
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly efficient heavy-metal extraction from water with carboxylated graphene nanoflakes†
Martin Rosillo-Lopez and
Christoph G. Salzmann *
*
Department of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: c.salzmann@ucl.ac.uk
First published on 20th March 2018
Abstract
Heavy metals such a lead or cadmium have a wide range of detrimental and devastating effects on human health. It is therefore of paramount importance to efficiently remove heavy metals from industrial wastewater streams as well as drinking water. Carbon materials, including graphene and graphene oxide (GO), have recently been advocated as efficient sorption materials for heavy metals. We show that highly carboxylated graphene nanoflakes (cx-GNF) outperform nano-graphene oxide (nGO) as well as traditional GO with respect to extracting Fe2+, Cu2+, Fe3+, Cd2+ and Pb2+ cations from water. The sorption capacity for Pb2+, for example, is more than six times greater for the cx-GNF compared to GO which is attributed to the efficient formation of lead carboxylates as well as strong cation–π interactions. The large numbers of carboxylic acid groups as well as the intact graphenic regions of the cx-GNF are therefore responsible for the strong binding of the heavy metal cations. Remarkably, the performance of the as-made cx-GNF can easily compete with previously reported carbon materials that have undergone additional chemical-functionalisation procedures for the purpose of heavy-metal extraction. Furthermore, the recyclability of the cx-GNF material with respect to Pb2+ loading is demonstrated as well as the outstanding performance for Pb2+ extraction in the presence of excess Ca2+ or Mg2+ cations which are often present under environmental conditions. Out of all the graphene materials, the cx-GNF therefore show the greatest potential for future application in heavy-metal extraction processes.
Introduction
Exposure to heavy metals causes a wide range of adverse health effects in humans.1 Lead poisoning, for example, can lead to kidney2 and bone damage,1 malfunction of the nervous system,3 psychosis,1 infertility,4 anaemia5 and cancer.6,7 Children in particular are susceptible to the effects of heavy-metal poisoning due to their under-developed blood–brain barrier.1 Yet, the global exposure levels of humans to heavy metals are on the rise. This is due, for example, to cadmium-based products such as nickel–cadmium batteries, airborne inorganic lead resulting from mines, smelters, battery plants and the glass industry, and contaminated wastewaters from a wide range of chemical processes in industry.1,8–10 The contamination of soil and water streams ultimately leads to the incorporation of heavy metals into the human food chain.1 The efficient removal of heavy metals from drinking water, industrial wastewater and the environment at large is therefore of paramount importance.Carbon materials have been at the vanguard of aqueous heavy-metal extraction over the last few years.11–31 This surge in interest arose after the isolation of graphene, a single layer of graphite,32 was first employed for metal-extraction processes.11,12 More recently, graphene oxide (GO), an oxidised form of graphene containing different functional groups such as epoxides, alcohols and carboxylic acids33 has been advocated as an alternative to graphene.34–36 The advantage of GO over graphene for metal extraction is its hydrophilic nature, tuneable pore sizes as well as a stronger chelation ability towards metals due to the functional groups. Considerable efforts have gone into chemically modifying GO to further enhance its metal extraction capability including the preparation of hybrid materials with ethylenediamine tetraacetic acid (EDTA),13,14 cyclodextrin,15 polypyrrole,16 polyethylenimine,17,18 silica19 and many more.20–23,31 Recently, some of the functionalised or doped GO materials have been utilised for capacitive-deionisation processes.26–30 However, the chemical functionalisation of GO is in general a lengthy and expensive step. It is therefore desirable to optimise and further investigate the metal-extraction properties of as-made carbon nanomaterials in order to provide low-cost adsorbents. In fact, very little research has so far been conducted using nano-graphene oxides (nGO), i.e. GO but with lateral dimensions below 100 nm, in heavy metal extraction. This is surprising because the chemical structure of GO, as described by the Lerf–Klinowski model,33 suggests that carboxylic acid groups, which should be most effective in chelating metals, are located on the edges of GO. Smaller GO sheets with large edge-to-basal-plane ratios should therefore have more carboxylic acid groups per unit mass and hence be well-suited for metal-extraction processes.
In this work, the metal-sorption capacity of highly edge-carboxylated graphene nanoflakes (cx-GNF)37 and nGO38 (both ∼30 nm in diameter) are benchmarked against conventional GO prepared via the modified Hummers method.39 The five heavy-metal cations under investigation are: Fe2+, Cu2+, Fe3+, Cd2+ and Pb2+. The chemical mechanisms of heavy-metal binding onto the carbon materials and the recyclability of cx-GNF towards loading and unloading of Pb2+ cations are investigated as well as the selectivity for Pb2+ cation extraction in the presence of large quantities of other cations typically present in drinking or industrial waste water such as magnesium and calcium.
Experimental
Preparation of cx-GNF, nGO and GO
The preparation of cx-GNF, nGO and GO is described in detail in ref. 37–39 as well as in Section 1 of the ESI.†Adsorption experiments with heavy metals
10 mg of cx-GNF, nGO or GO were combined with 10 mL of 0.1 M solutions of metal chloride or nitrate salts (FeCl2·4H2O, FeCl3·6H2O, CuCl2·2H2O, Cd(NO3)2·4H2O and Pb(NO3)2). The mixture was then sonicated for 3 × 10 minutes allowing the precipitate to settle out each time for 10 minutes before re-sonicating, resulting in a 60 min total reaction time. This was followed by filtration through a 200 nm polycarbonate membrane and washing with 3 × 10 mL of deionised water to remove any excess metal salt. The material on the membrane was collected and allowed to dry in a vacuum desiccator overnight. The dry powders were analysed by XPS and ATR-IR spectroscopy. For a kinetic study of the adsorption of Pb2+ ions onto the carbon materials, the corresponding mixtures were sonicated continuously for a range of different durations up to 100 minutes and worked up as previously described.Recyclability of cx-GNF for Pb2+ extraction
75 mg of Pb2+-loaded cx-GNF (Pb2+@cx-GNF) were sonicated in 10 mL of formic acid for <1 min. The dark brown dispersion was diluted with deionised water until pH 2 and then dialysed against deionised water via a SpectraPor 3 regenerated cellulose dialysis membrane (Spectrum Laboratories, MWCO 3.5 kDa). Once the conductivity of the surrounding water was below 5 μS cm−1 the dispersion was passed over an ion exchange resin (Amberlite IR120, Sigma-Aldrich). The dispersion was dialysed once more, concentrated in vacuo and freeze dried to regenerate the cx-GNF. The cx-GNF were then treated again with a 0.1 M Pb2+ solution in the same way as previously described.Adsorption of Pb2+ ions onto cx-GNF in the presence of 10 or 100 molar excess of Ca2+ or Mg2+ ions
5 mL of a 2 M Ca(NO3)2 or Mg(NO3)2 solution was combined with 5 mL of either a 0.2 M or 0.02 M Pb(NO3)2 solution to give 10 or 100 molar excesses of Ca2+ or Mg2+ ions. The solutions were then combined with 10 mg of cx-GNF and the precipitated GNFs were collected as described before.Adsorption of Pb2+ ions onto cx-GNF in commercial drinking water
The ability of the cx-GNF to extract Pb2+ cations from commercial drinking water was investigated by preparing a 5 mM solution of Pb(NO3)2 in Glaceau Smartwater which contains calcium and magnesium chloride as well as potassium bicarbonate. 10 mL of this solution were combined with 10 mg of cx-GNF and the GNFs were collected as described before.Sample characterisations
X-ray photoelectron spectroscopy (XPS) measurements were carried out on a Thermo Scientific K-Alpha XPS machine with a monochromated Al Kα source (E = 1486.6 eV), a double focusing 180 degree hemisphere analyser of ∼125 mm radius and detected with a 18 channel position-sensitive detector. A dual-beam flood gun (electrons and argon ions) was used to compensate for charge accumulation on the measured surfaces. All carbon samples were pressed onto an indium substrate before analysis. Survey scans were collected 3 times with a resolution of 1 eV and all elemental regions were scanned 10 times with a resolution of 0.1 eV. All scans were recorded with a 50 ms dwell time and 400 μm spot size. All elemental regions were calibrated against the C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C peak position at 285.0 eV from the C1s high-resolution spectrum. The metal-free cx-GNF data was fitted with two peaks, and a small degree of asymmetry was introduced to the C–C/CC peak. Yet, the C1s spectra of the M2+/3+@cx-GNF were fitted with three peaks.
C peak position at 285.0 eV from the C1s high-resolution spectrum. The metal-free cx-GNF data was fitted with two peaks, and a small degree of asymmetry was introduced to the C–C/CC peak. Yet, the C1s spectra of the M2+/3+@cx-GNF were fitted with three peaks.
FT-IR spectra were collected on a Bruker Tensor 27 FTIR spectrometer using the attenuated total-reflectance infrared spectroscopy mode (ATR-IR) fitted with a diamond crystal as the internal reflection element and a DLaTGS detector with 4 cm−1 resolution. For each measurement, a background spectrum was collected for 256 scans before recording the sample measurement which was collected for the same number of scans.
High-resolution solid-state 13C NMR spectra were recorded on a Bruker Avance 300 spectrometer with a 7.05 T wide-bore magnet at ambient probe temperature. The spectra were recorded at 75.5 MHz with a Bruker 4 mm double-resonance magic-angle spinning (MAS) probe using high-power proton decoupling (HPDEC). For the cx-GNF, the operating conditions were 13C 90° pulse duration = 3.7 μs, recycle delay = 120 s with 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 480 scans. Similarly, the operating conditions for the Pb-GNF were 13C 90° pulse duration = 3.0 μs, recycle delay = 120 s with 22528 scans. The cx-GNF and Pb2+@cx-GNF were packed into zirconia rotors of 4 mm external diameter and spun at 12 kHz or 8 kHz MAS frequency respectively. In each case tetramethylsilane (TMS) was calibrated against an aqueous solution of 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS, 0 ppm) and glycine (176.46 ppm), and the 13C chemical shifts are reported with respect to TMS.
480 scans. Similarly, the operating conditions for the Pb-GNF were 13C 90° pulse duration = 3.0 μs, recycle delay = 120 s with 22528 scans. The cx-GNF and Pb2+@cx-GNF were packed into zirconia rotors of 4 mm external diameter and spun at 12 kHz or 8 kHz MAS frequency respectively. In each case tetramethylsilane (TMS) was calibrated against an aqueous solution of 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS, 0 ppm) and glycine (176.46 ppm), and the 13C chemical shifts are reported with respect to TMS.
Determination of Pb2+ sorption capacities with optical absorbance spectroscopy
10 mg of cx-GNF, nGO or GO were combined with 10 mL of 5 mM Pb(NO3)2 solutions. The mixtures were then ultrasonicated, filtered and washed as previously described. The colourless filtrates as well as 10 mL of a 5 mM Pb2+ solution were topped up to 250 mL with deionised water. 4 mL of these solutions were acidified with 1 mL of 5 mM HCl, combined with a solution of 2,5-dimercapto-1,3,4-thiadiazole dipotassium salt (DMTD-K+)40 and topped up to 10 mL. The yellow solutions were then transferred into quartz cuvettes of 1 cm path length, and optical absorbance spectra were recorded between 300 and 500 nm on a PerkinElmer Lambda 365 spectrophotometer at steps of 1.0 nm and scan rate of 600 nm min−1. Finally, the Pb2+ sorption capacities of the carbon materials were calculated from the difference in the optical absorbances at 400 nm between the initial 5 mM solution and the solutions after the Pb2+ extraction.Results and discussion
Structural characterisation of the cx-GNF, nGO and GO starting materials
The chemical structures of cx-GNF, nGO and GO as well as the corresponding ATR-IR and XPS spectra are shown in Fig. 1a–f. The cx-GNF are a highly carboxylated graphenic nanomaterial37 as indicated by the very broad O–H stretching frequency peak between 3600 cm−1 and 2500 cm−1 in the IR spectrum (Fig. 1d). This is further corroborated by a strong peak at 289.3 eV in the XPS C1s region which is associated with C(III) species such as carboxylic acids (Fig. 1f).41 In contrast to the cx-GNF, nGO and GO show the presence of a peak at approximately 287 eV which is attributed to C(I) species such as alcohols and epoxides, and even C(II) species such as ketones.42,43 This peak is significantly more intense for GO than for nGO because these functional groups are typically found on the basal plane of oxidised graphenic materials, and GO has a larger basal-plane-to-edge ratio compared to nGO.38 Furthermore, the edges of nGO and GO are only sparingly decorated with carboxylic acids amongst other functional groups,33 and since nGO has more edges than GO, on account of nGO being 1–2 orders of magnitude smaller than GO, nGO will have more carboxyl groups than GO per unit mass; as evidenced by the C1s spectrum in Fig. 1f. By analogy, if a piece of paper is cut multiple times, the mass of the paper will remain the same after being cut, yet the overall length of the edges of the smaller cut-out sheets will exceed that of the uncut paper. Finally, for all three carbon materials, the peak attributed to graphenic carbon can be clearly seen at ∼285 eV. Full structural characterisations and investigations into the specific chemical properties of cx-GNF, nGO and GO are given in ref. 37–39 respectively.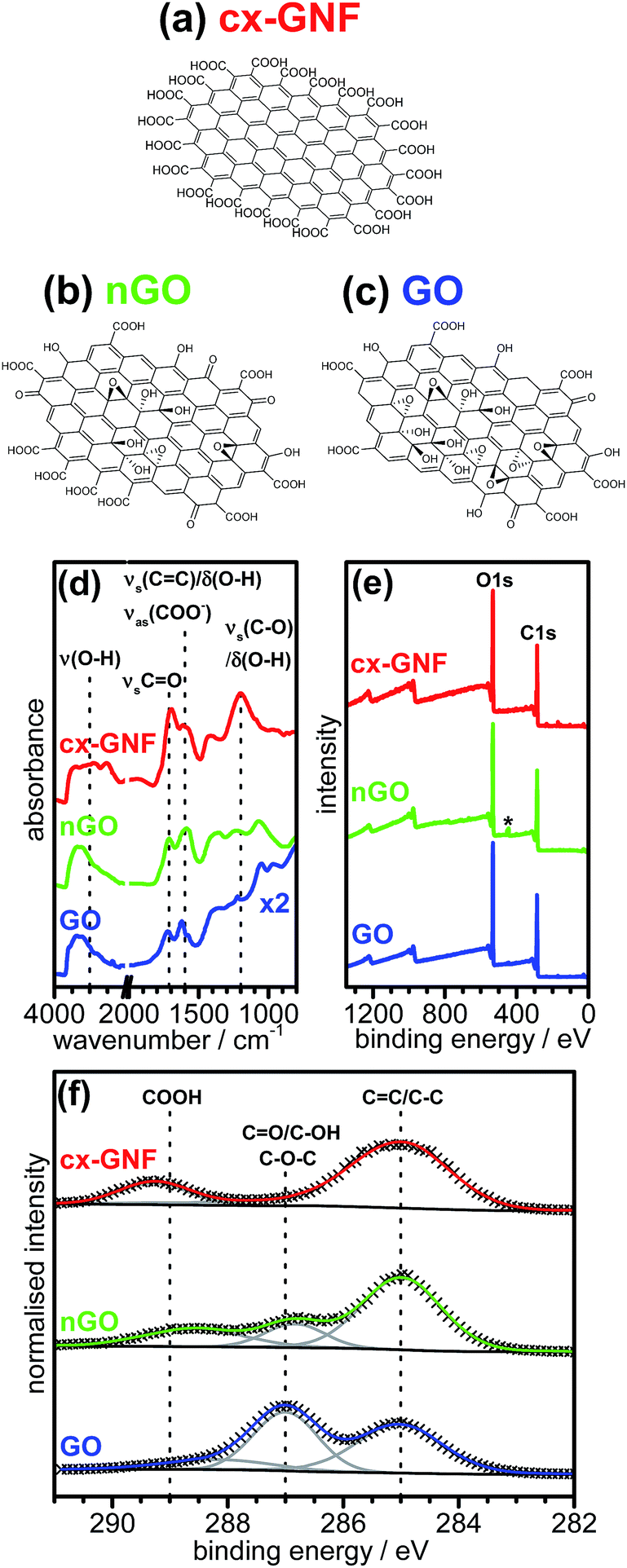 | ||
| Fig. 1 Schematic chemical structures of (a) cx-GNF, (b) nGO and (c) GO. The lateral dimensions of cx-GNF and nGO are approximately 30 nm whereas GO sheets can be up to several μm in diameter. (d) ATR-IR spectra, (e) XPS survey and (f) C1s regions of cx-GNF (red), nGO (green) and GO (blue). The peak marked with the asterisk in (e) is due to the indium substrate. | ||
Heavy-metal extraction from water with cx-GNF, nGO and GO
In order to quantify the relative sorption capacities of cx-GNF, nGO and GO towards heavy metals, the metal/carbon (M2+/3+/C) ratio of each metal–carbon composite, denoted as M2+/3+@cx-GNF, M2+/3+@nGO or M2+/3+@GO, respectively, or collectively as M2+/3+@carbon, was determined from the XPS survey spectra. Fig. 2a shows survey spectra of cx-GNF, nGO and GO after exposure to Pb2+ solutions. In the case of Pb2+@carbon, the Pb4f peak was used for the quantification of Pb2+. The XPS survey spectra of the other M2+/3+@carbon samples can be found in Fig. S1 of the ESI.† The determined M2+/3+/C ratios for the various heavy metals and carbon materials are shown in Fig. 2b. It is also noted that significant mass increases of the carbon materials were observed after some of the heavy-metal extractions. For example, the weight of the cx-GNF material increased by about 30 weight percent after the Pb2+ sorption experiment.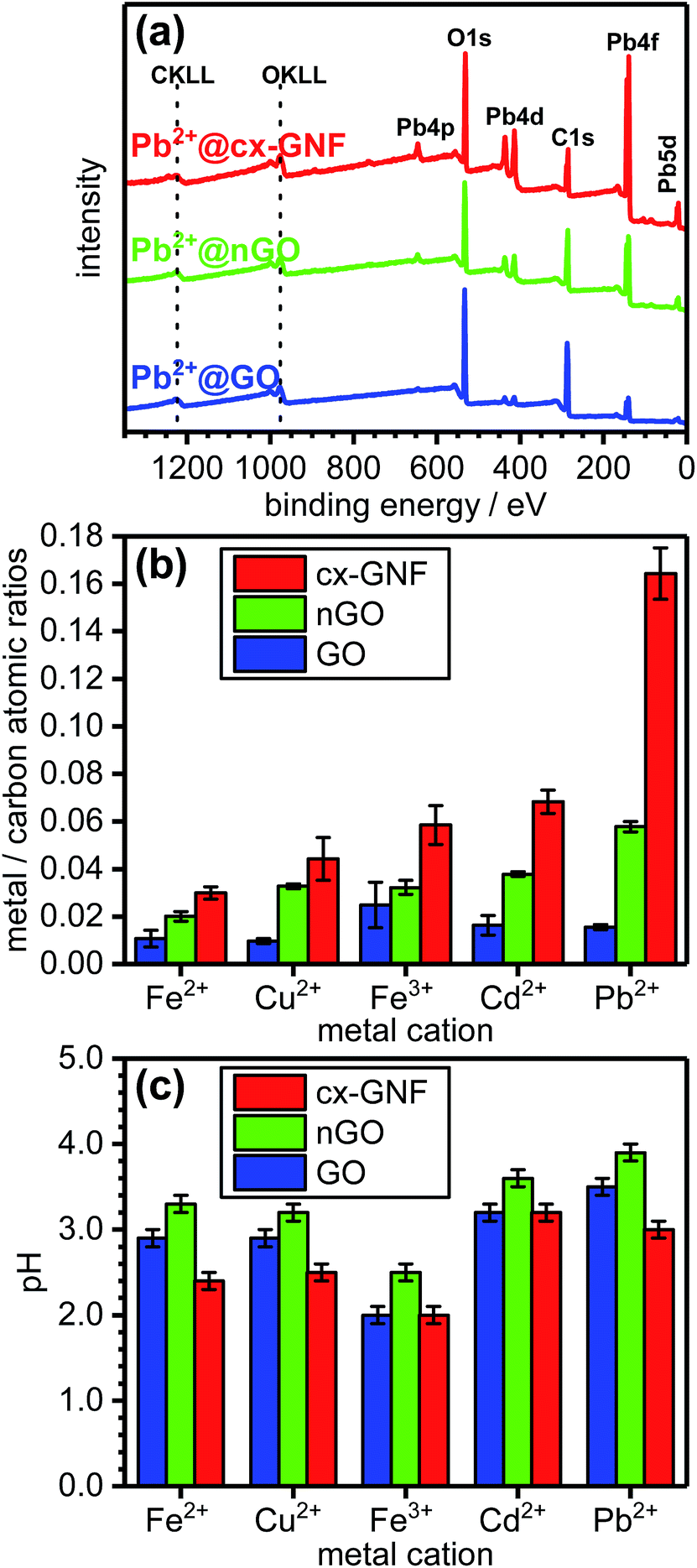 | ||
| Fig. 2 (a) XPS survey spectra of Pb2+@cx-GNF (red), Pb2+@nGO (green) and Pb2+@GO (blue). (b) Metal ion/carbon atomic ratios. (c) pH values of the Fe2+, Cu2+, Fe3+, Cd2+ or Pb2+ solutions treated with cx-GNF (red), nGO (green) and GO (blue), respectively. The pH of each of the mixtures was measured prior to filtration (10 mg of graphenic material in 10 mL of 0.1 M solutions of the respective metal salts). | ||
The ability of a carbon material to extract metal cations from solution is dependent upon a range of parameters including the ionic strength and pH of the mixture. In order to ensure comparable ionic strengths throughout the various adsorption experiments, 10 mg of each carbon material was treated with 10 mL of a 0.1 M solutions of the metal salt which means that the cations are in large excess. Consequently, the ionic strength of a mixture of cx-GNF, nGO or GO with a particular metal cation, is approximately constant.
Likewise, increasing the pH of a mixture is expected to lead to an increase in metal chelation. This arises as a result of a decrease in H+ ions in solution which in turn reduces the competition between the metal cations and H+ ions for the chelating ligand; which in the case of cx-GNF, nGO and GO, are the oxygen-containing functional groups. Furthermore, increasing the pH to basic conditions would lead to precipitation of metal hydroxide species44,45 and therefore give misleading results.
The pH of the M2+/3+@carbon mixtures was measured prior to filtration and the results are shown in Fig. 2c. It can be seen that for a particular metal ion, the pH of the M2+/3+@cx-GNF ≤ M2+/3+@GO < M2+/3+@nGO indicating that the cx-GNF are at a disadvantage for metal sorption with respect to GO and nGO for the reasons discussed earlier. The lower pH of the cx-GNF is expected given the highly carboxylated nature of the material and the fact that carboxylic acids are much stronger acids than alcohols and the other functional groups found at high concentrations on GO. Indeed, 1 mg mL−1 aqueous dispersions of cx-GNF, nGO and GO gave pH values of 2.5, 3.3 and 3.0 respectively. Interestingly, the pH of GO is slightly lower than nGO despite nGO containing more carboxylic acid groups. However, GO has many more acidic hydroxyl groups on its basal plane, and given that its structural instability in water generates even more acidic functional groups,46 this result is not necessarily surprising.
Despite the lower pH of the cx-GNF, they clearly outperform the other two carbon materials at extracting metals as evidenced by the M2+/3+/C ratios obtained from the XPS survey spectra shown in Fig. 2b. For example, the cx-GNF can bind about 10 times more Pb2+ than GO. This can be rationalised at this stage by consideration of the number of carboxylic acid groups per unit mass of each material. According to peak–area ratios from solid-state NMR measurements (COOH/graphenic carbon),37,38 the cx-GNF contain about four times more COOH groups than nGO, and about 10 times more COOH groups than GO.
Carboxylic acids are bidentate and generally stronger metal chelators compared to monodentate alcohols and epoxides. Indeed, it has been previously stated that COOH groups are the most efficient functional group present on graphenic materials for chelating Pb2+ cations.11 It is important to stress that despite nGO achieving better extraction results compared to GO, the pH of the M2+/3+@nGO mixtures were on average 0.4 units more basic than for M2+/3+@GO, allowing the nGO flakes to better coordinate to metals. Hence, the actual ability of nGO to remove heavy metals compared to GO may not be as pronounced as shown in Fig. 2b if the extractions were carried out at the same pH.
It is noteworthy that we deliberately did not use buffer solutions for these experiments and relied on the natural pH of the M2+/3+@carbon mixtures. This removes any doubt with respect to the effect ionic strength and foreign species might have in influencing the metal-sorption capability of the carbon materials.
A kinetic study of the Pb2+ adsorption onto the various carbon materials shows that the adsorption equilibria are reached very quickly for nGO and GO (Fig. S2 of the ESI†). The cx-GNF show a slight gradual increase in the Pb2+ uptake over a time period of 100 minutes. This could be due to the initial formation of dicarboxylate–lead complexes with COOH groups from different flakes which are successively replaced by monocarboxylate complexes as the sonication time progresses.
The fast equilibration of the Pb2+ adsorption onto nGO and GO illustrates that their poorer performance with respect to binding Pb2+ is not due to kinetic factors but to the intrinsically different interaction properties of the carbon materials with the heavy metals. Furthermore, the exfoliation process of the carbon materials upon sonication, which could potentially limit the accessible surface area, does not appear to be a limiting factor for the heavy metal extraction using nGO and GO.
The area-normalised C1s XPS spectra of the M2+/3+@cx-GNF are shown in Fig. 3. The most immediate observation are additional peaks at ∼286.7 eV which are present in all the M2+/3+@cx-GNF samples but absent for the pure cx-GNF. Furthermore, the areas of the C(III) peaks at ∼288.5 eV are larger and shifted towards lower binding energies for the M2+/3+@cx-GNF. The newly formed peaks at 286.7 eV suggest non-covalent cation–π interactions between the metal cations and the graphenic basal plane of the cx-GNF.47–49 This can be rationalised by considering the electrostatic attraction between the positively charged metal cations and the graphenic π-electrons which creates a local deshielding effect along the graphenic basal plane where the metal cations have adsorbed. This in turn confers a δ+ charge on the carbon atoms resulting in an increase in the binding energy of its core electrons. Hence, the observed decrease in the relative C1s C–C/CC peak intensity with respect to the C(III) peak. This effect is most noticeable for the Pb2+@cx-GNF sample which has the highest M2+/3+/C ratio of all the M2+/3+@cx-GNF (Fig. 2b), suggesting that Pb–π interactions are the most favourable out of all the heavy metals under these conditions, perhaps because Pb2+ has the lowest solvation enthalpy.50
 | ||
| Fig. 3 Area-normalised XPS C1s spectra of cx-GNF (cyan), Fe2+@cx-GNF (magenta), Cu2+@cx-GNF (orange), Fe3+@cx-GNF (green), Cd2+@cx-GNF (red) and Pb2+@cx-GNF (blue). | ||
To further prove that the trends observed in the XPS C1s spectra are due to cation–π interactions, solid state 13C-NMR spectra of the cx-GNF were collected before and after exposure to a Pb2+ solution (Fig. 4). The 13C-NMR spectra of the cx-GNF and Pb2+@cx-GNF both exhibit two peaks, one associated with COOH groups (∼170 ppm) and another for sp2 graphenic carbon (∼130 ppm).33 A distinct shift to higher ppm (downfield shift) is observed for both peaks in the spectrum of Pb2+@cxGNF, indicating more deshielded environments. The downfield shift observed for the sp2 carbon from 134 to 137 ppm is in good agreement with M–π interactions which induce a deshielding effect on the aromatic rings as described earlier.48 However, the downfield shift of the COOH group from 170 to 176 ppm is most likely the result of chelation between the Pb2+ ions and the carboxyl groups on the cx-GNF.51,52 It is noteworthy that the absence of C(I) species such as epoxides and alcohols between 60 and 70 ppm in the Pb2+@cx-GNF spectrum proves that the newly formed peaks at 286.7 eV in the XPS C1s regions of the M2+/3+@cx-GNF are due to M–π interactions and not to the formation of alcohol or epoxide groups. Interestingly, the M–π interactions were not observed in the XPS C1s spectra of M2+/3+@GO but were noticeable to a small extent for the M2+/3+@nGO materials (Fig. S3†). This could be because nGO nanomaterials have more intact aromatic sp2 basal planes compared to GO, as evidenced by the higher intensity of the sp2 CC peak in the solid state 13C-NMR spectra.37,38
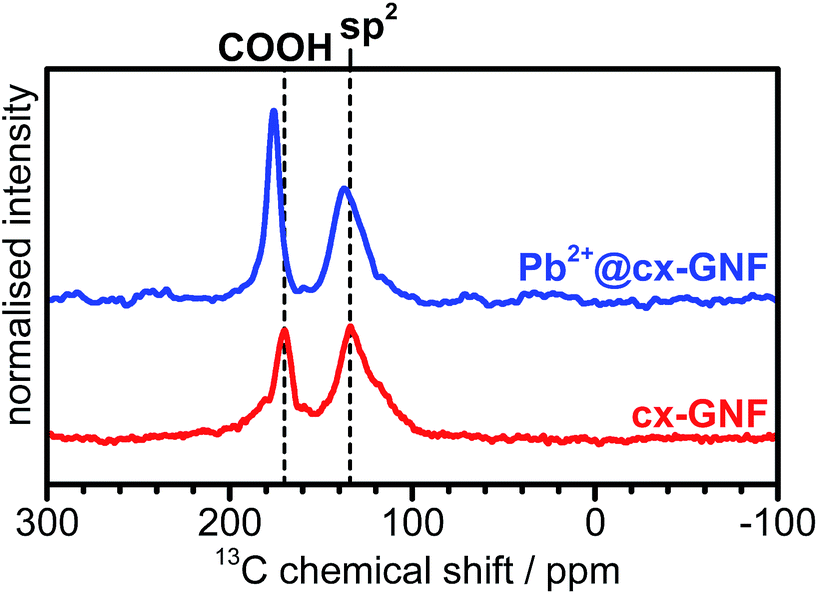 | ||
| Fig. 4 13C-NMR spectra of cx-GNF (red) and Pb2+@cx-GNF (blue). Vertical dashed lines denote peak positions of functional groups. | ||
Further evidence for metal chelation between the heavy-metal cations and the carboxylic acid groups of the cx-GNF is provided by the significant shift to lower binding energy of the XPS C1s C(III) peak from ∼289 eV to 288.5 eV (Fig. 3), consistent with the weakening of the CO bond in COOH to form an extended electron delocalised system in the metal carboxylate.53 This is further emphasized by the bathochromic shift in the CO stretching frequency from about 1715 cm−1 of the COOH groups in cx-GNF to 1558 cm−1 of the metal carboxylates in the M2+/3+@cx-GNF samples (Fig. 5). Interestingly, the effect is also most pronounced for Pb2+@cx-GNF, suggesting that Pb2+ ions interact most strongly with the cx-GNF both via M–π interactions as well as the metal–carboxylate chelation. Consequently, these two effects taken together explain the exceptionally high performance of the cx-GNF for extraction of Pb2+ compared with the other carbon materials as shown in Fig. 2b. It is noteworthy that the IR spectra of both the M2+/3+@nGO as well as M2+/3+@GO also show the same bathochromic shift (Fig. S4†) indicating that metal complexation has taken place, albeit to lesser extents.
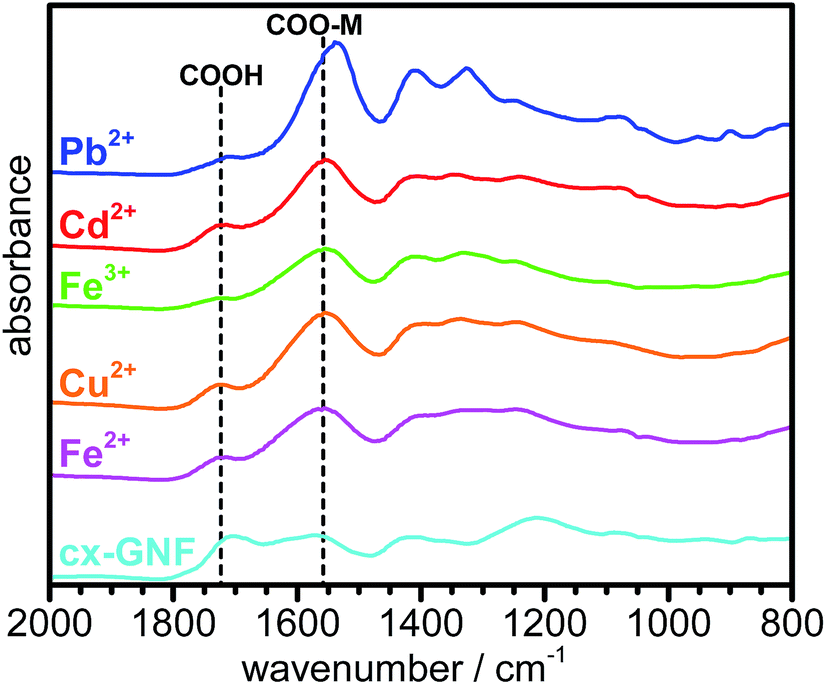 | ||
| Fig. 5 ATR-IR spectra of cx-GNF (cyan), Fe2+@cx-GNF (magenta), Cu2+@cx-GNF (orange), Fe3+@cx-GNF (green), Cd2+@cx-GNF (red) and Pb2+@cx-GNF (blue). | ||
Reversible loading and unloading of Pb2+ ions onto cx-GNF
The XPS spectra in Fig. 6 illustrate the reversible loading and unloading of Pb2+ ions onto the cx-GNF. Initially, the cx-GNF (Fig. 6a) were treated with 0.1 M Pb2+ solutions in the same way as described earlier yielding the corresponding Pb2+@cx-GNF materials (Fig. 6b). The Pb2+ ions were then removed by treatment with formic acid (Fig. 6c), leaving behind only a small trace of Pb2+ ions. A small quantity of calcium (3%) was also detected in the XPS survey spectrum which was most likely the result of the gradual uptake of Ca2+ ion traces in the deionised water during the multiple dialysis steps to remove the Pb2+ ions (see Experimental section). It is noteworthy that after the unloading of Pb2+ ions from the Pb2+@cx-GNF there is clear reversibility in the XPS C1s region back to the original cx-GNF in terms of the relative peak intensities. Finally, after removal of the Pb2+ ions, the cx-GNF were re-treated with 0.1 M Pb2+ solution as before which again yielded Pb2+@cx-GNF without any change in the loading efficiency as indicated by the Pb2+/C ratio (Fig. 6d). As expected due to the reversible behaviour, the final XPS C1s region was similar compared to the spectrum of the Pb2+@cx-GNF upon first loading of the cx-GNF with Pb2+ ions.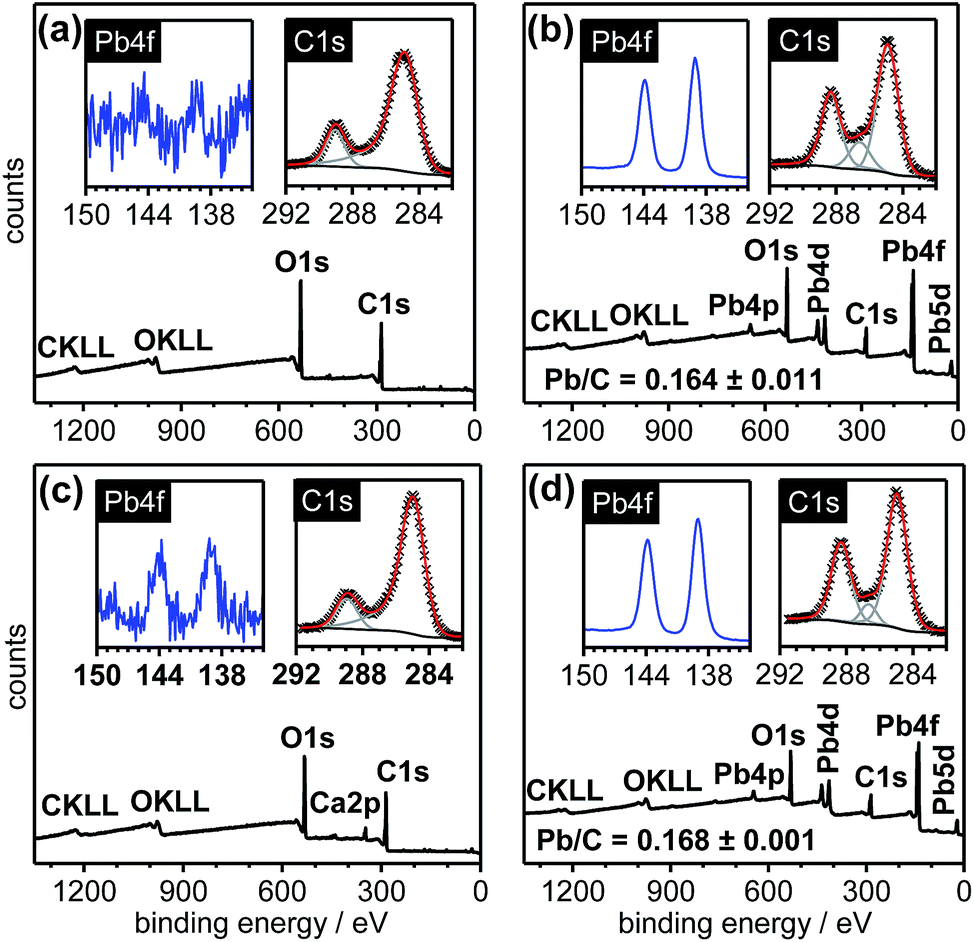 | ||
| Fig. 6 Reversible loading of Pb2+ ions onto cx-GNF. XPS survey spectra (outset), Pb4f regions (inset, left) and C1s regions (inset, right) of (a) cx-GNF before Pb2+ addition, (b) after Pb2+ addition, (c) treatment of (b) with formic acid to remove Pb2+ and (d) after reloading with Pb2+. | ||
Selectivity of cx-GNF towards Pb2+ cations in the presence of 10 and 100 molar equivalents of Ca2+ and Mg2+ cations
To assess the selectivity of the cx-GNF towards Pb2+ extraction in the presence of other cations, the cx-GNF were treated with mixtures containing lead ions as well as calcium or magnesium cations. Calcium and magnesium were chosen because these cations are the most common divalent metal cations found in drinking or industrial waste waters. The Mg2+ or Ca2+ cations were either in a 10 or 100 molar excess compared to the lead ions. In the presence of 10 mole equivalents of Ca2+ or Mg2+ ions per equivalent of Pb2+, only Pb2+ ions were removed from solution (Fig. 7, S5a and c† for XPS spectra) and in the same quantities as reported before in Fig. 2b where no other cations were present. When 100 mole equivalents were employed, there was still a significant selectivity towards the Pb2+ ions as shown in Fig. 7. A Pb2+/Mg2+ ratio of 6.5 and Pb2+/Ca2+ ratio of 2.0 was determined from the XPS survey spectra (Fig. S5b and d†).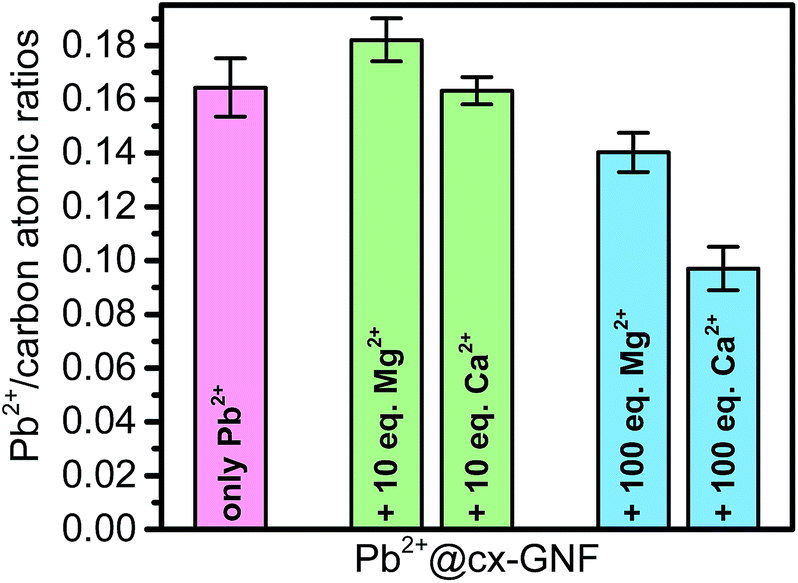 | ||
| Fig. 7 Pb2+/C atomic ratios determined from XPS survey spectra of cx-GNF treated with Pb2+ only (magenta), Pb2+ with 10 equivalents Ca2+ or Mg2+ ions (green), and Pb2+ with 100 equivalents Ca2+ or Mg2+ ions (blue). | ||
Upon adding 5 mM Pb(NO3)2 to commercial drinking water, which contains mainly CaCl2, MgCl2 and KHCO3, it was found that the Pb2+ extraction with cx-GNF was not affected by the presence of Mg2+ in the drinking water. Small amounts of Ca2+ could be identified in the XPS spectra of the cx-GNF material after the extraction process (cf. Fig. S6†). Yet, the Pb2+/Ca2+ atomic ratio was found to be 15 ± 3 illustrating that the presence of typical amounts of alkaline-earth metal ions in drinking water do not overall significantly affect the performance of the cx-GNF with respect to extracting Pb2+.
Determination of Pb2+ sorption capacities with optical absorbance spectroscopy
To determine the sorption capacities in milligrams of lead extracted per gram of carbon material, a photometric approach was used as described in the Experimental section. Calibration curves were recorded to ensure the Beer–Lambert law was obeyed at λ = 400 nm in the concentration range used in these experiments (Fig. S7†). To avoid unfavourably large absorbance values, more dilute 5 mM Pb(NO3)2 solutions were used instead of the earlier 0.1 M solutions. However, using XPS it was shown that the Pb2+/C atomic ratios of the Pb2+@carbon materials were the same regardless of whether 5 mM or 0.1 M Pb2+ solutions were used (Fig. S8† and 2b). This means that quantitative loading of the carbon materials with Pb2+ is achieved at both concentrations.The sorption capacities of the cx-GNF, nGO and GO for Pb2+ were calculated from the difference in the optical absorbances at 400 nm between the initial 5 mM solution and the solutions after the Pb2+ extraction (Fig. S9†); and found to be 659, 336 and 102 mg g−1, respectively, at pH values of the solutions of 2.2, 3.3 and 3.1. The Pb2+ sorption capacity of the cx-GNF is therefore more than six times that of conventional GO. Again, we note that under identical pH conditions, this value is likely to be even higher.
In addition to being the best material for Pb2+ extraction out of the investigated as-made carbon nanomaterials, the cx-GNFs can easily compete with most of the tailored chemically-functionalised carbon materials in terms of sorption capacity. Reviews of the heavy-metal sorption capacities of a wide range of chemically-modified carbon materials are given in ref. 23 and 31.
Finally, an attempt was made to determine the Pb2+ sorption capacity of activated charcoal (AC), but it was found to be negligible in comparison with the graphenic materials investigated here. Indeed, the Pb2+/C atomic ratio of Pb2+@AC was determined to be one order of magnitude below GO and therefore two orders of magnitude lower than for cx-GNF (Fig. S10†). This is in good agreement with Yang et al. who demonstrated the adsorption capacity of Cu2+ cations on GO is about ten times higher than activated charcoal.54
Conclusions
Carboxylated graphene nanoflakes are a highly efficient carbon material for the extraction of a wide range of heavy metals from water. They outperform the conventional GO and can easily compete with carbon nanomaterials that have been chemically-functionalised for the purpose of heavy-metal extraction.23,31 A remarkable affinity for Pb2+ ions even in the presence of large excesses of Mg2+ and Ca2+ ions has been demonstrated. Detailed insights into the chemical binding mechanisms of the heavy metal cations onto the cx-GNFs were gained highlighting the formation of metal carboxylates as well as M–π interactions as the two dominating modes for metal–carbon interactions. The high efficiency of the cx-GNF for heavy-metal extraction can therefore be attributed to the large number of carboxylic acid groups but also the intact graphenic areas on the basal plane. The loading and unloading of Pb2+ ions onto the cx-GNF was found to be completely reversible allowing for the cx-GNF to be readily recycled. Consequently, out of all the investigated as-made graphene materials, the cx-GNF are most suited for future applications in heavy-metal extraction processes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Royal Society for funding (UF150665), K. Holt for access to ATR-FTIR instrumentation and A. Aliev for recording solid state NMR spectra.References
- L. Jarup, Br. Med. Bull., 2003, 68, 167–182 CrossRef PubMed
.
- W. I. Mortada, M. A. Sobh, M. M. El-Defrawy and S. E. Farahat, Am. J. Nephrol., 2001, 21, 274–279 CrossRef CAS PubMed
- T. I. Lidsky and J. S. Schneider, Brain, 2003, 126, 5–19 CrossRef PubMed
- P. Apostoli, A. Bellini, S. Porru and L. Bisanti, Am. J. Ind. Med., 2000, 38, 310–315 CrossRef CAS PubMed
- J. Schwartz, P. J. Landrigan, E. L. Baker, W. A. Orenstein and I. H. Vonlindern, Am. J. Public Health, 1990, 80, 165–168 CrossRef CAS PubMed
- K. Steenland and P. Boffetta, Am. J. Ind. Med., 2000, 38, 295–299 CrossRef CAS PubMed
- E. K. Silbergeld, M. Waalkes and J. M. Rice, Am. J. Ind. Med., 2000, 38, 316–323 CrossRef CAS PubMed
- M. A. Barakat, Arabian J. Chem., 2011, 4, 361–377 CrossRef CAS
- M. Hua, S. Zhang, B. Pan, W. Zhang, L. Lv and Q. Zhang, J. Hazard. Mater., 2012, 211, 317–331 CrossRef PubMed
- L.-C. Hsu, C.-Y. Huang, Y.-H. Chuang, H.-W. Chen, Y.-T. Chan, H. Y. Teah, T.-Y. Chen, C.-F. Chang, Y.-T. Liu and Y.-M. Tzou, Sci. Rep., 2016, 6, 34250 CrossRef CAS PubMed
- Z. H. Huang, X. Y. Zheng, W. Lv, M. Wang, Q. H. Yang and F. Y. Kang, Langmuir, 2011, 27, 7558–7562 CrossRef CAS PubMed
- X. J. Deng, L. L. Lu, H. W. Li and F. Luo, J. Hazard. Mater., 2010, 183, 923–930 CrossRef CAS PubMed
- L. M. Cui, Y. G. Wang, L. Gao, L. H. Hu, L. G. Yan, Q. Wei and B. Du, Chem. Eng. J., 2015, 281, 1–10 CrossRef CAS
- C. J. Madadrang, H. Y. Kim, G. H. Gao, N. Wang, J. Zhu, H. Feng, M. Gorring, M. L. Kasner and S. F. Hou, ACS Appl. Mater. Interfaces, 2012, 4, 1186–1193 CAS
- L. L. Fan, C. N. Luo, M. Sun and H. M. Qiu, J. Mater. Chem., 2012, 22, 24577–24583 RSC
- S. K. Li, X. F. Lu, Y. P. Xue, J. Y. Lei, T. Zheng and C. Wang, PLoS One, 2012, 7, e43328 CAS
- N. Sui, L. N. Wang, X. H. Wu, X. H. Li, J. Sui, H. L. Xiao, M. H. Liu, J. Wan and W. W. Yu, RSC Adv., 2015, 5, 746–752 RSC
- Y. Liu, L. Xu, J. S. Liu, X. Y. Liu, C. H. Chen, G. Y. Li and Y. F. Meng, Chem. Eng. J., 2016, 285, 698–708 CrossRef CAS
- R. Sitko, B. Zawisza, E. Talik, P. Janik, G. Osoba, B. Feist and E. Malick, Anal. Chim. Acta, 2014, 834, 22–29 CrossRef CAS PubMed
- G. Y. Zhou, C. B. Liu, Y. H. Tang, S. L. Luo, Z. B. Zeng, Y. T. Liu, R. Xu and L. Chu, Chem. Eng. J., 2015, 280, 275–282 CrossRef CAS
- W. J. Hou, Y. M. Zhang, T. Liu, H. W. Lu and L. He, RSC Adv., 2015, 5, 8037–8043 RSC
- X. J. Hu, Y. G. Liu, H. Wang, A. W. Chen, G. M. Zeng, S. M. Liu, Y. M. Guo, X. Hu, T. T. Li, Y. Q. Wang, L. Zhou and S. H. Liu, Sep. Purif. Technol., 2013, 108, 189–195 CrossRef CAS
- I. Duru, D. Ege and A. R. Kamali, J. Mater. Sci., 2016, 51, 6097–6116 CrossRef CAS
- P. N. Alagappan, J. Heimann, L. Morrow, E. Andreoli and A. R. Barron, Sci. Rep., 2017, 7, 6682 CrossRef PubMed
- G. X. Zhao, X. M. Ren, X. Gao, X. L. Tan, J. X. Li, C. L. Chen, Y. Y. Huang and X. K. Wang, Dalton Trans., 2011, 40, 10945–10952 RSC
- P. Y. Liu, T. T. Yan, J. P. Zhang, L. Y. Shi and D. S. Zhang, J. Mater. Chem. A, 2017, 5, 14748–14757 CAS
- P. Y. Liu, T. T. Yan, L. Y. Shi, H. S. Park, X. C. Chen, Z. G. Zhao and D. S. Zhang, J. Mater. Chem. A, 2017, 5, 13907–13943 CAS
- M. Wang, X. T. Xu, J. Tang, S. J. Hou, M. S. A. Hossain, L. K. Pan and Y. Yamauchi, Chem. Commun., 2017, 53, 10784–10787 RSC
- M. M. Chen, D. Wei, W. Chu, T. Wang and D. G. Tong, J. Mater. Chem. A, 2017, 5, 17029–17039 CAS
- L. J. Liu, X. R. Guo, R. Tallon, X. K. Huang and J. H. Chen, Chem. Commun., 2017, 53, 881–884 RSC
- S. Wang, X. Li, Y. Liu, C. Zhang, X. Tan, G. Zeng, B. Song and L. Jiang, J. Hazard. Mater., 2017, 342, 177–191 CrossRef PubMed
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed
- A. Lerf, H. Y. He, M. Forster and J. Klinowski, J. Phys. Chem. B, 1998, 102, 4477–4482 CrossRef CAS
- H. Chang and H. Wu, Energy Environ. Sci., 2013, 6, 3483–3507 CAS
- G. Zhao, T. Wen, C. Chen and X. Wang, RSC Adv., 2012, 2, 9286–9303 RSC
- G. Zhao, J. Li, X. Ren, C. Chen and X. Wang, Energy Environ. Sci., 2011, 45, 10454–10462 CAS
- M. Rosillo-Lopez, T. J. Lee, M. Bella, M. Hart and C. G. Salzmann, RSC Adv., 2015, 5, 104198–104202 RSC
- M. Rosillo-Lopez and C. G. Salzmann, Carbon, 2016, 106, 56–63 CrossRef CAS
- J. Chen, B. W. Yao, C. Li and G. Q. Shi, Carbon, 2013, 64, 225–229 CrossRef CAS
- M. J. Ahmed and M. A. Mamun, Talanta, 2001, 55, 43–54 CrossRef
- H. P. Boehm, Carbon, 2002, 40, 145–149 CrossRef CAS
- T. C. Chiang and F. Seitz, Ann. Phys., 2001, 10, 61–74 CrossRef CAS
- S. Yumitori, J. Mater. Sci., 2000, 35, 139–146 CrossRef CAS
- G. X. Zhao, J. X. Li, X. M. Ren, C. L. Chen and X. K. Wang, Environ. Sci. Technol., 2011, 45, 10454–10462 CrossRef CAS PubMed
- R. Sitko, E. Turek, B. Zawisza, E. Malicka, E. Talik, J. Heimann, A. Gagor, B. Feist and R. Wrzalik, Dalton Trans., 2013, 42, 5682–5689 RSC
- A. M. Dimiev, L. B. Alemany and J. M. Tour, ACS Nano, 2013, 7, 576–588 CrossRef CAS PubMed
- D. A. Dougherty, Science, 1996, 271, 163–168 CAS
- S. Y. Jeong, S. H. Kim, J. T. Han, H. J. Jeong and G. W. Lee, Adv. Funct. Mater., 2012, 22, 3307–3314 CrossRef CAS
- M. Machida, T. Mochimaru and H. Tatsumoto, Carbon, 2006, 44, 2681–2688 CrossRef CAS
- J. Burgess, Metal ions in solution, Ellis Horwood, Chinchester, England, 1978 Search PubMed
- J. J. Pesek and J. F. Schneider, J. Inorg. Biochem., 1988, 32, 233–238 CrossRef CAS PubMed
- N. S. Sisombath, F. Jalilehvand, A. C. Schell and Q. Wu, Inorg. Chem., 2014, 53, 12459–12468 CrossRef CAS PubMed
- S. Park, K.-S. Lee, G. Bozoklu, W. Cai, S. T. Nguyen and R. S. Ruoff, ACS Nano, 2008, 2, 572–578 CrossRef CAS PubMed
- S. T. Yang, Y. L. Chang, H. F. Wang, G. B. Liu, S. Chen, Y. W. Wang, Y. F. Liu and A. N. Cao, J. Colloid Interface Sci., 2010, 351, 122–127 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra00823j |
| This journal is © The Royal Society of Chemistry 2018 |