–3 The main component of NOx that we refer to is NO which is generated from nitrogen and oxygen at an appropriate temperature.4 In the past 40 years, great efforts have been carried out for minimizing NO emission.From the thermodynamic view, the process (NO = 1/2N2 + 1/2O2; ΔG0f = −86 kJ mol−1)5 is favored, but it is kinetically hindered because of its high activation energy (364 kJ mol−1).6 To solve the afore-mentioned problem, a good deal of catalytic methods and catalysts have been studied. Currently, NO removal methods that we have learned can be roughly classified as two major technologies: NO decomposition and NO reduction. The former can be regarded as a “green” process since no additional toxic reactant, such as ammonia, is required and the NOx gases are decomposed into N2 and O2 as harmless components. Oppositely, the latter (e.g. NOx storage and reduction (NSR), selective catalytic reduction (SCR), etc.) usually requires the addition of a supplemental reductant which causes further problems, such as higher equipment cost and secondary pollution. At present, three principal types of catalysts active for NO removal are zeolites,7–9 noble metals10,11 and composite mixed oxides.12–16 However, the first, zeolites, are subject to hydrothermal deactivation and are only active at high temperature while the second, noble metals, are costly and easily agglomerated during the reaction. In contrast to the above two catalysts, composite mixed oxides, especially perovskite-type catalyst with the general formula of ABO3, have considered as a group of promising catalysts for NO removal as they are lower price, mixed valence states of the transition metals and higher stability.17–19
Lanthanum-based perovskites (LaBO3), some of which exhibit excellent performance for NO removal,20–22 have been extensively investigated. As far as we know, unmodified LaBO3 perovskites present strong limitations (e.g. comparatively small surface area and low catalytic activity at low temperatures)21,22 and therefore reliable technologies need to be developed. Among these technologies tackling aforementioned problems, we find that doping Ce into perovskite (e.g. La0.6Ce0.4CoO3) or CeO2 as a “support” (e.g. LaCoO3/CeO2) are effective. In addition, it have been reported that the solubility of Ce in LaBO3 perovskites is low and CeO2 appears in the mixed-oxides when Ce addition exceeds its solubility limit (S). The valence alternation ability of Ce3+/Ce4+ makes CeO2 a good oxygen reservoir which stores and releases oxygen under oxidizing and reducing conditions, respectively.23,24 In this review, CeO2 is focused because of its significant role in improving catalytic activity of LaBO3 perovskites catalysts. The roles played CeO2 by will be introduced by the following two topics: (i) CeO2 as separated phase (ii) CeO2 as the support.
2. Description of lanthanum-based perovskite-type catalysts
The perovskite-type oxides mentioned here adopt LaBO3 structure (Fig. 1), in which B-sites are occupied by small transition metals such as Fe, Mn.25–27 For the resulting compounds, each La cation is surrounded by 12 oxygen anions whereas each B cation is surrounded by 6 oxygen anions.28 In the ideal unit cell where the atoms are packed closely, La–O distance is equal to a/√2 (a is the cubic unit cell parameter) while B–O distance is a/2. Besides, La, B and O ions should satisfy the requirement that the value of tolerance factor (t)28–30 is equal to 1. The parameter is defined by the expression t = (rLa + rB)/{√2(rB + rO)}, where rLa, rB and rO correspond to the crystal radii for cations La and B and the anion O, respectively. However, the perovskite structure is also found for lower values of t in the range 0.75 to 1.28,30 The simple perovskite structure could be partially replaced by foreign ions with appropriate size difference, preserving matrix perovskite structure. In such cases, the cubical structure can be distorted into tetragonal, rhombohedral, or other lower symmetries.29,30 Besides the ionic radii requirement, electroneutrality is another condition. For example, the substitution of La3+ cation in LaMnO3 with Sr2+ is accompanied by a change of the oxidation state of Mn cation (Mn3+ → Mn4+) and exhibits oxidative non-stoichiometry.31 The substitution may stabilize an unusual oxidation state of the cation in position B and/or produce oxygen vacancies or structural defects.31–33 These excellent characteristics can enhance substantially the catalytic activity and therefore have frequently been exploited in perovskite-type catalysis.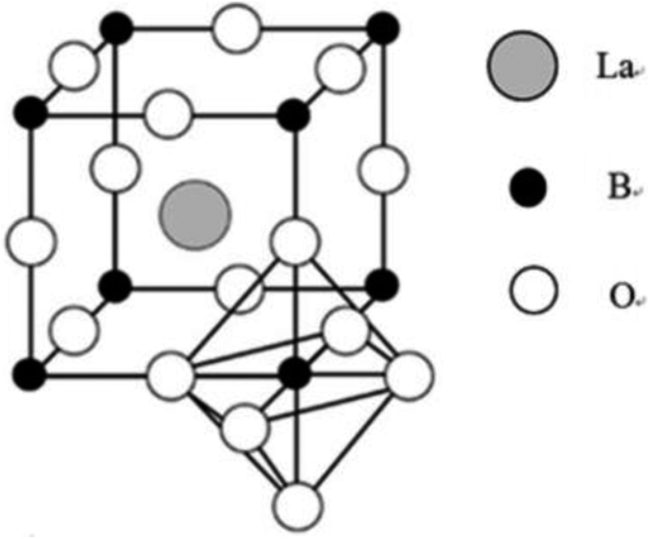 | ||
| Fig. 1 Crystal structure of the LaBO3 perovskite oxide.4 | ||
Ce often appears as a good promoter in perovskite lattice because of its two different valence states Ce4+ or Ce3+.4,34,35 The substitution of La3+ cation in LaBO3 with higher valence Ce4+ will affect indirectly the electronic state of the B-site ion. As mentioned above, Sr2+ are used to partially substitute the A-site ions in order to increase the basicity and to produce oxide anion vacancies by charge compensation. To get a better and clearer understanding about Ce doping, we take LaCoO3 as an example to explain the process of defect formation. Three reactions have been considered for Ce doping,36,37 the first two of which are for doping stoichiometric LaCoO3. The substitution of La3+ cation in LaCoO3 with Ce4+ leads to creation of La3+ vacancies (eqn (1)) or reduction of Co3+ to Co2+(eqn (2)).
 | (1) |
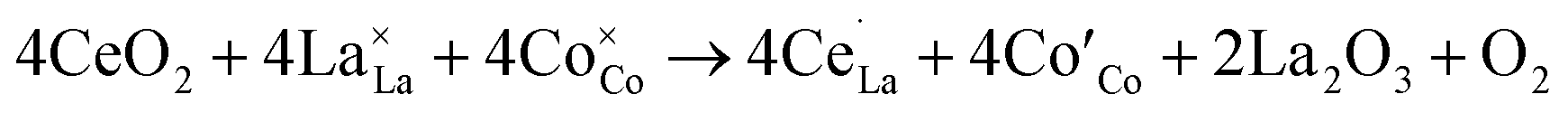 | (2) |
However, as prepared LaCoO3 calcined at ≥900 °C is typically slightly reductively non-stoichiometric.38 Therefore, the third reaction that Ce is doped into the LaCoO3 lattice containing oxygen lattice vacancies was considered. This means that those oxygen vacancies were refilled.
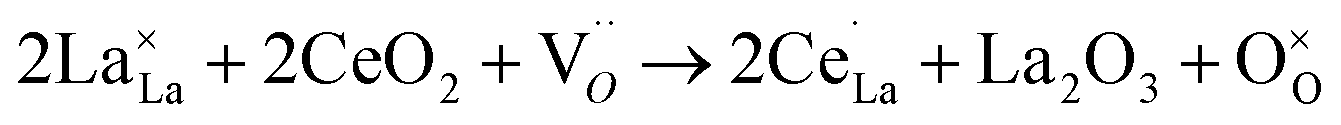 | (3) |
These defects caused by Ce doping lead to the fact that perovskite-type oxides show promising structural, surface and catalytic properties for catalyst application. In addition, supported lanthanum-based perovskite-type catalysts, e.g. LaBO3/CeO2, were also extensively studied.
3. CeO2 as separated phase
Among various perovskites, lanthanum-based samples represented by LaBO3 (B = Co, Fe, Mn) are known to be excellent catalysts for NO removal.21,22 Also, it has been reported that the Ce often appeared as a crucial component for perovskites because of its two different valence states Ce4+ or Ce3+, which is important to the catalytic reaction. This is the reason why people have been paying increasing attention to Ce-doped LaBO3 perovskites.For a low Ce content (which is below its solubility in perovskite structure), Ce can be incorporated into the LaBO3 lattice to form a solid solution. The valence change of B is complicated due to the addition of Ce. The substitution of La3+ in LaBO3 with Ce3+ result in no change of valence state of B ions (Bn+). The higher valence Ce4+ substitution lead to the fact that electroneutrality of the lattice is not maintained. As such, cation vacancies are formed and valence state of B ions is changed. No interstitial oxygen ions, however, are detected because perovskite lattice cannot accommodate them.39,40 For high Ce content (which exceed their solubility limit in perovskite structure), CeO2 are formed and segregated from perovskite oxides. The extra phase, CeO2, is either highly dispersed on the surface of Lal−xCexBO3 for comparatively low Ce content or found as larger particles for higher Ce content. The presence of CeO2 result in appearance of Ce vacancy sites or migration of (excess) B ions outside the perovskite lattice to yield ByOx or both.41 In this case, CeO2 are believed to play a key role in enhancement of catalytic performance.
3.1 Ce doped LaCoO3
The influence of Ce substitution on structural characteristics has been discussed in previous literatures.42–48 Based on the X-ray diffraction (XRD) results, Fig. 2, no peaks of CeO2 are detected when Ce addition was below 0.05, indicating that the doped Ce atoms might have embodied into the LaCoO3 lattice to form a solid solution. For x = 0 and 0.05, the structures were the rhombohedral LaCoO3-type (JCPDS 25-1060). As the content of doped Ce increasing, the other minor phase, CeO2, was also detected in addition to the major ABO3 perovskite phase. This is attributed to that Ce addition exceeds its solubility limit (x ≥ 0.1).45,46 Although no Co3O4 peaks were detected in the XRD diffraction spectroscopy, Co3O4 is proposed to be separated from perovskite oxides accompanying with the separation of CeO2 phase, which has been proved by previous literatures.43,45,47 According to Erdenee's study on samples with x = 0.2 and 0.4,43 the structure of LaCoO3 transforms from rhombohedral to cubic and peaks corresponding to Co3O4 and CeO2 were visible. The severe lattice distortion perhaps leads to form more oxygen vacancies.42 Table 1 lists the values of specific surface area (ssa) of La1−xCexCoO3 perovskites. It is observed that the increment of the ssa values of the obtained perovskites follows the sequence: LaCoO3 (ssa = 8.9 m2 g−1) < La0.7Ce0.3CoO3 (ssa = 9.8 m2 g−1) < La0.6Ce0.4CoO3 (ssa = 10.8 m2 g−1) < La0.8Ce0.2CoO3 (ssa = 11.0 m2 g−1) < La0.95Ce0.05CoO3 (ssa = 11.4 m2 g−1) < La0.9Ce0.1CoO3 (ssa = 12.6 m2 g−1). So we reach the conclusion that substituted samples have a larger ssa values than pure perovskites and the enhancement was not linear with substitution concentration. When Ce addition was below its solubility limit (x ≤ 0.1), it was noticed that the value of ssa significantly increase as pore volume was enlarged. However, with the increased proportion of Ce, their ssa decrease, which might be related to that the CeO2 was segregated out and filled in the pores of the perovskite.46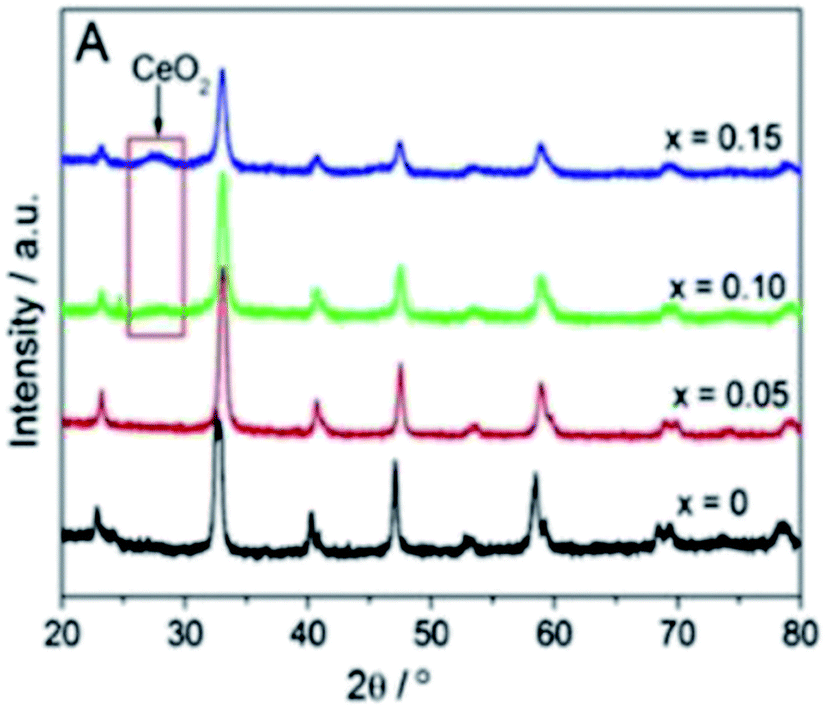 | ||
| Fig. 2 XRD patterns of the La1−xCexCoO3 perovskites (x = 0, 0.05, 0.10 and 0.15).46 | ||
| La1−xCexCoO3 | ||||||
|---|---|---|---|---|---|---|
| x = 0 | x = 0.05 | x = 0.1 | x = 0.2 | x = 0.3 | x = 0.4 | |
| SSA (m2 g−1) | 8.9 | 11.4 | 12.6 | 11.0 | 9.8 | 10.8 |
In addition to above-mentioned physical structures, the effect of Ce substitution on redox properties have also been discussed in this paper. Firstly, it has been reported that partial substitution with Ce can affect the surface characteristics which are always more important than lattice. The surface characteristics obtained from X-ray photoelectron spectroscopy (XPS) has been observed in the reports of Erdenee et al.43 and Wen et al.45 In their work, no change of La valence state (La3+) was detected when La was substituted with Ce. But they all agreed that the trivalent Co ion is partly changed. As has been reported by Wen et al.,45 when the doping ratio was below 0.1, tetravalent Ce ions were fully dissolved into the perovskite structure so that electronic unbalance was generated. As a result, a part of Co3+ in the B-site became Co2+ or La3+ vacancies were created as a charge compensation mechanism.36,37 When x was larger than 0.1, the average oxidation state of the Co cation increased because the existence of CeO2 might lead to deficiency of A-cation. Of course, they also showed XPS spectra of O 1s. According to ref. 43, 45 and 47, there are three different forms of oxygen including lattice oxygen (Olatt), adsorbed oxygen (Oads), and surface adsorbed water species. Wen et al.45 found that the La0.8Ce0.2CoO3 achieved the highest percentage of adsorption oxygen which participated in oxidation reactions. It is generally accepted that among Ce doped LaCoO3 samples, La0.8Ce0.2CoO3 shows the best activity.43,45 However, we cannot directly draw the conclusion that the improvement of catalytic activities are due to high amount of adsorbed oxygen, owing to the fact that although the perovskite LaCoO3 also gets high amount of adsorbed oxygen (Table 2), its activity is low. Additionally, it was proved that Ce ions were present in the trivalent and tetravalent form.43,46,47 The relative concentration of Ce3+, defined as Ce3+/(Ce3+ + Ce4+), was listed in Table 3. It varied from 19.2% to 21.0%, indicating that Ce4+ was dominant in all the samples. Secondly, a result of temperature programmed reduction of H2 (H2-TPR) experiment investigating effect of Ce addition on the redox ability of LaCoO3 is shown in Fig. 3. The literature46 also displayed the H2-TPR profiles of La1−xCexCoO3, in which two obvious reduction peaks were observed, which could be attributed to the reduction of Co3+ to Co2+ and Co2+ to Co0, respectively. As was seen from Fig. 3, two reduction peaks respectively located at about 400 °C and 600 °C. When a certain amount of Ce was introduced into the LaCoO3 perovskite structure, the two peaks moved forward to lower temperature direction correspondingly, indicating that Ce4+ insertion increased the catalyst reducibility. In other word, Ce-doped catalysts showed a better reducibility as the reduction temperatures of these catalysts were lower than pure LaCoO3. And the sample with La1−xCexCoO3 (x = 0.2) leads to the highest decrease in the reduction peak temperatures. This may be because (1) it creates an easier reducibility of the Co3+ into Co2+ and, (2) Ce4+ increases the number of cation vacancies within the lattice.
| La1−xCexCoO3 | ||||||
|---|---|---|---|---|---|---|
| x = 0 | x = 0.05 | x = 0.1 | x = 0.2 | x = 0.3 | x = 0.4 | |
| % | 46.6 | 42.2 | 43.5 | 48.4 | 39.8 | 38.3 |
| La1−xCexCoO3 | |||||
|---|---|---|---|---|---|
| x = 0 | x = 0.05 | x = 0.1 | x = 0.3 | x = 0.5 | |
| Ce3+/(Ce3+ + Ce4+) (%) | — | 20.6 | 21.0 | 20.3 | 19.2 |
 | ||
| Fig. 3 H2-TPR profiles of the La1−xCexCoO3 (0 ≤ x ≤ 0.4) catalysts.43 | ||
According to Wen's paper,45 the La1−xCexCoO3 samples synthesized through a citrate show the highest conversion (80%) at about 300 °C in a flow of NO + O2 + He when x was 0.2. However, the catalytic activities decreased at higher x values. They thought that these factors including the presence of CeO2 and the decrease of adsorbed oxygen amount caused the decrease of activity. Combined with his results of XRD, when x > 0.1, the peaks of CeO2 were observed. This means that small CeO2 appeared in the mixed-oxide. Thus, it is concluded that the existence of small CeO2 nano-particles is beneficial to the improvement of catalytic activities, but catalytic activities decreased because larger particles are formed at higher Ce content.
3.2 Ce doped LaFeO3
Like La1−xCexCoO3 samples, a series of La1−xCexFeO3 perovskite-type oxide catalysts are also characterized by XRD, BET, XPS and TPR to illustrate the influence of Ce substitution. In these literatures,49–51 it is found that the substitution of La by Ce can increase the value of ssa and change the reducibility of Fe cation and Fe4+/Fe3+ ratios. The influence of Ce substitution on LaFeO3 is very similar to that on LaCoO3. Therefore, changes in its physicochemical properties are not explained in this part, but effect of catalytic performances is paid more attention here.Giannakas et al.50 investigated and compared the catalytic performance of LaFeO3, La0.85Sr0.15FeO3, La0.8Sr0.1Ce0.1FeO3 and La0.8Ce0.2FeO3 which were prepared via a reverse micelles microemulsion route for NO reduction by CO, finding that the full sequence of catalytic activity of tested solids is La0.8Ce0.2FeO3 > La0.8Sr0.1Ce0.1FeO3 > La0.85Sr0.15FeO3 ≥ LaFeO3. They pointed that (1) the above sequence is in accordance with the sequence of increment of ssa of the solids and, (2) La0.8Ce0.2FeO3 and La0.8Sr0.1Ce0.1FeO3 show much higher catalytic activity than the solids containing no Ce, which may be caused by the presence of CeO2 phase in these two solids. Qin et al.51 also reported that catalytic reaction of La1−xCexFeO3 for NO reduction with CO and illustrated the influence of Ce substitution on SO2 resistance. It was found that although LaFeO3 exhibited an excellent catalytic performance without SO2 (100% NO conversion at 500 °C), catalytic reaction decreased drastically when SO2 gas was added to the CO + NO system(conversion rate decreased to 40–50% after approximately 100 min). Doping a certain amount of Ce into the LaFeO3 perovskite structure could obviously improve the SO2 resistance. For example, La0.6Ce0.4FeO3 sample maintained a conversion rate of 80% during the latter 270 min after addition of SO2 gas into the CO + NO reaction system. The reason was suggested to be the addition of Ce4+ which could prevent the adverse effect of SO2 on the LaFeO3 perovskite by absorbing SO2 and forming Ce(SO4)2. Thus, the presence of Ce4+ is the key factor to the improvement of SO2 resistance in LaFeO3 perovskite catalysts.
Recently, attapulgite (ATP) was considered as support for La1−xCexFeO3 catalysts, not only because of its inexpensive but also for its adsorption of many pollutants. A recent research52 on La1−xCexFeO3/ATP was conducted for testing its photocatalytic reduction of NO at low temperature. Following investigation on photo-SCR activity and stability of La1−xCexFeO3/ATP samples (Fig. 4) proves that the substitution of Ce is helpful for the improvement of catalytic performance. It is noteworthy that the conversion rate of NO reaches close to 80% when Ce doping amount is 0.3 at room temperature. Combined with XRD results, CeO2 is precipitated from the solid solution when the Ce doping ratio is 0.3. This implies that the existence of CeO2 is no harm for NOx reduction.
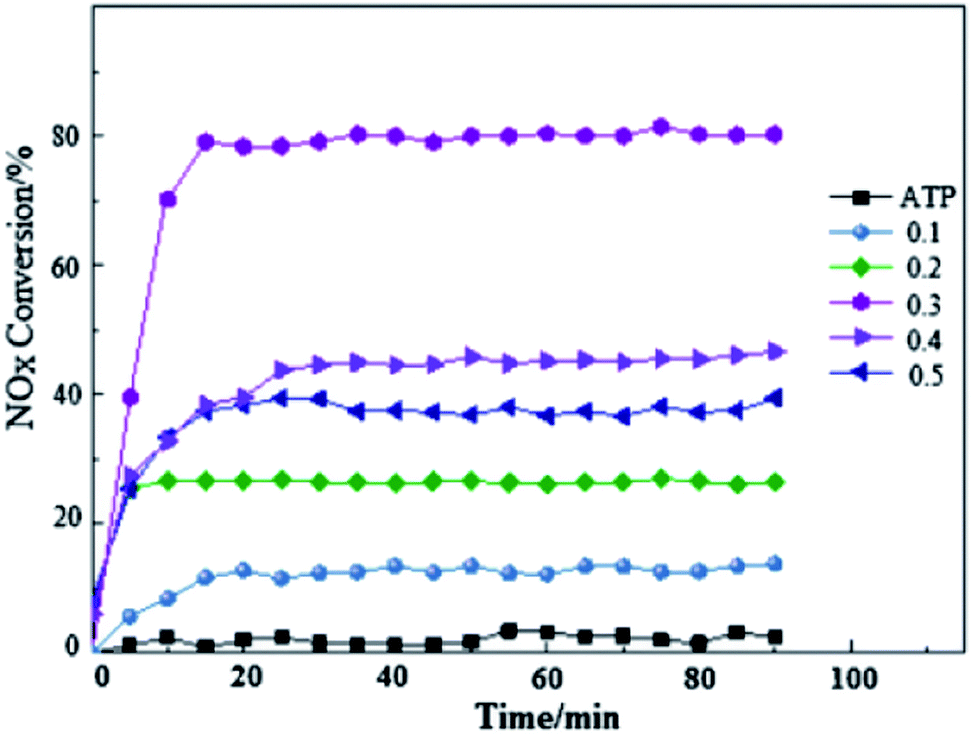 | ||
| Fig. 4 Photo-SCR denitrification activity and stability of La1−xCexFeO3/ATP (x = 0.1–0.5).52 | ||
Besides the NO removal, La1−xCexFeO3 was also considered as promising catalyst in some reactions. For example, Xiang et al.49 reported that the temperature of CH4 conversion at 90% was as low as 510 °C when x = 0.3. According to Ma's report,53 in the case of x = 0.5, the conversion of CO is about 68% at 530 °C.
3.3 Ce doped LaMnO3
As mentioned earlier, we have systematically studied the effect of Ce in La1−xCexCoO3 and La1−xCexFeO3 perovskites. Therefore, the properties of La1−xCexMnO3 are not discussed here, because the influences of Ce in La1−xCexMnO3 are similar to that in former two perovskites except that segregation of CeO2 is not accompanied by segregation of some manganese oxide according to cases.43,54–56 This may be ascribed to that manganese oxide can be dissolved in the segregated CeO2.54 In this part, the simultaneous substitution of Ce at the A-site and other ions (B′) at the B-site in a LaMnO3 lattice will be made concentrates mainly.For 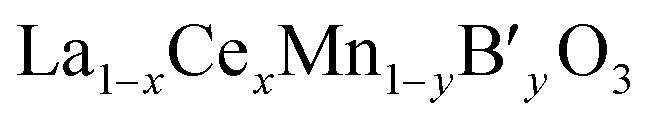 catalysts, the effect of the substitution of La by Ce in these catalysts is more complex than that in La1−xCexMnO3, since the Ce cation not only changes the reducibility of Mn cation (Mn3+ ↔ Mn4+), but also may affect the oxidation state of the B′ cation. In the case of
catalysts, the effect of the substitution of La by Ce in these catalysts is more complex than that in La1−xCexMnO3, since the Ce cation not only changes the reducibility of Mn cation (Mn3+ ↔ Mn4+), but also may affect the oxidation state of the B′ cation. In the case of 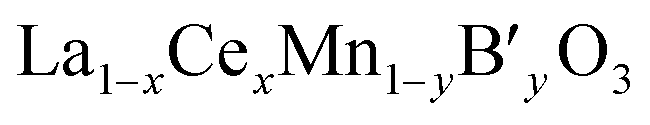 , the redox couple can be Ce4+/Ce3+, Mn4+/Mn3+and B′n+/B′(n−1)+ since oxidation state of B′ cation may be variable. According to He's paper,57 the reaction (Ce3+ + Cu2+ → Ce4+ + Cu+) taken place during the preparation process of the La0.8Ce0.2Mn0.6Cu0.4O3 catalysts. The presence of Cu+ and its redox equilibrium (Cu+ ↔ Cu2+) would facilitate the NO adsorption and reduction by CO. Similarly, Tarjomannejad et al.58 studied the LaMn1−xFexO3 and La0.8Ce0.2Mn0.3Fe0.7O3 perovskites catalysts, finding that the substitution of La3+ by Ce4+ result in decrease of Mn4+/Mn3+ and Fe4+/Fe3+ratios. Of course, the substituted samples can also lead to the formation of oxygen vacancies and the increase of Oads/Olatt ratio. An integration of all these factors resulted in the enhancement of catalytic activity (90% NO conversion at 323 °C). In addition, if the B′ cation exists in a permanent valence state, Ce4+/Ce3+and Mn4+/Mn3+ are the redox couples. The role of Ce is just to adjust the valence state of Mn cation or/and the ratio of Oads/Olatt. However, the catalytic performance of
, the redox couple can be Ce4+/Ce3+, Mn4+/Mn3+and B′n+/B′(n−1)+ since oxidation state of B′ cation may be variable. According to He's paper,57 the reaction (Ce3+ + Cu2+ → Ce4+ + Cu+) taken place during the preparation process of the La0.8Ce0.2Mn0.6Cu0.4O3 catalysts. The presence of Cu+ and its redox equilibrium (Cu+ ↔ Cu2+) would facilitate the NO adsorption and reduction by CO. Similarly, Tarjomannejad et al.58 studied the LaMn1−xFexO3 and La0.8Ce0.2Mn0.3Fe0.7O3 perovskites catalysts, finding that the substitution of La3+ by Ce4+ result in decrease of Mn4+/Mn3+ and Fe4+/Fe3+ratios. Of course, the substituted samples can also lead to the formation of oxygen vacancies and the increase of Oads/Olatt ratio. An integration of all these factors resulted in the enhancement of catalytic activity (90% NO conversion at 323 °C). In addition, if the B′ cation exists in a permanent valence state, Ce4+/Ce3+and Mn4+/Mn3+ are the redox couples. The role of Ce is just to adjust the valence state of Mn cation or/and the ratio of Oads/Olatt. However, the catalytic performance of 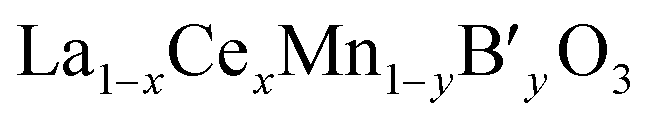 samples have no relation to whether the valence state is varied or not. Sometimes the B′ cation with a varied valence state can promote the catalytic reaction,57,58 sometimes inhibit. In the literature,59 the substitution of Mn by Co in La0.8Ce0.2MnO3 catalysts results in decreasing catalyst activity.
samples have no relation to whether the valence state is varied or not. Sometimes the B′ cation with a varied valence state can promote the catalytic reaction,57,58 sometimes inhibit. In the literature,59 the substitution of Mn by Co in La0.8Ce0.2MnO3 catalysts results in decreasing catalyst activity.
Besides, it has been reported that preparation methods interfere with the generation of CeO2 phase. In the paper,57 a considerable amount of CeO2 was found by XRD in the La0.8Ce0.2Cu0.4Mn0.6O3 and La0.8Ce0.2Ag0.4Mn0.6O3 samples synthesized by sol–gel method, but not in samples synthesized by the reverse microemulsion method.
4. CeO2 as the support
Aiming at resolving the problem that powder perovskites have much lower surface area, the scheme that perovskites are dispersed on thermally stable supports has been adopted and is being put into operation. Presently, perovskites are often supported on Al2O3, CeO2, attapulgite and other supports.60–64 Among a variety of support materials, CeO2 is one of the most popular supports because of its particular redox property. These supported perovskite oxides are usually prepared by impregnation methods and their performances depend closely on the support. In the paper,65 Zhang et al. investigated and compared the catalytic performance of LaMnO3/CeO2 and LaMnO3/TiO2 for the SCR of NO by NH3 in the presence of O2, finding that the former showed higher activity than the latter. The reason was suggested to be the difference of support. The phenomenon that TiO2 supporting led to a remarkable decrease in redox capacity, contrasting with the pure LaMnO3, has been also reported by Zhang et al.65 Thus, the conclusion that the catalytic performance of all supported samples can be higher than that of ABO3 perovskite alone is wrong.Another factor for catalytic performance of supported samples is preparation method. H2-TPR experiment has been done to investigate the structure and reducibility of the catalysts prepared by two methods.66 The result shown in Fig. 5 indicated that preparation method had relation to the amount of H2 consumed, and it was suggested that LaMnO3/CeO2 prepared by the dry impregnation (DI) method showed higher activity than that by the precipitation–deposition (PD) method. In order to find the reason, the catalysts were characterized by XRD, BET and EXAFS. In the result of XRD, the peaks corresponding to the perovskite were detected for LaMnO3/CeO2-PD catalysts but hardly detected for the LaMnO3/CeO2-DI catalysts, indicating that LaMnO3 perovskite phases were highly dispersed on the CeO2 support. The BET surface areas of LaMnO3/CeO2-DI (46 m2 g−1) was lower than that of LaMnO3/CeO2-PD (68 m2 g−1). The formation of a perovskite oxide phase for the LaMnO3/CeO2 catalysts was confirmed by the EXAFS studies. Combined with the H2-TPR study results, they propose that the higher activity may be ascribed to the strong interaction between the perovskite phase and the CeO2 support although this catalyst has smaller surface area. Oppositely, Alifanti et al.67 concluded that there was no interaction between the support and the elements of the perovskite.
 | ||
| Fig. 5 H2-TPR profiles of LaMnO3/CeO2 catalysts and LaMnO3.66 | ||
In addition, the morphology of CeO2 has also a great influence on the catalytic activity, which has been studied by Wang et al.68 It was found that the supported catalysts were more active than the unsupported catalyst (La0.8Ce0.2MnO3) and La0.8Ce0.2MnO3/CeO2 nanopolyhedra exhibited the higher catalytic activity than the other two morphologies of CeO2 supported samples. The results described above are possible due to higher specific surface area and larger number of oxygen vacancies.
In summary, the modification method of perovskite oxides is effective for improving their catalytic activity, even though the promotion mechanism is still not clearly explained. Such a supported perovskite-type catalysts can have wide application prospects for the possibility of increasing its activity by simple preparation methods.
5. Conclusion
For Ce doped LaBO3 (B = Co, Fe, Mn), all catalysts exhibit relatively larger specific surface areas compared with pure perovskites. The introduction of Ce changes the reducibility of B cations and Bn+/B(n−1)+ ratios and these factors induce the generation of crystal lattice defects which lead to higher catalytic activities. This review outlined the relationship between the existence of CeO2 phase and catalytic activity. When the Ce addition is below its solubility in perovskite structure (x ≤ S), Ce atoms can be totally incorporated in the perovskite lattice. Of course, Ce substitution at higher levels (x > S) can lead to the formation of separated CeO2 phase. CeO2 is either highly dispersed on the lattice surface or is found as larger particles. The former amorphous CeO2 cannot be detected by XRD, while the latter can be done.41 What is more, it is found that Ce content of perovskite-type catalysts exhibiting the best activity exceeds its solubility in perovskites, indicating the presence of CeO2. CeO2, however, does not show good catalytic activity by itself.45,69 This means that the effect of separated CeO2 on the catalytic performance is an indirect one, i.e., by regulating the Bn+/B(n−1)+ ratios and by forming ByOx phase. The interaction between different phases may also play a key role in improving catalytic activity to some extent. Nonetheless, it is found that as the content of doped Ce increasing, catalytic activity begins to decrease slightly. Thus, the CeO2 dispersing on lattice surface or small particles are beneficial for catalytic activity, but larger particles are adverse. As such, improving preparation methods to delay the generation and growth of CeO2 phase may be effective for further improvement of activity. But beyond that, more researches still need to go further in order to improve the catalytic efficiency of NO transformation. For example, effective support materials which can dramatically promote the activity of Ce-doped LaBO3 catalysts still need to be studied. On the basis of Ce-doped LaBO3, partial substitution of the A and/or B cations by cations on different oxidation states should also be attempted. Photocatalytic efficiency of La0.7Ce0.3FeO3/ATP that we mentioned previously can reach as high as 80% at room temperature. This make us get some great inspiration: in addition to improving catalysts, the reaction conditions must also be concerned.For LaBO3/CeO2 catalysts, promotion mechanism is still not clearly explained. However, comparing with CeO2 support or pure LaBO3 perovskite catalyst, the catalytic activities of the majority of supported catalysts are enhanced. The simple preparation methods provide a feasible scheme for improvement of catalytic activity of LaBO3 perovskites.
Conflicts of interest
Authors declare no conflict of interest.Acknowledgements
This work was financially supported by Natural Science Foundation of Xinjiang, China, Grant No. 2016D01A073. This work was also financially supported by Recruitment Program of Global Experts, and the Director Foundation of XTIPC, CAS, Grant No. 2015RC011.References
- J. Li, S. Wang, L. Zhou, G. Luo and F. Wei, Chem. Eng. J., 2014, 255, 126–133 CrossRef CAS.
- V. Ferrer, D. Finol, R. Solano, A. Moronta and M. Ramos, J. Environ. Sci., 2015, 27, 87–96 CrossRef PubMed.
- M. Li, V. G. Easterling and M. P. Harold, Appl. Catal., B, 2016, 184, 364–380 CrossRef CAS.
- N. Imanaka and T. Masui, Appl. Catal., A, 2012, 431, 1–8 CrossRef.
- A. Fritz and V. Pitchon, Appl. Catal., B, 1997, 13, 1–25 CrossRef CAS.
- S. Roy, M. S. Hegde and G. Madras, Appl. Energy, 2009, 86, 2283–2297 CrossRef CAS.
- M. Iwamoto and H. Hamada, Catal. Today, 1991, 10, 57–71 CrossRef CAS.
- M. Iwamoto, H. Furukawa, Y. Mine, F. Uemura, S. Mikuriya and S. Kagawa, J. Chem. Soc., Chem. Commun., 1986, 16, 1272–1273 RSC.
- T. Selleri, F. Gramigni, I. Nova and E. Tronconi, Appl. Catal., B, 2018, 225, 324–331 CrossRef CAS.
- R. Burch and M. D. Coleman, Appl. Catal., B, 1999, 23, 115–121 CrossRef CAS.
- T. Chafik, D. I. Kondarides and X. E. Verykios, J. Catal., 2000, 190, 446–459 CrossRef CAS.
- Z. Wang, Z. Qu, X. Quan, Z. Li, H. Wang and R. Fan, Appl. Catal., B, 2013, 134, 153–166 CrossRef.
- Z. Wang, Z. Qu, X. Quan and H. Wang, Appl. Catal., A, 2012, 411, 131–138 CrossRef.
- L. Chmielarz, P. Kustrowski, A. Rafalska-Lasocha and R. Dziembaj, Appl. Catal., B, 2005, 58, 235–244 CrossRef CAS.
- Q. Zhang, Y. Huang, S. Peng, Y. Zhang, Z. Shen, J. Cao, W. Ho, S. C. Lee and D. Y. H. Pui, Appl. Catal., B, 2017, 204, 346–357 CrossRef CAS.
- Y. Niu, T. Shang, S. Hui, X. Zhang, Y. Lei, Y. Lv and S. Wang, Fuel, 2016, 185, 316–322 CrossRef CAS.
- C. Zhou, Z. Feng, Y. Zhang, L. Hu, R. Chen, B. Shan, H. Yin, W. G. Wang and A. Huang, RSC Adv., 2015, 5, 28054–28059 RSC.
- Z. Li, M. Meng, Q. Li, Y. Xie, T. Hu and J. Zhang, Chem. Eng. J., 2010, 164, 98–105 CrossRef CAS.
- X. Guo, M. Meng, F. Dai, Q. Li, Z. Zhang, Z. Jiang, S. Zhang and Y. Huang, Appl. Catal., B, 2013, 142, 278–289 CrossRef.
- J. Chen, M. Shen, X. Wang, G. Qi, J. Wang and W. Li, Appl. Catal., B, 2013, 134, 251–257 CrossRef.
- J. Chen, M. Shen, X. Wang, J. Wang, Y. Su and Z. Zhao, Catal. Commun., 2013, 37, 105–108 CrossRef CAS.
- A. E. Giannakas, A. K. Ladavos and P. J. Pomonis, Appl. Catal., B, 2004, 49, 147–158 CrossRef CAS.
- R. Q. Long and R. T. Yang, Appl. Catal., B, 2000, 27, 87–95 CrossRef CAS.
- A. Logan and M. Shelef, J. Mater. Res., 1994, 9, 468–475 CrossRef CAS.
- S. Keav, S. K. Matam, D. Ferri and A. Weidenkaff, Catalysts, 2014, 4, 226–255 CrossRef.
- A. Ashok, A. Kumar, R. R. Bhosale, F. Almomani, S. S. Malik, S. Suslov and F. Tarlochan, J. Electroanal. Chem., 2018, 809, 22–30 CrossRef CAS.
- K.-Y. A. Lin, Y.-C. Chen and Y.-F. Lin, Chem. Eng. Sci., 2017, 160, 96–105 CrossRef CAS.
- M. A. Pena and J. L. G. Fierro, Chem. Rev., 2001, 101, 1981–2017 CrossRef CAS PubMed.
- V. M. Goldschmidt, Naturwissenschaften, 1926, 14, 477–485 CrossRef CAS.
- N. Labhasetwar, G. Saravanan, S. K. Megarajan, N. Manwar, R. Khobragade, P. Doggali and F. Grasset, Sci. Technol. Adv. Mater., 2015, 16, 036002 CrossRef PubMed.
- S. Ponce, M. A. Pena and J. L. G. Fierro, Appl. Catal., B, 2000, 24, 193–205 CrossRef CAS.
- R. Leanza, I. Rossetti, L. Fabbrini, C. Oliva and L. Forni, Appl. Catal., B, 2000, 28, 55–64 CrossRef CAS.
- G. Saracco, F. Geobaldo and G. Baldi, Appl. Catal., B, 1999, 20, 277–288 CrossRef CAS.
- B. Zhao, R. Ran, L. Sun, Z. Yang, X. Wu and D. Weng, Catal. Commun., 2018, 105, 26–30 CrossRef CAS.
- Z. Guguchia, T. Adachi, Z. Shermadini, T. Ohgi, J. Chang, E. S. Bozin, F. von Rohr, A. M. dos Santos, J. J. Molaison, R. Boehler, Y. Koike, A. R. Wieteska, B. A. Frandsen, E. Morenzoni, A. Amato, S. J. L. Billinge, Y. J. Uemura and R. Khasanov, Phys. Rev. B, 2017, 96, 094515 CrossRef.
- S. Khan, R. J. Oldman, C. R. A. Catlow, S. A. French and S. A. Axon, J. Phys. Chem. C, 2008, 112, 12310–12320 CAS.
- S. A. French, C. R. A. Catlow, R. J. Oldman, S. C. Rogers and S. A. Axon, Chem. Commun., 2002, 2706–2707 RSC.
- K. Huang, H. Y. Lee and J. B. Goodenough, J. Electrochem. Soc., 1998, 145, 3220–3227 CrossRef CAS.
- B. Tofield and W. Scott, J. Solid State Chem., 1974, 10, 183–194 CrossRef CAS.
- J. Topfer and J. B. Goodenough, J. Solid State Chem., 1997, 130, 117–128 CrossRef CAS.
- V. MathieuDeremince, J. B. Nagy and J. J. Verbist, Catalysis and Automotive Pollution Control III, Stud. Surf. Sci. Catal, 1995, 96, 393–404 CrossRef CAS.
- R. Zhang, N. Luo, B. Chen and S. Kaliaguine, Energy Fuels, 2010, 24, 3719–3726 CrossRef CAS.
- N. Erdenee, U. Enkhnaran, S. Galsan and A. Pagvajav, J. Nanomater., 2017, 9120586 Search PubMed.
- L. Forni, C. Oliva, F. P. Vatti, M. A. Kandala, A. M. Ezerets and A. V. Vishniakov, Appl. Catal., B, 1996, 7, 269–284 CrossRef CAS.
- Y. Wen, C. Zhang, H. He, Y. Yu and Y. Teraoka, Catal. Today, 2007, 126, 400–405 CrossRef CAS.
- J. Zhu, Y. Zhao, D. Tang, Z. Zhao and S. A. C. Carabineiro, J. Catal., 2016, 340, 41–48 CrossRef CAS.
- X. Zhu, S. Zhang, Y. Yang, C. Zheng, J. Zhou, X. Gao and X. Tu, Appl. Catal., B, 2017, 213, 97–105 CrossRef CAS.
- M. Ghasdi, H. Alamdari, S. Royer and A. Adnot, Sens. Actuators, B, 2011, 156, 147–155 CrossRef CAS.
- X.-P. Xiang, L.-H. Zhao, B.-T. Teng, J.-J. Lang, X. Hu, T. Li, Y.-A. Fang, M.-F. Luo and J.-J. Lin, Appl. Surf. Sci., 2013, 276, 328–332 CrossRef CAS.
- A. E. Giannakas, A. A. Leontiou, A. K. Ladavos and P. J. Pomonis, Appl. Catal., A, 2006, 309, 254–262 CrossRef CAS.
- Y. Qin, L. Sun, D. Zhang and L. Huang, Catal. Commun., 2016, 79, 53–57 CrossRef CAS.
- H. Zhang, X. Li, Y. Hui, L. Yu, Q. Xia, S. Luo and C. Yao, J. Mater. Sci.: Mater. Electron., 2017, 28, 9371–9377 CrossRef CAS.
- H. Q. Ma, H. M. Zhu, X. Tan, J. Y. Zhang and L. Chang, J. Rare Earths, 2004, 22, 357–360 Search PubMed.
- M. Alifanti, J. Kirchnerova and B. Delmon, Appl. Catal., A, 2003, 245, 231–243 CrossRef CAS.
- B. He, Q. Song, Q. Yao, Z. Meng and C. Chen, Korean J. Chem. Eng., 2007, 24, 503–507 CrossRef CAS.
- T. Nitadori, S. Kurihara and M. Misono, J. Catal., 1986, 98, 221–228 CrossRef CAS.
- H. He, M. Liu, H. Dai, W. Qiu and X. Zi, Catal. Today, 2007, 126, 290–295 CrossRef CAS.
- A. Tarjomannejad, A. Farzi, M. J. I. Gomez, A. Niaei, D. Salari and V. Albaladejo-Fuentes, Catal. Lett., 2016, 146, 2330–2340 CrossRef CAS.
- S. Wu, C. Song, F. Bin, G. Lv, J. Song and C. Gong, Mater. Chem. Phys., 2014, 148, 181–189 CrossRef CAS.
- A. Di Benedetto, G. Landi, V. Di Sarli, P. S. Barbato, R. Pirone and G. Russo, Catal. Today, 2012, 197, 206–213 CrossRef CAS.
- R. You, Y. Zhang, D. Liu, M. Meng, Z. Jiang, S. Zhang and Y. Huang, Chem. Eng. J., 2015, 260, 357–367 CrossRef CAS.
- C. Orak, S. Atalay and G. Ersoz, Sep. Sci. Technol., 2017, 52, 1310–1320 CrossRef CAS.
- X. Li, Y. Yin, C. Yao, S. Zuo, X. Lu, S. Luo and C. Ni, Particuology, 2016, 26, 66–72 CrossRef CAS.
- X. Yin, L. Hong and Z. L. Liu, J. Membr. Sci., 2006, 268, 2–12 CrossRef CAS.
- R. Zhang, W. Yang, N. Luo, P. Li, Z. Lei and B. Chen, Appl. Catal., B, 2014, 146, 94–104 CrossRef CAS.
- H. Einaga, W. Yoshida, C. Lee and K. Kusaba, Catal. Lett., 2016, 146, 2495–2503 CrossRef CAS.
- M. Alifanti, M. Florea and V. I. Parvulescu, Appl. Catal., B, 2007, 70, 400–405 CrossRef CAS.
- Y. Wang, Y. Xue, C. Zhao, D. Zhao, F. Liu, K. Wang and D. D. Dionysiou, Chem. Eng. J., 2016, 300, 300–305 CrossRef CAS.
- M. Alifanti, J. Kirchnerova and B. Delmon, Appl. Catal., A, 2003, 245, 231–244 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2018 |