
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation, in vitro and in vivo evaluation, and molecular dynamics (MD) simulation studies of novel F-18 labeled tumor imaging agents targeting focal adhesion kinase (FAK)†
Yu Fang ab,
Dawei Wanga,
Xingyu Xua,
Gila Davaa,
Jianping Liua,
Xiang Lia,
Qianqian Xuea,
Huan Wanga,
Jiangshan Zhangb and
Huabei Zhang*a
ab,
Dawei Wanga,
Xingyu Xua,
Gila Davaa,
Jianping Liua,
Xiang Lia,
Qianqian Xuea,
Huan Wanga,
Jiangshan Zhangb and
Huabei Zhang*a
aKey Laboratory of Radiopharmaceuticals of Ministry of Education, College of Chemistry, Beijing Normal University, No. 19 Xinjiekouwai Street, Haidian District, Beijing 100875, People's Republic of China. E-mail: hbzhang@bnu.edu.cn; Tel: +86-10-58822012
bCollege of Chemistry and Chemical Engineering, Anyang Normal University, No. 436 Xian'ge Avenue, Anyang 455000, People's Republic of China
First published on 14th March 2018
Abstract
Focal adhesion kinase (FAK) has been identified as a promising target in the early diagnosis and therapy of tumor. In this work, we obtained and evaluated another two novel pyrimidine-based F-18 labeled tumor imaging agents targeting FAK. Among them, the corresponding F-19 standards [19F]2, displayed inhibition of FAK with IC50 values of 57.1 nM (better than the results in our published work) and showed an good selectivity profile against some other kinds of cancer-related kinases. [18F]2 also had relatively good results in the in vivo biodistribution in S180-tumor-bearing mice, with tumor uptake of 5.40 ± 0.12 and 5.96 ± 0.09 % ID per g at 15 and 30 min post-injection, respectively. What's more, [18F]2 could be accumulated in tumor at 30 min post-injection, which could be observed from the coronal micro-PET images of mice bearing S180 tumor. In addition, the blocking study for the [18F]2 with PF-562271 (one of the well-known best selective FAK inhibitor), displayed distinct reduction in the uptake of the radiotracer in tumor at 30 min post-injection in mice, suggesting that the uptake of [18F]2 in tumor was due to FAK over-expression or high expression in tumor. And the results of the molecular dynamics (MD) simulations and the docking studies were in consistent with the changing trends of the interaction between the F-19 standards and the FAK. Finally, in order to further increase the uptake of the F-18 labeled tracer in tumor, the following points should arouse attention, which could also be considered as the new findings and contributions of this study to the field of the tumor imaging agents: (1) the F-18 labeled tumor radiotracers which have closer interaction with the FAK, should be further designed, via building of models such as 3D-QSAR model to make reasonable guidance to our drug design and consideration of some functional groups which have hydrogen-bonding or salt-bridge interactions with key residues in the kinase domain of FAK; (2) the F-18 radiotracers with better pharmacokinetic properties should be designed, via building of dynamic drug absorption and distribution model in different tissues, to predict whether the molecules have ideal absorption in tumor and low uptake in non-target tissues. The relevant study is being undertaken.
1. Introduction
Functional molecular imaging on the basis of radiotracers (imaging agents), has facilitated the diagnosis and management of illnesses such as cancer, heart diseases and brain disorders. PET/CT and SPECT/CT based on the radiotracers are subfields of nuclear imaging. They can identify disease state at an early stage of development of disease, often before symptoms occur or abnormalities can be detected with other tests such as X-ray, NMR and CT.Among them, PET/CT based on the positron isotope (such as F-18) labeled radiotracers, is the most sensitive imaging method recognized by modern medical field, which is of great significance for the early diagnosis, staging, prognosis judgment and treatment plan of cancer. PET/CT on the basis of 18F-FDG is most widely used in clinical diagnosis and evaluation of cancer. However, this kind of radiotracer has fatal drawbacks. For example, its high accumulation in inflammed and infected tissues can lead to false-positive results and its low uptake in tumors that are growing slowly can cause false-negative results. Therefore, there is an urgent need to develop new PET radiotracers with obvious targeting and tissue specificity for tumor.
Focal Adhesion Kinase (FAK), a kind of non-receptor tyrosine kinase,1,2 has been considered to be a pivotal pathogenic mediator of various diseases. Among them, cancer is the most widely studied disease.3 Many evidence has suggested that FAK participates in a variety of biological behaviors of tumor, with its over-expression or high expression in the vast majority of types of tumor cells.3–21 In addition, FAK over-expression or high expression has been shown to be correlated with worse outcome in most kinds of cancer patients with hepatocellular carcinoma, invasive breast carcinoma, intestinal-type gastric carcinoma, endometric cancer, acute myeloid leukemia, anaplastic astrocytoma tumor, sarcoma (such as osteosarcoma), ovarian carcinoma, neuroblastoma, thyroid cancer, prostate cancer, head and neck squamous cell carcinoma and so on.19–21
Therefore, people have developed many FAK inhibitors as anti-neoplastic drugs, among which some even in clinical phase II trial. By and large, small molecular FAK inhibitors fall into two categories:20,22,23 inhibitors which bind to the ATP-binding site,4,5,24–28 and inhibitors targeting the scaffolding function of FAK.29–31
However, as far as we know, most of all of the previous studies were just focused on the research of FAK inhibitors for the tumor therapy at the level of generic pharmaceuticals, except for the only two recent reports by our research group which were first dedicated to the development of FAK-targeted radiotracers for tumor imaging on the radiopharmaceutical level.22,23 In this work, we obtained another two F-18 labeled novel 5-bromo-N2-(4-(2-fluoro-pegylated (FPEG))phenyl)-N4-(4-methoxy-phenyl)-pyrimidine-2,4-diamine analogues and evaluated them as tumor imaging agents targeting FAK, in order to make some comparison with our previous work in the bioactivity of the radiotracers, where the left side chain of the target compounds changed from “4-(2-fluoro-pegylated (FPEG))-3,5-dimethoxyphenyl” in one of our previous reports22 to “4-(2-fluoro-pegylated (FPEG))phenyl”, and the FPEG chain on the left side of the target compounds changed from n = 1 in another one of our previous reports23 to n = 2 & n = 3 here.
2. Experimental
2.1. General remarks
All chemicals and reagents used in the study (including 4 and 6a–6b) were of commercial quality and were used as purchased. Proton nuclear magnetic resonance (1H NMR) spectra were acquired using a BRUKER AVANCE® III HD 400 Spectrometer in CDCl3 solutions at room temperature. Chemical shifts were given in chemical shift (δ) as parts per million (ppm) relative to tetramethylsilane (TMS). 13C NMR spectra were determined at 100 MHz on a JEOL® 400YH Spectrometer in DMSO-d6 with chemical shift (δ) given in parts per million (ppm) relative to TMS as internal standard and recorded at room temperature. The ESI-MS spectra were determined on a Waters Quattro Micro® Quadrupole Mass Spectrometer or on a Shimidzu® LCMS-2010 Quadrupole Mass Spectrometer. Thin layer chromatography was performed on glass plates coated with 60 GF254 silica. Plates were visualized using UV light (254 nm). We undertook the flash column chromatography via an Interchim Puriflash® 4100 medium pressure preparative chromatography apparatus with silica gel (Bonna-Agela® flash silica, 40–60 μm, 60 Å).Truncated human FAK(PTK2) [376-1052(end) amino acids of accession number NP_722560.1] was provided by Carna Biosciences, Inc. HTRF KinEASE®-TK kit was purchased from Cisbio Biosciences, Inc.
No-carrier-added (n.c.a.) 18F− was generated by the bombardment of an enriched [18O]water target (>97%) with a proton beam using an the Sumitomo HM-20 cyclotron at Cancer Hospital of Peking University (Beijing, China). QMA light ion-exchange cartridges and C-18 light Sep-Pak® cartridges were obtained from Waters (Milford, MA). We activated the Waters Sep-Pak® Accell™ Plus QMA Plus Light Cartridges (130 mg Sorbent per Cartridge, 37–55 μm particle size) with 1 N NaHCO3 (10 mL), followed by deionized water (10 mL), and the Waters Sep-Pak® C18 Plus Light Cartridges (130 mg Sorbent per Cartridge, 55–105 μm particle size) with methanol (10 mL) and deionized water (10 mL) before use. Radiopharmaceuticals were purified and their radiochemical purity were determined on a Shimadzu® LC-20AT HPLC apparatus equipped with a SPD-20A UV detector (λ = 254 nm) and Bioscan® flow count 3200 NaI/PMT γ-radiation scintillation detector. We performed the radiopharmaceutical HPLC separations on a Inertsil® ODS-3 C18 reverse phase semi-preparative column (GL Sciences, Inc. 5 μm, 10 mm × 250 mm), and we carried out the elution with a binary gradient system at a flow rate of 2.0 mL min−1. Their radiochemical purity determination were achieved on a Kromasil® 100-5C18 reverse phase column (AkzoNobel, 5 μm, 4.6 mm × 250 mm), and elution was with a binary gradient system at a flow rate of 2.0 mL min−1.
We purchased S180 ascites sarcoma mice from Beijing Vitalriver Animal Technology Co., Ltd., and Beijing Xinglong Animal Technology Co., Ltd. provided normal ICR mice (20–25 g, female). The measurement of the F-18 radioactivity of the tissues and organs of interest was performed on a PerkinElmer® 2480 WIZARD2 automatic γ-counter. The Micro-PET imaging was performed on a Siemens Inveon DPET 120. All protocols requiring the use of mice were approved by the Animal Care Committee of Beijing Normal University.
2.2. Synthesis
2.3. Radiochemistry
[18F]2 and [18F]3 were radiosynthesized and isolated as the procedure described in the literature:22 first, [18F]fluoride was transferred to a hot cell, passed through and trapped on a preconditioned anion exchange cartridge (QMA cartridge, preconditioned as those described in Section 2.1). The fixed 18F− was eluted from the QMA cartridge to a digestion high-pressure tank with a solution of 1.5 mL of (Kryptofix 2.2.2)/K2CO3 solution (a mixture of 13.5 mg of Kryptofix 2.2.2 in 1.25 mL CH3CN and 1.7 mg K2CO3 in 0.25 mL H2O). The solvents were removed under a sweep stream of nitrogen at 100 °C. Subsequently, 0.5 mL of dry CH3CN was added to the residue and evaporated to dryness for three times. Then, to the resulted dry Kryptofix 2.2.2/[18F] KF complex, a solution of the tosylate precursors 7a and 7b in dry CH3CN (0.5 mL) was added, respectively, and the mixture was stirred at 100 °C for 15 min and diluted with deionized H2O (10 mL) and loaded onto an activated C18 Sep-Pak cartridge (pretreated as those described in Section 2.1). The Sep-Pak cartridge was washed with deionized H2O (10 mL) again to remove unlabeled 18F− and the organic phase was then eluted with 2 mL of CH3CN for two times. The combined eluent was concentrated in vacuo, and the residue in 0.4 mL of CH3CN was injected into a semi-preparative HPLC. The preparation took 60 min, and the [18F]2 and [18F]3 were separated in a radiochemical yield of 9–16% (decay not corrected), respectively. The radiochemical purity of all tracers was greater than 98%.2.4. Octanol/water partition coefficient
The radioactivity of the n-octanol layer and that of the PBS layer for [18F]2 and [18F]3 were measured as previously described in our recent work:22 we added a 0.1 mL solution of each of the radiotracers (370 KBq, 10 μCi) in normal saline to a tube containing 0.9 mL of phosphate-buffered saline (0.05 M, PBS, pH 7.4) and 1.0 mL of n-octanol (each phase had been presaturated with the opposite phase overnight before use). The mixture was vortexed for 0.5–1.0 min and centrifuged at 5000 rpm for 5 min. Aliquots of both phases were transferred in triplicate with micropipettes to counting tubes and were assayed in a γ-counter after standing for a while. Then, the partition coefficient was calculated by dividing the radioactivity of the n-octanol layer with that of the PBS layer. This measurement was repeated three times.2.5. Stability test in vitro
The stability test in vitro of the radiotracers were performed following the procedure in our previous report:22 each of the radiotracers (370 KBq, 10 μCi) purified by the HPLC in 100 μL of normal saline was mixed with 500 μL of normal saline at 37 °C for 1 h and 2 h, respectively. Approximately 0.1 mL of the solution was analyzed by radio-HPLC (about the HPLC analysis condition, please refer to the above Section 2.1).The stability of each HPLC-purified radiotracer in mouse plasma was determined by incubating 100 μL of radiotracer (370 KBq, 10 μCi) in the solution of 500 μL murine plasma at 37 °C for 1 h and 2 h, respectively. Plasma proteins were precipitated by adding 200 μL of acetonitrile after centrifugation at 5000 rpm for 5 min at 4 °C. About 100 μL of the supernatant part was injected into radio-HPLC.
2.6. Bioactivity
We carried out the FAK inhibititory assays of the F-19 standards [19F]2 and [19F]3, and the profiling of the [19F]2 against five kinds of kinases (PYK2, JAK2, IGF-1R, EGFR and PDGFRβ) with the HTRF® (Homogeneous Time-Resolved Fluorescence Methodologies) kinEASE™-TK kit.
Before the compound testing, the optimization of the concentration of the enzyme used in the assay, as well as the enzymatic reaction time and the ATP concentration was performed.
Phosphorylation of the TK substrate–biotin was challenged with serially diluted compounds at 1/2-log concentrations starting at a top concentration of 100 μM (which was the final concentration for the enzymatic step (10 μL)). Each concentration was tested in triplicate.
During enzymatic reaction step, 4 μL compounds in kinase buffer and 2 μL TK substrate–biotin were incubated with 2 μL kinase in kinase buffer and 2 μL ATP in kinase buffer (with the above optimized concentration) was added to start the enzymatic reaction. The mixture was incubated for the above optimized time at room temperature.
The detection reagents [5 μL Eu3+-cryptate labeled TK-antibody in HTRF® detection buffer, and 5 μL steptavidin-XL665 in HTRF® detection buffer (0.125 μM, which was the final concentration for the assay (20 μL))] were mixed and added to the reaction system. The TR-FRET signal was proportional to the phosphorylation level and was detected by a plate reader (BMG FS) after being incubated for one hour. For each compound, the determination of the IC50 value was from a sigmoid dose–response curve using Graph-Pad Prism (GraphPad Software, San Diego, CA, USA).
Then, sixty pieces of tumor tissue slices (about 4 μm) from sixty S180 tumor bearing mice, and sixty pieces of normal muscle tissue slices (about 4 μm) from sixty normal mice, were used for immunohistochemistry for the identification of the expression level of FAK in S180 tumor.
For the immunohistochemistry, the working concentration of the FAK monoclonal antibody (purchased from Santa Cruz Biotechnology Co., LTd.) was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40, and the working concentration of the phospho-FAK (pY397FAK) polyclonal antibody (purchased from Upstate Bio technology Co., LTd.) was 1:800.
:40, and the working concentration of the phospho-FAK (pY397FAK) polyclonal antibody (purchased from Upstate Bio technology Co., LTd.) was 1:800.
The ready-to-use UltraSensitive™ SP kit (SP9000), the sensitizer DAB (diaminobenzidine) and the HE (hematoxylin–eosin) staining kit were purchased from Beijing Zhongshan company.
All the tumor tissue slices and normal muscle tissue slices were carried out for HE staining and immunohistochemical staining, with PBS instead of the FAK monoclonal antibody and the pY397FAK polyclonal antibody as the negative control. And the detailed procedures of the immunohistochemistry were according to the SP kit instructions.
The criteria of immunohistochemical results could be divided into four kinds of levels according to the rate of the FAK-positive/pY397FAK-positive cell and the intensity of the staining reaction of the tissue spices: (1) “negative (−)” means that the cell of the tissue slices were not stained, or the rate of the FAK-positive/pY397FAK-positive cell was lower than 25%; (2) “weak positive (+)” means that the cell of the tissue slices were stained as brown yellow, or the rate of the FAK-positive/pY397FAK-positive cell was between 25% and 50%; (3) “moderate positive (++)” means that the cell of the tissue slices were stained as pale yellow, or the rate of the FAK-positive/pY397FAK-positive cell was between 51% and 75%; (4) “strong positive (+++)” means that the cell of the tissue slices were stained as brown, or the rate of the FAK-positive/pY397FAK-positive cell was higher than 75%.
Each of the S180 tumor bearing mice was injected via the tail vein with a normal saline solution containing the 18F-labeled tracers (370 KBq per 100 μL). At 5, 15, 30, 60 and 120 min post injection, five mice for each time point were sacrificed by decapitation. The tissues and organs of interest were immediately collected, weighed and measured for 18F radioactivity in an automatic gamma counter. The percent dose per gram of wet tissue was calculated by a comparison of the tissue counts to suitably diluted aliquots of the injected material. The values were expressed as mean ± SD (n = 5).
For Micro-PET imaging study, the S180 tumor bearing mouse was injected intravenously with approximately 3.7 MBq of [18F]2 in 300 μL normal saline, anesthetized with 1.5% isoflurane in air (about 1.5 mL min−1) and fixed near the center of Micro-PET scanner with prone position. Static scans (for 30 min, 10 min scan) were obtained on a Siemens Inveon DPET 120. Then, the images were reconstructed and the images were exported.
For blocking studies in the in vivo biodistribution and micro-PET imaging studies, at one hour prior to the injection of the radiotracer [18F]1, a group of mice received PF-562271 in 5% Gelucire 44/14 in sterile water (Gattefosse) (dose: 33 mg kg−1, p.o.).
2.7. Computational chemistry
The structure of the NVP-TAE226 (the chemical structure of it was shown in the Section 2.5 of our previous report22), which was in the complex with FAK (the RCSB Protein Data Bank (PDB) deposition number of the X-ray single-crystal diffraction structures: 2JKK), was selected as the molecular template for sketching the structures of the F-19 standards in this work. For each of the compounds, the conformations resulting from molecular docking, were obtained via the previous structure sketching, molecular energy minimization, systematic conformational search using the Sybyl-X 1.3 molecular modeling software package (Tripos Associates, St. Louis, MO), AM1 optimization on Gaussian 09 and the molecular docking into the ATP binding site of FAK based on 2JKK.pdb with AutoDock 4.0. The results of the docking studies were viewed with the Sybyl-X 1.3.The conformations resulting from molecular docking were served as the initial conformations of ligands for the MD simulations, and the subsequent MD simulations were also undertaken following the protocols in the Section 4.8 of the literature.22
3. Results and discussion
3.1. Organic synthesis
Scheme 1 offers an overview of the synthetic route of the tosylate precursors 7a–7b and the fluoro-pegylated 5-bromopyrimidine derivatives [19F]2–[19F]3. The 5-bromo-2-chloro-N-(4-methoxyphenyl)pyrimidin-4-amine 5 was prepared by the stirring of the starting material 5-bromo-2,4-dichloropyrimidine 3 reacted with the 4-methoxyphenyl amine 4 under K2CO3 at room temperature. Subsequent treatment of the compound 5 with the different substituted aryl amines 6a–6b above in presence of p-toluenesulfonic acid (PTSA) yielded the desired tosylate precursors 7a–7b. Preparation of the F-19 standards [19F]2–[19F]3 of the corresponding radiotracers [18F]2–[18F]3 in this study was accomplished by the fluorination of the tosylate precursors 7a–7b with anhydrous KF and Kryptofix 2.2.2 in dry acetonitrile at 100 °C for just 15 min.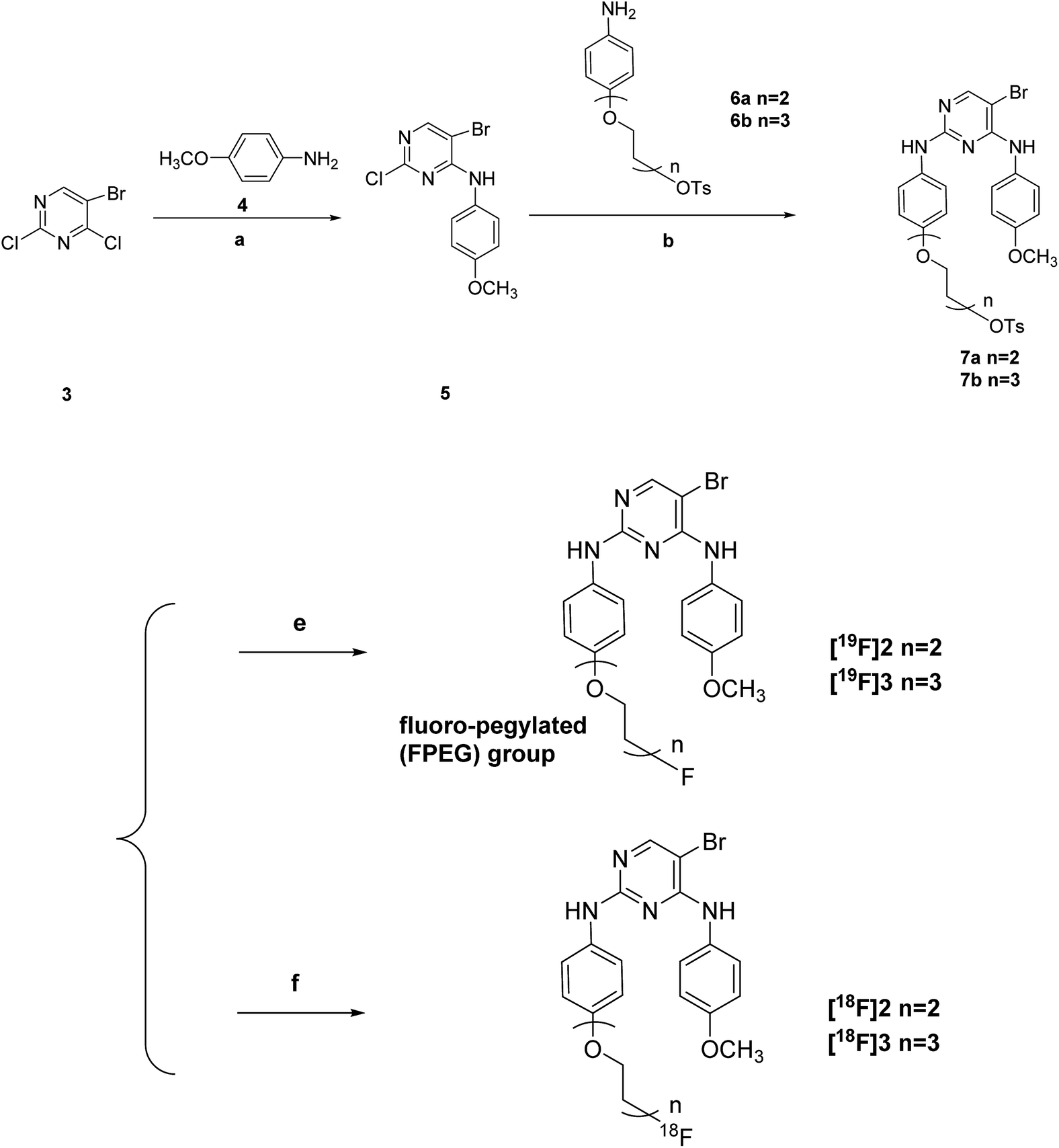 | ||
| Scheme 1 Reagents and conditions: (a) K2CO3, iPrOH:H2O = 1:1 (v/v), r.t.; (b) PTSA (p-toluenesulfonic acid), 1,4-dioxane, reflux; (c) anhydrous KF, Kryptofix 2.2.2, anhydrous acetonitrile, 100 °C; (d) 18F−, K2CO3, Kryptofix 2.2.2, anhydrous acetonitrile, 100 °C. | ||
3.2. Radiochemistry
The desired F-18 labeled tracers [18F]2 and [18F]3 were synthesized by the nucleophilic substitution of the tosylate precursors 7a and 7b under conventional conditions ([18F]fluoride/potassium carbonate and Kryptofix 2.2.2) in anhydrous acetonitrile at using a digestion high-pressure tank at 100 °C for 15 min, respectively (Scheme 1), affording [18F]2 and [18F]3 in 16% and 12% radiochemical yield (non-decay-corrected), respectively, and radiochemical purity of greater than 98% after solid phase extraction and HPLC purification. The total synthesis time was 60 min. The identity of the [18F]2 and [18F]3 was further confirmed by comparing theirs HPLC retention time with those of co-injected authentic nonradioactive F-19 standards [18F]2 and [18F]3 (Fig. 1), respectively, using analytical HPLC conditions described in the Section 2.1. Specific activity, estimated by comparing the UV peak intensity of purified F-18 labeled compounds with reference nonradioactive compounds, ranged from 34 to 52 GBq μmol−1 at the end of synthesis.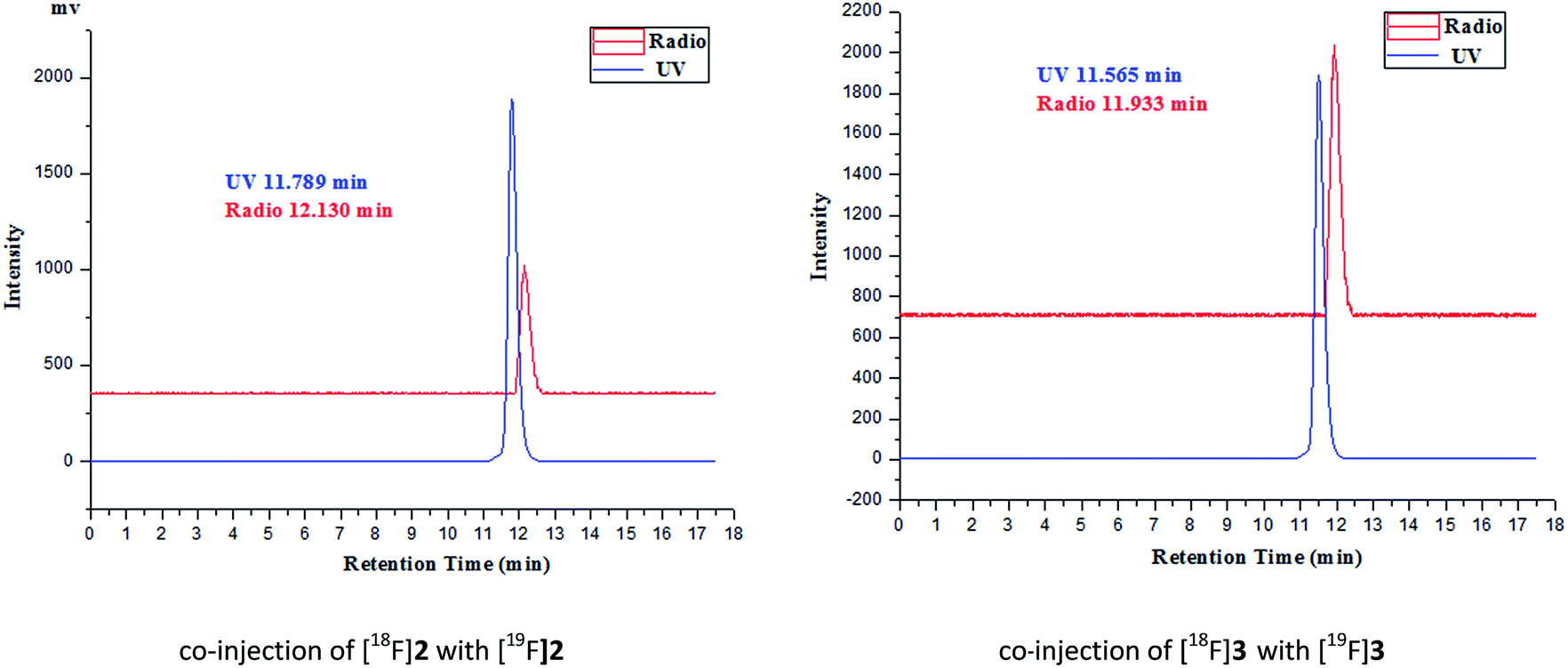 | ||
| Fig. 1 The HPLC chromatogram of F-18 labeled radiotracers and the corresponding F-19 standards. The HPLC profile data were exported from the *.lcd files by Shimadzu LCSolution Software and were graphically interpreted and coordinate translated by Origin 8.0. | ||
3.3. In vitro stability study
It could be seen from the radio-HPLC analysis that all the radiotracers were stable in both normal saline and murine plasma at 37 °C after 1 h and 2 h (Fig. S1) (see the ESI†). We chose two solvent conditions – normal saline and murine plasma in the in vitro stability study for the following reasons: first, since the radiotracers were injected into the body as a normal saline solution, their stability in normal saline should be observed. What's more, their in vitro stability in murine plasma should also be determined, because we often use mice as the primary animal model to carry out the in vivo biological evaluation. Therefore, if the radiotracers were not stable even in the murine plasma, they would be unstable after being injected into the blood of the mice and won't effectively reach the target organ. So the following biological evaluation would be meaningless.3.4. Bioactivity
| Compounds | IC50 ± SDb (nM) |
|---|---|
| a The preparation and the related biological evaluation results of [19F]1 and its corresponding F-18 labeled radiotracer [18F]1, have been described in our published work.23 (Note: the [19F]1 in this work was named 8a in our published work.23 The chemical structure of [19F]1 was shown in the ESI.) The in vitro inhibition test of [19F]1 against FAK, were performed in the same batch with the F-19 standards in this work.b Mean of three determinations. | |
| [19F]1 | 1085.4 ± 47.2 |
| [19F]2 | 57.1 ± 8.3 |
| [19F]3 | 1113.6 ± 60.9 |
| PF-562271 | 4.3 ± 0.3 |
Although it was similar to the results in our published work22 that the interaction between the F-19 standards ([19F]1, [19F]2 and [19F]3) and the FAK was also mainly affected by the length of the FPEG chain, there were some improvement in this study, comparing to our previous reports:22,23 (1) the FAK inhibitory potency improved by 19.0-fold when the FPEG chain on the F-19 standards was lengthened from n = 1 in our published work23 to n = 2 in this study; (2) the result of the [19F]2 were relatively good and had a modest IC50 of 57.1 nM, which were even better than the relatively good result (91.4 nM) in our published work.22 However, when we continued to lengthen the FPEG chain from n = 2 to n = 3, it also led to an dramatic 19.5-fold reduction of interaction between the F-19 standards and the FAK again.
The relatively best F-19 standard [19F]2 were also further profiled in vitro against other five kinds of cancer-related kinases (PYK2, JAK2, IGF-1R, EGFR and PDGFRβ), also using an HTRF® based kinase assay described in the Section 2.6.1, and with NVP-TAE226 (ref. 32) (one reported inhibitor of FAK developed by Novartis International AG) as the positive control substance.
We could observed from the Table 2 that the [19F]2 displayed an good selectivity profile against the tested kinases, with 62.4 folds selectivity against PYK2, 75.5 folds selectivity against EGFR, and more than 200 folds selectivity against IGF-1R and PDGFRβ, which were also even better than the results in our published work.22
| Kinase | IC50 ± SD (nM) for [19F]2a | IC50 ± SD (nM) for NVP-TAE226a | Reported IC50 (nM) for NVP-TAE226 (ref. 32) |
|---|---|---|---|
| a Mean of three determinations. | |||
| PYK2 | 3560.8 ± 625.6 | 5.7 ± 1.4 | 3.5 ± 2.1 |
| JAK2 | 11720.0 ± 1981.3 |
868.9 ± 152.6 | 840 ± not shown |
| IGF-1R | 16600.3 ± 2957.1 |
182.4 ± 40.1 | 140 ± 28.3 |
| EGFR | 4311.4 ± 562.1 | 1829.1 ± 466.2 | 1700 ± not shown |
| PDGFRβ | 32850.2 ± 4513.7 |
2321.0 ± 574.4 | 2600 ± 1600 |
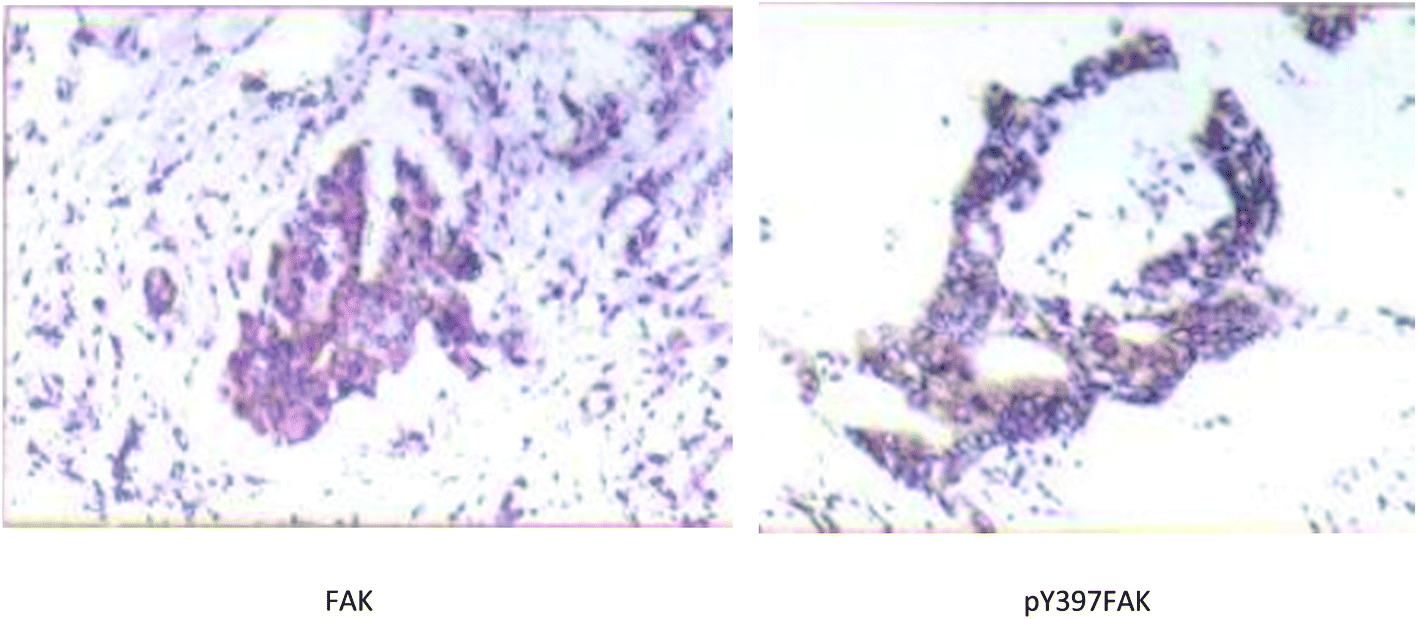 | ||
| Fig. 2 An example of strong positive FAK and pY397FAK staining in S180 tumor tissue slices (SP ×200). | ||
| Group | FAK | Moderate and strong positive expression rates | pY397FAK | Moderate and strong positive expression rates | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| − | + | ++ | +++ | − | + | ++ | +++ | |||
| Normal muscle tissue slices (n = 60) | 25 | 28 | 7 | 0 | 11.7% | 47 | 9 | 4 | 0 | 6.7% |
| S180 tumor tissue slices (n = 60) | 8 | 17 | 18 | 17 | 58.3% | 25 | 11 | 15 | 9 | 40.0% |
(1) The uptake of all of the three F-18 labeled compounds in tumor, accumulated to their respective high levels at 15 min post-injection (3.69 ± 0.51, 5.40 ± 0.12 and 2.80 ± 0.33 % ID per g for [18F]1, [18F]2 and [18F]3, respectively), and remained at analogously high levels at 30 min post-injection (3.71 ± 0.43, 5.96 ± 0.09 and 3.24 ± 0.07 % ID per g for [18F]1, [18F]2 and [18F]3, respectively). They reached to their peak value at 30 min post-injection (3.71 ± 0.43, 5.96 ± 0.09 and 3.24 ± 0.07 % ID per g for [18F]1, [18F]2 and [18F]3, respectively). After that, the value started to decline at 60 min post-injection and 120 min post-injection. However, for all the three tracers, their retention in tumor was relatively long, because the radioactivity was still more than half of the peak value at 120 minutes post-injection (3.11 ± 0.22, 3.86 ± 0.14 and 2.88 ± 0.44 % ID per g for [18F]1, [18F]2 and [18F]3, respectively. The statement of “relatively long retention in tumor” here was just referred to the page 3782 in one of the reports of the Qi's group33). And all the three radiotracers showed moderate clearance from blood and metabolism in kidney, which were conclude from the uptake of the three radiotracers in the blood and kidney from 5 min post-injection to 120 min post-injection.
(2) Not only did the length of the FPEG chain has a significant effect on the interaction between the F-19 standards in this work ([18F]1–[18F]3) (expressed as the in vitro inhibitory abilities of enzymatic activities of FAK in this article) and the FAK at the molecular level, but also it played a vital role in the biodistribution of the corresponding F-18 labeled radiotracers ([18F]1–[18F]3) in the S180-tumor-bearing mice models at the organ level. The uptake of the radiotracers in tumor improved from [18F]1 to [18F]2, with the addition of the FPEG chain from n = 1 to n = 2, which was mainly because, the enhanced FAK inhibitory activity of the F-19 standards reflected the increased interaction between the F-19 standards and the FAK, when the FPEG chain lengthened from n = 1 to n = 2. However, when the FPEG chain continued to lengthen, the interaction between the F-19 standards and the FAK was incredibly weakened, leading to the dramatical reduction of the uptake of [18F]3 in tumor and some of its target/non-target ratio (tumor/bone, tumor/muscle and tumor/blood ratio, e.g.) (even lower than those of [18F]1). Therefore, the stronger the interaction between the F-19 standards and the FAK, the higher the uptake of the corresponding F-18 labeled tracers in tumor in this work. And another reason for the relative highest uptake of [18F]2 at 30 min post-injection among the [18F]1–[18F]3. And another possible reason for the relative highest uptake of [18F]2 at 30 min post-injection among the [18F]1–[18F]3 is that, the [18F]2 showed faster clearance from the lung (one of the non-target organs) than those for [18F]1 and [18F]3, which was conclude from the uptake of the three radiotracers in the lung from 5 min post-injection to 30 min post-injection.
(3) For the [18F]2, we could observe distinct reduced uptake (about 88%) of the radiotracer in the tumor tissue (0.72 ± 0.05 % ID per g at 30 min post-injection of [18F]2) in mice that received PF-562271 (one of the reported best selective FAK inhibitor which was developed by Pfitzer Inc.) at one hour before the injection of radiotracer.
From the above analysis, it could be obtained that in order to further increase the uptake of the F-18 labeled tracer in tumor, the following properties of compounds should be improved: (1) the F-18 labeled tumor radiotracers which have closer interaction with the FAK (namely the stronger FAK inhibitory activity) should be further designed in the future. For example, we should build 3D-QSAR models with the experimental FAK IC50 data of the reported FAK inhibitors to make reasonable guidance for our drug design and quantitatively predict the FAK IC50 values of the molecular designed by us. What's more, some functional groups which have hydrogen-bonding or salt-bridge interactions with the key residues such as Asp564, Lys454 and Glu506 in the kinase domain of FAK, should be considered to improve the interaction of the drug molecules with the FAK; (2) in this work, the different speed of clearance from the lung affected the uptake of the radiotracers in tumor. This suggested that the F-18 radiotracers with better pharmacokinetic properties should be designed to improve uptake of the radiotracers in tumor and the speed of clearance from the non-target organs. Dynamic drug absorption and distribution model in different tissues (such as liver, blood, liver, tumor etc.) should be made. The general approach in pharmacokinetic and drug design studies, include calculation of many physical properties about pharmacokinetic properties (such as fat and water partition coefficient – logP, dipole moment, apparent volume of distribution – Vd, time to peak – Tmax, etc.) from the relevant data of the reported hundreds of small molecular tumor imaging agents, linear fitting of the physical properties to make the dynamic drug absorption and building of the distribution equation in different tissues. Then, we could use these equations to predict whether the molecules have ideal absorption in tumor and low uptake in non-target tissues, on the basis of the small molecules which have strong interaction with the FAK. These need to take a lot of work, and the relevant study is being carried out by our group.
 | ||
| Fig. 3 Coronal micro-PET images of mice bearing S180 tumor at 30 min post-injection of [18F]2 (A) and 30 min post-injection of [18F]2 after treatment of PF-562271 (a dose of 33 mg kg−1, p.o.) at one hour before the intravenous injection of radiotracer (blocking study) (B). | ||
What's more, Fig. 3B (blocking study) showed significantly reduced uptake of the radiotracer in the tumor tissue at 30 min post-injection in mice that received PF-562271 (the positive control drug) at one hour before the injection of radiotracer. Therefore, the micro-PET imaging study, in combination with the results of the above kinase profiling assay and our biodistribution data, indicated that the uptake of [18F]2 in tumor of the mouse model was due to FAK over-expression or high expression in tumor, and that [18F]2 might be a kind of selectively FAK-targeted tumor imaging agents.
3.5. MD simulations and docking studies
In this section, the molecular dynamics (MD) simulations were applied to further understand the changing trends of the interaction between the different F-19 standards and the FAK, and the uptake of the corresponding F-18 labeled tracers in tumor theoretically, in view of the fact that the MD simulations were carried out with taking the solvent effect on the conformational transitions of the complexes (be comprised of ligands such as the F-19 standards in this work and receptors such as FAK in this study) into full consideration, while the simple docking analysis ignored both the flexibility of the proteins (receptors) and the effect of solvation in water.First of all, the rationality of the MD simulations and the validity of the molecular conformation that was used as the initial conformation for the MD simulations, were proved, according to the Section 2.5 in our previous work.22
Then, we undertook the 40 ns MD simulations for all of the three F-19 standards in this work. It could be concluded from the Fig. 4 that, after equilibration, the RMSD values between the initial conformation and the one during the MD simulation for most of the complexes and ligands were less than 2.0 Å. In addition, the stronger the FAK inhibitory activity the F-19 standard had, the stronger the interaction between the F-19 standard and the FAK, the easier the complex system would enter into a state of equilibrium.
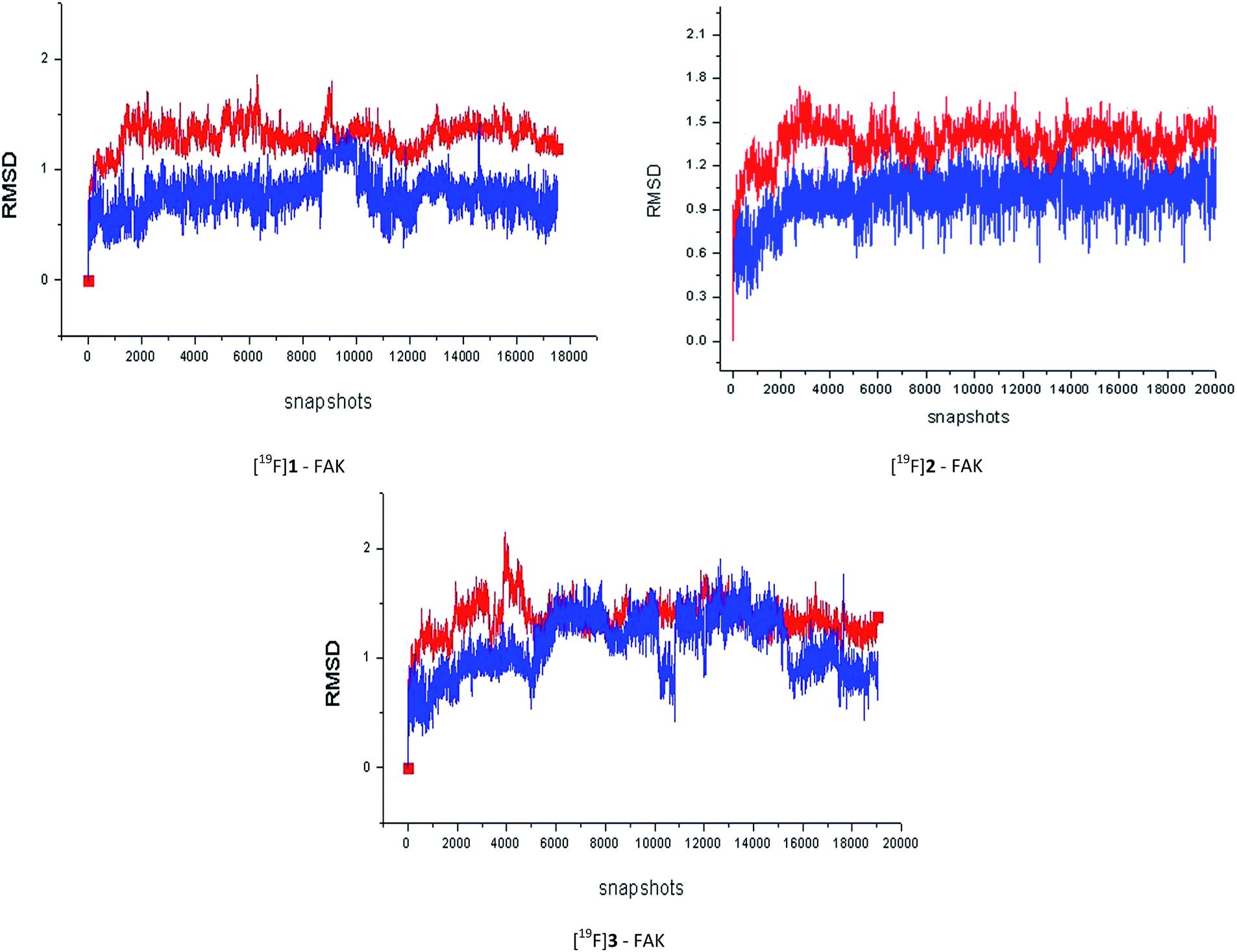 | ||
| Fig. 4 The root-mean-square deviations (RMSD) between the starting conformations and the ones during the MD simulation as a function of the simulation time, for the complex (red) and the ligand (blue) in the complexes respectively, which were calculated with the ptraj module of Amber 10 and graphically interpreted by Origin 8.0. The MD simulation production phase was undertaken for 40 ns with a 2 ps time step, namely, 20000 snapshots in sum. The calculating time for the MD simulation for each compound was about one month on the basis of our current computer cluster condition. | ||
Table 4 lists the components of molecular mechanics and solvation energies. The mean value of electrostatic energy (ΔEeletrostatic + ΔΔGPB) was 27.3 kcal mol−1 with an root-mean-square deviation of 1.6 kcal mol−1. For the van der Waals and hydrophobic interaction energies (ΔEvdW + ΔΔGSA), the mean value was −56.5 with an root-mean-square deviation of 2.1 kcal mol−1. The entropy (TΔS) had the a root-mean-square deviation of 1.0 kcal mol−1 with a mean value of −21.0 kcal mol−1. The average unsigned error between the experimental and the calculated energy was 0.45 kcal mol−1 and the root-mean-square deviation was 0.11 kcal mol−1. It could be seen from Fig. 5 that the calculated free energies reproduced the experimental ones relatively well and the rank order of calculated free energies and that of the experimental ones was generally consistent.
| Complex | ΔEeletrostatic | ΔEvdW | ΔEgas | ΔΔGPB | ΔΔGSA | ΔGMMPBSA | TΔS | ΔGbind,calc | ΔGbind,expb |
|---|---|---|---|---|---|---|---|---|---|
| a All energies are in kcal mol−1.b The ΔGbind,exp (experimental binding free energies) were estimated for the F-19 standards from the experimental IC50 values using the following relationship: ΔGbind,exp = RTlnKdissociated = RTln(IC50 + 0.5Cenzyme) ≈ RTlnIC50, where R was ideal gas constant, T was the temperature in kelvin (298 K was used in this work), and Cenzyme was the concentration of enzyme, which was a very small number after equilibration and could be omitted in most cases. |
|||||||||
| [19F]1-FAK | −10.87 | −46.48 | −57.35 | 38.42 | −8.93 | −27.86 | −20.13 | −7.73 | −8.17 |
| [19F]2-FAK | −13.12 | −49.23 | −62.35 | 41.86 | −9.69 | −30.18 | −20.66 | −9.52 | −9.87 |
| [19F]3-FAK | −14.76 | −44.47 | −59.23 | 40.29 | −10.74 | −29.68 | −22.09 | −7.59 | −8.15 |
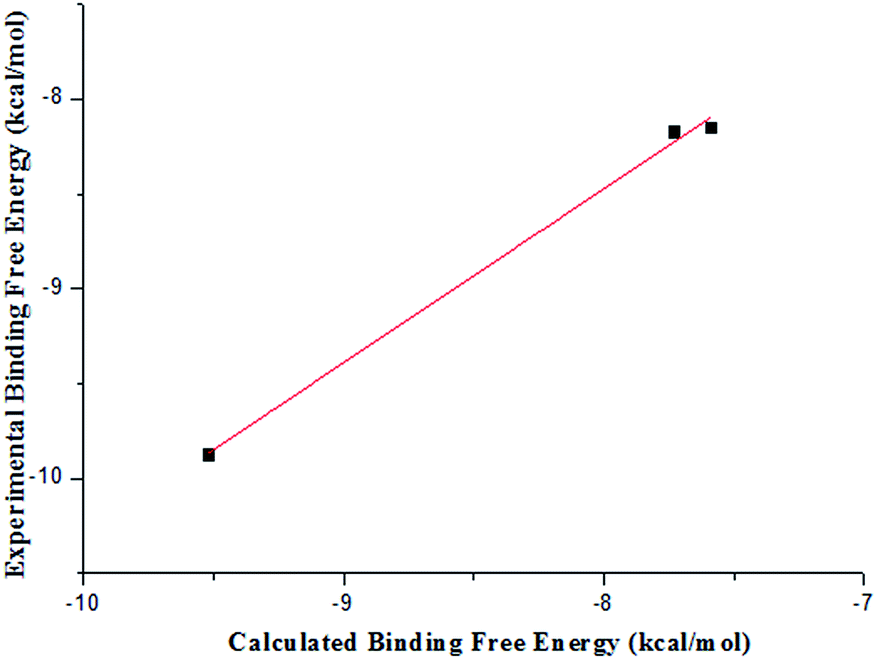 | ||
| Fig. 5 Comparison between the calculated and the experimental binding free energies estimated from IC50. | ||
In addition, some conclusions obtained from the docking studies (Fig. S2) (see the ESI†), could also help us to understand why the small changes of chain length could bring a significant difference in IC50 values:
In the binding pocket of FAK, there were four intermolecular hydrogen bonds between [19F]2 and the kinase domain of FAK, which were formed by one N-atom on the pyrimidine ring of [19F]2 with one “–NH–” group on the residue Cys 502 of FAK, the “–NH–” group on the pyrimidine ring of [19F]2 with the one O-atom on the residue Cys 502 of FAK, the second O-atom on the FPEG chain of [19F]2 with the “–NH–” group on the residue Glu 506 of FAK, and the F-atom on the FPEG chain of [19F]2 with one “–(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–OH” group on the residue Glu 506 of FAK, respectively.
O)–OH” group on the residue Glu 506 of FAK, respectively.
However, there were only two intermolecular hydrogen bonds between [19F]1 or [19F]3 and the kinase domain of FAK. For [19F]1 and FAK, the hydrogen bonds were formed by one N-atom on the pyrimidine ring of [19F]1 with one “–NH–” group on the residue Cys 502 of FAK, and the “–NH–” group on the pyrimidine ring of [19F]1 with the one O-atom on the residue Cys 502 of FAK, respectively. For [19F]3 and FAK, the hydrogen bonds were formed by one N-atom on the pyrimidine ring of [19F]3 with one “–NH–” group on the residue Cys 502 of FAK, and the F-atom on the FPEG chain of [19F]2 with one “–(CO)–OH” group on the residue Glu 506 of FAK, respectively.
So there were more intermolecular hydrogen bonds between [19F]2 and the kinase domain of FAK than those between [19F]1 or [19F]3 and the kinase domain of FAK, which might also contribute to the significant difference in IC50 values.
4. Conclusion
In this study, we prepared two 5-bromo-N2-(4-(2-fluoro-pegylated (FPEG))phenyl)-N4-(4-methoxyphenyl)-pyrimidine-2,4-diamine derivatives and evaluated them as the F-18 labeled radiotracers targeting FAK for tumor imaging.The corresponding F-19 standards inhibited the activity of FAK with IC50 values of 57.1 and 1113.6 nM. When the length of the FPEG chain was n = 2, the interaction between the F-19 standards and the FAK was relatively good, with the [19F]2 having FAK IC50 of 57.1 nM, which was better than the results in our published work.22 We also profiled the [19F]2 in vitro against some other kinds of cancer-related kinases, with 62.4 folds selectivity against PYK2, 75.5 folds selectivity against EGFR, and more than 200 folds selectivity against IGF-1R and PDGFRβ, which were also better than the results in our published work.22,23
High in vitro stability in normal saline solution and murine plasma were displayed by all the F-18 labeled compounds in this work. Among the two corresponding F-18 labeled compounds in this study, the [18F]2 with an FPEG chain of medium-length (n = 2) were also relatively good with modest tumor uptake of 5.40 ± 0.12 % ID per g and 5.96 ± 0.09 % ID per g at 15 and 30 min post-injection, respectively, in the studies on biodistribution in S180-tumor-bearing mice, which was also similar to the results in our published work.22 What's more, its tumor/muscle, tumor/bone and tumor/blood ratio at 15 min post-injection were 2.40, 1.64 and 1.92, respectively. And its tumor/muscle, tumor/bone and tumor/blood ratio at 30 min post-injection were 2.49, 1.55 and 1.94, respectively. In addition, coronal micro-PET images of mice bearing S180 tumor clearly confirmed that [18F]2 could be obviously accumulated in tumor at 30 min post-injection, which was better than the results of the imaging study in our previous report.23 Besides, for the [18F]2, both the biodistribution data and the micro-PET imaging study showed significantly reduced uptake of the radiotracer in the tumor tissue at 30 min post-injection in mice that received PF-562271 (one of the reported best selective FAK inhibitor which was developed by Pfitzer Inc. and inhibited the activity of FAK with IC50 value of 1.5 nM) at one hour before the injection of radiotracer, indicating that the uptake of [18F]2 in tumor of the mouse model was due to FAK expression, and that [18F]2 might be a kind of selectively FAK-targeted tumor imaging agents.
Furthermore, the results of the MD (molecular dynamics) simulations and the docking studies generally consisted with the changing trends of the interaction between the different F-19 standards and the FAK (expressed as the in vitro inhibitory abilities of enzymatic activities of FAK in this article), which was also in agreement with and had great effect on the changing trends of the uptake of the corresponding F-18 labeled tracers in tumor.
Finally, in order to further increase the uptake of the F-18 labeled tracer in tumor, the following points should arouse attention, which could also be considered as the new findings and contributions of this study to the field of tumor imaging agents: (1) the F-18 labeled tumor radiotracers which have closer interaction with the FAK (namely the stronger FAK inhibitory activity) should be further designed, via building of models such as 3D-QSAR model to make reasonable guidance for our drug design, and consideration of some functional groups which have hydrogen-bonding or salt-bridge interactions with key residues in the kinase domain of FAK; (2) the F-18 radiotracers with better pharmacokinetic properties should be designed to improve uptake of the radiotracers in tumor and the speed of clearance from the non-target tissues. Dynamic drug absorption and distribution model in different tissues (such as liver, blood, lung, tumor, etc.) should be built, to predict whether the molecules have ideal absorption in tumor and low uptake in non-target tissues, on the basis of the small molecules which have strong interaction with the FAK. The relevant study is being undertaken. These enlightenment may be useful guidance for us to develop better radiotracers for the early diagnosis of cancer.
Ethical statement
All animal procedures were performed in accordance with Management Regulations of Laboratory Animal of Beijing City, and the Guidelines for Care and Use of Laboratory Animals of Beijing Normal University.All protocols requiring the use of mice were approved by the Animal Ethics Committee of Beijing Normal University.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Major Scientific and Technological Special Project for “Significant New Drugs Development” (Grant No. 2014ZX09507007-001 and 2014ZX09507007-003); the National Science and Technology Support Program (Grant No. 2014BAA03B03) and the National Natural Science Foundation of China (Grant No. 21371026). We thank the Nuclear Medicine Department of Peking Cancer Hospital (Beijing, China) for providing the fluoride-18 nuclide, and also thank the Nuclear Medicine Department of Peking Union Medical College Hospital (Beijing, China) for providing the use of the micro-PET.Notes and references
- M. D. Schaller, C. A. Borgma, B. S. Cob, R. R. Vines, A. B. Reynolds and J. T. Parsons, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 5192–5196 CrossRef CAS.
- S. K. Hanks, M. B. Calalb, M. C. Harper and S. K. Patel, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 8487–8491 CrossRef CAS.
- P. Dao, N. Smith, C. Tomkiewicz-Raulet, E. Yen-Pon, M. Camacho-Artacho, D. Lietha, J. P. Herbeuval, X. Coumoul, C. Garbay and H. Chen, J. Med. Chem., 2015, 58, 237–251 CrossRef CAS PubMed.
- D. Lietha and M. J. Eck, PLoS One, 2008, 3, e3800 Search PubMed.
- W. G. Roberts, E. Ung, P. Whalen, B. Cooper, C. Hulford, C. Autry, D. Richter, E. Emerson, J. Lin, J. Kath, K. Coleman, L. Yao, L. Martinez-Alsina, M. Lorenzen, M. Berliner, M. Luzzio, N. Patel, E. Schmitt, S. LaGreca, J. Jani, M. Wessel, E. Marr, M. Griffor and F. Vajdos, Cancer Res., 2008, 68, 1935–1944 CrossRef CAS PubMed.
- P. Dao, R. Jarray, J. L. Coq, D. Lietha, A. Loukaci, Y. Lepelletier, R. Hadj-Slimane, C. Garbay, F. Raynaud and H. Chen, Bioorg. Med. Chem. Lett., 2013, 23, 4552–4556 CrossRef CAS PubMed.
- T. Heinrich, J. Seenisamy, L. Emmanuvel, S. S. Kulkarni, J. Bomke, F. Rohdich, H. Greiner, C. Esdar, M. Krier, U. Gradler and D. Musil, J. Med. Chem., 2013, 56, 1160–1170 CrossRef CAS PubMed.
- J. Zhang and S. N. Hochwald, Pharmacol. Ther., 2014, 142, 154–163 CrossRef CAS PubMed.
- W. G. Cance, E. Kurenova, T. Marlowe and V. Golubovskaya, Sci. Signaling, 2013, 6, 10 CrossRef PubMed.
- T. Lechertier and K. Hodivala-Dilke, J. Pathol., 2012, 226, 404–412 CrossRef CAS PubMed.
- W. W. Ma, Anticancer Agents Med. Chem., 2011, 11, 638–642 CrossRef CAS PubMed.
- W. G. Cance, J. E. Harris, M. V. Iacocca, E. Roche, X. Yang, J. Chang, S. Simkins and L. Xu, Clin. Cancer Res., 2000, 6, 2417–2423 CAS.
- G. W. McLean, N. O. Carragher, E. Avizienyte, J. Evans, V. G. Brunton and M. C. Frame, Nat. Rev. Cancer, 2005, 5, 505–515 CrossRef CAS PubMed.
- V. M. Golubovskaya, F. A. Kweh and W. G. Cance, Histol. Histopathol., 2009, 24, 503–510 CAS.
- N. A. Chatzizacharias, G. P. Kouraklis and S. E. Theocharis, Expert Opin. Ther. Targets, 2007, 11, 1315–1328 CrossRef CAS PubMed.
- E. K. Han and T. McGonigal, Anticancer Agents Med. Chem., 2007, 7, 681–684 CrossRef CAS PubMed.
- M. J. van Nimwegen and B. van de Water, Biochem. Pharmacol., 2007, 73, 597–609 CrossRef CAS PubMed.
- V. M. Golubovskaya, Anticancer Agents Med. Chem., 2010, 10, 735–741 CrossRef CAS PubMed.
- A. Schultze and W. Fiedler, Expert Opin. Invest. Drugs, 2010, 19, 777–788 CrossRef CAS PubMed.
- V. M. Golubovskaya, Front. Biosci., 2014, 19, 687–706 CrossRef CAS.
- A. Schultze and W. Fiedler, Anti-Cancer Agents Med. Chem., 2011, 11, 593–599 CrossRef CAS PubMed.
- Y. Fang, D. Wang, X. Xu, J. Liu, A. Wu, X. Li, Q. Xue, H. Wang, H. Wang and H. Zhang, Eur. J. Med. Chem., 2017, 127, 493–508 CrossRef CAS PubMed.
- D. Wang, Y. Fang, H. Wang, X. Xu, J. Liu and H. Zhang, RSC Adv., 2017, 7, 22388–22399 RSC.
- (a) R. Francois, S. Lu, D. He, M. Zajac-Kaye and S. A. Hochwald, Eur. J. Cancer, 2002, 48, S174 CrossRef; (b) S. F. Jones, G. Shapiro, J. C. Bendell, E. X. Chen, P. Bedard, J. M. Cleary, S. Pandya, K. J. Pierce, B. Houk, N. Hosea, K. S. Zandi, W. G. Roberts, S. M. Shreeve and L. L. Siu, J. Clin. Oncol., 2011, 29, 3002 Search PubMed.
- V. M. Golubovskaya, B. Ho, M. Zheng, A. Magis, D. Ostrov, C. Morrison and W. G. Cance, BMC Cancer, 2013, 13, 342–346 CrossRef CAS PubMed.
- K. R. Auger, K. N. Smitheman, S. Korenchuk, C. McHugh, R. Kruger, G. S. Van Aller, A. Smallwood, R. R. Gontarek, T. Faitg and N. Johnson, Eur. J. Cancer, 2012, 4, 118 CrossRef.
- V. M. Golubovskaya, C. Nyberg, M. Zheng, F. Kweh, A. Magis, D. Ostrov and W. G. Cance, J. Med. Chem., 2008, 51, 7405–7416 CrossRef CAS PubMed.
- (a) M. Iwatani, H. Iwata, A. Okabe, R. J. Skene, N. Tomita, Y. Hayashi, Y. Aramaki, D. J. Hosfield, A. Hori, A. Baba and H. Miki, Eur. J. Med. Chem., 2012, 61, 49–60 CrossRef PubMed; (b) N. Tomita, Y. Hayashi, S. Suzuki, Y. Oomori, Y. Aramaki, Y. Matsushita, M. Iwatani, H. Iwata, A. Okabe, Y. Awazu, O. Isono, R. J. Skene, D. J. Hosfield, H. Miki, T. Kawamoto, A. Hori and A. Bab, Bioorg. Med. Chem. Lett., 2013, 23, 1779–1785 CrossRef CAS PubMed.
- E. V. Kurenova, D. L. Hunt, D. He, A. T. Magis, D. A. Ostrov and W. G. Cance, J. Med. Chem., 2009, 52, 4716–4724 CrossRef CAS PubMed.
- (a) D. A. Ucar, E. Kurenova, T. J. Garrett, W. G. Cance, C. Nyberg, A. Cox, N. Massoll, D. A. Ostrov, N. Lawrence, S. M. Sebti, M. Zajac-Kaye and S. N. Hochwald, Cell Cycle, 2012, 11, 3250–3259 CrossRef CAS PubMed; (b) D. A. Ucar, A. T. Magis, D. H. He, N. J. Lawrence, S. M. Sebti, E. Kurenova, M. Zajac-Kaye, J. Zhang and S. N. Hochwald, Anticancer Agents Med. Chem., 2013, 13, 595–602 CrossRef CAS PubMed.
- V. Golubovskaya, N. L. Palma, M. Zheng, B. Ho, A. Magis, D. Ostrov and W. G. Cance, Anticancer Agents Med. Chem., 2013, 13, 532–545 CrossRef CAS PubMed.
- T. Liu, T. LaFortune, T. Honda, O. Ohmori, S. Hatakeyama, T. Meyer, D. Jackson, J. de Groot and W. Yung, Mol. Cancer Ther., 2007, 6, 1357–1367 CrossRef CAS PubMed.
- J. Xu, H. Liu, G. Li, Y. He, R. Ding, X. Wang, M. Feng, S. Zhang, Y. Chen, S. Li, M. Zhao, Y. Li, C. Qi and Y. Dang, Molecules, 2012, 17, 3774–3793 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H-NMR, 13C-NMR, 19F-NMR and ESI-MS spectra for some key intermediates and target compounds, the chemical structures of [19F]1 and [18F]1, and the biodistribution data of [18F]1–[18F]3 and so on, were included in the supplementary material. See DOI: 10.1039/c8ra00652k. |
| This journal is © The Royal Society of Chemistry 2018 |