
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFates of hemicellulose, lignin and cellulose in concentrated phosphoric acid with hydrogen peroxide (PHP) pretreatment†
Qing Wang ab,
Dong Tianab,
Jinguang Hucd,
Fei Shen*ab,
Gang Yangab,
Yanzong Zhangb,
Shihuai Dengab,
Jing Zhangab,
Yongmei Zengab and
Yaodong Huab
ab,
Dong Tianab,
Jinguang Hucd,
Fei Shen*ab,
Gang Yangab,
Yanzong Zhangb,
Shihuai Dengab,
Jing Zhangab,
Yongmei Zengab and
Yaodong Huab
aInstitute of Ecological and Environmental Sciences, Sichuan Agricultural University, Chengdu, Sichuan 611130, P. R. China. E-mail: fishen@sicau.edu.cn
bRural Environment Protection Engineering & Technology Centre of Sichuan Province, Sichuan Agricultural University, Chengdu, Sichuan 611130, P. R. China
cBiomolecular Materials, Department of Bioproducts and Biosystems, School of Chemical Engineering, Aalto University, PO Box 16300, FIN-00076 Aalto, Finland
dForest Products Biotechnology, Department of Wood Science, The University of British Columbia, Vancouver V6T 1Z4, BC, Canada
First published on 3rd April 2018
Abstract
Xylan, de-alkaline lignin and microcrystalline cellulose were employed as representative models of hemicellulose, lignin and cellulose in lignocellulosic biomass. These three model compounds, together with the real-world biomass, wheat straw were pretreated using the newly developed PHP pretreatment (concentrated phosphoric acid plus hydrogen peroxide) to better understand the structural changes of the recovered solid and chemical fractions in the liquid. Results showed that almost all xylan and higher than 70% lignin were removed from wheat straw, and more than 90% cellulose was recovered in the solid fraction. The pretreated model xylan recovered via ethanol-precipitation still maintained its original structural features. The degree of polymerization of soluble xylooligosaccharides in liquid was reduced, resulting in the increase of monomeric xylose release. Further xylose oxidization via the path of 2-furancarboxylic acid → 2(5H)-furanone → acrylic acid → formic acid was mainly responsible for xylan degradation. The chemical structure of de-alkaline lignin was altered significantly by PHP pretreatment. Basic guaiacyl units of lignin were depolymerized, and aromatic rings and side aliphatic chains were partially decomposed. Ring-opening reactions of the aromatics and cleavage of C–O–C linkages were two crucial paths to lignin oxidative degradation. In contrast to lignin, no apparent changes occurred on microcrystalline cellulose. The reason was likely that acid-depolymerization and oxidative degradation of cellulose were greatly prevented by the formed cellulose phosphate.
Introduction
Lignocellulosic biomass, including agricultural, herbaceous, and woody residues and typical energy crops, exhibits a great promise to provide sufficient sustainable resources to satisfy global energy demands and thus weaken the heavy dependence on petroleum-based liquid fuels.1 Realistic and scalable technologies are particularly needed to capture the energy in lignocellulosic biomass as aromatic, hydrocarbon, and alcohol transportation fuel because the transportation sector currently consumes approximately two-thirds of the world's petroleum production.2 As one of the most promising and eco-friendly alternatives to fossil fuels, ethanol produced from lignocellulosic biomass has been widely accepted for application owing to the low-cost feedstocks, abundant resources, no-competition with food, and limited net greenhouse gas emissions.3,4Lignocelluloses are mainly composed of cellulose, hemicellulose, and lignin with an extremely complicated structure, which is recalcitrant to microbial or enzymatic breakdown.5 Technically, ethanol production from lignocellulosic biomass by biological route contains three key steps, i.e., pretreatment, enzymatic saccharification, and fermentation.6 In these steps, converting such biomass into fermentable sugars via enzymatic saccharification is attractive compared with chemical hydrolysis due to its low energy consumption and less pollution.7 However, the biomass recalcitrance, including the steric hindrance of hemicellulose, the bonding effect of lignin and the crystalline structure of cellulose, has seriously restricted the cellulose accessibility to hydrolytic enzymes.8 In order to achieve an efficient glucose conversion from cellulose via enzymatic hydrolysis, pretreating lignocellulosic biomass is critical to overcome its structural recalcitrance.9 Thus, a suitable pretreatment method should be designed with at least three functionalities of breakdown of the cross-linked matrix, disruption of the hydrogen bonding network in crystalline cellulose, and increase of the porosity and available surface area of cellulose for subsequent enzymatic hydrolysis.10 Meanwhile, cellulose recovery, enzymatic hydrolysis efficiency, conversion at high solid consistency, possible inhibitors, by-products recovery and potential costs on energy and chemical input should be considered in practice.11
Up to now, various approaches, such as physical pretreatment (milling, extrusion, microwave, freeze), chemical pretreatment (acid, alkaline, ionic liquid, organosolv, ozonolysis), physico-chemical pretreatment (steam explosion, ammonia fiber explosion, CO2 explosion, liquid hot water, wet oxidation), and biological pretreatment, have been widely investigated on various feedstocks.9 As a novel pretreatment method, the concentrated phosphoric acid plus hydrogen peroxide (PHP) was firstly developed in previous investigations. It could accept a wide range of feedstock types (including softwoods, hardwoods, various agricultural stalks, bamboo, garden waste and their mixtures) with almost complete hemicellulose removal and 70–100% lignin removal.6 More than 90% cellulose can be recovered in the resulting solid with higher than 90% enzymatic hydrolysis efficiency.12 Especially, PHP pretreatment can be performed at mild conditions (40 °C, 2.0 h, and H3PO4/H2O2 loading of 70.2%/5.2%). Taking the pretreatment for wheat straw as an example, 299 g glucose could be harvested from 1.0 kg biomass (equivalent to 946.2 mg g−1 cellulose in wheat straw) via enzymatic hydrolysis.13 Moreover, 72.9% cellulose–glucose conversion was achieved with a low enzyme loading of 20 mg protei per /g cellulose (Cellic CTec2) at a high substrate consistency of 20%. Consequently, 112 g ethanol was produced from 1.0 kg wheat straw (dry basis) with an ethanol titer of 71.2 g L−1.14 Besides, a process for recycling phosphoric acid has been developed successfully in our previous reports,15,16 and more than 80% phosphoric acid can be recovered for reuse in the PHP pretreatment, therefore the cost of chemical input was greatly reduced. Based on the achieved results, the mechanisms of PHP pretreatment behind the efficient deconstruction of lignocellulose deserve to investigate systematically to better guide its application at large scale.
Although the phosphoric acid involved pretreatments were investigated widely, most of them employed diluted phosphoric acid as a catalyst in steam or hydrothermal pretreatment to accelerate the hemicellulose solubilization via acid depolymerization.15,16 It has also been reported when the concentrated phosphoric acid was solely used for pretreating lignocellulosic biomass at mild conditions, a large portion of hemicellulose could be removed through a dissolution manner.17,18 However, these two pretreatment methods involved with phosphoric acid could not work well on lignin removal. As for the pretreatment with H2O2 involvement, it has been proved that only H2O2 addition couldn't breakdown the recalcitrant structural of lignocellulosic biomass.6 AHP (alkaline hydrogen peroxide) and HPAC (hydrogen peroxide–acetic acid) have been successfully applied with the main goal of enhance the enzymatic hydrolysis through the efficient delignification process, due to the generated superoxide and hydroxyl radicals,19 while lignin removal via HPAC was likely related to the formed peracetic acid.20 By contrast, the high efficiency on breaking the recalcitrance for PHP pretreatment may be potentially attributed to the cooperative effect of polysaccharide disruption (by concentrated phosphoric acid) and acid-catalyzed lignin depolymerization/oxidation with H2O2. However, the mechanistic insights of PHP pretreatment are not clear, especially when compared with those of AHP and HPAC that already well investigated.
To better understand the possible reactions during PHP pretreatment, three model compounds, including microcrystalline cellulose (MCC), xylan, and de-alkaline lignin, were selected to represent biomass cellulose, hemicellulose and lignin, respectively. They were pretreated using PHP pretreatment with the comparison of wheat straw. The characteristic changes of recovered solid fraction of the corresponding model compound, and the main composition of dissolved fraction in PHP solution were investigated. The possible fates of hemicellulose, lignin and cellulose during PHP pretreatment were further suggested consequently.
Materials and methods
Model compounds and wheat straw
MCC from cotton linters (determined purity of 98.4%, product number: 435236) and xylan from Beechwood (determined purity of 93.2%, product number: V900513) were purchased from Sigma-Aldrich Ltd., in China. The de-alkaline lignin (no residual xylose and glucose can be detected, product number: 301113) was purchased from J&K Scientific Ltd., in China. Wheat straw was collected from the farm of Sichuan Agricultural University, Chengdu, China. The collected wheat straw was air-dried and milled through a 40-mesh sieve prior to pretreatment. Its main chemical composition was tested according to the standard method Tappi Standard T222 om-88,6 The content of cellulose (represented by glucan), hemicellulose (estimated downward by considering only the xylan content), lignin, acid-insoluble lignin, acid-soluble lignin, ethanol-extractives and ash was 31.6%, 16.2%, 18.5%, 2.7%, 13.3%, and 3.9%, respectively.Pretreatment
2.0 g model compounds were mixed with 20.0 g PHP solution (the fraction of the concentrated H3PO4 and H2O2 was 80% and 1.77%, respectively) in 50 mL glass bottles. Each bottle was sealed with a rubber stopper and hooped using an aluminium seal. The sealed bottles were shaken in an orbital shaker at 50 °C with 100 rpm for 1.0 h, 3.0 h and 5.0 h, respectively. The pretreatment was performed with 4 repetitions, in which 2 repetitions were employed to recover the solids, while the other 2 repetitions were employed to collect the liquid. At the selected pretreatment time, the liquid samples were extracted with diethyl ether and collected for composition analysis. The pretreatment was terminated via rapid dilution with distilled water or ethanol (99%, v/v), in which distilled water was employed to recover cellulose and lignin while ethanol was specially employed for recovering xylan. The recovered solids were further used for structural characterization. 8.0 g wheat straw was pretreated with 80.0 g PHP mixture in a 250 mL serum bottle. The pretreatment conditions were the same with those for model compounds. The pretreatment was quenched by rapid dilution with 450 mL distilled water. The pretreated wheat straw was washed using a centrifuge several times until the pH of the washes was higher than 5.5. The washed substrates were stored in a freezer (<−18 °C) for composition analysis and further calculation of cellulose recovery, xylan and lignin removal according to the method in reference.12,20Characterization of recovered solid fractions
The recovered solids from xylan, lignin, and cellulose pretreatment were analyzed by a FT-IR (Fourier transform infrared spectrometer) (SpectrumTwo, PerkinElmer, USA) to investigate the changes of chemical functional groups or bonds after pretreatment. The corresponding recovered solid was mixed thoroughly to guarantee the homogeneity. 4 mg recovered solid and 400 mg KBr were mixed and homogenized using an agate mortar, and a KBr-coated tablet was prepared via pressing them at 200 kg cm−2 for 5 min. IR spectra were recorded by a DTGS detector (deuterated triglycine sulfate detector) at 4 cm−1 resolution in range of 400–4000 cm−1, and 32 scans were taken per sample. An X-ray diffractometer (XRD) (Ultima IV with D/teX Ultra, Rigaku, Japan) was employed on the recovered solid from cellulose pretreatment to clarify the changes of degree of crystallinity. XRD patterns of the samples were recorded with Ni-filtered Cu-Kα radiation (λ = 0.1541 nm) generated at 40 kV and 40 mA with a scan speed of 0.02° s−1 ranging from 10° to 45°. Thermogravimetry analysis (TGA) (STA449, Netzsch, Germany) was performed on the recovered solid from lignin pretreatment to check the alteration of thermal stability, which can potentially reflect the deconstruction of lignin. The analysis was carried out in the N2 atmosphere with heating rate of 10°C min−1 in the temperature range of 20 °C to 900 °C.Composition in liquid fraction after pretreatment
Chemical composition of the liquid fraction after pretreatment was identified using a gas chromatograph-mass spectrometer (GC/MS) (GC: Clarus 680, MS: Clarus SQ 8 T, PerkinElmer, USA). Prior to the determination, the liquid samples were extracted using 20 mL diethyl ether, and 2.0 mL the extracted samples were filtered by 0.22 μm membrane for the analysis of GC/MS. The injector temperature of GC was set at 250 °C, and the split ratio was controlled as 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 with helium (≥99.999%) as carrier gas. A capillary column with nitroterephthalic acid modified polyethylene glycol (DB-FFAP, 30.0 m × 250 μm, Agilent, USA) were selected, and the temperature for column oven was programmed to start at 70 °C for 3 min, ramp 20°C min−1 to 240 °C, and hold for 40 min. The solvent delay for MS was 4.00 min while the source temperature was set at 240 °C and the electron energy was 70 eV. The potential chemical compounds in the samples were identified by TurboMassNIST 2011 Libraries. Actually, some standard substrates of the identified compounds can't be collected commercially, thus, the relative area (%), rather than accurate quantitation, was employed to check the changes of identified compounds with the increased pretreatment time.21–23
:1 with helium (≥99.999%) as carrier gas. A capillary column with nitroterephthalic acid modified polyethylene glycol (DB-FFAP, 30.0 m × 250 μm, Agilent, USA) were selected, and the temperature for column oven was programmed to start at 70 °C for 3 min, ramp 20°C min−1 to 240 °C, and hold for 40 min. The solvent delay for MS was 4.00 min while the source temperature was set at 240 °C and the electron energy was 70 eV. The potential chemical compounds in the samples were identified by TurboMassNIST 2011 Libraries. Actually, some standard substrates of the identified compounds can't be collected commercially, thus, the relative area (%), rather than accurate quantitation, was employed to check the changes of identified compounds with the increased pretreatment time.21–23
High-performance liquid chromatography (HPLC) (Flexar, PerkinElmer, USA) was employed to investigate the changes of the degree of polymerization (DP) of the liquid fraction after pretreatment of xylan. The liquid fraction was diluted properly, and 100 μL sample was injected into the HPLC. The xylooligosaccharides were separated by a sugar column (SH1011, Shodex, Japan) at 60 °C using the mobile phase of 0.05 mol L−1 H2SO4 with flow rate of 0.8 mL min−1. The separated sugars were detected using a refractive index detector at 50 °C.
Results and discussion
Fate of xylan during PHP pretreatment
As presented in Fig. 1, almost no xylan was recovered from wheat straw when PHP pretreatment was conducted within 1.0 h, suggesting the rapid and complete xylan removal occurred on wheat straw. This result was in accordance with the obtained results in previous work of PHP pretreatment on wheat straw.8,14 After the model xylan reacted with PHP, similarly, no xylan could be recovered in solid as the distilled water was employed for the recovery process, which may relate to the water solubility of model xylan. According to the above results, it could be deduced that the xylan in wheat straw underwent a transformation from insoluble state to soluble state, in which the concentrated H3PO4 may be responsible for hemicellulose solubilization by breaking up orderly hydrogen bonds among sugar chains, and hydrolyzing the natural xylan into low DP fragments, for example, xylooligosaccharides.17 Xylan as the dominant (62–72%) hemicellulose component in wheat straw, its complete removal potentially suggested breakdown of hemicellulose structure by PHP pretreatment.24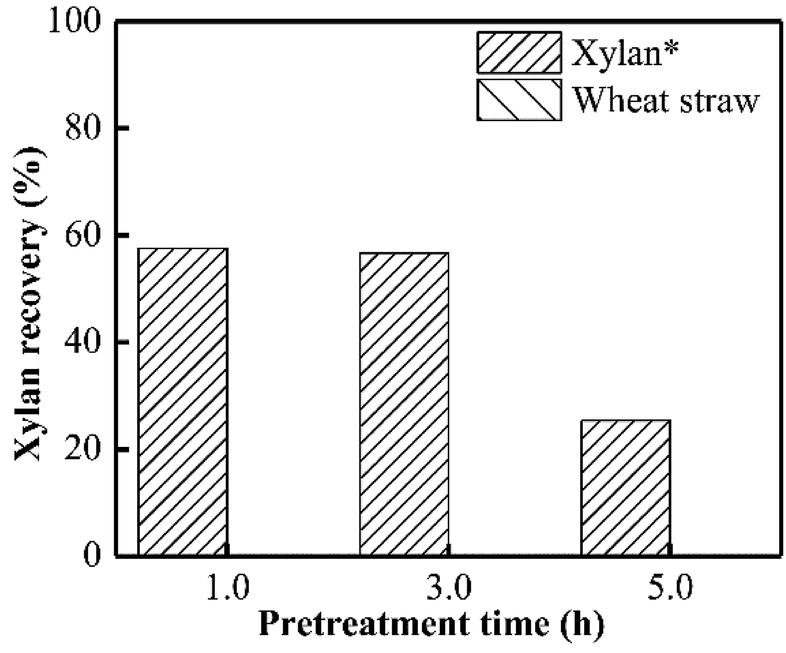 | ||
| Fig. 1 Xylan recovery after PHP-pretreatment on model xylan and wheat straw. *No xylan could be recovered from wheat straw and the model xylan once the distilled water was employed for solid recovery. Herein, the presented solid fraction was recovered via ethanol precipitation for the model xylan so that further investigations on the solid structure can be carried out. | ||
Xylan is usually insoluble in ethanol and thereby can be precipitated, which has been widely used for hemicellulose recovery.25 Thus, as ethanol was employed here to recover the xylan, approximately 56.7% could be precipitated and separated as the pretreatment time was 3.0 h. Prolonging the pretreatment time to 5.0 h significantly decreased the xylan recovery to 25.3% (see Fig. 1).
FT-IR spectra of the recovered solid at 5.0 h was investigated and compared with the original model xylan (see Fig. 2). Overall, FT-IR spectrum of the recovered solid at 5.0 h was almost similar with that of the model xylan, suggesting the recovered solid keep the basic structure of xylan after pretreatment. In addition, the adsorption band appeared at 1046 cm−1 was attributed to C–O, C–C stretching or C–OH bonding, and 3439 cm−1 was attributed to ether linkage. These are related to the featured linkages of xylose unit in xylan.26–28 Their adsorption intensities in the recovered solid became weaker compared with the original model xylan, suggesting the linkages of xylose unit in xylan were cleaved to some extent, as well as the potential DP reduction. Besides, the adsorption band at 896 cm−1 generally relates to β-D-pyran-type xylose frequency vibration and ring frequency vibration,29 and the weakened vibration after PHP pretreatment indicated that the potential deconstruction happened on the basic unit of xylan structure, which may relate to the formed oxidability in PHP system.
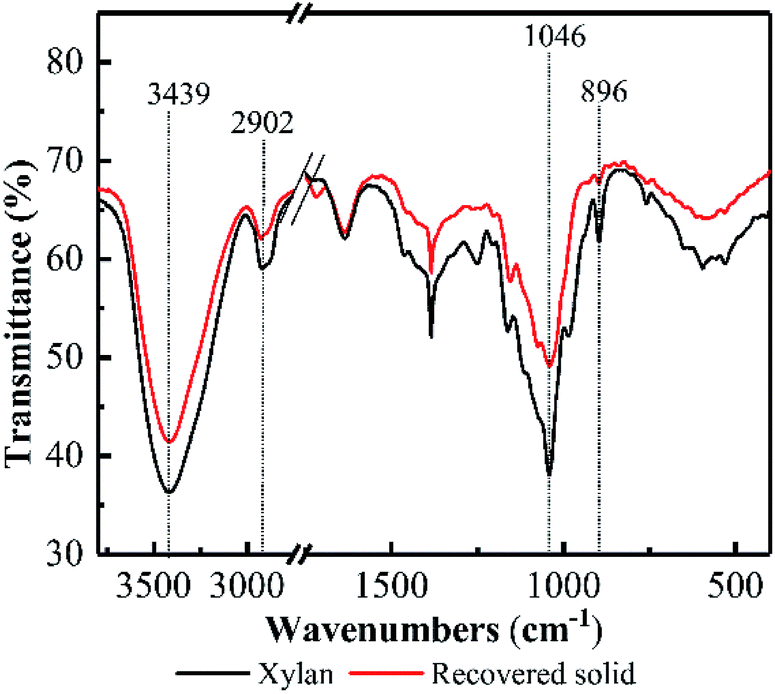 | ||
| Fig. 2 FT-IR spectra of the model xylan before/after PHP pretreatment. | ||
To further identify the possible products after PHP pretreatment on model xylan, the distribution of xylooligosaccharides in liquid fraction were detected with HPLC (Fig. S1†). According to the separation principle of the employed column (SH1011, Shodex, Japan), the signal of xylooligosaccharides with the higher DP (or molecular weight) will appear at the shorter retention time. 4 typical peaks, that can be identified as xylooligosaccharides, appeared before 11.5 min, and the peak at 12.0 min was identified as the monomeric sugar of xylose. It could be easily found that the relatively higher DP xylooligosaccharides (the former 3 peaks) were in the relatively lower levels, and their distribution percentage was decreased as the pretreatment time was prolonged from 1.0 h to 5.0 h. By contrast, the content of xylose obviously increased as the pretreatment time prolonged. These results indicated that the xylan depolymerization occurred during the PHP pretreatment. Besides, the peaks appeared at 15–16 min could be identified as formic acid. The increased peak area implied the degradation of xylan potentially happened during the pretreatment, which was consistent with the above FT-IR result that the vibration at 896 cm−1 became much weaker.
As presented in Fig. 1, there was 40–70% xylan retained in PHP solution, in which part of xylan may be degraded via the formed oxidation. To propose the main chemical path for the xylan degradation in PHP pretreatment, the main chemical compounds in the liquid fraction were identified using GC-MS (see Table 1).
| No. | Identified compounds | Formula | Area (%) | ||
|---|---|---|---|---|---|
| 1.0 h | 3.0 h | 5.0 h | |||
| 1 | Acetic acid | C2H4O2 | 9.3 | 7.1 | 7.5 |
| 2 | Furfural | C5H4O2 | 56.8 | 31.7 | 33.0 |
| 3 | Formic acid | CH2O2 | 15.1 | 53.4 | 52.4 |
| 4 | 5-Methyl-furfural | C6H6O2 | 1.9 | 0.3 | 0.4 |
| 5 | Acrylic acid | C3H4O2 | 2.0 | 1.0 | 1.4 |
| 6 | 2(5H)-Furanone | C4H4O2 | 2.9 | 0.8 | 1.0 |
| 7 | Glutaconic anhydride | C5H4O3 | 6.8 | 3.9 | 3.2 |
| 8 | Levulinic acid | C5H8O3 | 4.0 | 1.1 | 0.5 |
| 9 | 2-Furancarboxylic acid | C5H4O3 | 1.1 | 0.9 | 0.7 |
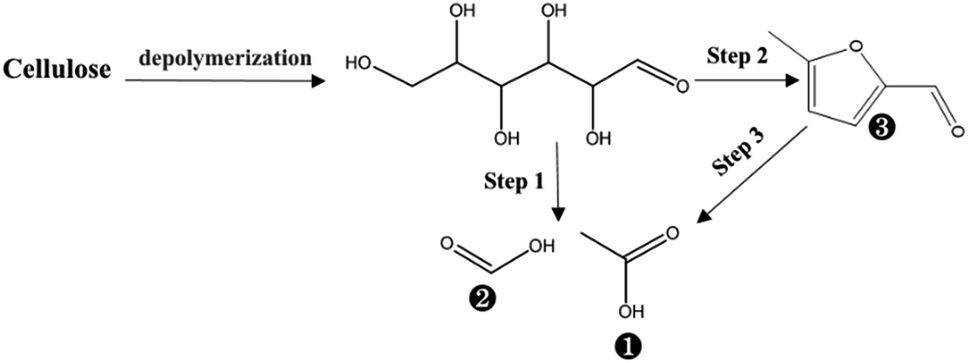
Overall, 9 compounds were identified via the library of GC/MS (NIST 2011), in which the 5-methyl-furfural and the levulinic acid can be regarded as the derived products from the C6 sugar impurity in xylan.30 An identified compound with the highest abundance was furfural in the liquid fraction, and its abundance decreased with prolonging PHP pretreatment time, implying the potential dehydration happened on the derived xylose from xylan depolymerization.31,32 The relatively higher abundance were acetic acid and formic acid, moreover, the increase of their abundances could be observed during the PHP pretreatment, further suggesting that the oxidation in PHP was mainly responsible for the degradation of furfural. In PHP pretreatment, the performance of oxidation mainly resulted from H2O2, and the produced free radicals of O˙ and OH˙.33,34 Meanwhile, stronger oxidizers of peracetic acid and performic acid were substantially detected during PHP pretreatment on lignocellulosic biomass (data not shown). The peroxyacids can be hydroxylated to form electrophilic hydroxonium ion (HO+) with special reactivities on lignin, however, carbohydrates could not offer the reactive sites for electrophilic attack of HO+, thereby no degradation was observed on them.35,36 Thus, H2O2 itself and the derived free radicals of O˙, and OH˙ were mainly responsible for the degradation xylose in PHP pretreatment, rather than the formed HO+. According to the signal abundances at 1.0 h, 3.0 h and 5.0 h, the possible degradation path for the derived xylose from the xylan could be proposed as the following steps.
Xylose produced by acid-hydrolysis of xylan was dehydrated to furfural according to the putative mechanism from FT-IR and HPLC results (Step 1).37 As for the produced furfural, it could generate 2-furancarboxylic acid via oxidation of O˙ (Step 2), followed by oxidation to 2(5H)-furanone via H2O2 or 2OH˙ (Step 3).38 The produced 2(5H)-furanone can be potentially converted into acrylic acid and formic acid via acid hydrolysis on the furan ring and further oxidation of O˙ (Step 4). As for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C in the acrylic acid, it can be further oxidized to form acetic acid and formic acid as the end-products (Step 5). Besides, the furfural degradation could be achieved by the other path based on the analysis of products in PHP reaction. The furan ring of furfural could be opened via acid hydrolysis and further oxidation to open the CC by OH˙ to glutaconic acid (in fact, it was detected as glutaconic anhydride because of the easy dehydration of glutaconic acid at determination conditions of GC) (Step 6 and Step 7). Acrylic acid and acetic acid can be formed via oxidation of glutaconic acid (Step 8). The achieved acrylic acid can also be divided into 2 end-products of acetic acid and formic acid by the oxidization of Step 5.
C in the acrylic acid, it can be further oxidized to form acetic acid and formic acid as the end-products (Step 5). Besides, the furfural degradation could be achieved by the other path based on the analysis of products in PHP reaction. The furan ring of furfural could be opened via acid hydrolysis and further oxidation to open the CC by OH˙ to glutaconic acid (in fact, it was detected as glutaconic anhydride because of the easy dehydration of glutaconic acid at determination conditions of GC) (Step 6 and Step 7). Acrylic acid and acetic acid can be formed via oxidation of glutaconic acid (Step 8). The achieved acrylic acid can also be divided into 2 end-products of acetic acid and formic acid by the oxidization of Step 5.
Based on the proposed steps, the degradation of xylan in PHP was summarized in Fig. 3. The relatively higher formic acid abundance in liquid fraction comparing with acetic acid meant that the oxidation path of xylose → 2-furancarboxylic acid → 2(5H)-furanone → acrylic acid → formic acid would be mainly responsible for xylan degradation. As reported in previous work,13 the optimized pretreatment time for lignocellulosic biomass (wheat straw) was 2.0 h, which implied more than 60% xylan can be recovered in solid for achieving the high-valuable products of xylooligosaccharides. In addition, the xylose in liquid fraction at 1.0 h, 3.0 h, and 5.0 h occupied approximately 57.6%, 56.7%, and 25.3% respectively, which meant about 42.4–74.7% xylan was degraded. The types and contents of by-products suggested that PHP pretreatment was promising for producing high-value platform chemicals (i.e., furfural) from lignocellulose, which deserves further investigation.
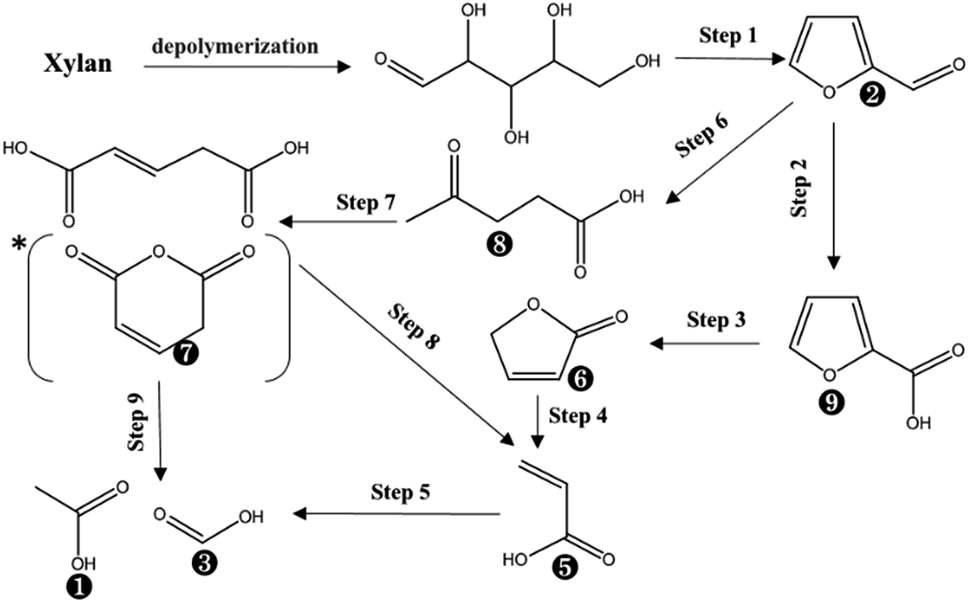 | ||
| Fig. 3 The suggested chemical degradation path of the xylan during PHP pretreatment; *it was detected as glutaconic anhydride for easy dehydration glutaconic acid at determination conditions of GC; the labelled numbers near the chemical structure formula are consistent with the listed no. in Table 1. | ||
Fate of lignin during PHP pretreatment
According to Fig. 4, approximately 70% lignin in wheat straw could be removed when PHP pretreatment was performed for 1.0 h, and only 20% lignin can be recovered after 5.0 h. This result was consistent with the published work on wheat straw with PHP pretreatment.12 By contrast, the de-alkaline lignin could be recovered by 6.5% as PHP pretreatment was in 1.0 h, and prolonging the pretreatment time to longer than 3.0 h greatly reduced the lignin yield to zero. The lower lignin recovery value for de-alkaline lignin sample was mainly attributed to its lower structural stability. Obviously, the integrity of lignin structure was broken during the preparation process for de-alkaline lignin. For example, some parts of lignin side chain and β-O-4 linkages were lost after alkali fractionation, as well as the complete breaking of lignin–carbohydrate complexes and native ester bonds.39 In addition, it also could be directly observed the pretreated de-alkaline lignin was easily separated by filtration with longer pretreatment time comparing with that of 1.0 h, suggesting the de-alkaline was dissolved by PHP solution. As reported in previous work, the lignin in lignocellulosic biomass could not be efficiently removed (lower than 10%) when only concentrated phosphoric acid or hydrogen peroxide was employed.6,17 As aforementioned, the electrophilic hydroxonium ion (HO+) could be hydroxylated from peroxyacids, and can specially oxidize lignin, which was responsible for lignin removal during PHP pretreatment.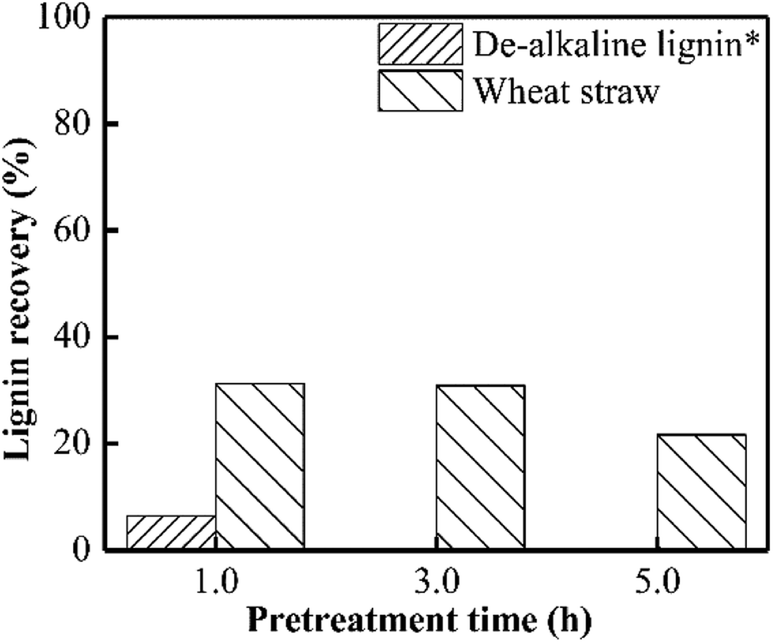 | ||
| Fig. 4 Lignin recovery after PHP pretreatment on model de-alkaline lignin and wheat straw; *no solid could be recovered after PHP pretreatment on model de-alkaline lignin for 3.0 h or 5.0 h. | ||
To further elucidate what really happened on lignin during PHP pretreatment, TGA was performed on the de-alkaline lignin and the recovered solid. DTG curves reflect the weight loss rate of substrate with respect to the temperature of thermal degradation, which greatly depends on the structure and composition of the investigated substrate. Lignin molecular structure is composed of mostly aromatic rings with various branches. These complex chemical structures resulted in a wide range of degradation temperature from 100 to 800 °C.40 As indicated in Fig. 5, the initial weight loss of lignin degradation occurred at 25–125 °C due to evaporation of the absorbed water. Obviously, the weight loss rate for the recovered solid was higher compared with the de-alkaline lignin. This result suggested the hydrophobicity of lignin was decreased greatly after pretreatment, consequently, the hydrophilicity of the recovered solid could be potentially enhanced, which may be potentially attributed to linkage breaking of monomers in lignin, such as –COOH, –OH, –CO–, via the oxidation in PHP pretreatment. The relatively high weight loss appeared in the temperature range of 160–500 °C, in which the degradation of methoxide and hydroxy carboxyl groups on the lignin was an important contributor.41 The maximum degradation temperature of the de-alkaline lignin was estimated at 336 °C, and it was postponed to 347 °C for the recovered solid. Moreover, the weight loss rate of recovered solid was lower than that of the de-alkaline lignin, which implied CH3O–, –OH and –COOH groups on lignin were reduced due to the oxidation in PHP pretreatment. Besides, an obvious weight loss can be observed in the temperature range of 680–790 °C, which was attributed to the decomposition of the aromatic and ether bonds among phenylpropane units.42 A distinct weight loss appeared in the de-alkaline lignin, however, it was almost not observed in the recovered solid, indicating a noteworthy reduction of aromatic compounds. It appears that ring-opening reaction among aromatics or formation of phenolic compounds with small molecular weight was responsible for the reduction of aromatic compounds in lignin. In addition, almost no solid was recovered at 3.0 h and 5.0 h, which could further support the above conclusion.
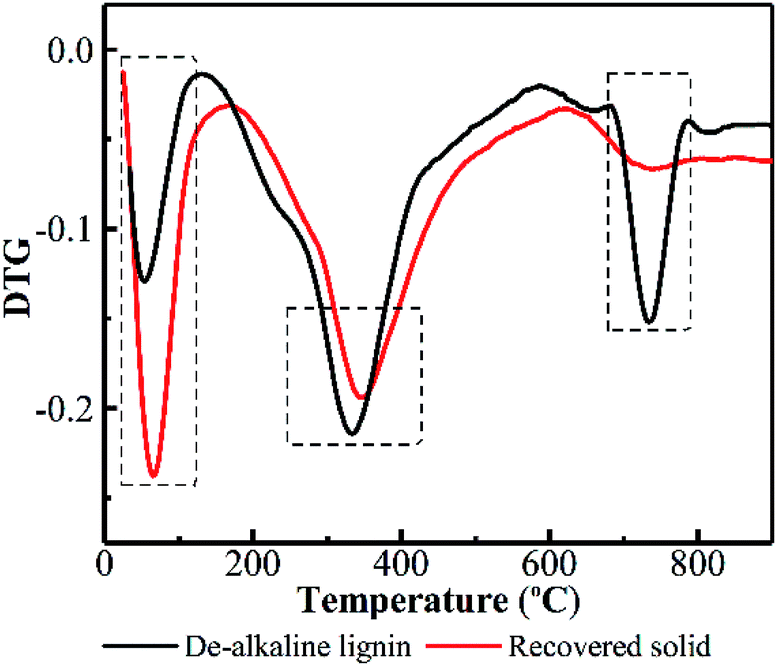 | ||
| Fig. 5 DTG curve of the de-alkaline lignin before/after PHP pretreatment. | ||
Based on the TGA results, the decreases of hydroxide radical, carboxyl, carbonyl and aromatic compounds content in lignin could be deduced, as well as the oxidization of aliphatic chains of lignin. FT-IR was employed to further elucidate the response of PHP pretreatment to lignin degradation (see Fig. 6). The typical spectral bands of the stretching vibrations for phenolic OH, aliphatic OH, and C–H bonds were observed in de-alkaline lignin, which was consistent with the reported references.43 Overall, the spectral bands appeared in the recovered solid were weakened compared with the de-alkaline lignin. In detail, the absorptions at 1268 cm−1 and 815 cm−1 in the de-alkaline lignin were attributed to C–O stretching and C–H out-of-plane vibrations in positions 2, 5 and 6 of guaiacyl units, respectively.44 However, they almost disappeared in the recovered solid, suggesting the original structure of guaiacyl units in lignin was destroyed after PHP pretreatment. The spectral bands at 1600 cm−1, 1512 cm−1, and 1424 cm−1 relates to the structure of aromatic rings,44 and the weakened absorption intensities in the recovered solid implied the structures related to aromatic rings in lignin were loosen to some extent by PHP pretreatment, however, the aromatic compounds still existed in the recovered solid because of the obvious absorption at 1600 cm−1. The abundant branches in lignin, such as the C–O deformation at Cα and aliphatic ether, appeared at 1033 cm−1,45 and the vanished absorption here in the recovered solid proved the aliphatic chains at the side of the basic lignin units were also decomposed. Besides, C–H stretching of methyl and methylene groups in aliphatic chain and aromatic methoxyl groups were mainly responsible for the bands at 2841–2847 cm−1.43 Moreover, C–H deformations and stretching sourced from the aromatic groups and the aliphatic ethers, respectively, at 1125 cm−1 were also observed in the de-alkaline lignin.43,46 By contrast, the absorption intensity of 1125 cm−1 almost could not be detected in the recovered solid, and the greatly reduced transmittance at 2841–2847 cm−1 supported the deductions of decomposing the aliphatic chains and loosening aromatic rings by PHP pretreatment. Additionally, these results also well responded to the almost vanished weight loss at 550–900 °C in the DTG curves.
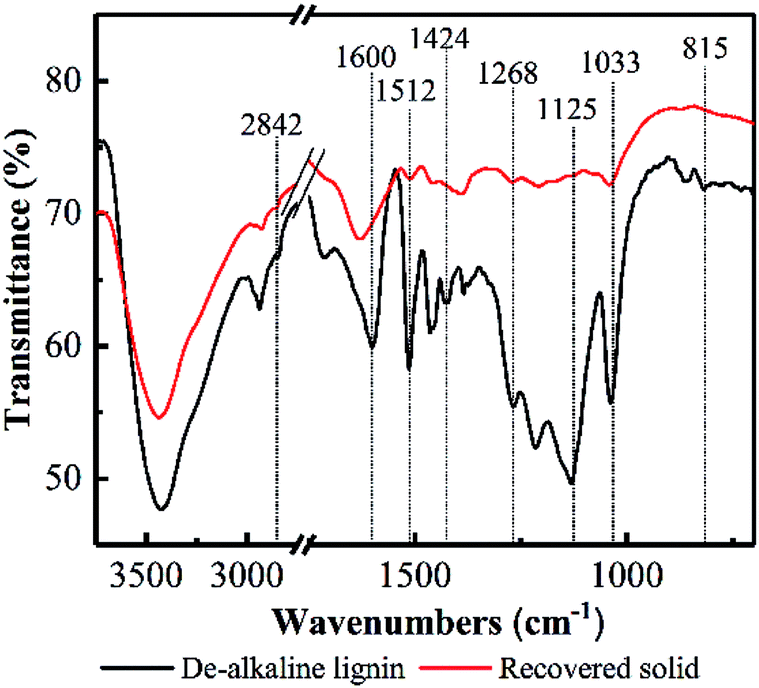 | ||
| Fig. 6 FT-IR spectrum of the de-alkaline before/after PHP pretreatment. | ||
According to the results in TGA and FT-IR, the lignin degradation by PHP pretreatment can be substantially confirmed, in which depolymerization and ring-opening reaction were two main drivers. Furthermore, GC/MS were employed to identify the derived products in liquid fraction to reveal the possible decomposition path of lignin in PHP pretreatment (see Table 2).
| No. | Identified compounds | Formula | Area (%) | ||
|---|---|---|---|---|---|
| 1.0 h | 3.0 h | 5.0 h | |||
| 1 | Propanoic acid, 2-hydroxy-methyl ester | C4H8O3 | 6.5 | 5.0 | 5.8 |
| 2 | Acetic acid | C2H4O2 | 23.7 | 19.5 | 23.1 |
| 3 | Formic acid | CH2O2 | 26.8 | 19.9 | 22.5 |
| 4 | Guaiacol | C7H8O2 | 3.6 | 2.3 | 2.9 |
| 5 | Butylated hydroxytoluene | C15H24O | 7.5 | 7.0 | 7.8 |
| 6 | 2-Hydroxy-gamma-butyrolactone | C4H6O3 | 3.8 | 2.5 | 1.4 |
| 7 | 2-tert-Butyl-p-cresol | C11H16O | 3.4 | 0.5 | 0.4 |
| 8 | Vanillic acid, methyl ester | C9H10O4 | 1.1 | 1.4 | 1.6 |
| 9 | Vanillic acid, ethyl ester | C10H12O4 | 1.4 | 2.3 | 5.5 |
| 10 | Benzoic acid | C7H6O2 | 4.5 | 2.9 | 2.4 |
| 11 | n-Hexadecanoic acid | C16H32O2 | 2.4 | 2.6 | 2.3 |
| 12 | Octadecanoic acid | C18H36O2 | 0.8 | 1.2 | 0.9 |
| 13 | Dehydroabietic acid | C20H28O2 | 0.6 | 20.2 | 12.9 |
| 14 | 3-Hydroxy-4-methoxybenzoic acid | C8H8O4 | 12.8 | 12.2 | 9.9 |
Overall, 14 compounds were identified in liquid fraction after PHP pretreatment on de-alkaline lignin. The resultant abundance of the corresponding peak for each identified compound indicated 8 monomers with the single aromatic ring from the lignin were recognized with the abundance of 31.1–38.0% in total, in which isovanillic acid, butylated hydroxytoluene, guaiacol, benzoic acid and vanillic acid ethyl ester were the dominant compounds. Obviously, the lignin depolymerization via breaking the linkage of C–O–C (β-O-4, α-O-4) could be achieved by PHP pretreatment, which can be supported by the results from FT-IR with the weakened absorptions at 1600 cm−1, 1512 cm−1, 1424 cm−1, and 1268 cm−1. As the aforementioned statements, the existing H2O2, and the derived free radicals, such as O˙, OH˙ in PHP may be responsible for depolymerization via the oxidation.34 Overall, the total abundance of these 8 single aromatic ring compounds was reduced with the prolonged pretreatment time, 38.0% (1.0 h) → 31.9% (5.0 h), suggesting the further degradation happened on the formed monomers from lignin depolymerization. The acetic acid and formic acid were another 2 dominant products in liquid fraction with the abundance range of 19.5–23.7% and 19.9–26.8%, respectively. The potential degraded products of 2-hydroxy-propanoic acid methyl ester and 2-hydroxy-gamma-butyrolactone were also be detected with a relatively higher abundance. The abundance of acetic acid and formic acid was relatively stable with increasing pretreatment time, implying they may be the end-products during the lignin degradation in PHP pretreatment. As for 2-hydroxy-propanoic acid methyl ester and 2-hydroxy-gamma-butyrolactone, their abundances tended to be reduced at longer pretreatment time and thereby can be regarded as 2 potential intermediates. In PHP pretreatment, the produced HO+ from peroxyacids was electron-deficient and easily reacted with the electron-rich sites in lignin, such as olefinic and aromatic ring structure, resulting in the lignin macromolecule to degrade via the oxygen insertion of Baeyer–Villiger oxidation.47 According to the transformation from the single aromatic ring compounds to acetic acid and formic acid, it could be inferred that the ring-opening reaction substantially occurred in degrading the derived single aromatic ring compounds. Besides, it was also reported that the aliphatic C–H bonds can be hydroxylated by peroxyacids via the oxygen insertion (Baeyer–Villiger oxidation). Thus, acetic acid and formic acid can also be sourced from the oxidization on side aliphatic chains (Step 8 in Fig. 7). In addition, the compounds with C amount > 16, such as dehydroabietic acid, octadecanoic acid, and n-hexadecanoic acid, can be detected, especially dehydroabietic acid with an abundance of 20.0% at 3.0 h. This result suggested the direct ring-opening happened simultaneously on lignin without lignin depolymerization. Of course, the produced long-chain acids, for example, octadecanoic acid, n-hexadecanoic acid can be further hydroxylated and degraded into acetic acid and formic acid via oxygen insertion on the aliphatic C–H bonds (Step 7 in Fig. 7).
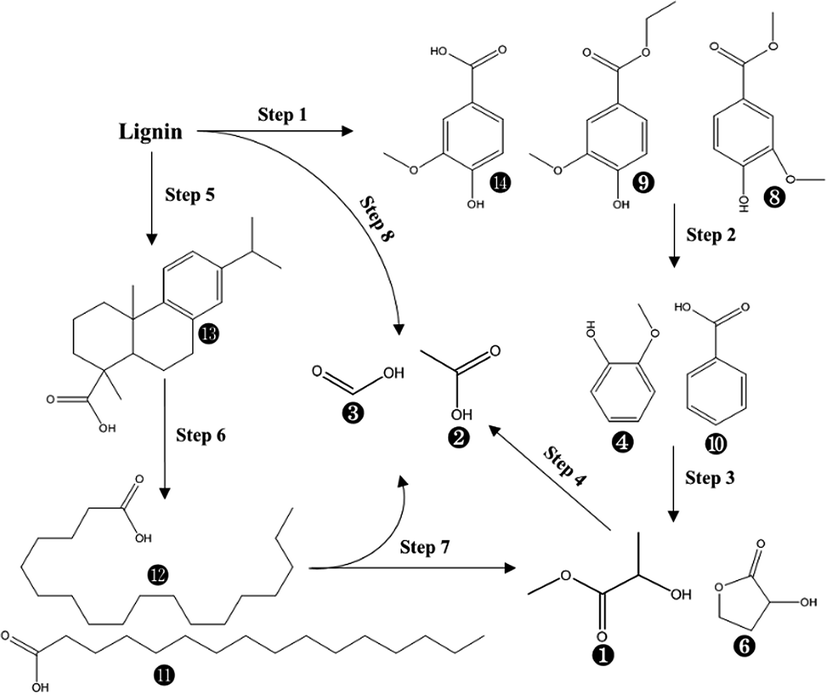 | ||
| Fig. 7 The suggested chemical degradation path of the de-alkaline lignin in PHP pretreatment; the labelled numbers near the chemical structure formula are consistent with the listed no. in Table 2. | ||
Based on the discussion above, the possible degradation path for lignin in PHP pretreatment was suggested in Fig. 7. Two paths were mainly responsible for the lignin degradation via oxidization, in which lignin was depolymerized into phenolic compounds (single aromatic ring) resulting in acetic acid and formic acid through hydroxylating aliphatic C–H bonds on the side chains, as well as ring-opening of aromatics in the derived phenolic compounds. On the other hand, the direct ring-opening of aromatics on lignin fragments resulted in the formation the long C-chain carboxylic acids (C amount > 16).
Fate of cellulose during PHP pretreatment
The main purpose of PHP pretreatment is to enhance the enzyme accessibility via overcoming the lignocellulose recalcitrance (acetyl groups, lignin barrier, and cellulose crystalline structure), while recovering the cellulose component as much as possible. According to Fig. 8, cellulose recovery did not exhibit a significant difference between the wheat straw and the model MCC. Moreover, although a slight decrease in the cellulose recovery could be observed, more than 90% cellulose could still be recovered as the pretreatment time was prolonged to 5.0 h. Previously published work also indicated the relatively higher cellulose recovery after PHP pretreatment on various feedstocks.6 These results suggested no strong chemical degradation on cellulose by PHP pretreatment, which was totally different with that of xylan and lignin.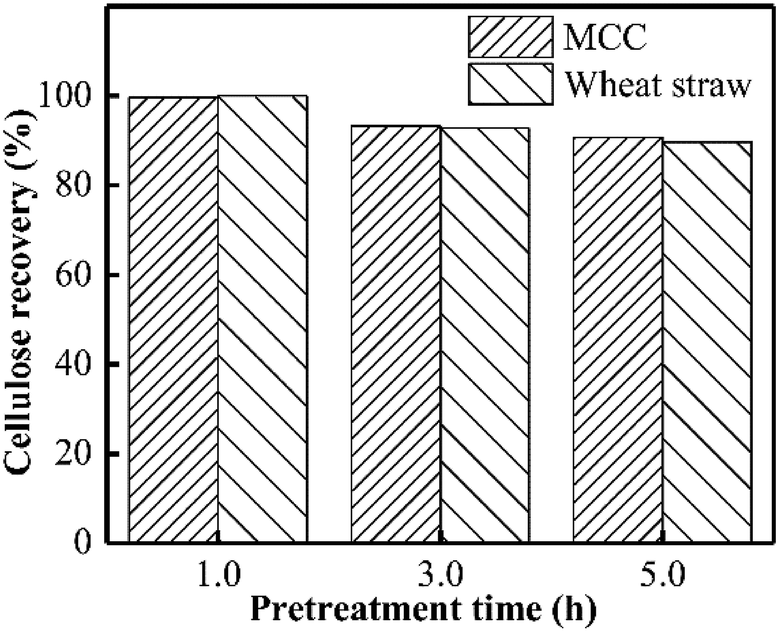 | ||
| Fig. 8 Cellulose recovery after PHP pretreatment on model MCC and wheat straw. | ||
Based on XRD determination (Fig. 9), the degree of crystallinity of wheat straw was higher than that of the PHP-pretreated wheat straw due to the removal of the amorphous hemicellulose and lignin.12 By contrast, the degree of crystallinity of MCC (64.6%) and the pretreated MCC (64.4%, 5 h) did not exhibit significant changes, implying the recovered solid from PHP pretreatment may still maintain the basic feature structure. Furthermore, no significant changes on FT-IR spectra of pretreated MCC could be detected compared with the unpretreated MCC, again suggested the degradation of cellulose did not happen seriously during PHP pretreatment.
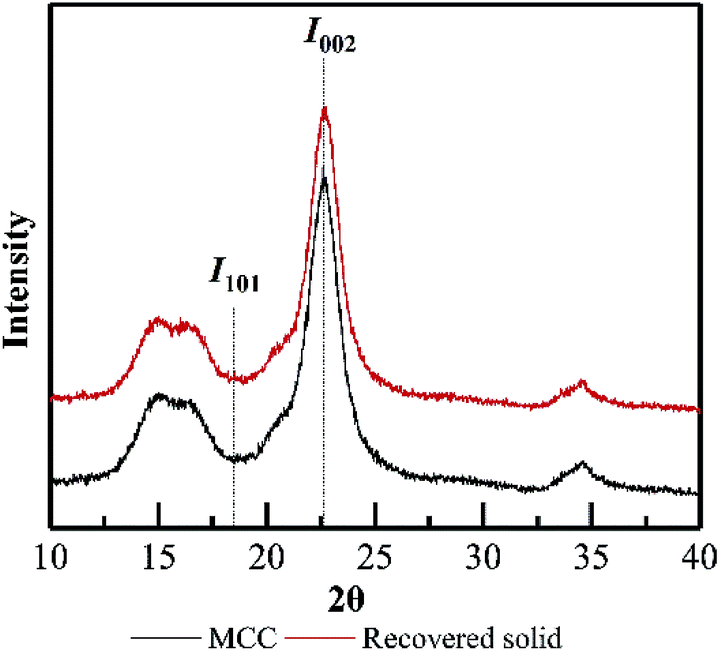 | ||
| Fig. 9 XRD results on MCC and the recovered solid after PHP pretreatment. | ||
As proved above, the intensive oxidation dominated the degradation of xylan and lignin, however, cellulose almost remained its original structure without obvious degradation, in which a potential protection from the oxidizing cellulose may exist in PHP pretreatment. As an efficient cellulose solvent, cellulose dissolution in phosphoric acid is generally attributed to esterification between –OH groups on cellulose and H3PO4 to form cellulose phosphate, which prevents the formation of hydrogen-bond formation among the –OH groups on cellulose chain.48 Consequently, the crystalline region will be broken by the action of the concentrated phosphoric acid.
Additionally, an absorption band at 1244 cm−1 in the FT-IR appeared on recovered MCC (see Fig. 10), attributing to PO stretching vibration,48 proved the existence of cellulose phosphate ester. Based on these facts, it could be deduced that some hydroxyl groups on the surface of MCC was reacted with H3PO4 to form cellulose phosphate during the PHP pretreatment. A large quantity of –OH (electronegativity) from bonded H3PO4 may cover the cellulose surface, resulting in a layer with negative charges. The resultant layer may make the cellulose surface nucleophilic in the PHP solution. Thus, the produced free radicals, such as O˙, OH˙, will be potentially blocked, and the possible oxidation degradation from free radicals can be weakened. Meanwhile, as for the electrophilic hydroxonium ion (HO+) from peroxy acids, its oxidizability would rapidly disappear due to the charge neutralization by the negative charges on the layer. Besides, the formed ester bond in cellulose phosphate may exhibit a priority to hydrogen ions comparing with the β-1,4-glucosidic bonds, therefore, the acid hydrolysis on the cellulose phosphate via cleaving these glucosidic bonds could be potentially avoided. Thus, it could be deduced the formed cellulose phosphate may act as a coat covered on cellulose to prevent the possible degradation and depolymerization. The corresponding mechanisms deserved further investigation in the later work.
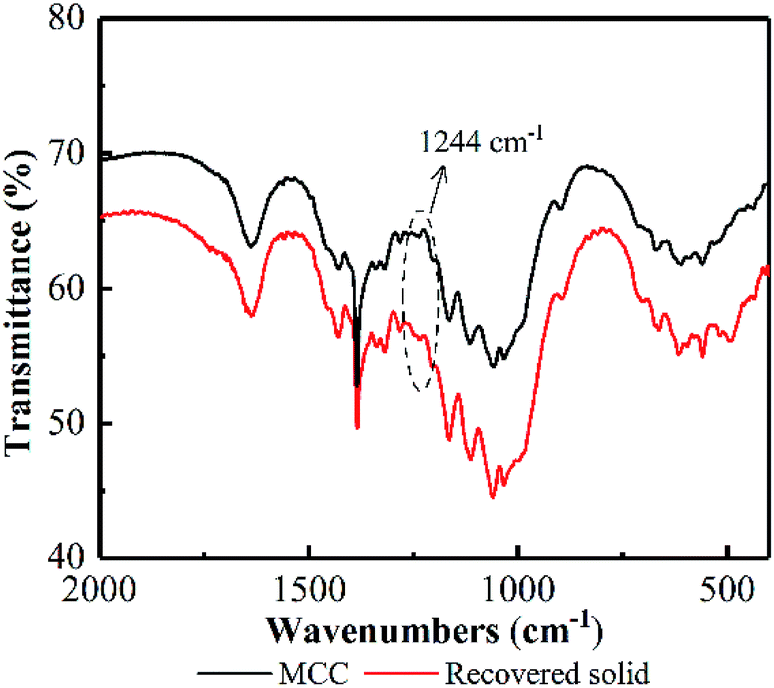 | ||
| Fig. 10 FT-IR spectrum of MCC before/after PHP pretreatment. | ||
According to the discussion above, the formation of cellulose phosphate will prevent crystalline part in cellulose from oxidation degradation and acid hydrolysis. As for the amorphous cellulose, the depolymerization by acid hydrolysis and degradation via oxidation may happen in the PHP pretreatment, which was similar with the process of xylan. Based on Table 3, GC/MS determination indicated only 3 compounds were detected in the liquid fraction, in which acetic acid was the main product with the abundance higher than 51.6%, followed by formic acid and 5-methyl-furfural. Thus, the cellulose degradation can be suggested as the following path (see Fig. 11), which generally happened on cellulose in oxidation or the concentrated acid conditions.49 Besides, the cellulose loss can be maintained below 10%, even though the pretreatment time was prolonged to 5.0 h, partially proving the acid depolymerization or oxidation degradation substantially happened on the amorphous cellulose, rather than the crystalline cellulose.
| No. | Identified compounds | Formula | Area (%) | ||
|---|---|---|---|---|---|
| 1.0 h | 3.0 h | 5.0 h | |||
| 1 | Acetic acid | C2H4O2 | 70.8 | 69.4 | 51.6 |
| 2 | Formic acid | CH2O2 | 26.6 | 28.2 | 45.3 |
| 3 | 5-Methyl-furfural | C6H6O2 | 2.5 | 2.4 | 3.1 |
 | ||
| Fig. 11 The possible degradation of cellulose during PHP pretreatment; the labelled numbers near the chemical structure formula are consistent with the listed no. in Table 3. | ||
Conclusions
As the dominant fraction, xylan in hemicellulose of wheat straw was almost completely removed by PHP pretreatment, as well as more than 70% lignin. The acid-depolymerization and oxidative degradation were proved to be two main contributors to xylan removal, in which it was decomposed into xylooligosaccharides, even into xylose, and further oxidization on xylose into acetic acid and formic acid. Lignin removal mainly underwent two oxidation degradation paths including ring-opening of aromatics and cleavage of C–O–C linkages. More than 90% cellulose were recovered from pretreatment, suggesting a special protection from cellulose degradation may exist in PHP pretreatment, and the formed cellulose phosphate was mainly responsible for hindering acid-depolymerization and oxidative degradation on cellulose.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Department of Science and Technology of Sichuan Province [grant number: 2017HH0047], and the National Natural Science Foundation of China [grant number: 21306120].References
- T. Y. Nguyen, C. M. Cai, R. Kumar and C. E. Wyman, ChemSusChem, 2015, 8, 1716–1725 CrossRef CAS PubMed.
- L. R. Lynd, M. S. Laser, D. Bransby, B. E. Dale, B. Davison, R. Hamilton, M. Himmel, M. Keller, J. D. McMillan, J. Sheehan and C. E. Wyman, Nat. Biotechnol., 2008, 26, 169–172 CrossRef CAS PubMed.
- M. Taha, M. Foda, E. Shahsavari, A. Aburto-Medina, E. Adetutu and A. Ball, Curr. Opin. Biotechnol., 2016, 38, 190–197 CrossRef CAS PubMed.
- H. Zabed, J. N. Sahu, A. N. Boyce and G. Faruq, Renewable Sustainable Energy Rev., 2016, 66, 751–774 CrossRef CAS.
- S. Haghighi Mood, A. Hossein Golfeshan, M. Tabatabaei, G. Salehi Jouzani, G. H. Najafi, M. Gholami and M. Ardjmand, Renewable Sustainable Energy Rev., 2013, 27, 77–93 CrossRef CAS.
- Q. Wang, Z. Wang, F. Shen, J. Hu, F. Sun, L. Lin, G. Yang, Y. Zhang and S. Deng, Bioresour. Technol., 2014, 166, 420–428 CrossRef CAS PubMed.
- S. Sun, S. Sun, X. Cao and R. Sun, Bioresour. Technol., 2016, 199, 49–58 CrossRef CAS PubMed.
- E. Shirkavand, S. Baroutian, D. J. Gapes and B. R. Young, Renewable Sustainable Energy Rev., 2016, 54, 217–234 CrossRef CAS.
- L. Narayanasamy and T. Murugesan, Environ. Sci. Technol., 2014, 33, 482–489 CAS.
- N. Mosier, C. Wyman, B. Dale, R. Elander, Y. Y. Lee and M. Ladisch, Bioresour. Technol., 2005, 96, 673–686 CrossRef CAS PubMed.
- M. Galbe and G. Zacchi, Biomass Bioenergy, 2012, 46, 70–78 CrossRef CAS.
- Q. Wang, J. Hu, F. Shen, Z. Mei, G. Yang, Y. Zhang, Y. Hu, J. Zhang and S. Deng, Bioresour. Technol., 2016, 199, 245–257 CrossRef CAS PubMed.
- J. Qiu, Q. Wang, F. Shen, G. Yang, Y. Zhang, S. Deng, J. Zhang, Y. Zeng and C. Song, Appl. Biochem. Biotechnol., 2017, 181, 1123–1139 CrossRef CAS PubMed.
- J. Qiu, L. Ma, F. Shen, G. Yang, Y. Zhang, S. Deng, J. Zhang, Y. Zeng and Y. Hu, Bioresour. Technol., 2017, 238, 174–181 CrossRef CAS PubMed.
- J. Zeng, Z. Tong, L. Wang, J. Y. Zhu and L. Ingram, Bioresour. Technol., 2014, 154, 274–281 CrossRef CAS PubMed.
- W. Wu, V. Rondon, K. Weeks, P. Pullammanappallil, L. O. Ingram and K. T. Shanmugam, Bioresour. Technol., 2018, 251, 171–180 CrossRef CAS PubMed.
- Y. H. P. Zhang, S. Y. Ding, J. R. Mielenz, J. B. Cui, R. T. Elander, M. Laser, M. E. Himmel, J. R. McMillan and L. R. Lynd, Biotechnol. Bioeng., 2007, 97, 214–223 CrossRef CAS PubMed.
- N. Sathitsuksanoh, Z. Zhu and Y.-H. P. Zhang, Cellulose, 2012, 19, 1161–1172 CrossRef CAS.
- E. D. Dutra, F. A. Santos, B. R. A. Alencar, A. L. S. Reis, R. d. F. R. de Souza, K. A. d. S. Aquino, M. A. Morais Jr and R. S. C. Menezes, Biomass Convers. Biorefin., 2018, 8, 225–234 CrossRef CAS.
- S. G. Wi, E. J. Cho, D. S. Lee, S. J. Lee, Y. J. Lee and H. J. Bae, Biotechnol. Biofuels, 2015, 8, 228 CrossRef PubMed.
- S. Karagöz, T. Kawakami, A. Kako, Y. Iiguni and H. Ohtani, RSC Adv., 2016, 6, 46108–46115 RSC.
- H. R. Yuan, S. Y. Xing, H. T. L. Hu, T. Lu and Y. Chen, J. Anal. Appl. Pyrolysis, 2015, 112, 325–332 CrossRef CAS.
- Y. Fu, K. Kato, H. Ohtani and Y. Chen, J. Wood Chem. Technol., 2014, 34, 1–7 CrossRef CAS.
- R. C. Sun, J. Tomkinson, Y. X. Wang and B. Xiao, Polymer, 2000, 41, 2647–2656 CrossRef CAS.
- D. Barana, A. Salanti, M. Orlandi, D. S. Ali and L. Zoia, Ind. Crops Prod., 2016, 86, 31–39 CrossRef CAS.
- Y. Peng and S. Wu, J. Anal. Appl. Pyrolysis, 2010, 88, 134–139 CrossRef CAS.
- R. Sun, J. M. Lawther and W. B. Banks, Carbohydr. Polym., 1996, 29, 325–331 CrossRef CAS.
- D. L. Sills and J. M. Gossett, Biotechnol. Bioeng., 2012, 109, 353–362 CrossRef CAS PubMed.
- M. Kacuráková, P. Capek, V. Sasinková, N. Wellner and A. Ebringerová, Carbohydr. Polym., 2000, 43, 195–203 CrossRef.
- J. J. Bozell, L. Moens, D. C. Elliott, Y. Wang, G. G. Neuenscwander, S. W. Fitzpatrick, R. J. Bilski and J. L. Jarnefeld, Resour., Conserv. Recycl., 2000, 28, 227–239 CrossRef.
- J. B. Binder, J. J. Blank, A. V. Cefali and R. T. Raines, ChemSusChem, 2010, 3, 1268–1272 CrossRef CAS PubMed.
- O. Yemis and G. Mazza, Bioresour. Technol., 2011, 102, 7371–7378 CrossRef CAS PubMed.
- V. G. Kul'Nevich and L. A. Badovskaya, Russ. Chem. Rev., 1975, 44, 1256–1279 Search PubMed.
- R. Ma, Y. Xu and X. Zhang, ChemSusChem, 2015, 8, 24–51 CrossRef CAS PubMed.
- J. Gierer, Wood Sci. Technol., 1986, 20, 1–33 CrossRef CAS.
- R. Ma, M. Guo and X. Zhang, ChemSusChem, 2014, 7, 412–415 CrossRef CAS PubMed.
- L. X. Zhang, H. Yu, H. B. Yu, Z. Chen and L. Yang, Chin. Chem. Lett., 2014, 25, 1132–1136 CrossRef CAS.
- V. V. Poskonin, L. A. Badovskaya, L. V. Povarova and R. I. Ponomarenko, ChemInform, 2010, 31, 1 CrossRef.
- P. Peng, F. Peng, J. Bian, F. Xu and R. C. Sun, Wood Sci. Technol., 2012, 46, 871–885 CrossRef CAS.
- H. Yang, R. Yan, H. Chen, D. H. Lee and C. Zheng, Fuel, 2007, 86, 1781–1788 CrossRef CAS.
- M. Zhang, F. L. P. Resende, A. Moutsoglou and D. E. Raynie, J. Anal. Appl. Pyrolysis, 2012, 98, 65–71 CrossRef CAS.
- D. Ferdous, a K. Dalai, S. K. Bej and R. W. Thring, Can. J. Chem. Eng., 2001, 79, 913–922 CrossRef CAS.
- B. Biswas, R. Singh, J. Kumar, A. A. Khan, B. B. Krishna and T. Bhaskar, Bioresour. Technol., 2015, 213, 319–326 CrossRef PubMed.
- M. S. Jahan, D. A. N. Chowdhury, M. K. Islam and S. M. I. Moeiz, Bioresour. Technol., 2007, 98, 465–469 CrossRef CAS PubMed.
- A. Tejado, C. Peña, J. Labidi, J. M. Echeverria and I. Mondragon, Bioresour. Technol., 2007, 98, 1655–1663 CrossRef CAS PubMed.
- R. Singh, A. Prakash, S. K. Dhiman, B. Balagurumurthy, A. K. Arora, S. K. Puri and T. Bhaskar, Bioresour. Technol., 2014, 165, 319–322 CrossRef CAS PubMed.
- N. D. Patil, S. G. Yao, M. S. Meier, J. K. Mobley and M. Crocker, Org. Biomol. Chem., 2015, 13, 3243–3254 CAS.
- G. Li, J. Zeng, H. Gao and X. Li, Int. J. Food Prop., 2011, 14, 978–987 CrossRef CAS.
- M. Niu, Y. Hou, S. Ren, W. Wang, Q. Zheng and W. Wu, Green Chem., 2015, 17, 335–342 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Four supplementary figures are available there. See DOI: 10.1039/c8ra00764k |
| This journal is © The Royal Society of Chemistry 2018 |