DOI:
10.1039/C8RA00761F
(Paper)
RSC Adv., 2018,
8, 8898-8909
Nitrogen-doped graphene-activated metallic nanoparticle-incorporated ordered mesoporous carbon nanocomposites for the hydrogenation of nitroarenes†
Received
25th January 2018
, Accepted 13th February 2018
First published on 26th February 2018
Abstract
Herein, nanoscale metallic nanoparticle-incorporated ordered mesoporous carbon catalysts activated by nitrogen-doped graphene (NGr) were fabricated via an efficient multi-component co-assembly of a phenolic resin, nitrate, acetylacetone, the nitrogen-containing compound 1,10-phenanthroline, and Pluronic F127, followed by carbonization. The obtained well-dispersed nitrogen-doped graphene-activated transition metal nanocatalysts possess a 2-D hexagonally arranged pore structure with a high surface area (∼500 m2 g−1) and uniform pore size (∼4.0 nm) and show excellent activity for the selective hydrogenation–reduction of substituted nitroarenes to anilines in an environmentally friendly aqueous solution. The high catalytic performance and durability is attributed to the synergistic effects among the components, the unique structure of the nitrogen-doped graphene layer-coated metallic nanoparticles, and electronic activation of the doped nitrogen.
1. Introduction
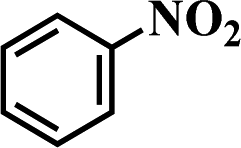
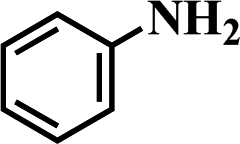
Structurally diverse anilines constitute major building blocks and key intermediates for the production of drugs, pharmaceuticals, pigments, dyes, and agrochemicals.1,2 The typical route for their production is the selective reduction of the corresponding nitro-derivatives. The traditional non-catalytic reduction processes (usually carried out using stoichiometric reducing agents such as Fe/HCl, Zn, Sn, Al, and sulfur compounds)3,4 require a stoichiometric excess of reagents, which eventually produce a large amount of inevitable waste and are therefore not considered to be environmentally benign and economic processes. In comparison, catalytic reduction using heterogeneous metal catalysts is a well-established technique and often regarded as the method of choice for the reduction of nitroarenes to anilines (which can be performed over homogeneous or heterogeneous catalysts using various reducing reagents such as hydrosilanes, sodium borohydride, formic acid, hydrazine derivatives, and hydrogen).5–10 However, the vast majority of catalytic reduction processes requires the use of abundant volatile organic solvents such as methanol, ethanol, toluene, tetrahydrofuran, and so on, which are expensive and have serious toxicity issues associated with them. In the actual production, the use of organic solvents makes the separation process more complicated with higher energy consumption. In addition, the reduction of aromatic nitro compounds often stops at an intermediate stage, yielding hydroxylamines, hydrazines, azoarenes, or azoxyarenes as byproducts.11–13 Obviously, the most environmentally benign, atomic efficient, and compatible route for the production of aromatic amines is the direct and quantitative hydrogenation of nitroarenes over highly active and selective catalysts with hydrogen molecules in the absence of a solvent or in environmentally friendly solvents (an aqueous medium), which has attracted widespread research interest.
Catalysis is a key technology for achieving more benign processes in the chemical, pharmaceutical, and materials industries. In terms of industrial applications, the careful design of heterogeneous catalysts instead of homogeneous catalysts is prevailing due to the ease of their separation and recycling. Compared with the traditional bulk catalysts, nanocatalysts are considered to be a bridge between homogeneous and heterogeneous catalysts,14,15 which, in general, show high catalytic activity and selectivity. However, for the reduction of –NO2, especially in the presence of other reducible substituents, such as aldehydes, ketones, nitriles, alkenes, and alkynes, noble metals are often not chemoselective, and this still remains a challenge. The applications of the most common commercially available palladium, platinum, ruthenium or rhodium catalysts are hindered by these issues, and these catalysts must be modified with suitable additives to improve their selectivity, but mostly at the cost of activity.16–19 A breakthrough obtained by the Corma group, who reported a heterogeneous gold-based catalyst that can selectively hydrogenate nitroarenes with functional groups, has provided new alternatives for the selective hydrogenation of nitroarenes.20 However, it remains a great challenge to realize commercialization because of the factors affecting the supply and volatile price. Thus, there still exists a significant interest in novel inexpensive, active, and selective catalysts, especially in those based on non-noble metals, despite numerous developments of hydrogenation catalysts. In terms of the selective catalytic reduction of nitro compounds, nitrogen-containing nanostructured carbon materials that possess enhanced chemical, electrical, and functional properties are potentially of significant technological interest for the development of catalytic systems. However, regarding the nitrogen-doped activated carbon or carbon nanotubes (CNTs) without any loaded metals, which have been proven to be effective catalysts for the reduction of 4-nitrophenol21 and nitrobenzene,22 strong reductants, such as NaBH4 and hydrazine hydrate, have been inevitably used rather than molecular hydrogen, but these have inferior activity and limited reaction scope, which may be because carbon material with highly graphitized sp2 carbon is too chemically inert to decompose molecular hydrogen.23,24 Furthermore, nitrogen-doped nanostructured ordered mesoporous carbon materials M–N–C (where M refers to transition metals, typically Ni, Fe, and Co),25–30 a type of metal and nitrogen bi-modified carbon material that usually evolves from the pyrolysis of metal complexes with macrocyclic N4 ligands or a nitrogen-containing precursor, with an interpenetrated and regular mesopore structure have recently attracted increasing attention because of their potential cost and environmental advantages; moreover, they exhibit highly selective hydrogenation of numerous structurally diverse nitroarenes that is proceeded under industrially viable conditions with high conversion rates. These materials present fascinating features such as intrinsic electrical properties, good chemical and thermal stability, and functional properties and have been widely investigated in heterogeneous catalysis as potential alternatives to platinum and palladium. However, it is still a difficult task to organize different nano objects with tunable sizes, morphologies, and functions into integrated and modified nanostructures for the development of novel nanosystems that exhibit high performances in catalysis. Most of the catalysts prepared by pyrolysis at high temperatures (600–900 °C) are composed of exposed nanoparticles ranging from a few to tens of agglomerates that are easy to lose during the reaction; therefore, exploration of effective methods to control the nucleation and growth of supported metallic particles and design size-variable and nitrogen-modified catalysts is crucial for the development of high-performance heterogeneous catalysts in industry.
To overcome the abovementioned limitations, we have speculated that the combination of N-doped ordered mesoporous carbon and first-row transition metals may lead to successful integration of the properties of the two components in new hybrid materials, which may present important features in catalysis for the selective hydrogenation of nitroarenes. Herein, encouraged by recent investigations on the use of cobalt-based or iron-based catalysts,31,32 we report a protocol to prepare novel nanoscale non-noble metal catalysts supported on nitrogen-doped ordered mesoporous carbon via a chelate together with a nitrogen-containing compound-assisted multicomponent co-assembly method. The obtained transition metal-based catalysts possess a 2-D hexagonally arranged pore structure with a high surface area, uniform pore volume, and moderate pore size that is not affected by the addition of nitrogen. Significantly, the catalysts present uniform, well-dispersed metal nanoparticles that are partially embedded in the carbon framework and the remaining part is exposed to the mesoporous channels. As expected, the pyrolysis of metal–phen complexes leads to the formation of nanoscale metal particles surrounded by a modified nitrogen-doped graphene layer (NGr) that are favorable for providing anchoring sites as well as regulating the chemical and electronic properties of noble metals. Modification of the surface takes place and significantly decreases the size of the metal particles of the phen-treated materials as compared to the case of the material prepared without phen; usually, remarkable differences in the catalyst activity are observed. Therefore, using different nitrogen ligands, the performance of the heterogeneous catalyst can be tuned, and both the chelate and nitrogen-containing ligand indirectly control the activity and selectivity of the given reaction.
2. Experimental
2.1. Chemicals and reagents
The poly(ethylene oxide)-block-poly(propylene oxide)-block-poly-(ethylene oxide) triblock copolymer Pluronic F127 (Mw = 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 g mol–1, PEO106PPO70PEO106) was purchased from Sigma-Aldrich. Ni(NO3)2·6H2O, Fe(NO3)3·9H2O, Co(NO3)2·6H2O, phenol, formalin solution (37 wt%), sodium hydroxide, hydrochloric acid, ethanol, 1,10-phenanthroline monohydrate (phen), acetylacetone (acac), and nitroarenes were purchased from Sinopharm Chemical Reagent Co., Ltd., which were of analytical grade and used as received without purification. Deionized water was used in all the experiments.
600 g mol–1, PEO106PPO70PEO106) was purchased from Sigma-Aldrich. Ni(NO3)2·6H2O, Fe(NO3)3·9H2O, Co(NO3)2·6H2O, phenol, formalin solution (37 wt%), sodium hydroxide, hydrochloric acid, ethanol, 1,10-phenanthroline monohydrate (phen), acetylacetone (acac), and nitroarenes were purchased from Sinopharm Chemical Reagent Co., Ltd., which were of analytical grade and used as received without purification. Deionized water was used in all the experiments.
2.2. General procedure for catalyst preparation
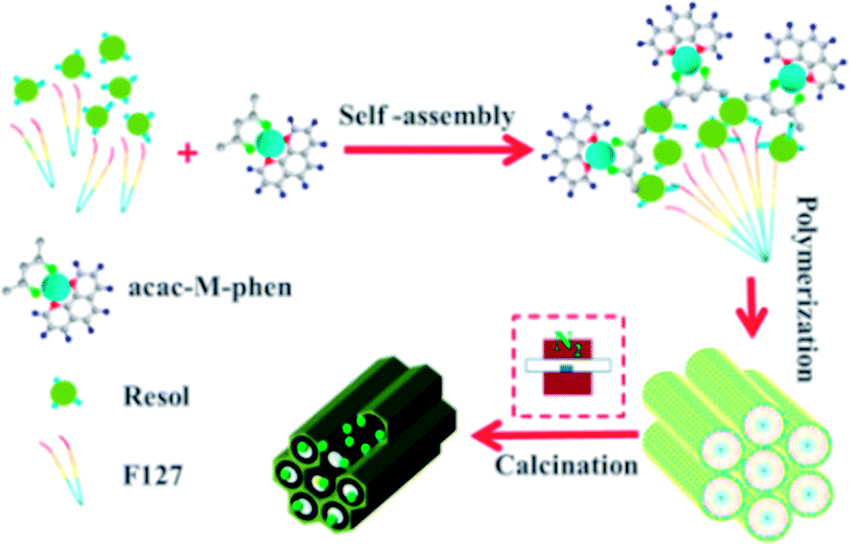
Preparation of mesoporous carbon composites. Soluble phenolic resin precursors (20 wt% in ethanol) were prepared using phenol and formaldehyde via a base-catalyzed process according to a previously reported procedure.33 The nitrogen-doped graphene-activated metallic nanoparticle-incorporated ordered mesoporous carbon catalysts were synthesized by a chelate together with a nitrogen-containing compound-assisted multicomponent co-assembly of phenolic resin, nitrate, acetylacetone, a nitrogen-containing compound, and Pluronic F127, followed by carbonization, as shown in Scheme 1. The typical preparation process is as follows: 1.0 g of Pluronic F127 was dissolved in 14.0 g of absolute ethanol at 40 °C in a water bath under constant stirring. Then, 5.0 g of the resol precursor solution (20 wt% in ethanol) was added to the abovementioned mixture and stirred for another 10 min. After this, appropriate amounts of nitrates (0.15 g Ni(NO3)2·6H2O, Co(NO3)2·6H2O or 0.2 g Fe(NO3)3·9H2O was used) dissolved in 3.0 g of ethanol were added. Subsequently, 0.12 g (acac/nitrates were in a 2:1 molar ratio) of acac was added followed by the addition of 0.10 g of 1,10-phenanthroline monohydrate (phen/nitrates were in a 1:1 molar ratio). For comparison, samples were also prepared without loading a metal or without adding the nitrogen containing compound 1,10-phenanthroline. After further stirring for 30 min, the mixture was cast into Petri dishes, followed by the evaporation of ethanol for several hours at 40 °C in a hood. The resulting sticky films were transferred to an electric thermostatic drying oven for thermopolymerization at 100 °C for 24 h. The obtained dark brown composite films were scraped off, followed by pyrolysis in a tube furnace at the defined temperature at the ramp rate of 1 °C min−1 (500 °C, 600 °C, 700 °C, 800 °C or 900 °C) for 3 h under a N2 atmosphere to decompose the triblock copolymer templates, carbonize the resol precursors and the nitrogen-containing ligand, and in situ generate metallic nanocrystallites surrounded by nitrogen-doped graphene. All the materials were designated as M/NGr@OMC-T (where M refers to the corresponding metallic species; T represents the calcination temperature, °C).
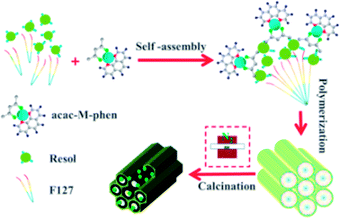 |
| | Scheme 1 Preparation of M/NGr@OMC catalysts. | |
2.3. Catalyst characterization
Powder X-ray diffraction (XRD) was performed via a Rigaku D/MAX-2200 apparatus using Cu Kα radiation (λ = 0.1542 nm) operated at a voltage of 40 kV and a current of 40 mA. Moreover, 2θ scans were performed from 0.5° to 5° at 0.5° min−1 and from 10° to 90°at 8° min−1. The crystallite sizes of the metal nanoparticles were calculated using the Scherrer equation. N2 sorption was carried out using a Micromeritics ASAP 2020 sorptometer at −196 °C. The sample was degassed in vacuum at 200 °C for 8 h before the measurement. The specific surface area (SBET) was evaluated using the Brunauer–Emmett–Teller (BET) method. Pore size distribution was analyzed using the adsorption branch and the Barrett–Joyner–Halenda (BJH) method. The pore size (Dp) was obtained from the maximum of the pore distribution curve. The pore volume (Vp) was taken at the P/P0 = 0.990 single point. TEM images were obtained using the JEOL JEM-2010F field emission microscope. The sample was prepared by placing a drop of the ethanol solution of a well-ground catalyst powder onto a carbon-coated copper grid followed by evaporation of ethanol. XPS spectra were obtained using an ESCALAB 250Xi spectrometer equipped with monochromatized Al Kα radiation (hν = 1486.6 eV) operated at ca. 1 × 10−9 torr. The spectra were calibrated using the binding energy of the C 1 s peak at 284.6 eV. The surface N/C molar ratios of the catalysts were obtained from the XPS spectra. The actual amounts of the C and N elements in the final M/NGr@OMC-T catalysts were determined using the Perkin Elmer 2400 CHN elemental analyzer. The actual metal contents in the M/NGr@OMC-T catalysts were measured by inductively coupled plasma atomic emission spectrometry (ICP-AES) conducted using a Perkin Elmer emission spectrometer. Thermogravimetric analysis (TGA) curves were obtained using a thermogravimetric analyzer (Netzsch STA 4449 F3) from 25 °C to 900 °C at an air flow of 80 mL min−1 at the heating rate of 5 °C min−1. Raman spectra were obtained at ambient temperature using a 785 nm HPNIR excitation laser via a Renishaw Raman spectrometer equipped with an Olympus microscope and a CCD detector. The laser power on the sample was 15 mW, and a total of 20 acquisitions were taken for each spectrum.
2.4. Catalytic reactions and product analyses
2.4.1. General procedure for the hydrogenation of nitroarenes. All hydrogenation experiments were carried out in 100 mL autoclaves. To avoid unspecific hydrogenation caused by the reaction devices, all catalytic reactions were carried out either in glass vials, which were placed inside the autoclave, or in Teflon vessel-fitted autoclaves to avoid direct contact between the reactants and reaction devices. The magnetic stirring bar and the corresponding nitro compound (1 mmol) were transferred into the glass vial, and then, 2 mL deionized water was added. After this, 20 mg of catalyst was added, and the vial was fitted with a septum, cap, and needle. The autoclave was flushed with hydrogen twice at a 30 bar pressure, and then, it was pressurized to a 50 bar hydrogen pressure. During the reaction, the inside temperature of the autoclave was measured to be 100 °C, and this temperature was used as the reaction temperature. After the completion of the reaction, the autoclave was immediately cooled down to room temperature by flowing cold water. The remaining hydrogen was discharged, and the samples were removed from the autoclave. To individual vials, n-hexadecane (100 μL) was added as an external standard, and the reaction mixture from the vial was completely transferred to a beaker. After this, the mixture was extracted and diluted with ethyl acetate (10–15 mL) followed by filtration and then analyzed by gas chromatography and gas chromatography-mass spectrometry (GC-MS, Shimadzu GCMS-QP2010 Plus). Conversion and yields were determined by GC-FID (Varian CP-3800) with a capillary column (column VF-1 ms, 15 m, 0.25 mm, 0.25 μm) and a flame ionization detector (FID). Quantitative and qualitative analysis of all the amines were conducted by GC and GC-MS analysis.
2.4.2. Procedure for catalyst recovery and utilization. Under similar experimental conditions as described in Section 2.4.1, after completion of the reaction, the autoclave was cooled down to room temperature. The remaining hydrogen was discharged completely, and the substrate was removed from the autoclave. The catalyst from the reaction mixture was filtered off, washed with water and then ethyl acetate followed by drying without any other treatment, and then used for the next cycle.
3. Results and discussion
3.1. Structure identification of the M/NGr@OMC-T catalysts
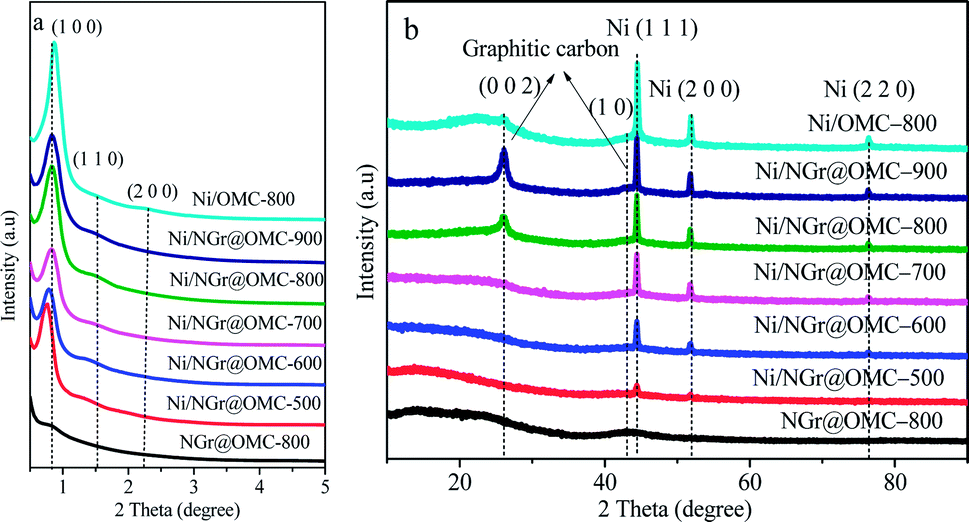
The low-angle XRD patterns corresponding to the samples calcined at different temperatures are shown in Fig. 1a. Most materials retained their particularly ordered structure, showing three diffraction maxima (100), (110), and (200), indicating an ordered 2D hexagonal mesoporous structure (space group p6mm). It should be noted that no diffraction peaks were observed for the samples without loaded metal nanoparticles; this indicated the disorder of their mesoporous structure; moreover, in this case, phen was used as a ligand without acac (not shown here). This result demonstrated that the addition of metal ion as well as acac strongly influenced the co-assembly self-organization of resol and F127 with the ligands in the solvent evaporation process. The formation of metal nanoparticles with semi-exposed morphology in the nitrogen-doped ordered mesoporous composites was mainly attributed to the chelating effect of the acac molecules and phen molecules with the metal ion. It should be noted that an increase in the calcination temperature from 500 °C to 900 °C resulted in a diffraction maximum position shift to higher 2θ. In contrast, the Ni/NGr@OMC-500 sample showed an apparent diffraction peak at 2θ = 0.77° with a d-spacing of 11.5 nm. This diffraction peak could be indexed to the (100) reflection of the hexagonal structure. With a further increase in the calcination temperature to 800 °C, the intensity of the (100) reflection shifted to the low diffraction angle of 0.83°, and the higher order weak (110) and (200) reflections were also observed. Correspondingly, the d100-spacing was decreased from 11.5 to 10.6 nm, which corresponded to the cell parameters (a0) of 13.3 nm and 12.2 nm, respectively (Table 1). The higher d value for the Ni/NGr@OMC-500 sample indicates a lower carbon network contraction degree during carbonization, which can be attributed to the presence of nickel metallic nanoparticles embedded into the carbon matrix.
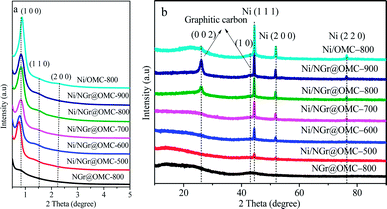 |
| | Fig. 1 (a) Low-angle XRD patterns and (b) wide-angle XRD patterns of the nickel nanoparticle-incorporated nitrogen-doped ordered mesoporous carbon nanocomposites synthesized at different temperatures. | |
Table 1 Textural properties of the prepared materials
| Sample |
d100 (nm) |
a0a (nm) |
SBET (m2 g−1) |
Vp (cm3 g−1) |
Dp (nm) |
WTb (nm) |
| a0 is the cell parameter calculated from the SXRD patterns (a0 = 2d100/√3). Wall thickness (WT) values calculated from WT = a0 − Dp. |
| NGr@OMC |
10.0 |
11.5 |
269 |
0.16 |
3.7 |
7.8 |
| Ni/NGr@OMC-500 |
11.5 |
13.3 |
444 |
0.34 |
4.7 |
8.6 |
| Ni/NGr@OMC-600 |
11.2 |
12.9 |
453 |
0.35 |
4.5 |
8.4 |
| Ni/NGr@OMC-700 |
10.7 |
12.3 |
483 |
0.35 |
4.5 |
7.8 |
| Ni/NGr@OMC-800 |
10.6 |
12.2 |
520 |
0.37 |
4.1 |
8.1 |
| Ni/NGr@OMC-900 |
10.5 |
12.1 |
444 |
0.36 |
4.1 |
8.0 |
| Ni/OMC-800 |
10.0 |
11.5 |
439 |
0.30 |
4.1 |
7.4 |
| Co/NGr@OMC-500 |
10.6 |
12.2 |
486 |
0.36 |
4.1 |
8.1 |
| Fe/NGr@OMC-500 |
10.7 |
12.3 |
445 |
0.39 |
4.2 |
8.1 |
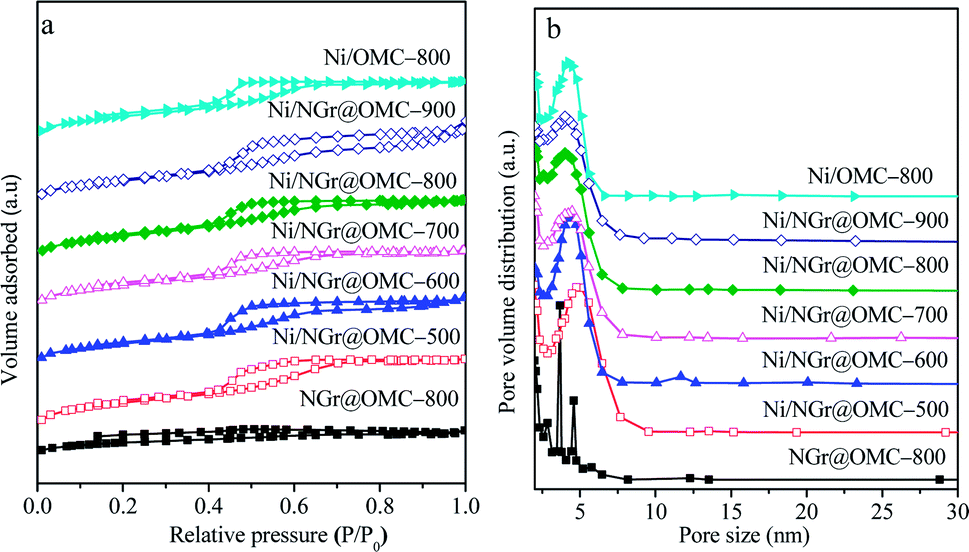
The N2 sorption analysis (Fig. 2a) of M/NGr@OMC-T showed typical type IV isotherms with clear hysteresis loops and narrow pore size distributions, attributed to the existence of well-developed meso- and micropores formed by the removal of the F127 template and burn-out of C, H, and O from the phenolic resin framework during the pyrolysis procedure.34 These results suggest that the obtained M/NGr@OMC-T samples possess similar pore structures and features. The pore size distribution curves (Fig. 2b) derived from the adsorption branches using the BJH model revealed the presence of relatively uniform mesopores with mean mesopore sizes ranging from 3.8 to 4.3 nm, which slightly declined with an increase in the calcination temperature. The calculated values of pore wall thicknesses (WT) were in the range of 7.4–8.6 nm, less than the sizes (15–30 nm) of Ni particles as calculated by XRD. Consequently, it is reasonable to deduce that these Ni particles partially penetrate into the mesopore walls and are partially exposed to the mesoporous channels; this was also evidenced by the TEM observations. As listed in Table 1, the M/NGr@OMC-T composites displayed high BET surface areas (SBET) of 269–520 m2 g−1, large pore volume (Vp) of 0.34–0.40 cm3 g−1, and pore sizes (Dp) of 3.5–4.5 nm.
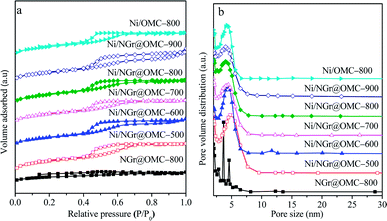 |
| | Fig. 2 (a) N2 sorption isotherms and (b) BJH pore size distributions of the nickel nanoparticle-incorporated nitrogen-doped ordered mesoporous carbon nanocomposites synthesized at different temperatures. | |
3.2. Characterization of metal dispersion and component contents
To more accurately understand the composition of the nickel nanoparticles (NPs) in the catalyst, wide angle X-ray diffraction (XRD) investigations were carried out (Fig. 1b). Herein, two remarkable peaks at 2θ = 26° and 43° corresponding to the (002) and (1 0) diffraction peaks of graphitic carbons were observed in the wide-angle XRD patterns of the materials Ni/NGr@OMC-800 and Ni/NGr@OMC-900 (Fig. 2b), which indicated the crystallization of the carbon frameworks in the resulting products and incorporation of graphite carbon inside the mesochannels. On the other hand, when the calcination temperature was lower than 800 °C, these peaks disappeared, showing a significant difference. This demonstrates that different temperatures have a significant influence on the crystalline form of graphitic carbons. It should be noted that the much weaker peaks at 2θ = 26° and 43° were observed for the sample without added phen; this indicated that the combined pyrolysis of phen strongly promoted the formation of graphitic carbon, which played a key role in the hydrogenation of nitroarenes. The growth of particles within the Ni/NGr@OMC series was also supported by the wide angle XRD and TEM results. Wide-angle XRD patterns of the materials after being pyrolyzed at 500–900 °C showed three resolved diffraction peaks (Fig. 1b), which could be assigned to the (1 1 1), (2 0 0), and (2 2 0) reflections of metallic nickel. This result indicates that the main form of nickel in the catalyst is metallic nickel. Furthermore, as estimated from the line broadening of the (1 1 1) reflection by the Scherrer equation, the sizes of the nanocrystalline Ni particles were 11.7, 15.4, 18.4, 21.5, and 24.8 nm for the samples Ni/NGr@OMC-500, Ni/NGr@OMC-600, Ni/NGr@OMC-700, Ni/NGr@OMC-800, and Ni/NGr@OMC-900, respectively. This is contrary to the case of the M–Nx/C materials prepared by the simplified pyrolysis of metal–phen or pyrolysis of metal–phen on supports such as Al2O3 or active carbon reported in literature, which have a wide size distribution of particles or agglomerates in the range 20–80 nm and even larger structures up to 800 nm.10a,b Furthermore, it is difficult to uniformly incorporate the precursor compounds into the inner pores of the carbon support by this method, and active metal–nitrogen (M–Nx) moieties are usually formed exclusively on the external surface of the support, resulting in insufficient catalytic activity for hydrogenation. TEM images of all Ni/NGr@OMC nanocomposites showed stripe-like and hexagonally arranged pore morphology over large domains, confirming an ordered mesostructure with a 2-D hexagonal pore symmetry (Fig. 3b–g). As shown in the TEM image of the sample Ni/NGr@OMC-500 series (Fig. 3b), uniform Ni nanoparticles, with a mean diameter of ∼12 nm, were highly dispersed in the carbon matrix. When the temperature was increased to 800 °C for the Ni/NGr@OMC-800 sample, uniform Ni nanoparticles of about 22 nm were obtained without aggregation of large particles. In comparison, agglomerates in the range 20–80 nm and even larger structures up to 200 nm were present for Ni/OMC-800 (Fig. 3g). With the enlargement of the particle size, these Ni nanoparticles can penetrate the carbon walls and get stuck in two and even three mesopores, but are still confined in the carbon matrix. High-resolution TEM (HRTEM) images (Fig. 3h) clearly show that the well-crystallized Ni nanoparticles are embedded in the graphite carbon matrix and surrounded by NGr with a homogeneous dispersion. Furthermore, TEM images show that the Ni nanoparticles in the obtained composites have unique semi-exposure morphology, with one part of the nanoparticle being partially exposed to pore channels and the other part being tightly trapped in the carbon framework. In addition, the confinement effect of the channels within OMC may help to limit the migration distance of metal elements, inhibit their aggregation, and promote the formation of Ni (Fe or Co)–NGr moieties during the heat-treatment process.
 |
| | Fig. 3 TEM images of the nickel nanoparticle-incorporated nitrogen-doped ordered mesoporous carbon nanocomposites synthesized at different temperatures: (a) NGr@OMC-800, (b) Ni/NGr@OMC-500, (c) Ni/NGr@OMC-600, (d) Ni/NGr@OMC-700, (e) Ni/NGr@OMC-800, (f) Ni/NGr@OMC-900, (g) Ni/OMC-800, and (h) HRTEM of Ni/NGr@OMC-800. | |
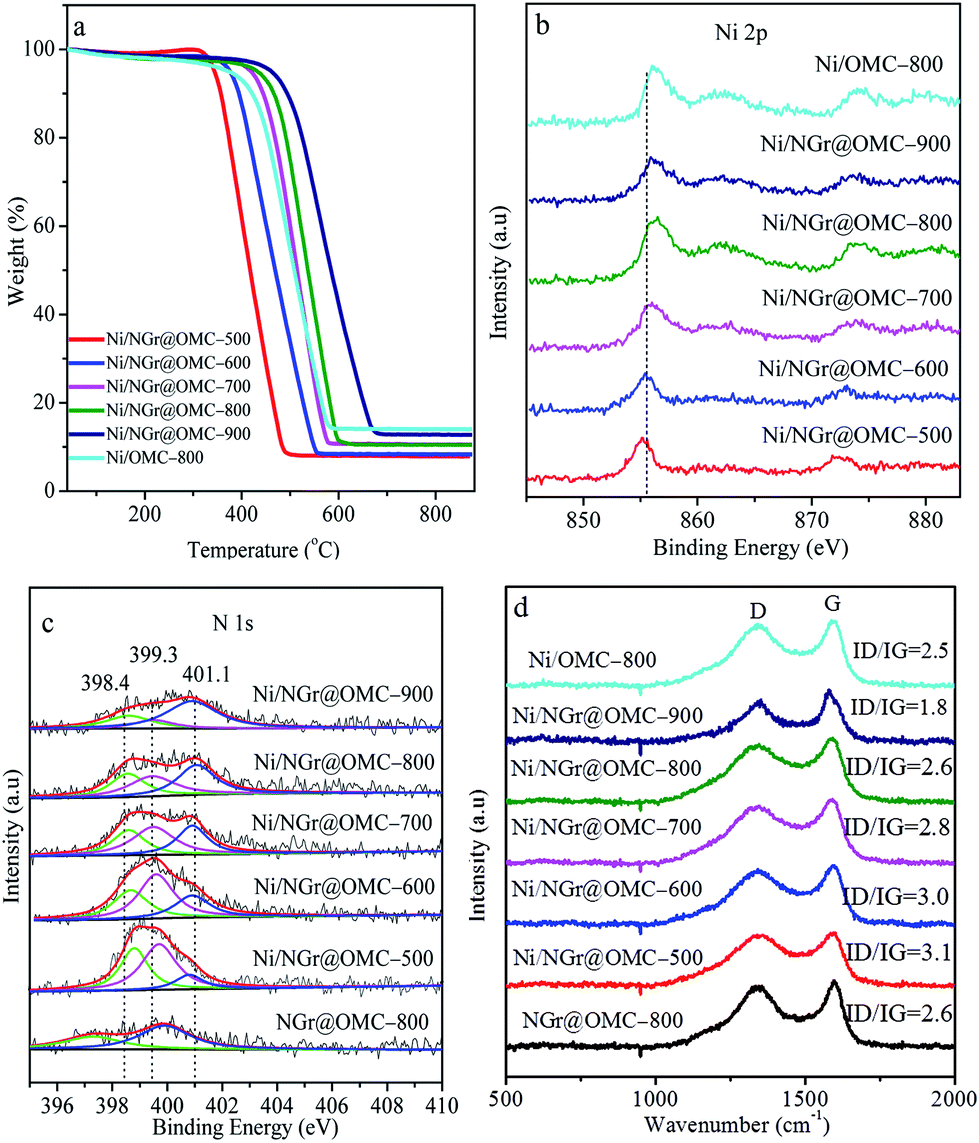
TGA curves obtained in air for the nitrogen-doped ordered mesoporous carbon/nanoparticle metal composites display similar curves, except for their distinct weight losses (Fig. 4a). Weight loss observed below 200 °C was caused by desorption of the physisorbed water. Significant weight losses occurred in the range of 400–600 °C, independent of the content of Ni, suggesting carbon combustion. Slight weight-increasing phenomena were visible above 600 °C, resulting from the oxidation of metallic Ni nanoparticles in air with the NiO residue. The mass ratios of the residual NiO composites to the materials were determined to be 14.0, 7.9, 8.4, 10.4, 10.5, and 14.0 wt% for the samples Ni/OMC-800, Ni/NGr@OMC-500, Ni/NGr@OMC-600, Ni/NGr@OMC-700, Ni/NGr@OMC-800, and Ni/NGr@OMC-900, respectively, which were consistent with the mass ratios of Ni to composites, calculated to be 12.2, 6.9, 7.3, 8.9, 9.2, and 11.7 wt%, respectively, based on the Ni content obtained from the ICP measurement (Table 3). It should be noted that the combustion temperature of carbons shifted to higher temperatures for the Ni/NGr@OMC-500, Ni/NGr@OMC-600, Ni/NGr@OMC-700, Ni/NGr@OMC-800, and Ni/NGr@OMC-900 samples because a higher degree of graphitization in the carbon walls hindered the burning of carbon.35 It was noted that a similar phenomenon was found when the sample Ni/OMC-800 was compared with the sample Ni/NGr@OMC-800.
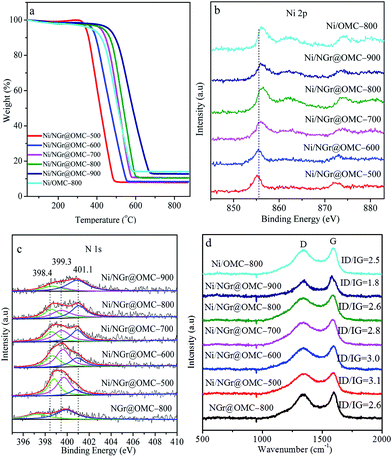 |
| | Fig. 4 (a) TGA curves, (b) Ni 2 p XPS spectra, (c) N 1 s XPS spectra, and (d) Raman spectra of the nickel nanoparticle-incorporated nitrogen-doped ordered mesoporous carbon nanocomposites synthesized at different temperatures. | |
Table 2 Surface elemental analysis of the materials calculated by XPS
| Sample |
C (wt%) |
N (wt%) |
Ni (wt%) |
N/metal (mol mol−1) |
| NGr@OMC |
87.9 |
1.7 |
— |
— |
| Ni/NGr@OMC-500 |
80.0 |
3.6 |
3.3 |
4.5 |
| Ni/NGr@OMC-600 |
81.3 |
3.3 |
4.1 |
3.9 |
| Ni/NGr@OMC-700 |
81.8 |
2.6 |
5.0 |
2.2 |
| Ni/NGr@OMC-800 |
79.6 |
2.4 |
6.6 |
1.9 |
| Ni/NGr@OMC-900 |
80.6 |
1.6 |
8.8 |
1.5 |
| Ni/OMC-800 |
80.0 |
— |
8.8 |
— |
| Co/NGr@OMC-800 |
79.5 |
1.8 |
6.3 |
1.2 |
| Fe/NGr@OMC-800 |
84.3 |
1.7 |
4.3 |
3.1 |
Table 3 Actual elemental content of the materials measured by elemental analysis
| Sample |
C (wt%) |
N (wt%) |
Ni (wt%) |
N/metal (mol mol−1) |
| NGr@OMC |
82.0 |
0.8 |
— |
— |
| Ni/NGr@OMC-500 |
75.3 |
3.0 |
6.5 |
1.9 |
| Ni/NGr@OMC-600 |
78.6 |
2.2 |
6.9 |
1.3 |
| Ni/NGr@OMC-700 |
79.1 |
1.6 |
8.5 |
0.8 |
| Ni/NGr@OMC-800 |
81.0 |
1.2 |
8.6 |
0.6 |
| Ni/NGr@OMC-900 |
82.4 |
0.9 |
10.7 |
0.4 |
| Ni/OMC-800 |
84.2 |
— |
11.2 |
— |
| Co/NGr@OMC-800 |
80.3 |
1.3 |
6.3 |
0.9 |
| Fe/NGr@OMC-800 |
79.7 |
1.1 |
6.0 |
0.7 |
3.3. XPS characterization
Due to the superposition of C and N signals, no conclusive information on the location of nitrogen in the samples could be obtained by XRD or TEM analysis. Thus, to obtain further insight into the structure of the catalyst and especially the role of nitrogen from the organic ligand, XPS investigations on the bonding of nitrogen and nickel were carried out (Fig. 4b and c). The calcination temperature plays a key role in determining the type and status of the N species. In the NGr@OMC-800 sample, without any loaded metal, two N1s peaks occurred at 397.3 eV and 400.1 eV, which were assigned to pyridinic nitrogen and graphite N centers, respectively.36 The former peak might be a reflection of N within the free ligand, whereas the latter arises from graphitization of the ligand N atoms. On the other hand, in Ni/NGr@OMC-500 and, to a more pronounced extent, in the most active catalyst Ni/NGr@OMC-800, these peaks are shifted to higher binding energies (398.4 eV and 401.1 eV). The N 1 s peak 401.1 eV, which can be assigned to nitrogen in a graphite-like structure, gains intensity and undergoes a weak shift to a higher binding energy after pyrolysis at higher temperatures. This result indicated that N was incorporated into the support lattice. Compared to the NGr@OMC-800 sample without any loaded metal, a new N1s peak at ∼399.3 eV was observed for nitrogen species in contact with the deposited Ni atoms on top. It was assigned to the so-called distorted NiNx center,37 in which the Ni–N distances were slightly different at different calcination temperature; this gave rise to peak splitting and shifting to lower binding energies with an increase in the calcination temperature. It is probable that similar NiNx centers are formed with the progressing pyrolysis on the surface of the emerging nickel particles. Furthermore, the Ni XPS spectra presented a Ni 2p3/2 primary peak corresponding to Ni2+, as indicated by the binding energy of 855.7 eV and the presence of a satellite peak, which strongly coordinated with the N atoms in the NGr layer, and a shift to higher values with the increasing calcination temperature was also observed. Loss of nitrogen was observed with the increasing pyrolysis temperature in both the near-surface region and the bulk, the surface N/Ni ratio decreased from around 4.5 in the catalyst Ni/NGr@OMC-500 to 1.5 in Ni/NGr@OMC-900 (Table 2), and the bulk N/Ni ratio decreased from around 1.9 in the catalyst Ni/NGr@OMC-500 to 0.4 in Ni/NGr@OMC-900, (Table 3). Significantly, in the less active catalyst Ni/NGr@OMC-900, the characteristic NiNx peak centred around 399.3 eV decreased in intensity, leaving behind signals with higher proportions of pyridinic (398.6 eV) and graphite enclosed nitrogen (401.1 eV). These results indicated decomposition of the NiNx centers during pyrolysis when the calcination temperature was over 900 °C, accompanied by partial sublimation and incorporation of nitrogen into the support structure. It should be noted that no Ni–N centers are found when the catalyst is prepared in the same way but without any phenanthrene, in which the nanoparticles with a larger and uneven size distribution are formed. A comparison of the characterization and catalytic results suggests that these particular NiNx centers formed in a narrow range of pyrolysis temperatures around 800 °C govern the unique catalytic activity. This general composition is in agreement with the elemental analysis. The observed carbon layers might be generated from the metal precursor (acac) and/or the ligand (phen), whereas carbonization of nitrogen-containing ligands should lead to nitrogen-doped graphene-type structures (NGr). The Raman spectra of the Ni-based catalysts are presented in Fig. 4d, in which the G-band at ∼1590 cm−1 indicates in-plane vibration of sp2 carbon atoms, whereas the D band at ∼1350 cm−1 is a defect-induced Raman feature representing the imperfect crystalline structure of carbon.37b Compared with that of Ni/OMC-800, the ID/IG value showed an increase from 2.5 to 2.6 for Ni/NGr@OMC-800; this indicated the formation of many more structural defects on the N-doped OMC. Moreover, the decrease in the intensity ratios of the D and G bands (ID/IG) suggest that the heat treatment at elevated temperatures (from 500 °C to 900 °C) will increase the graphitization degree and decrease the N defect density.
By utilization of the similar effective complexation between acac, phen, and Co2+ or Fe3+ ions, a series of size-tunable Co-based and Fe-based nanoparticle-incorporated nitrogen-doped mesoporous carbon nanocomposites can be synthesized using cobalt nitrate and iron nitrate at a constant ratio as the metallic source and conducting calcination at 800 °C in N2. The catalysts were characterized by XRD, XPS, TGA, Raman spectroscopy, BET, and TEM (Fig. S1–S8†). The results demonstrated that the catalysts had similar ordered mesoporous structures and components; however, compared with those in the Ni/NGr@OMC-800 sample, the metallic nanoparticles in the Co/NGr@OMC-800 and Fe-NGr@OMC-800 samples were slightly smaller. Specifically, TEM images of the Co/NGr@OMC-800 and Fe/NGr@OMC-800 samples confirmed that cobalt nanoparticle species and iron nanoparticle species with a uniform size were well dispersed in the nitrogen-doped ordered mesoporous carbon matrix (Fig. S6†). Moreover, the TEM images showed that the cobalt or iron nanoparticles were homogeneously embedded in the mesoporous carbon frameworks and partially exposed to the pore channels; this was similar to the case of the Ni/NGr@OMC-800 sample. The wide-angle XRD patterns showed that the well-resolved diffraction peaks could be assigned to cobalt for the sample Co/NGr@OMC-800 (Fig. S1b†), whereas the mixed phases of Fe, α-Fe2O3, γ-Fe2O3, and Fe3C were found for the sample Fe/NGr@OMC-800 (Fig. S2b†), and among them, iron was the dominant component.
The formation of metal nanoparticles with the semi-exposed morphology incorporated with NGr in the ordered mesoporous composites was mainly attributed to the chelating effect of the acac and phen molecules. This feature seemed plausible to be explained by the following aspects: in the ethanol solution of resol, F127, and metal ions, the resol molecules with several phenolic hydroxyl groups not only interacted with the EO segments of F127 through the formation of hydrogen bonds,16a,24 but also coordinated with metal ions to form metal–phenol complexes such that the resol precursors could not undergo simple thermopolymerization around the triblock copolymers to generate a rigid hydrocarbon network with three-connected benzene rings via the formation of covalent bonds. Upon addition of acac and phen, metal ions were chelated with acac and phen due to strong complexation, as shown in Scheme 1. The resol molecules could be smoothly assembled and thermopolymerized around the surfactant micelles during solvent evaporation without perturbation from Ni(II) ions to generate (acac)x–M–(Phen)y/resol/P127 composites of a long-range ordered array structure. During pyrolysis above 500 °C in N2, the F127 template molecules were first decomposed, leaving highly ordered aligned mesopores, and the phenolic resin was then carbonized into a rigid carbon framework accompanied by the in situ growth of metal nanoparticles via slow decomposition of (acac)x–M–(Phen)y complexes; this resulted in the formation of metal nanoparticles accompanied by the formation of nitrogen-doped graphene-incorporated mesoporous carbon materials. Without the addition of acac and phen molecules, the metallic ion species in the phenolic resin can readily migrate, aggregate, and grow during both thermosetting and pyrolysis treatments because of the low melting points of the nitrates, leading to the formation of large particles up to with a wide range from 10–80 nm. With the addition of acac and phen, the nanoclusters of metal oxides formed at the early stage of pyrolysis can be capped with the acac molecules that prevent their fast growth; on the other hand, the in situ formed nitrogen-doped graphene also helps to inhibit the growth and migration of metal nanoparticles. The promoting effect of nitrogen atoms, leading to a smaller particle size and basicity provided by NGr, may be responsible for the higher catalytic activity.
3.4. Catalytic reaction
All the prepared materials were tested for their hydrogenation activity towards the industrially important substrate nitrobenzene in an aqueous solution under industrially feasible conditions. Interestingly, the reaction rate of hydrogenation is dependent on the amount of water present. When we used dry organic solvents, for example, tetrahydrofuran (THF) or ethanol, the reaction times had to be prolonged to achieve full conversion. This stimulated us to perform the reaction simply in water without any additional organic solvent present. To our delight, the catalyst activity was even higher in pure water than that in THF or ethanol. Parameters, such as pyrolysis temperature of the catalysts, type of ligands, and type of supported metals, were systematically investigated (Table 4). A blank experiment with almost no detectable products could exclude the device itself that catalysed the reaction (Table 4, entry 1). Neither the homogeneous nickel complexes (Table 4, entry 2) nor the precursor gel containing incorporated Ni2+-phen without pyrolysis were active (Table 4, entry 3). However, after pyrolysis, the resulting catalysts showed different activities. The carbon carrier itself did not show any activity; this indicated that the metal nanoparticles acted as catalytic active centers (Table 4, entry 4). Using the benchmark substrate Ni/NGr@OMC-800, nitrobenzene was hydrogenated to obtain aniline in an excellent yield (86%) (Table 4, entry 9); thus, Ni/NGr@OMC-800 exhibited almost triple the activity of the sample Ni/OMC-800 without nitrogen-doped activated graphene (Table 4, entry 5); it must be noted that the lower catalytic activity of Ni/OMC-800 may be caused by the much larger particle size and the absence of nitrogen-doped activated graphene. These observations also appear to indicate that the doped nitrogen atoms play a crucial role in our system. The versatile nitrogen can provide more surface nucleation sites and promote the formation of some individual sections around the N-rich sites,38 allowing efficient anchoring of metal NPs.39 The pyrolysis temperature also plays a critical role in the activity. It is interesting to note that the Ni/NGr@OMC-800 catalyst with a lower N content shows better activity than the sample with pyrolysis temperature in the range from 500 to 700 °C (Table 4, entry 6–8); this better activity is probably due to the higher degree of crystallinity of graphite carbon in Ni/NGr@OMC-800, which also has a higher graphite N content, and not caused by the defect sites on the surface of catalysts since the sample with a lower pyrolysis temperature in the range from 500 to 700 °C with a higher content of defect sites (Fig. 4b) is not the best catalyst. These results indicate that graphite N in NGr of the catalysts is critical for impelling the reaction rather than the amount of N atoms in nitrogen-doped graphene-activated catalysts, in accordance with the XRD and XPS results. Upon further increasing the pyrolysis temperature to 900 °C, the activity of the resulting catalyst decreased (Table 4, entry 10); this might have been caused by metal particle aggregation to provide less active sites. Obviously, it is generally accepted that the dispersion and the mean size of NPs may greatly affect the catalytic activity.40 However, the yield of aniline was still higher as compared to that obtained in the corresponding reaction using Ni/OMC-800 or the sample with a pyrolysis temperature in the range from 500 to 700 °C as catalysts. These results strongly demonstrate that NGr in the catalysts is indispensable for the hydrogenation of nitroarenes. In comparison, the Co/NGr@OMC-800 and Fe/NGr@OMC-800 catalysts showed slightly lower activity than Ni/NGr@OMC-800 (Table 4, entry 11 and 12).
Table 4 Catalytic performance of catalysts for the chemoselective hydrogenation of nitrobenzenea

|
| Entry |
Catalyst |
Reducing agent |
Conv. (%) |
Sel. (%) |
| Reaction conditions: catalyst (20 mg), nitrobenzene (1 mmol), water (2 mL), 100 °C, 1 h. Reducing agent: 5 MPa H2. |
| 1 |
No catalyst |
H2 |
0 |
0 |
| 2 |
Ni–phen |
H2 |
0 |
0 |
| 3 |
Ni–phen-resol-F127 |
H2 |
0 |
0 |
| 4 |
NGr@OMC-800 |
H2 |
<1 |
>99 |
| 5 |
Ni/OMC-800 |
H2 |
33 |
>99 |
| 6 |
Ni/NGr@OMC-500 |
H2 |
32 |
>99 |
| 7 |
Ni/NGr@OMC-600 |
H2 |
38 |
>99 |
| 8 |
Ni/NGr@OMC-700 |
H2 |
45 |
>99 |
| 9 |
Ni/NGr@OMC-800 |
H2 |
86 |
>99 |
| 10 |
Ni/NGr@OMC-900 |
H2 |
62 |
>99 |
| 11 |
Co/NGr@OMC-800 |
H2 |
66 |
>99 |
| 12 |
Fe/NGr@OMC-800 |
H2 |
61 |
>99 |
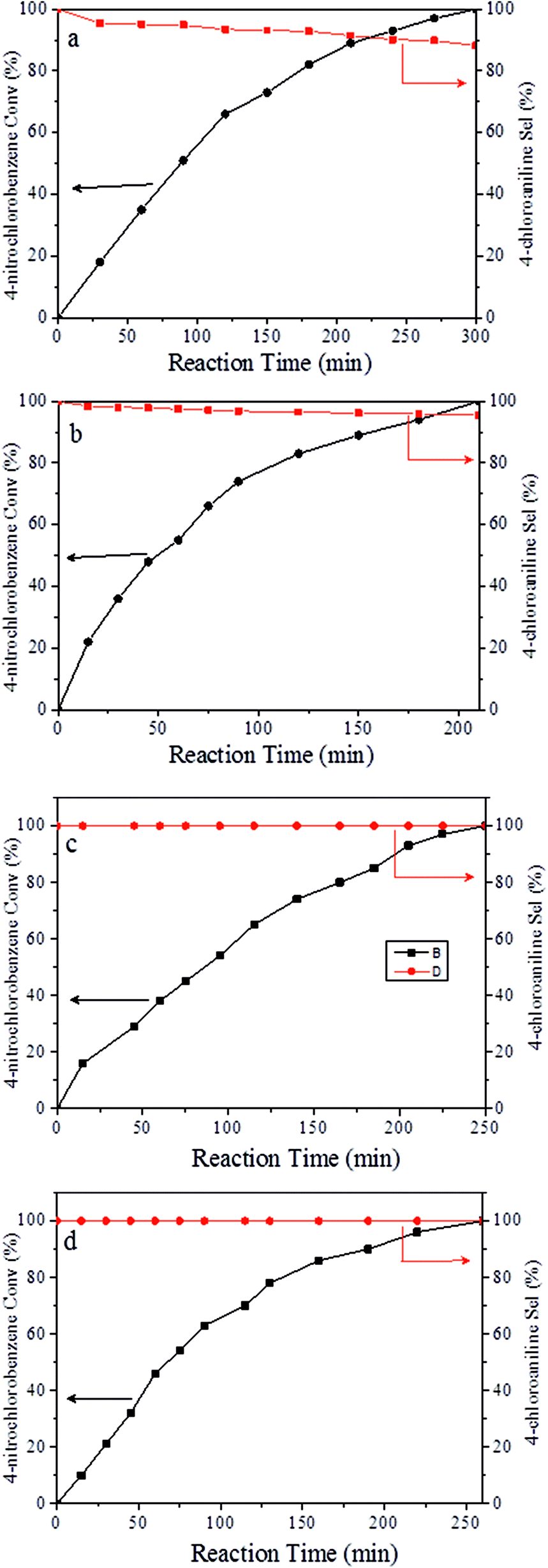
It should be noted that the chemoselective hydrogenation of substituted nitroarenes with halogen groups is highly desired. For example, the unwanted C–Cl bond hydrogenolysis is difficult to fully avoid. Herein, substituted nitrobenzenes with –Cl (an electron-withdrawing substituent) at the para position were chosen as the reactants. The kinetic profile of the reaction revealed that 4-chloronitrobenzene (p-CNB) was smoothly converted into 4-chloroaniline without a significant formation of dangerous intermediates, e.g., hydroxylamine or nitroso compounds were undetectable (Fig. 5); this was because these steps for the conversion of the two intermediates were relatively fast. The p-CNB conversion via hydrogenation with H2 in the absence of a catalyst was below the detection limit. Compared with the Ni/OMC-800 catalyst (Fig. 5a), the Ni/NGr@OMC-800 catalyst (Fig. 5b) modified with nitrogen-doped graphene displayed a higher conversion activity as well as higher selectivity for the hydrogenation of p-CNB to generate the corresponding amine product. Nickel and platinum have been proven to have poor selectivity for the hydrogenation of halogenated nitro compounds. Dehalogenation occurs for both the Ni/OMC-800 catalyst and Ni-NGr@OMC-800 catalyst, but the selectivity of the Ni/OMC-800 catalyst (88.2%) is much lower than that of the Ni-NGr@OMC-800 catalyst (95.6%). To our great delight, when the Co-NGr@OMC-800 catalyst (Fig. 5c) and Fe/NGr@OMC-800 catalyst (Fig. 5d) were used for the hydrogenation of p-CNB, the unwanted by products in p-CNB hydrogenation, which included aniline (produced by dehalogenation), N-ethyl chloroaniline, bis-dichlorophenyldiazene, and dichloroazoxybenzene (caused by coupling reactions), were undetectable in the process of the reaction or after the reaction.41 During the reaction, the –Cl bond remained intact, thereby avoiding the undesired hydrodechlorination. The Ni/NGr@OMC-800 catalyst exhibited higher conversion as well as higher selectivity towards chloronitrobenzene reduction as compared to the Ni/OMC-800 catalyst without modification by the NGr species, avoiding C–Cl bond scissions even at a total conversion, and no further decrease in the yield of chloroaniline products was observed, even upon extending the reaction duration after the complete conversion of chloronitrobenzene. The electron-enriched chemical state of nitrogen-doped graphene species, which was proved by results that the XPS position of the Ni species shifted to higher binding energies by increasing the calcination temperature, was caused by the synergistic effect with transition metal species and nitrogen-doped graphene species, which inhibited dechlorination. However, chemisorption of hydrogen on N-doped graphene inhibited the active hydrogen species from attacking the C–X bond or produced specific absorption toward C–X groups, which therefore inhibited the dehalogenation of halonitrobenzene. In this process, the dihydrogen molecule is formally cleaved into a hydride and a proton. The hydride atom is bound to the metal-based nanoparticle, whereas the proton is attached to the basic N atoms in close proximity to the nanoparticle in the liquid phase.32,42
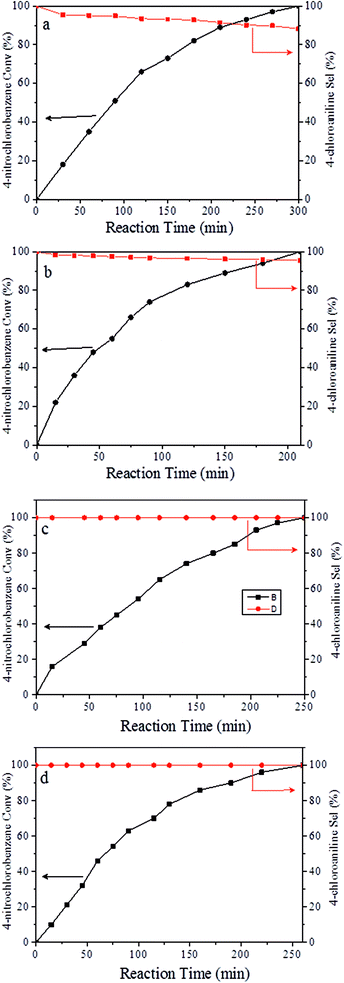 |
| | Fig. 5 Catalytic results for the selective hydrogenation of p-CNB over (a) Ni/OMC-800, (b) Ni/NGr@OM-800, (c) Co/NGr@OM-800, and (d) Co/NGr@OM-800. Reaction conditions: catalyst (20 mg), p-CNB (1 mmol), water (2 mL), and 100 °C. Reducing agent: 5 MPa H2. | |
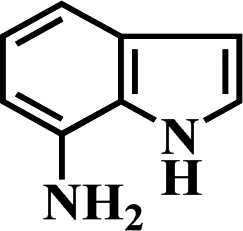
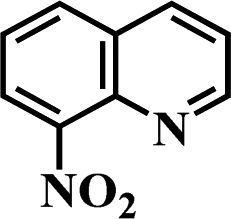
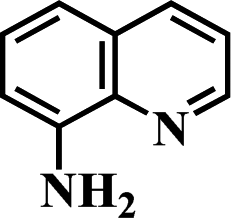
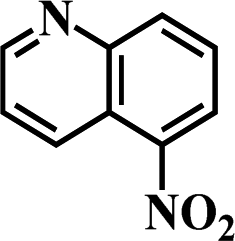
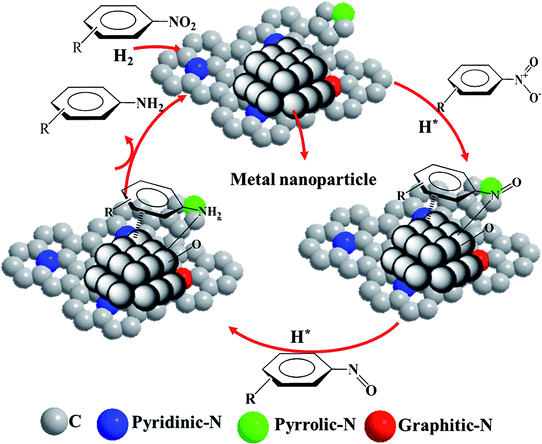
After the successful hydrogenation of the benchmark reaction, we extended Ni/NGr@OMC-800 to various substituted nitroarenes to demonstrate the generality of this reaction under the optimized conditions, and the results are summarized in Table 5. It can be seen that the chemoselective reduction of all the functionalized nitroarenes can be completed at 100 °C in an aqueous solution with perfect chemoselectivity (>99%). The substituted nitrobenzenes, having electron-donor or electron-acceptor groups, such as –CH3, –CF3, CH3O–, –OH, –NH2, –NO2, and so on (Table 5, entries 1–15), were also furnished with excellent yields. The Ni/NGr@OMC-800 catalyst also had a high activity towards catalyzing the reduction of esters, alkynes, and nitriles with hydrogen. This feature is advantageous because it can realize the selective reduction of the nitro-group in the presence of other reducible functionalities (Table 5, entries 16 and 17). Notably, the catalyst could also promote heterocyclic nitroaromatic reduction in yields of >99% without being deactivated by N (Table 5, entries 18 and 19). For example, 8-aminoquinolines, which are important nitrogen-containing heterocycles widely used in bio-active molecules and agrochemicals and have been developed as significant bidentate directing groups or as auxiliary ligands in various kinds of organic reactions, especially in the area of C–H bond functionalization,43 can be efficiently and selectively reduced to the corresponding aniline product in an aqueous solution, where green production is critical for the requirements of product quality. A possible reaction mechanism for the hydrogenation of nitroarenes catalyzed by the M/NGr@OMC-T catalyst is proposed in Fig. 6. The nitro group is first reduced to the nitroso group, followed by reduction by two H* to form the hydroxylamine intermediate; then, hydroxylamine is further reduced to the amino group. During the process, NGr plays a positive role in improving the catalytic activity; this is ascribed to metal nanoparticles being activated by the surrounded NGr as the absorption of reactants on the surface of NGr is a critical step for catalyzing the reactions. This reaction mechanism is analogous to that reported in literature.6b,9b,44
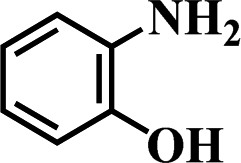
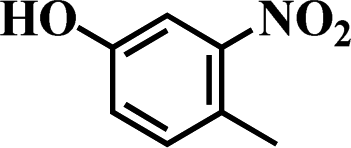
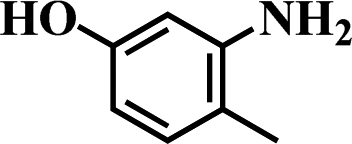
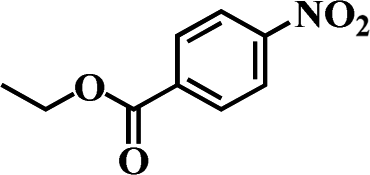
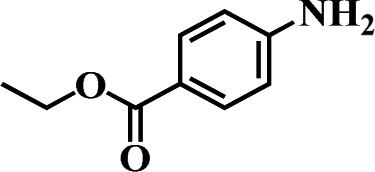
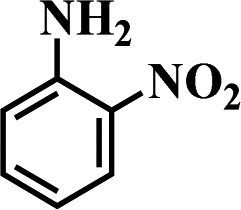
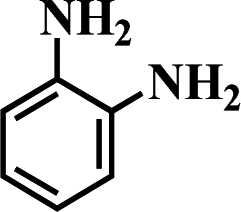
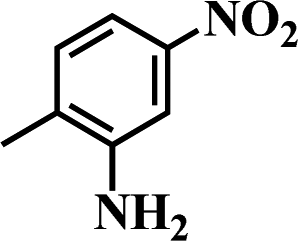
Table 5 Selective hydrogenation of various substituted nitroarenes to the corresponding anilines over Ni/NGr@OMC-800
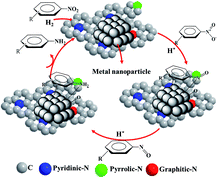 |
| | Fig. 6 A possible reaction mechanism for the hydrogenation of nitroarenes catalyzed by the M/NGr@OMC-T catalyst. | |
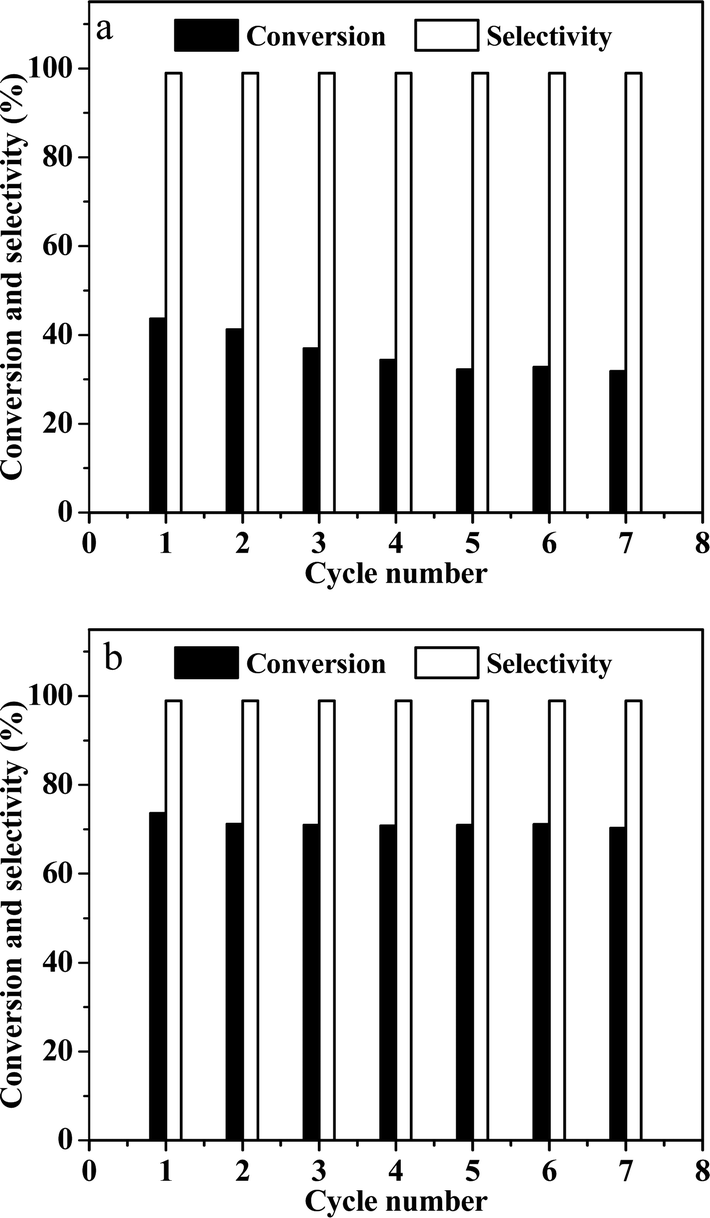
Finally, the recyclability of catalysts was explored (Fig. 7). In the case of the Ni/OMC-800 catalyst (Fig. 7a), a slight decrease in the conversion was detected in the first to fourth runs, whereas for the Ni-NGr@OMC-800 catalyst (Fig. 7b), after an initial slight decrease in the first run, the activity remained almost constant. ICP analysis of the liquid phase after each run revealed no metal leaching for both catalysts (detection limit < 1 ppm), which, coupled with ICP analyses, clearly demonstrated that the catalysts maintained their original composition even after recycling.
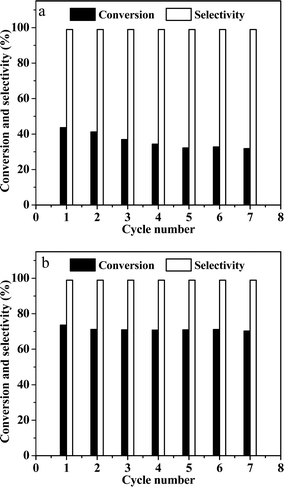 |
| | Fig. 7 (a) Reuse of Ni-OMC-800, 1 h up to the 7th cycle; (b) reuse of Ni-NGr-OMC-800, 1 h up to the 7th cycle. Reaction conditions: catalyst (20 mg), nitrobenzene (1 mmol), water (2 mL), 100 °C, and 5 MPa H2. | |
4. Conclusions
Herein, we have demonstrated a facile chelate-assisted multicomponent co-assembly for the synthesis of ordered mesoporous carbons with embedded and well-dispersed transition metal nanoparticles modified with nitrogen-doped graphene as novel catalysts for the reduction of various functionalized nitroarenes to the corresponding aniline in an aqueous solution. The recycling tests and characterizations demonstrate that the catalysts are stable and can be reused for the hydrogenation of nitroarenes. The excellent catalytic properties of the catalysts can be attributed to the remarkably synergistic effect between the metal nanoparticles and nitrogen-doped graphene. This study can stimulate significant interest in the development of novel non-noble catalysts for other important hydrogenation reactions.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported by the Open Project of State Key Laboratory of Advanced Special Steel of Shanghai University (SKLASS2015-Z052), the National Basic Research Program of China (973 Program, No. 2014CB643403), the National Natural Science Foundation of China (No. 51574164), and the Basic Major Research Program of Science and Technology Commission Foundation of Shanghai (No. 14JC1491400).
Notes and references
- H. U. Blaser, H. Steiner and M. Studer, ChemCatChem, 2009, 1, 210–221 CrossRef CAS.
- N. Ono, The Nitro Group in Organic Synthesis, Wiley-VCH, New York, 2001 Search PubMed.
- R. F. Kolvar and E. F. Armond, US pat., 3975444, 1976.
- R. S. Downing, P. J. Kunkelyer and H. van Bekkum, Catal. Today, 1997, 37, 121–136 CrossRef CAS.
-
(a) K. L. Zhu, M. P. Shaver and S. P. Thomas, Chem. Sci., 2016, 7, 3031–3035 RSC;
(b) S. Fountoulaki, V. Daikopoulou, P. L. Gkizis, I. Tamiolakis, G. S. Armatas and I. N. Lykakis, ACS Catal., 2014, 4, 3504–3511 CrossRef CAS.
-
(a) C. L. Wang, R. Ciganda, L. Salmon, D. Gregurec, J. Irigoyen, S. Moya, J. Ruiz and D. Astruc, Angew. Chem., Int. Ed., 2016, 55, 3091–3095 CrossRef CAS PubMed;
(b) F. Yang, C. Chi, C. X. Wang, Y. Wang and Y. F. Li, Green Chem., 2016, 18, 4254–4262 RSC.
-
(a) I. Sorribes, G. Wienhöfer, C. Vicent, K. Junge, R. Llusar and M. Beller, Angew. Chem., Int. Ed., 2012, 51, 7794–7798 CrossRef CAS PubMed;
(b) R. V. Jagadeesh, K. Natte, H. Junge and M. Beller, ACS Catal., 2015, 5, 1526–1529 CrossRef CAS;
(c) R. V. Jagadeesh, D. Banerjee, P. B. Arockiam, H. Junge, K. Junge, M. M. Pohl, J. Radnik, A. Brückner and M. Beller, Green Chem., 2015, 17, 898–902 RSC;
(d) K. J. Datta, A. K. Rathi, M. B. Gawande, V. Ranc, G. Zoppellaro, R. S. Varma and R. Zboril, ChemCatChem, 2016, 8, 2351–2355 CrossRef CAS;
(e) G. Wienhöfer, I. Sorribes, A. Boddien, F. Westerhaus, K. Junge, H. Junge, R. Llusar and M. Beller, J. Am. Chem. Soc., 2011, 133, 12875–12879 CrossRef PubMed.
- N. García, P. García-García, M. A. Fernández-Rodríguez, R. Rubio, M. R. Pedrosa, F. J. Arnáiz and R. Sanza, Adv. Synth. Catal., 2012, 354, 321–327 CrossRef.
-
(a) M. Tian, X. L. Cui, M. Yuan, J. Yang, J. T. Ma and Z. P. Dong, Green Chem., 2017, 19, 1548–1554 RSC;
(b) Z. K. Zhao, H. L. Yang, Y. Li and X. W. Guo, Green Chem., 2014, 16, 1274–1281 RSC;
(c) C. J. Jiang, Z. Y. Shang and X. H. Liang, ACS Catal., 2015, 5, 4814–4818 CrossRef CAS;
(d) B. Yang, Q. K. Zhang, X. Y. Ma, J. Q. Kang, J. M. Shi and B. Tang, Nano Res., 2016, 9, 1879–1890 CrossRef CAS;
(e) C. F. Zhang, J. M. Lu, M. R. Li, Y. H. Wang, Z. Zhang, H. J. Chen and F. Wang, Green Chem., 2016, 18, 2435–2442 RSC;
(f) H. G. Huang, X. G. Wang, X. Li, C. J. Chen, X. J. Zou, W. Z. Ding and X. G. Lu, Green Chem., 2017, 19, 809–815 RSC.
-
(a) F. A. Westerhaus, R. V. Jagadeesh, G. Wienhöfer, M. M. Pohl, J. Radnik, A. E. Surkus, J. Rabeah, K. Junge, H. Junge, M. Nielsen, A. Brückner and M. Beller, Nat. Chem., 2013, 5, 537–543 CrossRef CAS PubMed;
(b) F. Chen, A. E. Surkus, L. He, M. M. Pohl, J. Radnik, C. Topf, K. Junge and M. Beller, J. Am. Chem. Soc., 2015, 137, 11718–11724 CrossRef CAS PubMed.
- C. Yu, B. Liu and L. Hu, J. Org. Chem., 2001, 66, 919–924 CrossRef CAS PubMed.
- A. Saha and B. Ranu, J. Org. Chem., 2008, 73, 6867–6870 CrossRef CAS PubMed.
- X.-B. Lou, L. He, Y. Qian, Y.-M. Liu, Y. Cao and K.-N. Fan, Adv. Synth. Catal., 2011, 353, 281–286 CrossRef CAS.
-
(a) P. Lara, O. Rivada-Wheelaghan, S. Conejero, R. Poteau, K. Philippot and B. Chaudret, Angew. Chem., Int. Ed., 2011, 50, 12080–12084 CrossRef CAS PubMed;
(b) X. L. Cui, K. Liang, M. Tian, Y. Y. Zhu, J. T. Ma and Z. P. Dong, J. Colloid Interface Sci., 2017, 501, 231–240 CrossRef CAS PubMed;
(c) X. Sun, A. I. Olivos-Suarez, D. Osadchii, M. J. V. Romero, F. Kapteijn and J. Gascon, J. Catal., 2018, 357, 20–28 CrossRef.
-
(a) R. Bronger, T. Dung Le, S. Bastin, J. García-Antón, C. Citadelle, B. Chaudret, P. Lecante, A. Igau and K. Philippot, New J. Chem., 2011, 35, 2653–2660 RSC;
(b) F. W. Zhang, C. Zhao, S. Chen, H. Li, H. Q. Yang and X. M. Zhang, J. Catal., 2017, 348, 212–222 CrossRef CAS;
(c) M. Du, Q. Liu, C. Huang and X. Qiu, RSC Adv., 2017, 7, 35451–35459 RSC.
-
(a) S. Zhang, C. R. Chang, Z. Q. Huang, J. Li, Z. M. Wu, Y. Y. Ma, Z. Y. Zhang, Y. Wang and Y. Q. Qu, J. Am. Chem. Soc., 2016, 138, 2629–2637 CrossRef CAS PubMed;
(b) J. Li, X. Y. Shi, Y. Y. Bi, J. F. Wei and Z. G. Chen, ACS Catal., 2011, 1, 657–664 CrossRef CAS;
(c) G. Vilí, D. Albani, M. Nachtegaal, Z. P. Chen, D. Dontsova, M. Antonietti, N. López and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2015, 54, 11265–11269 CrossRef PubMed;
(d) K. Shen, L. Chen, J. L. Long, W. Zhong and Y. W. Li, ACS Catal., 2015, 5, 5264–5271 CrossRef CAS.
-
(a) F. Cárdenas-Lizana, C. Berguerand, I. Yuranov and L. Kiwi-Minsker, J. Catal., 2013, 301, 103–111 CrossRef;
(b) Z. Y. Sun, Y. F. Zhao, Y. Xie, R. T. Tao, H. Y. Zhang, C. L. Huang and Z. M. Liu, Green Chem., 2010, 12, 1007–1011 RSC;
(c) H. S. Wei, X. Y. Liu, A. Q. Wang, L. L. Zhang, B. T. Qiao, X. F. Yang, Y. Q. Huang, S. Miao, J. Y. Liu and T. Zhang, Nat. Commun., 2014, 5, 5634 CrossRef CAS PubMed;
(d) A. Shukla, R. K. Singha, T. Sasaki and R. Bal, Green Chem., 2015, 17, 785–790 RSC;
(e) M. Makosch, W. I. Lin, V. Bumbálek, J. Sá, J. W. Medlin, K. Hungerbühler and J. A. van Bokhoven, ACS Catal., 2012, 2, 2079–2081 CrossRef CAS;
(f) X. B. Lin, M. Wu, D. Y. Wu, S. Kuga, T. Endoe and Y. Huang, Green Chem., 2011, 13, 283–287 RSC.
-
(a) P. Tomkins, E. Gebauer-Henke, W. Leitner and T. E. Müller, ACS Catal., 2015, 5, 203–209 CrossRef CAS;
(b) F. Q. Leng, I. C. Gerber, P. Lecante, S. Moldovan, M. Girleanu, M. R. Axet and P. Serp, ACS Catal., 2016, 6, 6018–6024 CrossRef CAS;
(c) J. H. Kim, J. H. Park, Y. K. Chung and K. H. Park, Adv. Synth. Catal., 2012, 354, 2412–2418 CrossRef CAS.
-
(a) P. F. Luo, K. L. Xu, R. Zhang, L. Huang, J. Wang, W. H. Xing and J. Huang, Catal. Sci. Technol., 2012, 2, 301–304 RSC;
(b) S. Furukawa, K. Takahashi and T. Komatsu, Chem. Sci., 2016, 7, 4476–4484 RSC;
(c) I. Nakamula, Y. Yamanoi, T. Imaoka, K. Yamamoto and H. Nishihara, Angew. Chem., Int. Ed., 2011, 50, 5830–5833 CrossRef CAS PubMed;
(d) S. F. Cai, H. H. Duan, H. P. Rong, D. S. Wang, L. S. Li, W. He and Y. D. Li, ACS Catal., 2013, 3, 608–612 CrossRef CAS.
-
(a) A. Corma and P. Serna, Science, 2006, 313, 332–334 CrossRef CAS PubMed;
(b) M. Boronat, P. Concepcion, A. Corma, S. Gonzalez, F. Illas and P. Serna, J. Am. Chem. Soc., 2007, 129, 16230–16237 CrossRef CAS PubMed;
(c) A. Grirrane, A. Corma and H. Garcia, Science, 2008, 322, 1661–1664 CrossRef CAS PubMed.
- X. K. Kong, Z. Y. Sun, M. Chen, C. L. Chen and Q. W. Chen, Energy Environ. Sci., 2013, 6, 3260–3266 CAS.
-
(a) Y. J. Gao, D. Ma, C. L. Wang, J. Guan and X. H. Bao, Chem. Commun., 2011, 47, 2432–2434 RSC;
(b) S. C. Wu, G. D. Wen, R. Schlögl and D. S. Su, Phys. Chem. Chem. Phys., 2015, 17, 1567–1571 RSC.
- E. C. Lee, Y. S. Kim, Y. G. Jin and K. Chang, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 073415 CrossRef.
- L. Kong, A. Enders, T. S. Rahman and P. A. Dowben, J. Phys.: Condens. Matter, 2014, 26, 443001 CrossRef PubMed.
- Y. J. Ren, H. S. Wei, G. Z. Yin, L. L. Zhang, A. Q. Wang and T. Zhang, Chem. Commun., 2017, 53, 1969–1972 RSC.
- W. W. Lin, H. Y. Cheng, J. Ming, Y. C. Yu and F. Y. Zhao, J. Catal., 2012, 291, 149–154 CrossRef CAS.
- J. W. Wang, J. W. Liu, N. T. Yang, S. S. Huang, Y. H. Sun and Y. Zhu, Nanoscale, 2016, 8, 3949–3953 RSC.
- F. W. Zhang, C. Zhao, S. A. Chen, H. Li, H. Q. Yang and X. M. Zhang, J. Catal., 2017, 348, 212–222 CrossRef CAS.
- R. V. Jagadeesh, G. Wienhöfer, F. A. Westerhaus, A. E. Surkus, M. M. Pohl, H. Junge, K. Junge and M. Beller, Chem. Commun., 2011, 47, 10972–10974 RSC.
- R. V. Jagadeesh, A. E. Surkus, H. Junge, M. M. Pohl, J. Radnik, J. Rabeah, H. Huan, V. Schünemann, A. Brückner and M. Beller, Science, 2013, 342, 1073–1076 CrossRef CAS PubMed.
- Z. Z. Wei, J. Wang, S. J. Mao, D. F. Su, H. Y. Jin, Y. H. Wang, F. Xu, H. R. Li and Y. Wang, ACS Catal., 2015, 5, 4783–4789 CrossRef CAS.
- D. Formenti, C. Topf, K. Junge, F. Ragaini and M. Beller, Catal. Sci. Technol., 2016, 6, 4473–4477 CAS.
- Z. K. Sun, B. Sun, M. H. Qiao, J. Wei, Q. Yue, C. Wang, Y. H. Deng, S. Kaliaguine and D. Y. Zhao, J. Am. Chem. Soc., 2012, 134, 17653–17660 CrossRef CAS PubMed.
- P. Gao, A. Q. Wang, X. D. Wang and T. Zhang, Chem. Mater., 2008, 20, 1881–1888 CrossRef CAS.
- Y. P. Zhai, Y. Q. Dou, X. X. Liu, S. S. Park, C.-S. Ha and D. Y. Zhao, Carbon, 2011, 49, 545–555 CrossRef CAS.
- F. Jaouen, J. Herranz, M. Lefèvre, J. P. Dodelet, U. I. Kramm, I. Herrmann, P. Bogdanoff, J. Maruyama, T. Nagaoka, A. Garsuch, J. R. Dahn, T. Olson, S. Pylypenko, P. Atanassov and E. A. Ustinov, ACS Appl. Mater. Interfaces, 2009, 1, 1623–1639 CAS.
-
(a) X. Y. Dai, Z. Chen, T. Yao, L. R. Zheng, Y. Lin, W. Liu, H. X. Ju, J. F. Zhu, X. Hong, S. Q. Wei, Y. E. Wu and Y. D. Li, Chem. Commun., 2013, 53, 11568–11571 RSC;
(b) R. F. Nie, H. H. Yang, H. F. Zhang, X. L. Yu, X. H. Lu, D. Zhou and Q. H. Xia, Green Chem., 2017, 19, 3126–3134 RSC.
- B. Coq and F. C. Figueras, Chem. Rev., 1998, 178–180, 1753–1783 Search PubMed; A. M. Doyle, S. K. Shaikhutdinov, S. D. Jackson and H. J. Freund, Angew. Chem., Int. Ed., 2003, 42, 5240–5243 CrossRef CAS PubMed.
- X. Lepró, E. Terrés, Y. Vega-Cantú, F. J. Rodríguez-Macías, H. Muramatsu, Y. A. Kim, T. Hayahsi, M. Endo, R. M. Torres and M. Terrones, Chem. Phys. Lett., 2008, 463, 124–129 CrossRef.
- X. C. Zou, X. C. Huang, A. Goswami, R. Silva, B. R. Sathe, E. Mikmekova and T. Asefa, Angew. Chem., Int. Ed., 2014, 53, 4372–4376 CrossRef CAS PubMed.
- D. He, H. Shi, Y. Wu and B.-Q. Xu, Green Chem., 2007, 9, 849–851 RSC.
- P. R. Chen, F. K. Yang, A. Kostka and W. Xia, ACS Catal., 2014, 4, 1478–1486 CrossRef CAS.
-
(a) H. Chen, P. H. Li, M. Wang and L. Wang, Org. Lett., 2016, 18, 4794–4797 CrossRef CAS PubMed;
(b) C. J. Whiteoak, O. Planas, A. Company and X. Ribasa, Adv. Synth. Catal., 2016, 358, 1679–1688 CrossRef CAS;
(c) M. Corbet and F. De Campo, Angew. Chem., Int. Ed., 2013, 52, 9896–9898 CrossRef CAS PubMed.
- J. Xia, G. He, L. Zhang, X. Sun and X. Wang, Appl. Catal., B, 2016, 180, 408–415 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Catalyst characterization of Co/NGr@OMC-800 and Fe/NGr@OMC-800 catalysts; XRD, TGA, Raman spectrum, N2 sorption, TEM and XPS spectra. See DOI: 10.1039/c8ra00761f |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Xueguang Wang
a,
Xueguang Wang